Introduction
Nephrolithiasis is one of the common and important
problems in daily urological practice. Among patients in different
regions, calcium oxalate (CaOx) is an important component involved
in the formation of kidney stone (1). CaOx stone has become an urgent issue
due to its high incidence and recurrence rate (2). It had been demonstrated that oxalate
metabolism was closely related to the occurrence and development of
calcium oxalate stones disease (3). When oxalate homeostasis is
disordered, a large amount of oxalate will accumulate in tissues,
especially in kidney (4). It is
already revealed that oxalate could result in the death of renal
interstitial cells and basolateral cells (5). Additionally, a recent study
demonstrated that cellular dysfunction and damage in HK-2 cells
could be induced by oxalate exposure (6). Hence, it is fairly necessary to
perform an in-depth exploration for molecular mechanisms of calcium
oxalate kidney stones formation.
Several studies have demonstrated that the
nucleotide-binding domain and leucine-rich repeat-containing family
pyrin domain-containing 3 (NLRP3) inflammasome was involved in the
onset and progression of calcium oxalate stones (7,8).
NLRP3 inflammasome, a kind of cytoplasmic protein, is composed of
NLRP3 receptor, apoptosis-associated speck-like protein (ASC) and
procaspase-1 (9). Both
intracellular and extracellular stimulations could activate the
NLRP3 inflammasome by pathogen-associated molecular patterns and
damage-associated molecular patterns (10). It has been demonstrated that
procaspase-1 could be cleaved into its active form caspase-1 by the
activation of NLRP3 inflammasome, which induces the maturity and
secretion of IL-1β and IL-18 (11,12).
In addition, reactive oxygen species (ROS) production have been
proved to be involved in the activation of NLRP3 inflammasome
(13). A previous study conducted
by the authors demonstrated that calcium oxalate monohydrate could
induce the activation of NLRP3 inflammasome and change the adhesion
of renal tubular epithelium to calcium oxalate crystals (14). However, the type and mechanism of
NLRP3 inflammasome-mediated calcium oxalate stones formation still
need deeper investigation.
Currently, cell pyroptosis was proved as programmed
cell death characterized by nuclear pyknosis and DNA fragmentation
(15). Previous studies have
illustrated that oxalate crystals-induced injury was a kind of
programmed cell death (16,17).
The classical pathway of pyroptosis was described as a pathway
centered on caspase-1 activation, which was mediated by
inflammasome (18). Gasdermin D
(GSDMD), a member of gasdermin protein family, is the substrate
protein of all inflammatory caspases (18). It was previously reported that the
activated GSDMD was cleaved into GSDMD-N by inflammatory caspase
(such as caspase-1), which was involved in the formation of
membrane pores (18,19). Through the membrane pores mediated
by GSDMD-N, extracellular materials could enter the cells and the
intracellular components also be released into intercellular
environment, which will subsequently cause the swelling and
deformation of cells (20).
Although a number of studies explained the association between
oxalate calcium crystals and NLRP3-GSDMD pathway and its potential
therapeutic targets (21,22), ultrastructural characteristics and
intensive mechanisms of GSDMD-induced cell pyroptosis in oxalate
calcium crystals formation are still not completely clear.
Materials and methods
Cell culture
HK-2 cells were purchased from Shanghai Institutes
for Biological Sciences (Shanghai, China) and cultured in high
glucose DMEM supplemented with 10% fetal bovine serum and 1%
penicillin/streptomycin (all purchased from Gibco; Thermo Fisher
Scientific, Inc.). Cells were incubated in 37°C and 5%
CO2. A total of 20 mM primary solution of oxalate
(MilliporeSigma; Merck KGaA) was prepared in phosphate buffered
saline (PBS), and applied within 1 week with a pH number of
7.2–7.4. HK-2 cells were treated with oxalate at different
concentrations (0.1, 0.2, 0.4, 0.6, 0.8, 1.0 and 2.0 mM) for 24 h,
and then cells were separately collected or treated for further
experiments. Necrosulfonamide (NSA) (MedChemExpress), an inhibitor
of GSDMD-N, was diluted in dimethyl sulphoxide (DMSO) (Tianjin
Solomon Biotechnology Co., Ltd.) and pretreated HK-2 cells at the
concentrations of 5 and 15 µM for 2 h.
Cell Counting Kit-8 (CCK-8) assay
HK-2 cell viability was examined through CCK-8
assay. HK-2 cells (4×103) were seeded in 96-well plates
and then treated with oxalate at different concentrations (0.1,
0.2, 0.4, 0.6, 0.8, 1.0 and 2.0 mM) for 24 h. After the cells were
incubated with CCK-8 solution (10 µl; GlpBio Technology) for 2 h,
the optical density was measured by a microplate reader at 450 nm
absorption wavelength.
Lactate dehydrogenase (LDH)
release
The concentration of LDH in culture medium after
oxalate treatment was detected by LDH release assay. N-acetyl
cysteine (NAC) (MilliporeSigma; Merck KGaA), a scavenger of ROS,
was prepared in PBS and pretreated HK-2 cells for 2 h. HK-2 cells
(1×105) were incubated with oxalate in 6-well plates at
37°C for 24 h at different concentrations (0.1, 0.2, 0.4, 0.6, 0.8,
1.0 and 2.0 mM) after the pretreatment with NAC or NSA for 2 h at
37°C. The concentration of LDH in medium was examined by LDH-kit
(Nanjing Jiancheng Bioengineering Institute) according to the
manufacturer's instructions. The optical density was detected by a
microplate reader at 450 nm absorption wavelength.
Detection of ROS production
HK-2 cells (1.5×104) were seeded in
24-well plates and incubated with oxalate (0.8 mM) for 24 h at 37°C
after pretreatment with NSA. ROS production in HK-2 cells was
detected using 10 µM dichlorofluorescein diacetate (DCF-DA)
(Beyotime Institute of Biotechnology) following the manufacturer's
protocol. Finally, the detection of DCF fluorescence was carried
out at 488 nm excitation and 525 nm emission wavelengths using a
fluorescence microscope.
TUNEL staining
Oxalate-induced nuclear DNA damage was detected by
TUNEL Apoptosis Assay Kit (Beyotime Institute of Biotechnology)
according to the manufacturer's instructions. HK-2 cells
(1.5×104) were seeded in 24-well plates and incubated
with oxalate (0.8 mM) for 24 h at 37°C after pretreatment with NSA
for 2 h or after NLRP3 gene silencing. Cells were washed with PBS
three times and fixed with 4% paraformaldehyde for 30 min at room
temperature, followed by permeabilization with 0.1% Triton X-100
for 5 min. After washing, cells were stained with 50 µl TUNEL
working solution for 60 min at 37°C, and nuclei were stained with 5
µg/ml DAPI for 5 min at room temperature. After washing, one drop
of Antifade Mounting Medium (Beijing Solarbio Science &
Technology Co., Ltd.) was added and then the coverslip was added.
Oxalate-induced nuclear DNA damage was detected by TUNEL Apoptosis
Assay Kit (cat. no. C1088; Beyotime Institute of Biotechnology)
according to the manufacturer's instructions. The cellular
fluorescence intensities were observed through a confocal
microscope and 4 fields of view for every group were recorded.
Calcein-AM/PI staining
Oxalate-induced cell death in HK-2 cells was
detected by Calcein-AM/PI Double Stain Kit (Tianjin Solomon
Biotechnology Co., Ltd.) according to the manufacturer's protocol.
HK-2 cells (1.5×104) were seeded in 24-well plates and
incubated with oxalate (0.8 mM) for 24 h at 37°C after pretreatment
with NSA for 2 h or after NLRP3 gene silencing. Thereafter, cells
were stained with 2 µM Calcein-AM and 5 µM PI for 15 min at 37°C,
and nuclei were stained with 5 µg/ml DAPI for 5 min at room
temperature. Oxalate-induced cell death in HK-2 cells was detected
by Calcein-AM/PI Double Stain Kit (cat. no. CA1630; Tianjin Solomon
Biotechnology Co., Ltd.) according to the manufacturer's protocol.
The cellular fluorescence intensities were observed through a
confocal microscope.
Small interfering RNA (siRNA)
knockdown experiments
Double-stranded siRNA targeting NLRP3 gene and
negative control (NC) siRNA were purchased from Shanghai GenePharma
Co., Ltd. HK-2 cells were transfected using siRNA mixed with
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Subsequently, HK-2 cells
(1×105) were incubated with oxalate (0.8 mM) in 6-well
plates for 24 h at 37°C. Then, HK-2 cells were transfected using
100 pM siRNA mixed with 5 µl Lipofectamine 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) for 6 h at 37°C according to the
manufacturer's protocol. Subsequent experiments were performed 48 h
after transfection. The sequences used were as follows: NLRP3-siRNA
forward, 5′-GGAGAGACCUUUAUGAGAATT-3′ and reverse,
5′-UUCUCAUAAAGGUCUCUCCTT-3′; and negative control (NC) forward,
5′-UUCUCCGAACGUGUCACGUTT-3′ and reverse,
5′-ACGUGACACGUUCGGAGAATT-3′.
Reverse transcription-quantitative PCR
(RT-qPCR)
The total RNA from HK-2 cells was extracted by
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and
cDNA was then synthesized using reverse transcription system kit
(cat. no. K1691; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. RT-qPCR was performed using the SYBR
Premix Ex Taq II (Takara Biotechnology Co., Ltd.) following the
manufacturer's protocols. The specific RT-qPCR primer sequences
were as follows: NLRP3 forward, 5′-CGTGAGTCCCATTAAGATGGAGT-3′ and
reverse, 5′-CCCGACAGTGGATATAGAACAGA-3′; caspase-1 forward,
5′-TTTCCGCAAGGTTCGATTTTCA-3′ and reverse,
5′-GGCATCTGCGCTCTACCATC-3′; IL-1β forward,
5′-AGCTACGAATCTCCGACCAC-3′ and reverse,
5′-CGTTATCCCATGTGTCGAAGAA-3′; and GAPDH forward,
5′-GAAGGTGAAGGTCGGAGT-3′ and reverse, 5′-GAAGATGGTGATGGGATTTC-3′.
The thermocycling conditions for RT-qPCR were as follows: 95°C
initial denaturation for 3 min, followed by 45 cycles of
denaturation at 95°C for 30 sec, annealing at 50–60°C for 30 sec
and extension at 72°C for 30 sec. The amplification and analysis
were performed on a real-time PCR system (Applied Biosystems™ 7500
Real-Time PCR System; Applied Biosystems; Thermo Fisher Scientific,
Inc.). All of the data were analyzed with the 2−ΔΔCq
method (23) and normalized using
GAPDH cDNA as internal control.
Western blot analysis
Proteins from HK-2 cells or mice kidney tissue were
lysed in RIPA lysis buffer (Beijing Solarbio Science &
Technology Co., Ltd.) containing the protease inhibitor cocktail
(Roche Diagnostics), and protein concentrations were determined
referring to bovine serum albumin standard using the bicinchoninic
acid method. The same amounts of proteins (50 µg) were separated on
10–12% gels using sodium dodecyl sulfate-polyacrylamide gel
electrophoresis and transferred to nitrocellulose membranes,
followed by western blotting as previously described (14). Membranes were washed using TBST
containing 0.1% Tween 20. After blocking with 5% skimmed milk for 1
h at room temperature, the membranes were incubated with primary
antibodies in 4°C overnight. After extensive washing, blots were
incubated with secondary antibodies for 1 h at room temperature.
The bands were developed using Pierce™ ECL Western blotting
substrate (Thermo Fisher Scientific, Inc.). Primary antibodies were
against GSDMD (cat. no. 20770-1-AP; 1:500; Proteintech Group,
Inc.), GSDMD-N (cat. nos. DF12275 and DF13758; 1:1,000; Affinity
Biosciences), NLRP3 (cat. no. ab263899; 1:1,000; Abcam), Caspase-1
(cat. no. ab179515; 1:1,000; Abcam), IL-1β (cat. no. ab283818;
1:1,000; Abcam), IL-18 (cat. no. 10663-1-AP; 1:2,000; Proteintech
Group, Inc.), osteopontin (OPN) (cat. no. sc-21742; 1:200; Santa
Cruz Biotechnology, Inc.) and GAPDH (cat. no. sc-365062; 1:200;
Santa Cruz Biotechnology, Inc.) as a reference protein. Anti-rabbit
(cat. no. SC-2357; 1:2,000) and anti-mouse (cat. no. SC-516102;
1:2,000) IgG secondary antibodies were purchased from Santa Cruz
Biotechnology, Inc.
Transmission electron microscopy
(TEM)
After incubation with oxalate (0.8 mM) for 24 h,
HK-2 cells transfected with NLRP3-siRNA or scrambled-siRNA were
collected and fixed in 2.5% glutaraldehyde for 2 h at room
temprature. After washing three times with PBS, postfixation
staining was performed using 1% osmium tetroxide for 1 h at room
temperature. Furthermore, cells were dehydrated in gradient ethanol
solution and treated with propylene oxide, Spurr's low viscosity
resin for 18 h. Cells were further treated with pure resin for 24 h
and embedded in Beem capsules. Resin blocks were hardened at 70°C
for 2 days. Ultra-thin slices (70 nm) were prepared and stained
with 1% lead citrate and 0.5% uranyl acetate after the processes of
dehydration, embedding, infiltration and slicing following the
manufacturer's protocol. Ultrathin sections were observed using
HT7700 Transmission Electron Microscopy (Hitachi, Ltd.).
Animal experiments
C57BL/6 (6-week old; mean weight, 20 g) male mice
were purchased from the Vital River Laboratories (Beijing, China).
GSDMD−/− mice were obtained from the Chen's Lab
in Tianjin University. GSDMD−/− mice were
obtained from heterozygous GSDMD+/− mice
intercross confirmed by genotyping. All mice were maintained and
treated in accordance with the Animal Ethics Committee at the
Second Hospital of Tianjin Medical University (Tianjin, China;
approval no. KY2022K103). All mice were housed under 25°C and 30%
humidity conditions with an equal light/dark cycle, and raised with
free access to specialized feed and water. The oxalate nephropathy
model was induced through intraperitoneal injection of 0.8%
glyoxylic acid (Gly) (Shanghai Macklin Biochemical Co., Ltd.). Two
groups of 10 mice each were randomly divided into the control group
and the Gly group. The mice were sacrificed through cervical
dislocation after Gly treatment for 7 days. The kidney, plasma and
urine specimens were collected after treatment for 7 days. The
kidneys were washed twice with 0.9% normal saline and were dried on
the absorbent paper. Subsequently, the kidneys were weighed with a
1/10,000 analytical balance. All samples were stored at −80°C or
fixed for subsequent experiments.
Histological staining and biochemical
examination
The cross section of the kidney specimen was
obtained after the kidney tissues were fixed, embedded and
positioned. Mouse kidney tissues were fixed in 4% paraformaldehyde
overnight at room temperature and paraffin embedded. Sections (5
µm) were stained with hematoxylin for 5 min and eosin for 3 min at
room temperature, and then observed by light microscope (Nikon
Corporation). The sections were stained with haematoxylin and eosin
(H&E) for basic histological examination. The oxalate crystals
were observed through Von Kossa staining (cat. no. G3282; Beijing
Solarbio Science & Technology Co., Ltd.). For tissue
immunostaining, mouse kidneys were dissected, fixed with buffered
4% paraformaldehyde overnight at 4°C, cryoprotected in 30% sucrose
solution overnight, and finally embedded in optimal cutting
temperature compound. The immunohistochemistry (IHC) staining was
performed with specific primary antibodies against the target
proteins for 1 h at room temperature. Primary antibodies were
against NLRP3 (cat. no. 68102-1-lg; 1:50; Proteintech Group, Inc.),
Caspase-1 (cat. no. ab138483; 1:100; Abcam), GSDMD (cat. no.
20770-1-AP; 1:50; Proteintech Group, Inc.), GSDMD-N (cat. no.
DF13758; 1:50; Affinity Biosciences), IL-1β (cat. no. ab283818;
1:100; Abcam), IL-18 (cat. no. 10663-1-AP; 1:50; Proteintech Group,
Inc.) and osteopontin (OPN) (cat. no. sc-21742; 1:50, Santa Cruz
Biotechnology, Inc.). Anti-rabbit (cat. no. SC-2357; 1:5,000; Santa
Cruz Biotechnology, Inc.) and anti-mouse (cat. no. SC-516102;
1:5,000; Santa Cruz Biotechnology, Inc.) IgG secondary antibodies
were applied. Serum creatinine was detected using Creatinine
Quantitative Colorimetric kit (cat. no. BC4910; Beijing Solarbio
Science & Technology Co., Ltd.) according to the manufacturer's
instructions.
Quantification of image
The intensity of fluorescence or autoradiogram in
cellular/histological staining and western blotting assays was
quantified with the software ImageJ (version 1.8.0; National
Institutes of Health). Quantified data were normalized with those
of the control, and then were plotted as bar charts.
Statistical analysis
Statistical analysis was performed through
statistical software (SPSS v20.0; IBM Corp.) and the experimental
data were analyzed using an independent unpaired t-test or one-way
ANOVA. LSD test was used as a post hoc test for comparison between
groups following one-way ANOVA. Each experiment was conducted for
4–6 repeats and the data are presented as the mean ± standard
error. All results with two-side P<0.05 were considered to
indicate a statistically significant difference.
Results
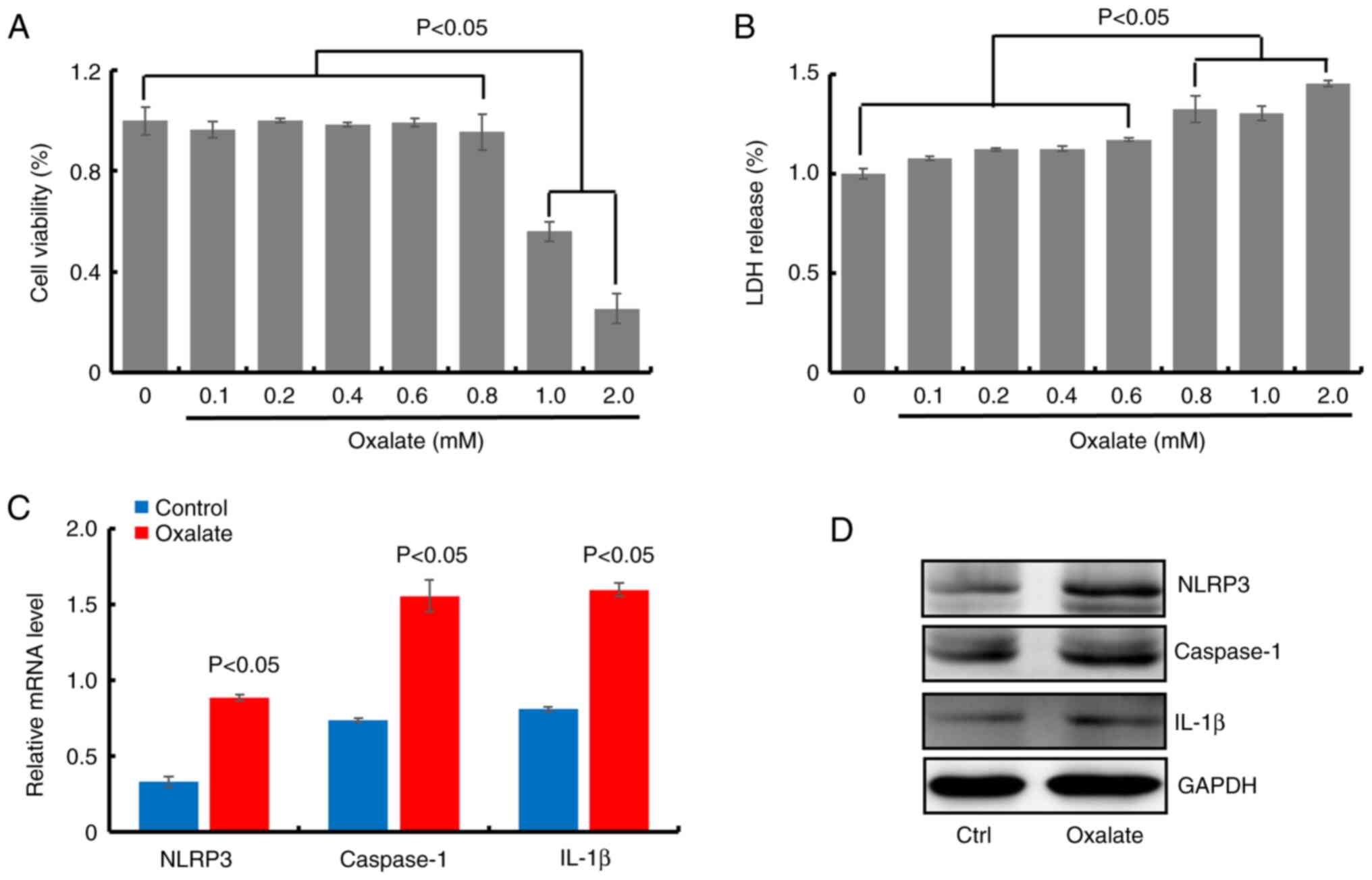
Oxalate induces the formation of NLRP3
inflammasome in HK-2 cells
In order to confirm the role of NLRP3 inflammasome
in the occurrence and development of calcium oxalate renal stones,
the viability in HK-2 cells treated with oxalate at different
concentrations (0.1, 0.2, 0.4, 0.6, 0.8, 1.0 and 2.0 mM) was
firstly evaluated. The bar graph revealed that oxalate did not
reduce the viability of HK-2 cells at concentrations less than 1.0
mM (at 0.1, 0.2, 0.4, 0.6 and 0.8 mM) after treatment for 24 h
(Fig. 1A). Meanwhile, the LDH
levels in cell medium in HK-2 cells treated with oxalate at
different concentrations (0.1, 0.2, 0.4, 0.6, 0.8, 1.0 and 2.0 mM)
were measured. As illustrated in Fig.
1B, oxalate treatment increased LDH release in a dose-dependent
manner, compared with the control cells, especially at higher
oxalate concentration (0.8, 1.0 and 2.0 mM), suggesting cell
membrane injury in oxalate-treated HK-2 cells. Thus, 0.8 mM was
selected as treatment concentration of oxalate in the present
study. Furthermore, the transcription and translation levels of
NLRP3, caspase-1 and IL-1β were detected in HK-2 cells with 0.8 mM
oxalate treatment. The results of RT-qPCR and western blot assays
revealed that the mRNA and protein levels of NLRP3, caspase-1 and
IL-1β significantly increased in oxalate-treated HK-2 cells,
compared with the control cells (Figs.
1C and D and S1). These
results suggested that NLRP3 inflammasome was formed in
oxalate-treated HK-2 cells.
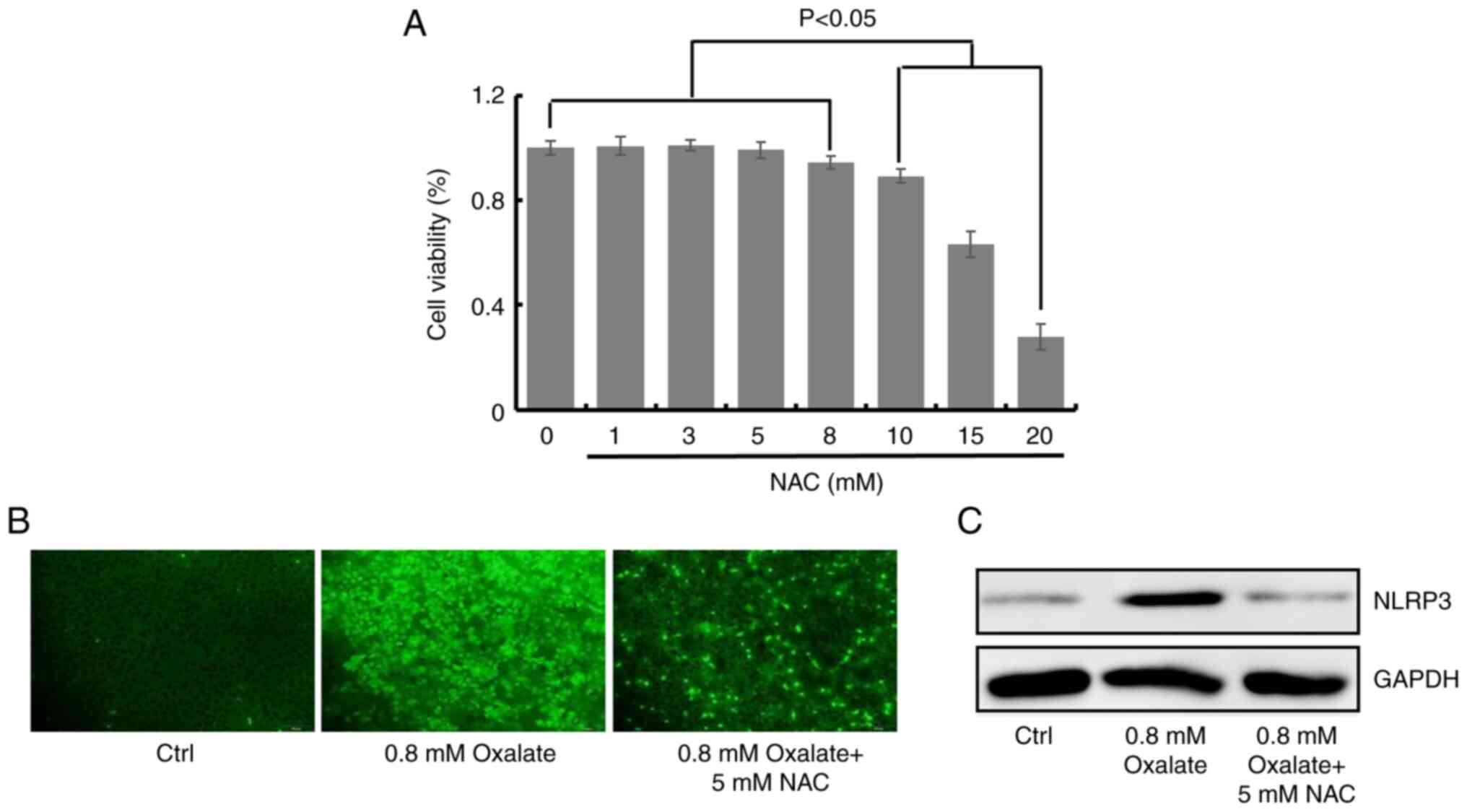
Oxalate-induced activation of NLRP3
inflammasome is mediated by ROS production
ROS is known to be involved in the activation of
NLRP3 inflammasome (24). To
identify the possible contribution of ROS production in
oxalate-induced activation of NLRP3 inflammasome, the ROS
production in HK-2 cells after oxalate treatment was detected. NAC,
a scavenger of the production of ROS, was non-cytotoxic to HK-2
cells at 1, 3, 5 and 8 mM, compared with the untreated control
cells (Fig. 2A). The fluorescence
images demonstrated that 0.8 mM oxalate could significantly induce
ROS production, while 5 mM NAC decreased ROS level in
oxalate-treated HK-2 cells (Figs.
2B and S2). In addition, the
protein contents of NLRP3 were detected to define the role of ROS
in activation of NLRP3 inflammasome through NAC treatment. As
revealed in Fig. 2C, NAC (5 mM)
could inhibit the expression level of NLRP3 induced by oxalate
treatment in HK-2 cells (Figs. 2C
and S3). Collectively, the
aforementioned results demonstrated that oxalate-induced formation
of NLRP3 inflammasome was mediated by ROS production.
Oxalate can induce injury of HK-2
cells mediated by NLRP3
A previous study suggested that oxalate could induce
rat renal tubular epithelial cells injury, which was a type of
programmed cell death (16).
Different from apoptosis, classical pyroptosis pathway depends on
the sheared GSDMD activated by NLRP3-caspase-1 signaling, which
participated in oxalate induced renal injury and renal crystals
(21). In the present study,
western blot analysis results revealed that the protein levels of
NLRP3 and caspase-1 were significantly increased in oxalate-treated
HK-2 cells (Figs. 3A and S4). Meanwhile, the NLRP3 and caspase-1
concentrations could be prevented after NLRP3 gene silencing in
HK-2 cells (Figs. 3A and S4). Additionally, LDH release was
decreased in NLRP3-knocked down HK-2 cells treated with oxalate
compared with scrambled control, which suggested disrupted
integrity of cell membrane (Fig.
3B). Pyroptosis is characterized by nuclear pyknosis, cell
swelling and membrane rupture (25). By TUNEL staining and Calcein-AM/PI
staining, it was observed that oxalate could induce the
fragmentation of nuclear DNA and the death of HK-2 cells (Figs. 3C and D, S5 and S6). At the same time, it was revealed
that the oxalate-induced morphological changes in HK-2 cells could
be protected by NLRP3 gene silencing (Figs. 3C and D, S5 and S6). Based on the aforementioned results,
it has been suggested that the oxalate-induced injury in HK-2 cells
was a kind of programmed cell death, which was mediated by NLRP3
signaling and characterized with the fragmentation of nuclear DNA
and the disruption of cell membrane integrity, and all the features
were consistent with pyroptosis.
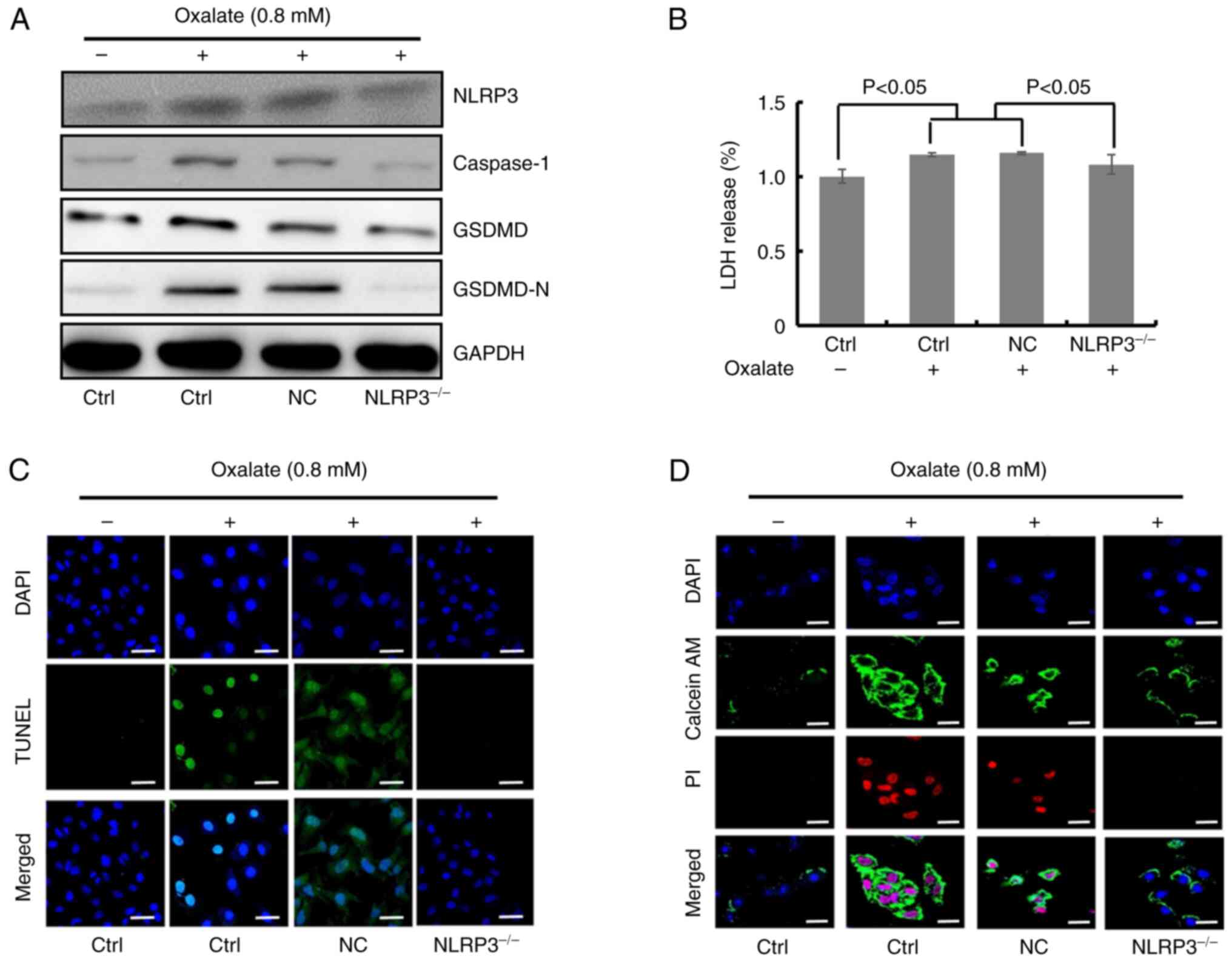 | Figure 3.NLRP3 knockdown ameliorates
oxalate-caused injury of HK-2 cells. (A) Western blot detection of
NLRP3, caspase-1, GSDMD and GSDMD-N protein contents in HK-2 cells
treated with 0.8 mM oxalate for 24 h after transfection with
NLRP3-siRNA. (B) Relative lactate dehydrogenase levels in HK-2
cells transfected with NLRP3-siRNA upon 0.8 mM oxalate treatment
for 24 h (n=6). (C) Images of two colors (blue and green) and
merged colors in HK-2 cells with TUNEL staining were observed by
confocal microscopy. HK-2 cells with NLRP3 silence were treated
with 0.8 mM oxalate for 24 h followed by staining. Scale bars, 100
µm. (D) Images of three colors (blue, green and red) and merged
colors in HK-2 cells with Calcein-AM/PI staining were recorded
through confocal microscopy. HK-2 cells transfected with
NLRP3-siRNA or scrambled-siRNA were dealt with 0.8 mM oxalate for
24 h. Scale bars, 100 µm. NLRP3, leucine-rich repeat-containing
family pyrin domain-containing 3; GSDMD, gasdermin D; siRNA, small
interfering RNA; Ctrl, control; NC, negative control. |
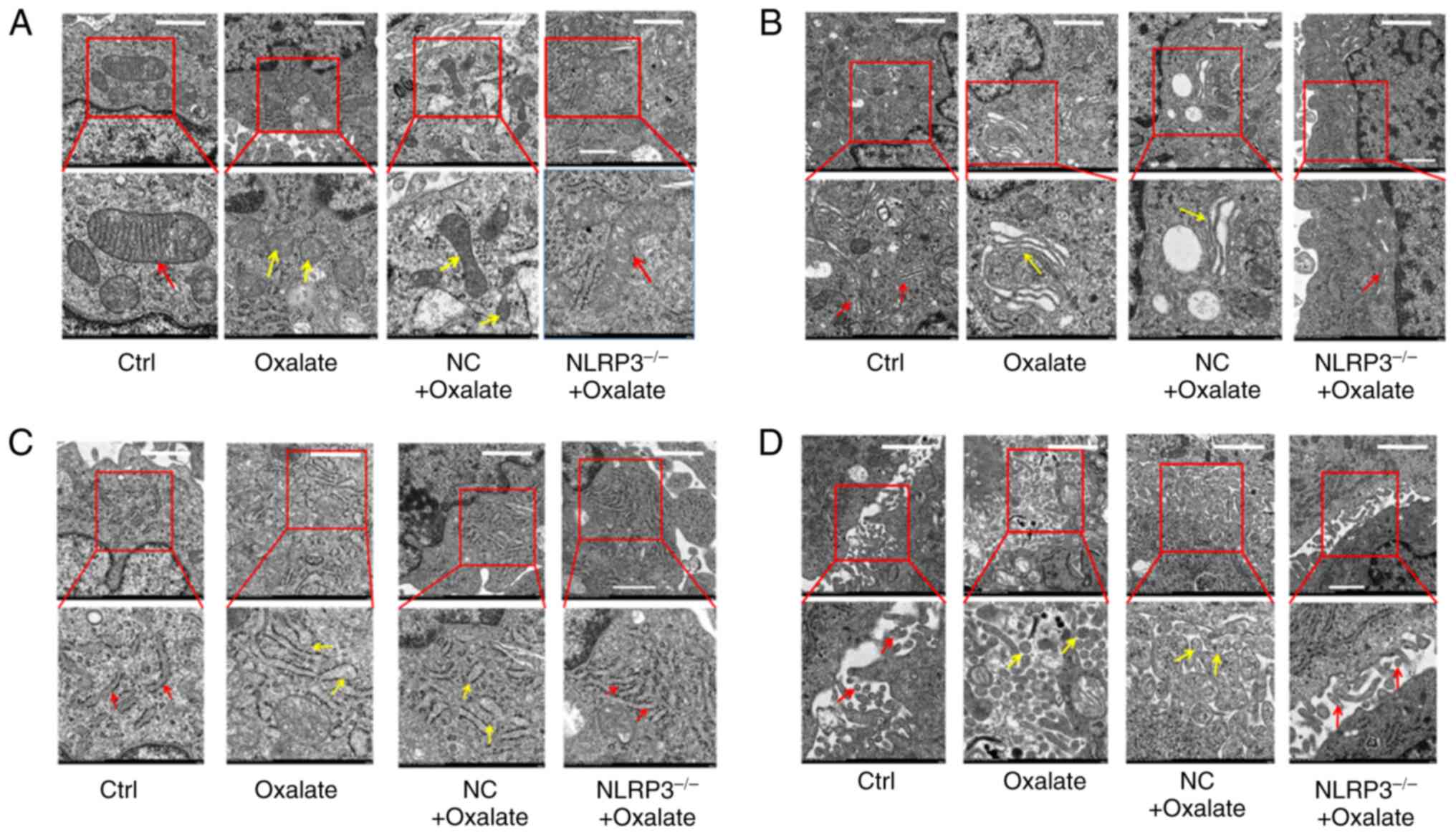
Ultrastructural changes of
oxalate-induced pyroptotic injury in HK-2 cells
Previous studies have demonstrated the fragmentation
of nuclear DNA and the disruption of cell membrane integrity in
oxalate-induced HK-2 cells. Numerous morphological changes are
typical features of pyroptosis (26). In order to explore the
ultrastructural changes of oxalate-induced injury in HK-2 cells,
the HK-2 cells with different treatment through TEM were observed.
The TEM images revealed that the structures of several organelles
in the oxalate-treated HK-2 cells were changed. As demonstrated in
electron micrograph, mitochondrial cristae was disrupted and
disappeared (Fig. 4A), Golgi
apparatus became swollen and hypertrophic (Fig. 4B), rough endoplasmic reticulum was
slender and shallow (Fig. 4C) and
microvilli appeared thick and short (Fig. 4D). Furthermore, all these injuries
of organelles could be ameliorated in HK-2 cells subjected to NLRP3
gene silencing (Fig. 4A-D). In
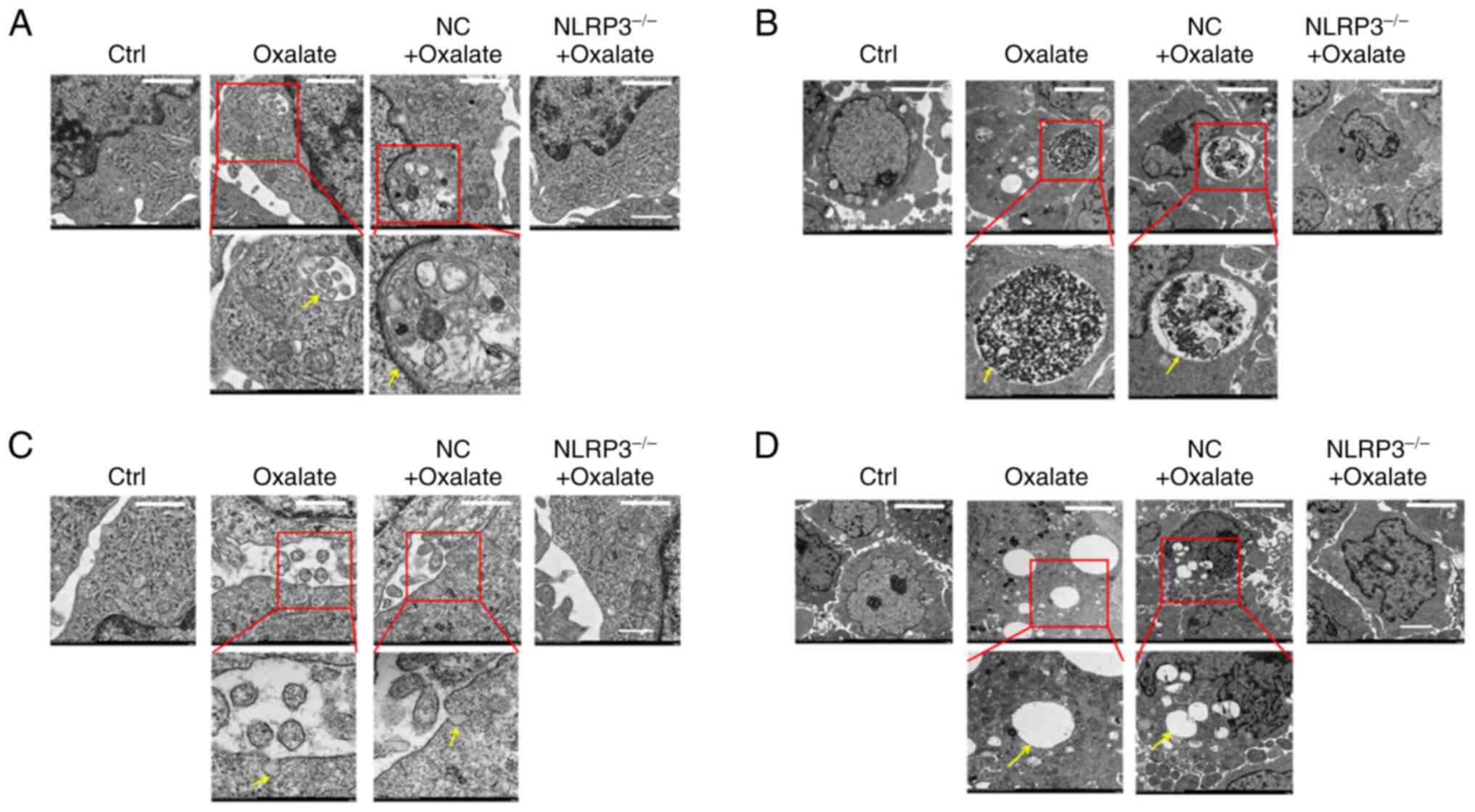
addition, certain particular ultra-structures in HK-2 cells after
oxalate treatment were also observed. As displayed in Fig. 5, autophagosomes, oxalate crystal
phagosomes, vacuoles and membrane pores appeared in cytoplasm and
cell membrane of HK-2 cells. Moreover, these abnormal structures in
oxalate treated HK-2 cells could be repaired by NLRP3 gene
silencing (Fig. 5A-D). These
findings suggested that oxalate induced a series of ultrastructural
injuries in HK-2 cells organelles, similar to typical changes of
pyroptosis.
GSDMD is involved in the
oxalate-induced renal cell injury and oxalate crystals formation in
vitro and in vivo
The aforementioned experiments had confirmed that
the features of oxalate-induced injury in HK-2 cells, which was
consistent with pyroptosis. It has been reported that GSDMD can be
cleaved into GSDMD-N to induce cell pyroptosis (18). In order to clarify the role of
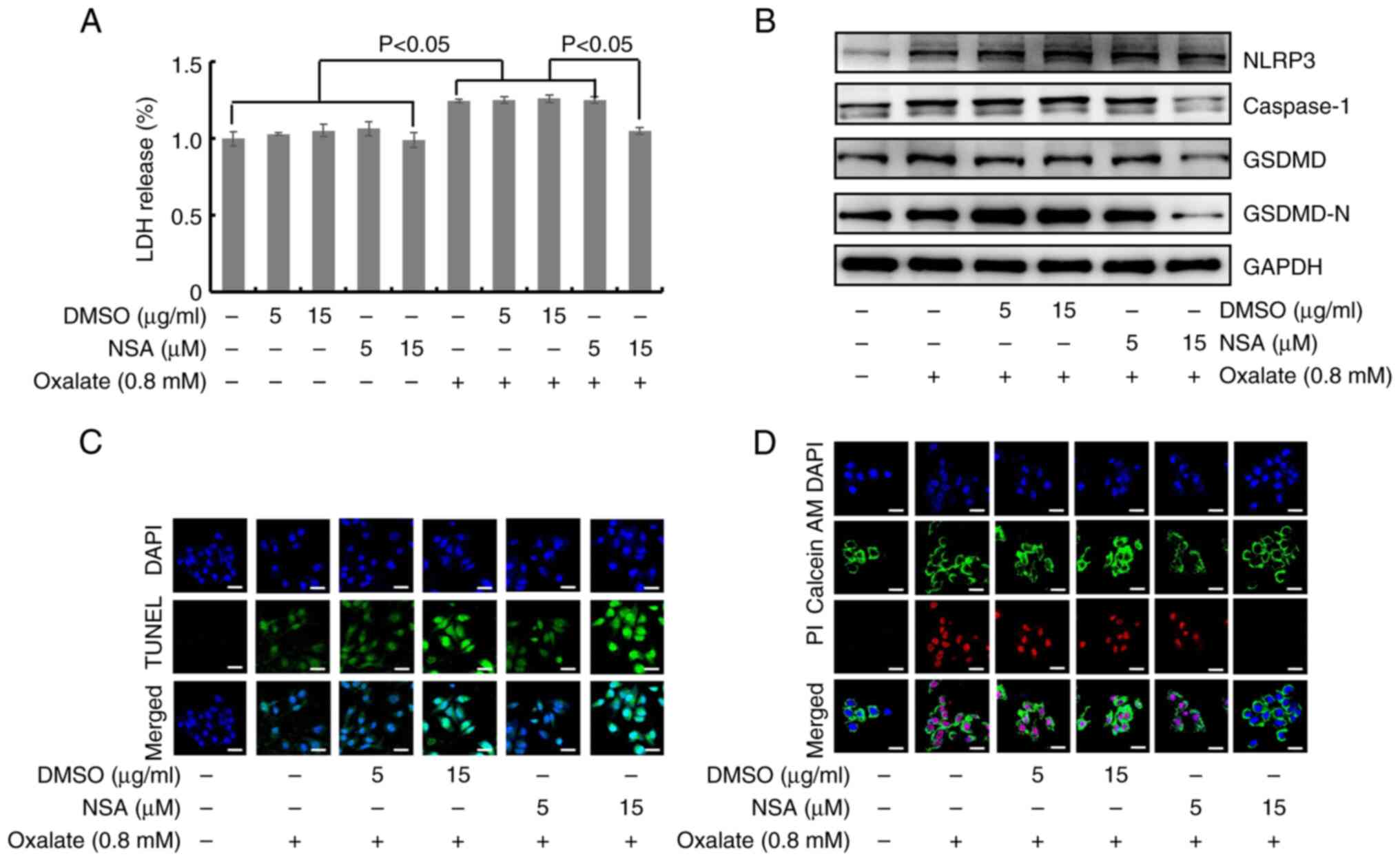
GSDMD in oxalate-induced HK-2 cell injury, the influences of NSA on
oxalate-stimulated HK-2 cells in vitro were detected. NSA,
is an inhibitor of the protein function of GSDMD-N which inhibits
GSDMD-N oligomerization. As illustrated in Fig. 6A, there was not toxicity in HK-2
cells incubated with 5 and 15 µM NSA or DMSO solvent. However, NSA
could prevent the oxalate-induced LDH-release in HK-2 cells at 15
µM (Fig. 6A). Meanwhile, western
blot results revealed that oxalate could significantly increase the
expression levels of NLRP3, caspase-1, GSDMD and GSDMD-N, and 15 µM
NSA treatment could inhibit the protein concentration of GSDMD,
GSDMD-N and caspase-1 (Figs. 6B
and S7). Additionally, TUNEL
staining results demonstrated that NSA could not alleviate the
fragmentation of nuclear DNA in HK-2 cells after oxalate treatment
(Figs. 6C and S8), but NSA could decrease the
oxalate-stimulated PI positive HK-2 cells through Calcein-AM/PI
staining (Figs. 6D and S9). Based on the aforementioned results,
it was concluded that GSDMD and GSDMD-N were involved in
oxalate-induced pyroptosis of HK-2 cells, which could be prevented
by NSA.
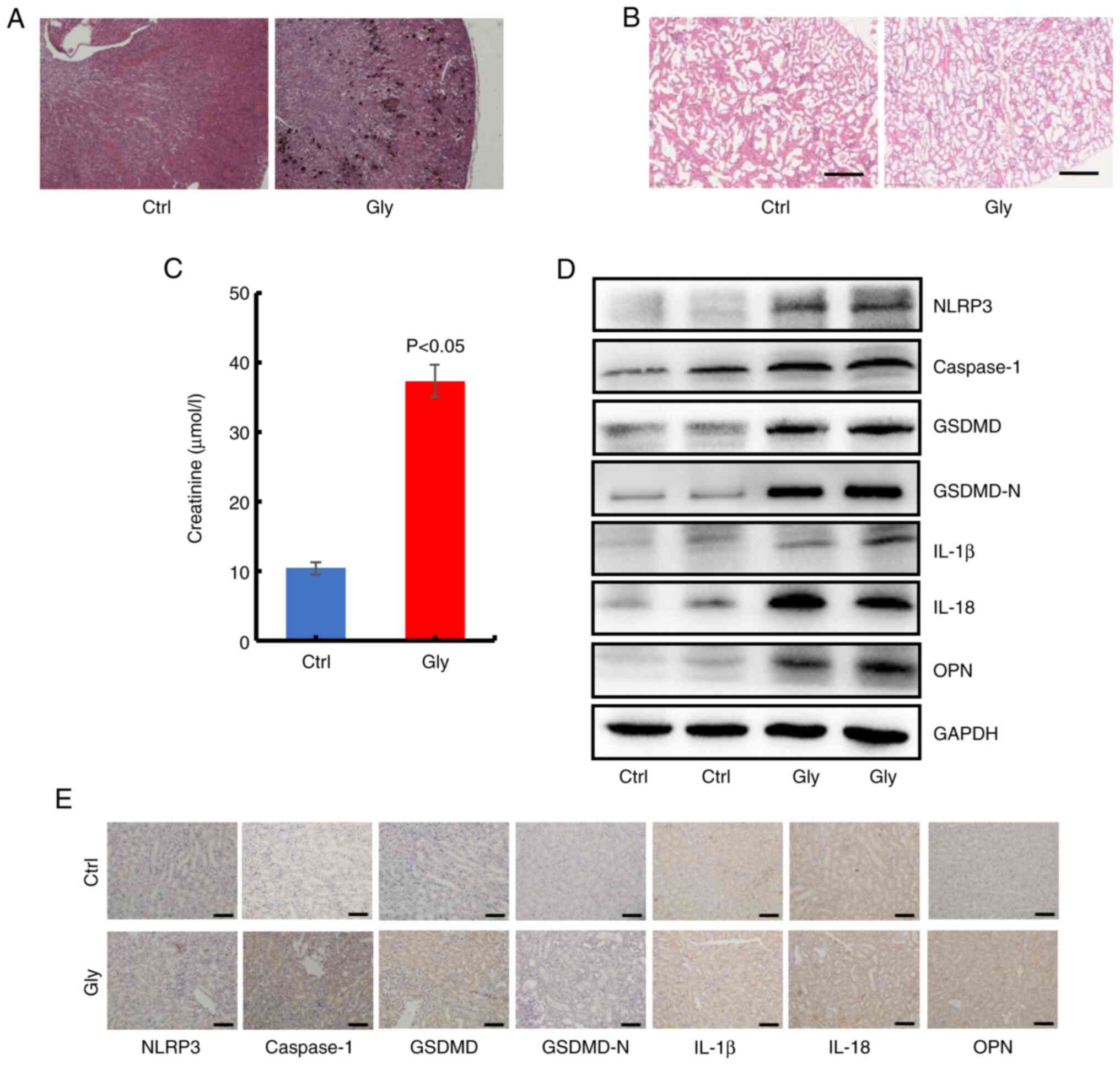
To confirm the role of GSDMD in oxalate calcium
crystals formation in vivo, oxalate crystals animal model in
C57BL/6 mice was first constructed using Gly. The Von Kossa
staining showed obvious oxalate crystals in kidney of mice treated
with Gly, compared with the untreated mice (Fig. 7A). Histological examination
revealed considerable injury in Gly-treated mice through H&E
staining, including epithelial cell swelling, renal tubule edema
and neutrophil infiltration (Fig.
7B). In addition, the results of biochemical examination
revealed higher blood creatinine in oxalate crystal mice (Fig. 7C). Moreover, western blot analysis
for harvested kidneys revealed that a series of proteins of the
NLRP3-GSDMD pathway (NLRP3, caspase-1, GSDMD, IL-1β, IL-18 and OPN)
were significantly increased in Gly-treated mice (Figs. 7D and S10). Furthermore, the similar
alterations of these proteins were verified through IHC staining of
the collected kidneys (Figs. 7E
and S11). Collectively, these
in vivo findings indicated that NLRP3-GSDMD signaling was
involved in oxalate-stimulated renal pyroptotic injury.
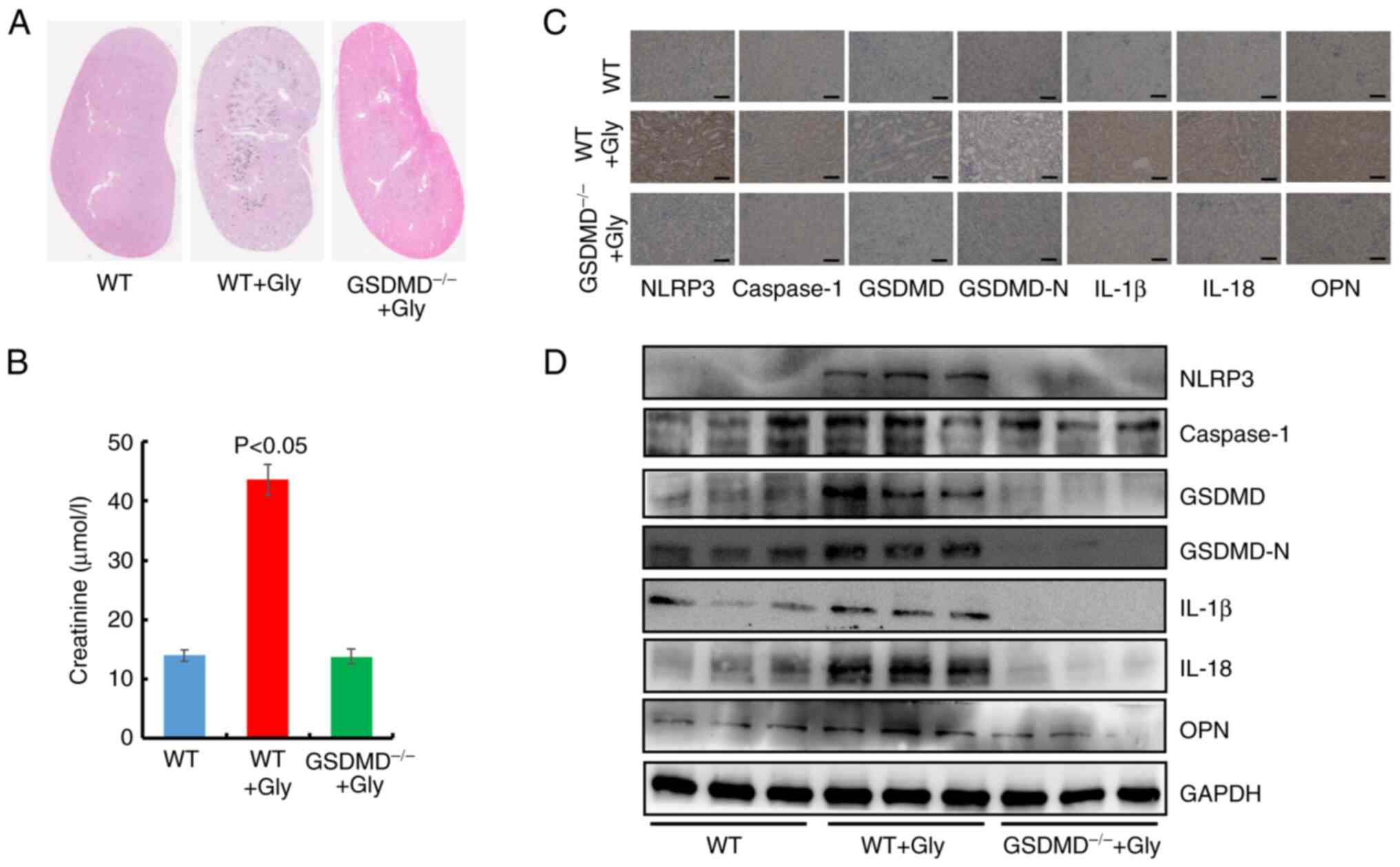
Furthermore, oxalate crystals models in C57BL/6
wild-type (WT) mice and GSDMD−/− mice were
established. As revealed in Fig.
8A, oxalate crystals were significantly decreased in
GSDMD−/− mice with glyoxylic acid, compared with
WT mice. Simultaneously, the level of blood creatinine was
decreased in GSDMD−/− mice compared with the WT
mice treated with Gly (Fig. 8B).
After the mice were sacrificed, IHC staining and western blotting
were performed to detect the kidney protein levels in NLRP3-GSDMD
pathway (Figs. 8C and D, S12 and S13). These results exhibited that GSDMD
deficiency decreased the protein concentrations of NLRP3,
caspase-1, IL-1β, IL-18 and OPN in GSDMD−/− mice
in comparison with those in WT+Gly mice, suggesting GSDMD prevented
Gly-induced oxalate crystal formation and the activation of
NLRP3-GSDMD signaling in vivo. Collectively, the
aforementioned findings illustrated the crucial impact of GSDMD on
oxalate crystal formation.
Discussion
Based on previous studies, 5–15% of the population
suffers from kidney stones throughout their lifetime, and >80%
of kidney stones are calcium oxalate stones (27,28).
Numerous studies have been performed to explore the pathogenesis of
calcium oxalate stones in recent years. At present, a few factors
are considered to contribute to calcium oxalate kidney stones,
including lifestyle, hereditary and metabolism (1,3).
Meanwhile, accumulating studies suggested oxidative stress and
inflammatory injury was associated with calcium oxalate stones
(29,30). Furthermore, a number of different
studies demonstrated that calcium oxalate crystals could result in
injury in renal tubular epithelial cells (31,32).
A previous study conducted by the authors demonstrated that oxalate
could reduce the cell viability in rat renal tubular epithelial
cells (14). It was proved that
ROS-mediated NLRP3 inflammasome activation was involved in the
onset and progression of calcium oxalate stones (21,33).
In the present study, it was revealed that oxalate-induced
activation of NLRP3 inflammasome and overexpression of caspase-1
could be inhibited by NAC. Consistent with previous studies, the
present results confirmed that oxalate could induce the activation
of NLRP3 pathway in HK-2 cells, which was mediated by ROS
production. However, the type and mechanism of oxalate-treated HK-2
cell injury had not been fully clarified.
A previous study demonstrated that
inflammasome-dependent pyroptosis was a kind of programmed cell
death (18). Both pyroptosis and
apoptosis constitute programmed cell death mechanisms, but with the
in-depth studies on the mechanism and ultrastructural feature of
pyroptosis, the differences of pyroptosis and apoptosis were
gradually realized (26). A small
number of studies investigated the molecular mechanisms of
NLRP3-mediated pyroptosis in calcium oxalate kidney stones
(21,22); however, the specific morphological
characteristic of pyroptosis in oxalate-induced crystals was still
not reported. Liu et al (34) identified a series of characteristic
morphological changes of pyroptosis in human primary gingival
epithelial cells, including swollen cells, large bubbles in the
cytoplasm, membrane pores formation and structural changes in other
organelles. Similarly, the results of the present study revealed
that oxalate could induce autophagosome formation, mitochondrial
damage and rough endoplasmic reticulum damage in HK-2 cells. In
addition, several studies also demonstrated that ultrastructural
changes of pyroptosis were induced by different factors in
macrophages and cancer cells (18,35).
Membrane pore was the most typical feature to distinguish
pyroptosis from apoptosis (25).
In the present study, the results exhibited the formation of
membrane pores and cytosolic vacuoles in oxalate-induced injury in
HK-2 cells, in accordance with a previous study in which calcium
oxalate crystals-treated dendritic cells were investigated
(7). Apart from the aforementioned
ultrastructural changes, the present study also uncovered that
oxalate crystals were devoured and microvilli in the free surface
of oxalate-treated HK-2 cells. Analogous subcellular morphological
changes were detected in calcium oxalate crystals-treated bone
marrow-derived dendritic cells and tubular epithelial cells
(7). In summary, the features of
oxalate-induced HK-2 cell injury were attributed to NLRP3
inflammasome-mediated pyroptosis. The ultrastructural changes
demonstrated in the present study partially explained HK-2 cell
injury caused by oxalate from the perspective of
micromorphology.
Pyroptosis mainly includes the caspase-1-mediated
classical pyroptosis pathway and caspase-4/5/11-mediated
non-classical pyroptosis pathway (25). In the classical pyroptosis pathway,
various factors firstly activate recognition receptors (such as
NLRP3 and NLRP1) on cell membrane, and subsequently, the
corresponding inflammasome cleaves the procaspase-1 into its mature
form caspase-1 (15). GSDMD, a
member of gasdermin protein family, is the performer of cell
pyroptosis. Caspase-1 could cleave GSDMD into its mature form
GSDMD-N, which mediates the formation of membrane pores (18). Meanwhile, multiple GSDMD-N
molecules are involved in the formation of a membrane pore
(20). A previous study confirmed
the role of GSDMD and GSDMD-N in cell pyroptosis (19). In the present study, it was also
demonstrated that oxalate treatment could activate NLRP3-GSDMD
signaling and increase the levels of GSDMD and GSDMD-N in HK-2
cells. Furthermore, inhibitor of GSDMD-N could ameliorate
oxalate-induced cell pyroptosis in vitro and in vivo.
Surprisingly, it was initially observed that GSDMD deletion
dramatically suppressed oxalate crystals accumulation in the kidney
of GSDMD−/− mice treated with Gly, which suggested the
significance of GSDMD in formation of oxalate crystals. Moreover,
GSDMD itself was likely to possess a crucial function in oxalate
crystal formation. Collectively, these findings illustrated that
GSDMD was involved in the onset and progression of calcium oxalated
kidney stone. Nevertheless, the direct role and intensive mechanism
of GSDMD in oxalated-induced crystal formation and cell injury will
be further investigated.
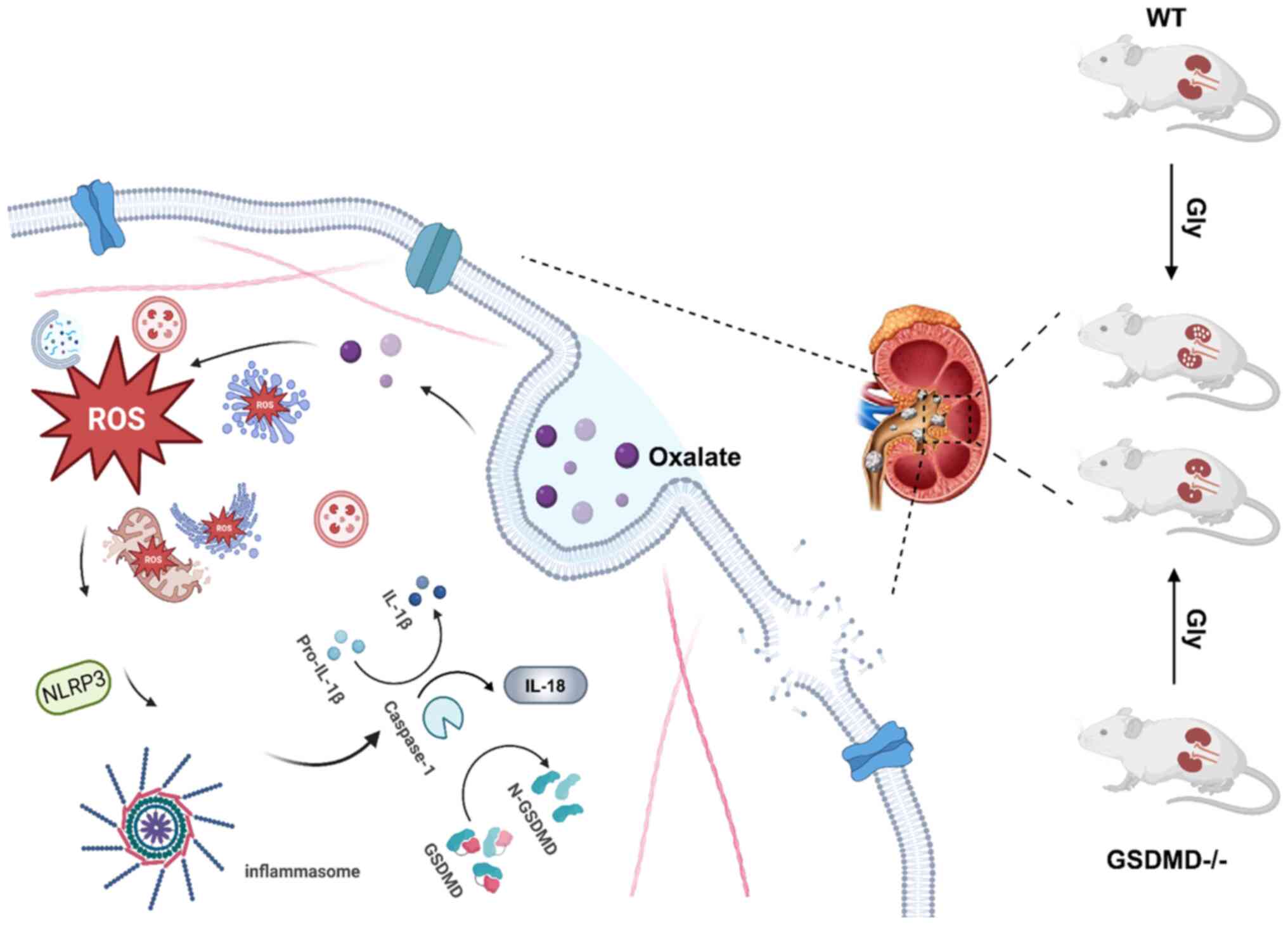
In conclusion, the present study confirmed that
ROS-mediated NLRP3-GSDMD signaling pathway was involved in
oxalate-induced injury in HK-2 cells. Importantly, the results
deciphered the ultrastructural injury in oxalate-treated HK-2
cells, which was consistent with the typical cell pyroptosis.
Moreover, GSDMD and its cleavage form GSDMD-N were associated with
the oxalate-induced disruption of membrane integrity in HK-2 cells.
Particularly, GSDMD knock down obviously inhibited formation of
oxalate calcium crystals in mice, which suggested that plays an
important role in the oxalate-induced renal injury and formation of
oxalate calcium crystals (Fig. 9).
The findings of the present study provided a new target for
prevention and treatment of oxalate nephropathy and oxalate calcium
stones. However, the intensive mechanisms of GSDMD-mediated renal
injury in formation of oxalate calcium still need to be
investigated. Moreover, determining how to repair this injury will
contribute to inhibit the formation of oxalate calcium
crystals.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 82070725), the Science and
Technology Project of Tianjin (grant no. 17ZXMFSY00060), the Key
Laboratory Fund Project of the Second Hospital of Tianjin Medical
University (grant no. 2017ZDSYS14) and the Education Commission
Research Project of Tianjin (grant no. 2017KJ208).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YC, LC and SQ designed the experiments. SY, HK, QW,
SC and XW performed the experiments. YC, SY and HK conceived the
project and analyzed the data. YC, LC and SQ wrote and revised the
paper. SQ provided funding. YC and SQ confirm the authenticity of
all the raw data. All authors contributed to the article, and read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
The maintenance, treatment and experiments regarding
the mice were approved by the Animal Ethics Committee at the Second
Hospital of Tianjin Medical University (Tianjin, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Thongprayoon C, Krambeck AE and Rule AD:
Determining the true burden of kidney stone disease. Nat Rev
Nephrol. 16:736–746. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fontenelle LF and Sarti TD: Kidney stones:
Treatment and prevention. Am Fam Physician. 99:490–496.
2019.PubMed/NCBI
|
|
3
|
Mitchell T, Kumar P, Reddy T, Wood KD,
Knight J, Assimos DG and Holmes RP: Dietary oxalate and kidney
stone formation. Am J Physiol Renal Physiol. 316:F409–F413. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Crivelli JJ, Mitchell T, Knight J, Wood
KD, Assimos DG, Holmes RP and Fargue S: Contribution of dietary
oxalate and oxalate precursors to urinary oxalate excretion.
Nutrients. 13:622020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Knoll T, Steidler A, Trojan L, Sagi S,
Schaaf A, Yard B, Michel MS and Alken P: The influence of oxalate
on renal epithelial and interstitial cells. Urol Res. 32:304–309.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Song Q, Liao W, Chen X, He Z, Li D, Li B,
Liu J, Liu L, Xiong Y, Song C and Yang S: Oxalate activates
autophagy to induce ferroptosis of renal tubular epithelial cells
and participates in the formation of kidney stones. Oxid Med Cell
Longev. 2021:66303432021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mulay SR, Kulkarni OP, Rupanagudi KV,
Migliorini A, Darisipudi MN, Vilaysane A, Muruve D, Shi Y, Munro F,
Liapis H and Anders HJ: Calcium oxalate crystals induce renal
inflammation by NLRP3-mediated IL-1β secretion. J Clin Invest.
123:236–246. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Joshi S, Wang W, Peck AB and Khan SR:
Activation of the NLRP3 inflammasome in association with calcium
oxalate crystal induced reactive oxygen species in kidneys. J Urol.
193:1684–1691. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim YG, Kim SM, Kim KP, Lee SH and Moon
JY: The role of inflammasome-dependent and inflammasome-independent
NLRP3 in the kidney. Cells. 8:13892019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Komada T and Muruve DA: The role of
inflammasomes in kidney disease. Nat Rev Nephrol. 15:501–520. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Darisipudi MN, Thomasova D, Mulay SR,
Brech D, Noessner E, Liapis H and Anders HJ: Uromodulin triggers
IL-1β-dependent innate immunity via the NLRP3 inflammasome. J Am
Soc Nephrol. 23:1783–1789. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu P, Zhang Z and Li Y: Relevance of the
pyroptosis-related inflammasome pathway in the pathogenesis of
diabetic kidney disease. Front Immunol. 12:6034162021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kelley N, Jeltema D, Duan Y and He Y: The
NLRP3 inflammasome: An overview of mechanisms of activation and
regulation. Int J Mol Sci. 20:33282019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qi S, Wang Q, Xie B, Chen Y, Zhang Z and
Xu Y: P38 MAPK signaling pathway mediates COM crystal-induced
crystal adhesion change in rat renal tubular epithelial cells.
Urolithiasis. 48:9–18. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shi J, Gao W and Shao F: Pyroptosis:
Gasdermin-mediated programmed necrotic cell death. Trends Biochem
Sci. 42:245–254. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu J, Yang K, Jin Y, Liu Y, Chen Y, Zhang
X, Yu S, Song E, Chen S, Zhang J, et al: H3 relaxin protects
against calcium oxalate crystal-induced renal inflammatory
pyroptosis. Cell Prolif. 53:e129022020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ding T, Zhao T, Li Y, Liu Z, Ding J, Ji B,
Wang Y and Guo Z: Vitexin exerts protective effects against calcium
oxalate crystal-induced kidney pyroptosis in vivo and in vitro.
Phytomedicine. 86:1535622021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
He WT, Wan H, Hu L, Chen P, Wang X, Huang
Z, Yang ZH, Zhong CQ and Han J: Gasdermin D is an executor of
pyroptosis and required for interleukin-1β secretion. Cell Res.
25:1285–1298. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli
VG, Wu H and Lieberman J: Inflammasome-activated gasdermin D causes
pyroptosis by forming membrane pores. Nature. 535:153–158. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sborgi L, Rühl S, Mulvihill E, Pipercevic
J, Heilig R, Stahlberg H, Farady CJ, Müller DJ, Broz P and Hiller
S: GSDMD membrane pore formation constitutes the mechanism of
pyroptotic cell death. EMBO J. 35:1766–1778. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Song Z, Zhang Y, Gong B, Xu H, Hao Z and
Liang C: Long noncoding RNA LINC00339 promotes renal tubular
epithelial pyroptosis by regulating the miR-22-3p/NLRP3 axis in
calcium oxalate-induced kidney stone. J Cell Biochem.
120:10452–10462. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gan XG, Wang ZH and Xu HT: Mechanism of
miRNA-141-3p in calcium oxalate-induced renal tubular epithelial
cell injury via NLRP3-mediated pyroptosis. Kidney Blood Press Res.
47:300–308. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou R, Yazdi AS, Menu P and Tschopp J: A
role for mitochondria in NLRP3 inflammasome activation. Nature.
469:221–225. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vande Walle L and Lamkanfi M: Pyroptosis.
Curr Biol. 26:R568–R572. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
D'Arcy MS: Cell death: A review of the
major forms of apoptosis, necrosis and autophagy. Cell Biol Int.
43:582–592. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang S, Du P, Zhang N, Liu J, Tang X, Zhao
Q and Yang Y: Oligomeric proanthocyanidins protect against HK-2
cell injury induced by oxalate and calcium oxalate monohydrate
crystals. Urolithiasis. 44:203–210. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Moe OW: Kidney stones: Pathophysiology and
medical management. Lancet. 367:333–344. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Khan SR, Canales BK and
Dominguez-Gutierrez PR: Randall's plaque and calcium oxalate stone
formation: Role for immunity and inflammation. Nat Rev Nephrol.
17:417–433. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lv P, Liu H, Ye T, Yang X, Duan C, Yao X,
Li B, Tang K, Chen Z, Liu J, et al: XIST inhibition attenuates
calcium oxalate nephrocalcinosis-induced renal inflammation and
oxidative injury via the miR-223/NLRP3 pathway. Oxid Med Cell
Longev. 2021:16761522021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fong-Ngern K, Vinaiphat A and
Thongboonkerd V: Microvillar injury in renal tubular epithelial
cells induced by calcium oxalate crystal and the protective role of
epigallocatechin-3-gallate. FASEB J. 31:120–131. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu H, Ye T, Yang X, Liu J, Jiang K, Lu H,
Xia D, Peng E, Chen Z, Sun F, et al: H19 promote calcium oxalate
nephrocalcinosis-induced renal tubular epithelial cell injury via a
ceRNA pathway. EBioMedicine. 50:366–378. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu L, Gan X, Liu X and An R: Calcium
oxalate crystals induces tight junction disruption in distal renal
tubular epithelial cells by activating ROS/Akt/p38 MAPK signaling
pathway. Ren Fail. 39:440–451. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu J, Wang Y, Meng H, Yu J, Lu H, Li W,
Lu R, Zhao Y, Li Q and Su L: Butyrate rather than LPS subverts
gingival epithelial homeostasis by downregulation of intercellular
junctions and triggering pyroptosis. J Clin Periodontol.
46:894–907. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yu J, Li S, Qi J, Chen Z, Wu Y, Guo J,
Wang K, Sun X and Zheng J: Cleavage of GSDME by caspase-3
determines lobaplatin-induced pyroptosis in colon cancer cells.
Cell Death Dis. 10:1932019. View Article : Google Scholar : PubMed/NCBI
|