Introduction
The prevalence of neurodegenerative diseases, such
as Parkinson's disease and Alzheimer's disease (AD) among elderly
individuals, is increasing yearly (1). AD has been considered a global public
health threat, and is the most common cause of dementia. The Global
Burden of Disease study recently forecasted that the number of
individuals with dementia would increase from 57.4 million cases
globally in 2019 to 152.8 million cases in 2050 (2). The classical neuropathological
hallmarks of AD include amyloid-β (Aβ) plaque accumulation and
neuro-fibrillary tangles aggregated by hyperphosphorylated tau
protein (3). In general terms,
amyloid precursor protein (APP) is cleaved by β-secretase, and
γ-secretase can support the generation and maturation of Aβ
peptides, which range between 38 and 43 amino acids (4). Prospective studies have reported that
accumulation of different species of Aβ peptides contributed to
glial activation and increased release of pro-inflammatory
cytokines, chemokines and reactive oxygen species, all of which
induce subsequent pathological events in AD, such as
neuroinflammation and oxidative stress (5,6). Aβ
triggers a cascade that causes neuron death, synapse loss and
further cognitive decline (7).
However, the validity of advanced or ongoing anti-amyloid therapies
for AD is controversial due to the inefficiency in ameliorating
cognitive outcomes in patients with AD (8).
Glia and immune cell-activated neuroinflammation is
initiated by Aβ deposition, leading to neuron loss and cell
apoptosis (9). Sphingosine kinase
(SphK) is the predominant isoform responsible for the
phosphorylation of sphingosine to produce sphingosine 1-phosphate
(S1P), which acts as a pivotal lipid signaling regulator in the
process of cell apoptosis, senescence and inflammation (10,11).
SphK1 and SphK2 are well-identified isoforms of SphK and exhibit
different properties and biological outcomes due to their different
intracellular localization and tissue distribution (12). Notably, activation of intracellular
SphK1 promotes the generation of S1P, which is necessary for the
phosphorylation of IκB kinase and activation of the canonical NF-κB
inflammatory signaling pathway (13). Emerging research has indicated
underlying mechanisms and pathways of the SphK1/S1P axis involving
neuroinflammation within different neurological disorders (14,15).
One study revealed that SphK1 and its product S1P promoted the M1
microglia polarization and enhanced the production of
pro-inflammatory cytokines in injured spinal cord tissue of rats,
which ultimately amplificated the inflammation cascade, including
nuclear phosphorylation of p38 MAPK and NF-κB p65 (14). However, another study demonstrated
that elevated SphK1 activity in neurons restored Aβ phagocytosis of
microglia and resolution of neuroinflammation via acetylation of
cyclooxygenase 2 (COX2) (15).
Therefore, the role and underlying mechanism of SphK1 in Aβ-related
neuroinflammation remain to be fully elucidated.
Panax notoginseng is a traditional Chinese
medicine with beneficial functions in promoting blood circulation
and alleviating limb swelling (16). In addition, it has been used in
several clinical trials to treat vascular dementia, cognitive
decline and brain disorders in acute stroke (17,18).
Notoginsenoside R1 (NGR1), one of the ingredients isolated from
P. notoginseng saponins (PNSs), has been reported to possess
anti-inflammatory and anti-oxidative stress properties (19,20).
Chen et al (19)
demonstrated that NGR1 alleviated IL-1β-induced inflammation and
oxidative stress in vivo and in vitro by activating
the nuclear factor erythroid 2-related factor 2/heme oxygenase 1
signaling pathway. In addition, NGR1 attenuated isoflurane-induced
neurological disorders, including cognitive decline, memory loss
and neuroinflammation, in rats (20). Notably, NGR1 exhibited a
neuroprotective effect, including increasing memory function,
improving learning ability and alleviating neuronal
hyperexcitability via the regulation of voltage-gated sodium
channels (Nav) proteins, in a mouse model of AD (21). These existing results implied that
NGR1 might exert a neuroprotective effect on AD-related
neuroinflammation and other neurological injuries. However, to the
best of our knowledge, the specific mechanism of NGR1 in Aβ-induced
neuroinflammation in AD progression remains unclear.
Given the beneficial role of NGR1 in ameliorating
AD-related neurological disorders, the present study assessed
whether NGR1 treatment protects against Aβ25-35-induced
neuron apoptosis and inflammation and the underlying mechanism. The
present study first examined Aβ-protein fragment 25–35
(Aβ25-35)-induced cell apoptosis and NF-κB inflammatory
signaling pathway activation in PC12 rat adrenal chromaffin cell
tumor cells co-incubated with Aβ25-35 and different
concentrations of NGR1. In addition, the present study used small
interfering RNA (siRNA/si) to knock down SphK1 to indicate how
neuroinflammation improvement by NGR1 depended on the regulation of
the SphK1/NF-κB signaling axis in PC12 cells.
Materials and methods
Experimental reagents
NGR1 (batch number B21099; purity ≥98%) was acquired
from Shanghai Yuanye Biotechnology Co., Ltd. For cell treatment,
NGR1 was dissolved in DMSO (MilliporeSigma) and diluted in cell
culture medium to keep the DMSO content below 0.1% of the total
volume of the cell culture medium The preparation of
Aβ25-35 (MilliporeSigma) has been described in our
previous study (22). Briefly,
Aβ25-35 was first dissolved to a concentration of 1 mM
in deionized water and then aged for 72 h at 37°C in a humidified
chamber to induce its aggregation. When apparent white flocculent
aggregation occurred after incubation for 72 h, the aggregated
solution was dissolved in DMSO to a concentration of 250 µM and
stored at −20°C before being added to the culture medium to examine
the cell viability at the final desired concentration (range, 5–30
µM), and to perform related molecular experiments at a final
concentration of 20 µM. The final concentration of DMSO in the
culture medium was <0.01% (vol/vol).
Cell experiments
PC12 rat pheochromocytoma cells (The Cell Bank of
Type Culture Collection of The Chinese Academy of Sciences) were
maintained in Ham's F-12 medium (Gibco; Thermo Fisher Scientific,
Inc.) with 5% (vol/vol) heat-inactivated FBS (Gibco; Thermo Fisher
Scientific, Inc.), 15% (vol/vol) horse serum (Gibco; Thermo Fisher
Scientific, Inc.) and 1% penicillin-streptomycin solution (100 U/ml
penicillin and 100 µg/ml streptomycin; Gibco; Thermo Fisher
Scientific, Inc.) in a humidified incubator (5% CO2; 95%
air; 37°C). For analysis of the effects of NGR1 on
Aβ25-35-induced cell injury, PC12 cells were stimulated
with 20 µM Aβ25-35 peptide alone or combined with
different concentrations of NGR1 (50, 100, 250, 500 and 1,000
µg/ml) for 24 h at 37°C. For analysis of the effects of SphK1
inhibitor II (SKI–II) on Aβ25-35-induced cell injury,
PC12 cells were cultured with 20 µM Aβ25-35 peptide
alone or combined with 10 µM SKI–II (HY-13822; MedChemExpress) with
or without NGR1 (1,000 µg/ml) for 24 h at 37°C based on previous
studies (23,24). PC12 cells incubated at 37°C for 24
h with only culture medium were used as the control group. A
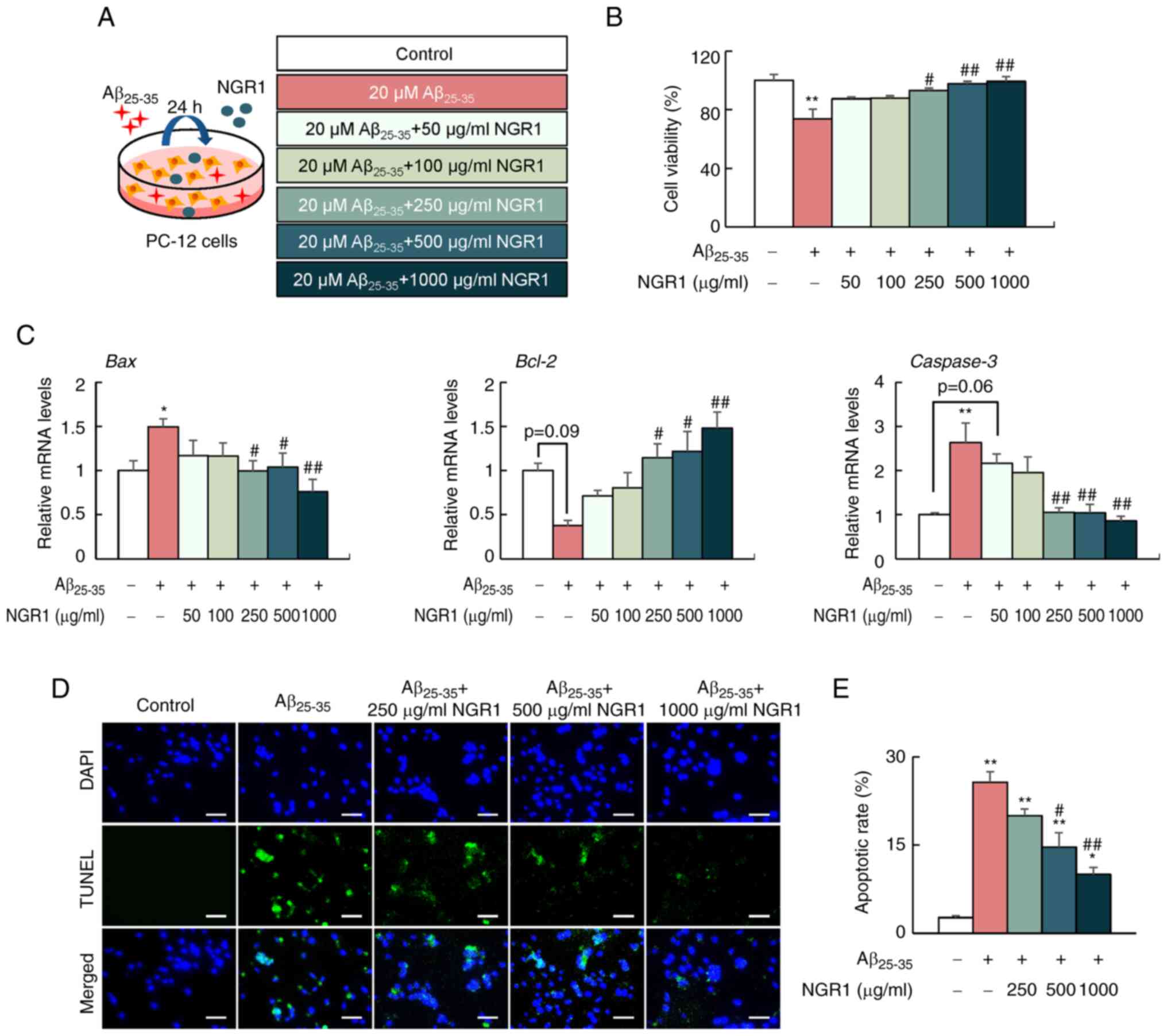
schematic diagram of cell treatments is shown in Fig. 1A. Cell viability was examined using
an MTT assay according to the manufacturer's instructions as
previously described (25).
RNA extraction and quantitative PCR
(qPCR)
Total RNA from PC12 cells was isolated using Biozol
reagent (Biomiga), and cDNA was synthesized using a cDNA Reverse
Transcriptase Kit (Vazyme Biotech Co., Ltd.) according to the
manufacturer's instructions. The temperature protocol for reverse
transcription was as follows: 37°C for 15 min and 98°C for 5 min.
qPCR was performed using SYBR Green Real-time PCR Master Mix
(Vazyme Biotech Co., Ltd.) on an Eppendorf Master Cycler ep
RealPlex4 (Eppendorf SE) as previously described (26). The reaction conditions were as
follows: 2-min pre-denaturation at 95°C, followed by 40 cycles of
15 sec at 95°C and 30 sec at 60°C. GAPDH was used as a
housekeeping gene for data standardization. The gene expression was
calculated using the 2−ΔΔCq method as described
previously (27). The primer
sequences are listed in Table
I.
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Name | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| r. Bax |
GAACTGGACAACAACATGGA |
GCAAAGTAGAAAAGGGCAAC |
| r.
Bcl-2 |
GGGTCATGTGTGTGGAGAG |
AGCCAGGAGAAATCAAACAG |
| r.
Caspase3 |
GAGCTTGGAACGCGAAGAAAA |
ACACAAGCCCATTTCAGGGTA |
| r.
SphK1 |
CGCCTGGGCAACACCGATAA |
GGCTACATAGGGGTTTCTGG |
| r.
MMP-3 |
ACCTATTCCTGGTTGCTGCT |
CAGGTCTGTGGAGGACTTGT |
| r.
MMP-9 |
AGTGCCCTTGAACTAAGGCT |
GCCTCCACTCCTTCCTAGTC |
| r.
GAPDH |
ATGGAGAAGGCTGGGGCTCACCT |
AGCCCTTCCACGATGCCAAAGTTGT |
TUNEL staining
A TUNEL BrightGreen apoptosis detection kit (Vazyme
Biotech Co., Ltd.) was used to detect apoptotic cells as described
in our previous study (22).
Briefly, PC12 cells were cultured on coverslips until they reached
70–80% confluence. Subsequently, the PC12 cells were treated with
20 µM Aβ25-35 peptide alone or co-incubated with
different concentrations of NGR1 ranging between 50 and 1,000 µg/ml
for 24 h at 37°C. After fixation with 4% paraformaldehyde at room
temperature for 15 min and permeabilization with 0.2% Triton X-100
(MilliporeSigma) for 10 min at room temperature, the apoptotic cell
nuclei were stained with green fluorescein, while all cell nuclei
were labeled with blue DAPI, as observed by confocal microscopy
(IX83; Olympus Corporation). The fluorescence data were analyzed
and quantified using ImageJ Fiji 2.9.0 (National Institutes of
Health). The apoptotic cell rate was measured as the ratio of
TUNEL-positive nuclei over DAPI-stained nuclei as follows:
Apoptotic rate (%)=[(number of TUNEL-positive nuclei)/(number of
DAPI-stained nuclei)] ×100 (%).
Immunoblotting
Proteins from PC12 cells were extracted in RIPA
lysis buffer (MilliporeSigma) containing protease and phosphatase
inhibitors (Roche Diagnostics). According to the manufacturer's
instructions, protein concentrations were quantified using a BCA
protein assay kit (Thermo Fisher Scientific, Inc.). The proteins
(10 µg/lane) were equally loaded onto 10% SDS gels and transferred
to methanol-prewetted PVDF membranes (MilliporeSigma). The
membranes were blocked for 1 h at room temperature with a 0.2% KPL
detector block (SeraCare Life Sciences; LGC Limited) and then
incubated with primary antibodies at 4°C overnight. The primary
antibodies used were anti-SphK1 (cat. no. abs135525; Absin
Bioscience, Inc.), anti-phospho-NF-κB p65 (Ser536) (cat. no. 3033),
anti-NF-κB p65 (cat. no. 3034), GAPDH (cat. no. 2118) and β-actin
(cat. no. 3700) (Cell Signaling Technology, Inc.) at a dilution of
1:1,000. After washing with tris-buffered saline with 0.1%
Tween® 20 detergent, the membrane was incubated with HRP
anti-rabbit or -mouse antibody at a dilution ratio of 1:10,000 at
room temperature for 1 h (cat. no. 7074 and 7076, Cell Signaling
Technology, Inc.). The band visualization was performed used an ECL
chemiluminescence kit according to the manufacturer's instructions
(E422, Vazyme Biotech Co., Ltd.). The procedures of western
blotting were described previously (28). The intensity of the bands in the
autoradiograms was examined using ImageJ Fiji 2.9.0 (National
Institutes of Health).
siRNA-mediated gene silencing and
transfection
PC12 cells were maintained in 6- or 12-well plates
until the cells were ~70% confluent. For SphK1 siRNA interference,
the cells were transfected with 50 nM si-SphK1 or si-Control
(Sangon Biotech Co., Ltd.) for 6 h at 37°C and then incubated with
20 µM Aβ25-35 peptide alone or co-cultured with 1,000
µg/ml NGR1 for 24 h at 37°C. The cells in the negative control
group were maintained at 37°C for 30 h without any treatment and
transfection. The si-SphK1 target sequences were as follows:
5′-GGACUUGGAGAGUGAGAAATT-3′ (sense) and 5′-UUUCUCACUCUCCAAGUCCTT-3′
(antisense). The scrambled si-Control sequences were as follows:
5′-UUCUCCGAACGUGUCACGUTT-3′ (sense) and 5′-ACGUGACACGUUCGGAGAATT-3′
(antisense). Transfection of PC12 cells with the specific siRNA
against SphK1 and scrambled si-Control was performed using
Lipofectamine® 3000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) in Opti-MEM (Gibco; Thermo Fisher Scientific,
Inc.) as described previously (22,29).
Statistical analysis
All data are presented as the mean ± SEM.
Differences among the mean values were assessed using one-way ANOVA
followed by the Tukey's post hoc test using Prism 9 (Dotmatics).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of varying concentrations of
NGR1 on the viability of PC12 cells
Previous studies have reported the cell toxicity of
Aβ25-35 peptide (10–30 µM) in PC12 and SH-SY5Y cells
(30–32). Our previous study demonstrated that
treatment with Aβ25-35 peptide (10–30 µM) alone reduced
the cell viability at 24, 48 and 72 h (22). Consistently, the present study
demonstrated that Aβ25-35 peptide administration
contributed to a significant decrease of cell viability in the dose
range of 10–30 µM at 24 h compared with the control group (Fig. S1A). No significant cell toxicity
was detected in PC12 cells treated with NGR1 at concentrations
ranging between 50 and 1,500 µg/ml. By contrast, 2 mg/ml NGR1
treatment for 24 h significantly decreased the cell viability
(Fig. S1B). Notably, addition of
NGR1 increased the suppressed cell viability caused by 20 µM
Aβ25-35 administration in a dose-dependent manner from
250 to 1,000 µg/ml after 24 h (Fig.
1B). Thus, NGR1 concentrations ranging between 250 and 1,000
µg/ml were used in subsequent experiments.
NGR1 decreases apoptosis in
Aβ25-35-treated PC12 cells
The influence of NGR1 on apoptosis-related gene
expression in PC12 cells was analyzed by reverse
transcription-qPCR. As shown in Fig.
1C, 20 µM Aβ25-35 treatment significantly increased
the mRNA expression levels of Bax and Caspase-3,
which are classical apoptosis promoters. By contrast, the mRNA
expression levels of Bcl-2, an essential regulator in cell
apoptosis inhibition, were decreased in the
Aβ25-35-treated group compared with the control group;
however, this difference was not significant (Fig. 1C). However, NGR1 supplementation
(250–1,000 µg/ml) reduced the mRNA expression levels of Bax
and Caspase-3 but increased the mRNA expression levels of
Bcl-2 in the PC12 cells co-incubated with 20 µM
Aβ25-35 peptide at 24 h in a dose-dependent manner
(Fig. 1C). In addition, TUNEL
staining was performed to further verify the effect of NGR1 on
apoptosis (Fig. 1D and E).
Aβ25-35 administration (20 µM) increased the percentage
of TUNEL-positive cells compared with the control group (Fig. 1D and E). By contrast, NGR1
treatment (500–1,000 µg/ml) dose-dependently decreased the
apoptotic cells labeled by TUNEL in the PC12 cells co-treated with
Aβ25-35 peptide (Fig. 1D
and E).
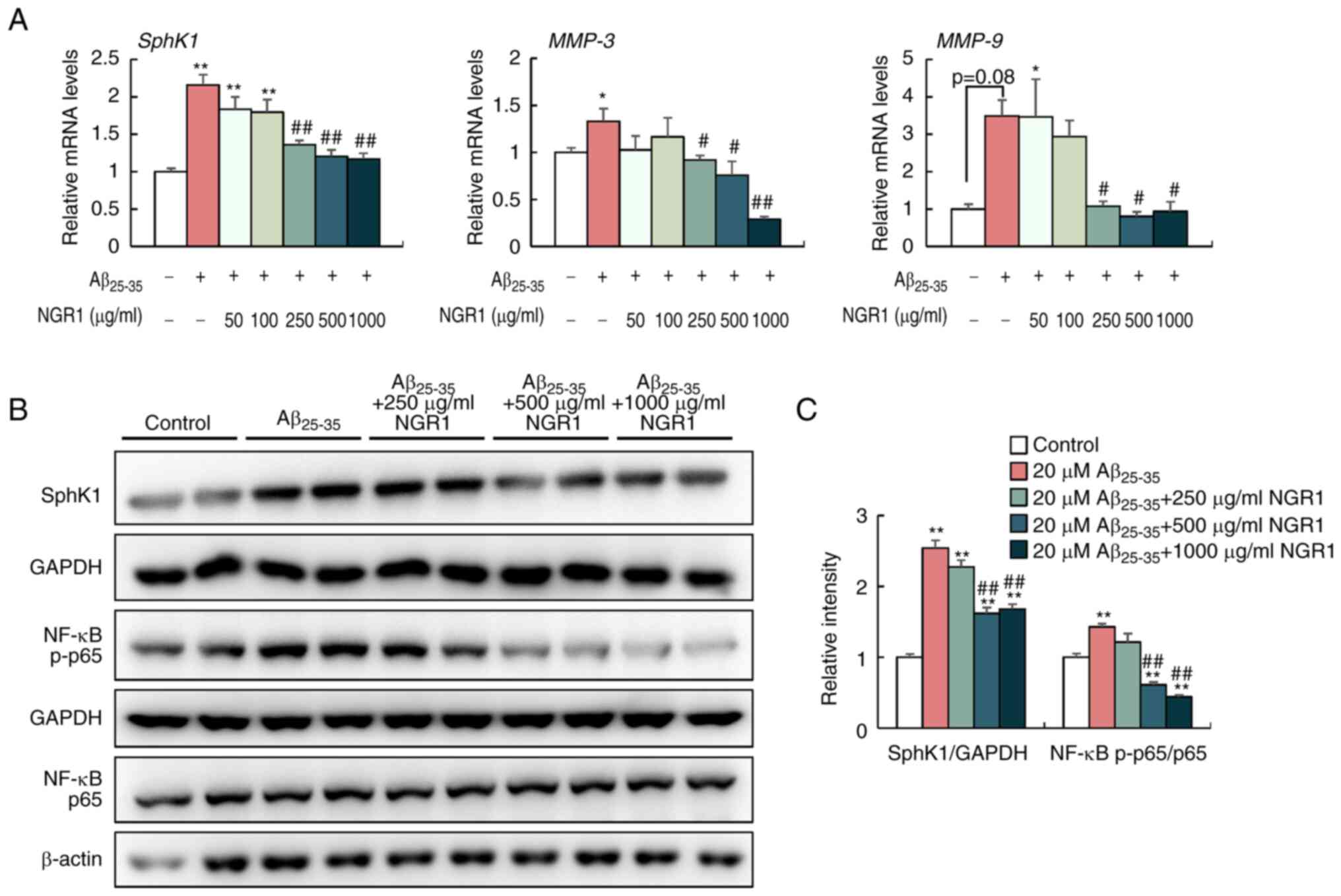
NGR1 inhibits
Aβ25-35-induced SphK1/NF-κB signaling activation in PC12
cells
Intracellular SphK1 is considered to modulate NF-κB
activation and is responsible for the inflammatory cascade
following different inflammatory stimuli (33,34).
To elucidate the anti-inflammatory mechanism of NGR1 in
Aβ25-35-treated PC12 cells, the present study examined
the gene and protein expression levels of SphK1 and the ratio of
NF-κB p-p65/p65 in PC12 cells co-incubated with 20 µM
Aβ25-35 and different concentrations of NGR1 (Fig. 2). As shown in Fig. 2A, Aβ25-35 treatment
increased the mRNA expression levels of SphK1 compared with
the control group, whereas NGR1 supplementation (ranging between
250 and 1,000 µg/ml) significantly reduced SphK1 gene
expression in PC12 cells. Furthermore, in a dose-dependent manner,
NGR1 treatment (250–1,000 µg/ml) significantly decreased the
protein levels of SphK1 and decreased the ratio of p-p65/p65 in
PC12 cells compared with the 20 µM Aβ25-35-treated group
(Fig. 2B and C). Additionally,
MMP-3 and MMP-9 are pivotal inflammatory components in the
progression of AD, as they promote the aggregation of Aβ and tau
proteins in the brain (35).
Consistently, Aβ25-35 treatment increased the mRNA
expression levels of MMP-3 and MMP-9 in vitro, while
NGR1 administration (250–1,000 µg/ml) prominently decreased the
mRNA expression levels of MMP-3 and MMP-9 in a
dose-dependent manner (Fig.
2A).
SphK1/NF-κB signaling is involved in
the anti-inflammatory effect of NGR1 in Aβ25-35-treated
PC12 cells
To clarify the role of SphK1/NF-κB signaling in the
anti-inflammatory effect of NGR1 in Aβ25-35-treated PC12
cells, 10 µM SKI–II, a SphK1 inhibitor, was co-administrated with
20 µM Aβ25-35 peptide and 1,000 µg/ml NGR1 for 24 h
(Fig. 3). SphK1 inhibition also
significantly decreased the mRNA expression levels of Bax
and Caspase-3 but increased the mRNA expression levels of
Bcl-2 in PC12 cells to a similar extent compared with 1,000
µg/ml NGR1 supplementation compared with Aβ25-35
treatment alone (Fig. 3A). At the
same time, a decreased percentage of TUNEL-positive apoptotic cells
was observed in the SKI–II-treated group and SKI–II and NGR1 group
compared with the 20 µM Aβ25-35 peptide treatment group
in PC12 cells (Fig. 3B).
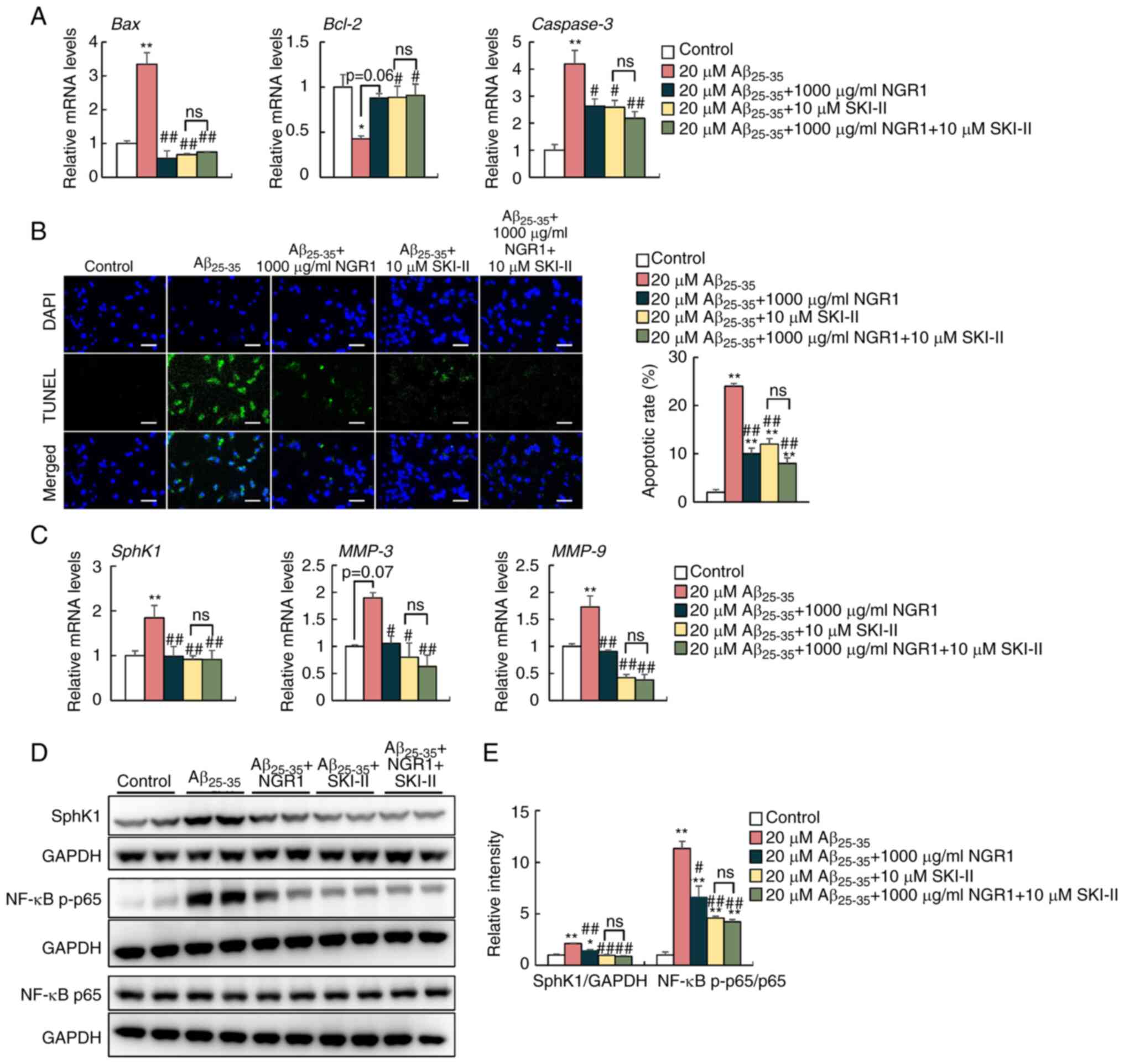 | Figure 3.Effects of pharmacological inhibition
of Sphk1 on Aβ25-35-induced apoptosis and inflammation
in PC12 cells. (A) mRNA expression levels of Bax, Bcl-2 and
Caspase3 in PC12 cells treated with 20 µM Aβ25-35
and 1,000 µg/ml NGR1 with or without 10 µM SKI–II. n=6. (B)
Representative confocal images and apoptotic rate quantifications
of TUNEL-positive (green) apoptotic cells and DAPI-labeled nuclei
(blue) of PC12 cells. Scale bar, 50 µm. n=3. (C) mRNA expression
levels of SphK1, MMP-3 and MMP-9 in PC12 cells. n=6.
(D) Protein levels of SphK1, NF-κB p-p65 and total NF-κB p65 in
PC12 cells. (E) Semi-quantification of the protein levels of SphK1,
NF-κB p-p65 and total NF-κB p65 and their corresponding loading
controls in PC12 cells. n=4. Data are presented as the mean ± SEM.
*P<0.05, **P<0.01 vs. untreated control group;
#P<0.05, ##P<0.01 vs. 20 µM
Aβ25-35 group. Aβ25-35, amyloid-β-protein
fragment 25–35; NGR1, notoginsenoside R1; ns, not significant; p-,
phosphorylated; SKI–II, SphK1 inhibitor II; SphK1, sphingosine
kinase 1. |
SphK1 inhibition by SKI–II treatment decreased the
mRNA expression levels of MMP-3 and MMP-9 in PC12
cells compared with the Aβ25-35 peptide-treated group
(Fig. 3C). Similar to treatment
with 1,000 µg/ml NGR1 and Aβ25-35, western blotting
demonstrated that SKI–II treatment significantly decreased the
protein levels of SphK1 and decreased the ratio of p-p65/p65 in
PC12 cells compared with those of the 20 µM Aβ25-35
peptide-treated group (Fig. 3D and
E). However, 1,000 µg/ml NGR1 and 10 µM SKI–II combined
treatment did not further inhibit the activation of the NF-κB
signaling pathway (Fig. 3D and E).
Overall, these data illustrated the involvement of SphK1 in the
inhibitory effect of NGR1 on inflammation induced by
Aβ25-35 in PC12 cells.
Knockdown of SphK1 relieves
inflammation induced by Aβ25-35 in PC12 cells
To further verify whether the activation of SphK1
mediates NGR1-alleviated inflammation, scrambled siRNA and si-SphK1
were transfected into PC12 cells separately, and an untreated and
untransfected negative control group was established (Figs. 4 and S2), and SphK1 was knocked down (Fig. S2). As shown in Fig. 4A, SphK1 knockdown decreased mRNA
expression levels of Bax and Caspase-3, and increased
mRNA expression levels of Bcl-2 in PC12 cells compared with
cells treated with Aβ25-35 alone in the si-Control
group, which indicated that SphK1 knockdown abolished the apoptosis
induced by Aβ25-35. Consistently, a decreased proportion
of TUNEL-labeled apoptotic cells was observed in the 20 µM
Aβ25-35-treated si-SphK1 group when compared with the 20
µM Aβ25-35-treated si-Control group (Fig. 4B). In addition, SphK1 knockdown
significantly decreased the mRNA expression levels of MMP-3
and MMP-9 and decreased the ratio of p-p65/p65 in PC12 cells
compared with the 20 µM Aβ25-35-treated si-control group
(Fig. 4C-E). However, knockdown of
SphK1 combined with Aβ25-35 and NGR1 treatment did not
exert an effect on mRNA expression levels of Bax, Caspase-3
and Bcl-2, and the proportion of TUNEL-stained apoptotic
cells compared with combination treatment of NGR1 and
Aβ25-35 in the si-control groups (Fig. 4A and B). In addition, knockdown of
SphK1 combined with Aβ25-35 and NGR1 treatment also
decreased the mRNA level of MMP-3 and MMP-9 and
inhibited NF-κB p65 activation compared with those of the PC12
cells treated with Aβ25-35 alone in the si-Control
group, but to a similar extent compared with combined treatment
with NGR1 and Aβ25-35 in the si-control group (Fig. 4C-E). However, these data
demonstrated that knockdown of SphK1 could effectively suppress
Aβ-induced apoptosis and inflammation in PC12 cells.
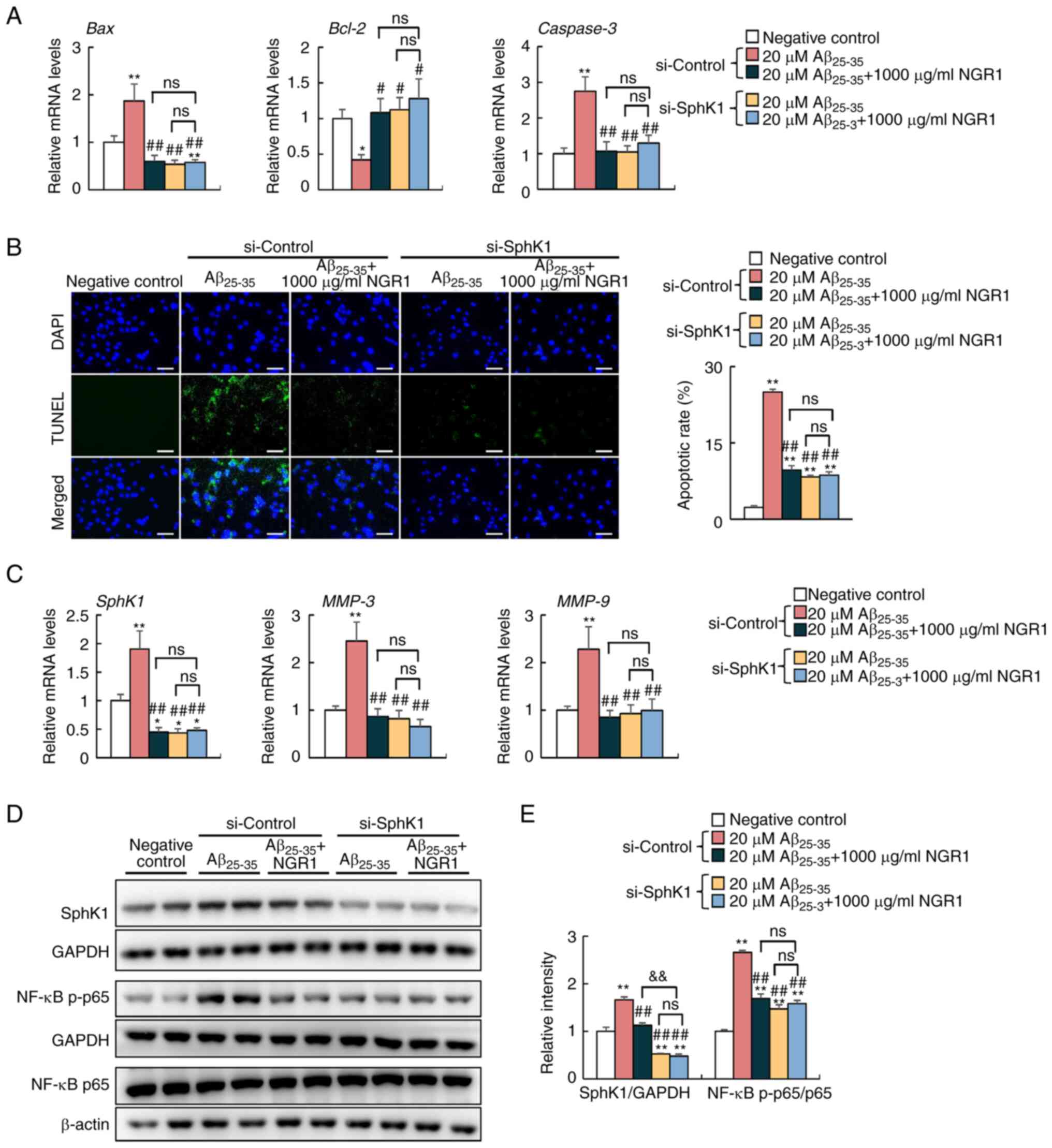 | Figure 4.Effects of siRNA-mediated knockdown
of Sphk1 on Aβ25-35-induced apoptosis and inflammation
in PC12 cells. (A) mRNA expression levels of Bax, Bcl-2 and
Caspase3 in PC12 cells transfected with si-Control and
si-Sphk1. N=6. (B) Representative confocal images and apoptotic
rate quantifications of TUNEL-positive (green) apoptotic cells and
DAPI-labeled nuclei (blue) of PC12 cells. Scale bar, 50 µm. N=3.
(C) mRNA expression levels of SphK1, MMP-3 and MMP-9
in PC12 cells. N=6. (D) Protein levels of SphK1, NF-κB p-p65 and
total NF-κB p65 and their corresponding loading controls in PC12
cells. (E) Semi-quantification of the protein levels of SphK1,
NF-κB p-p65 and total NF-κB p65 in PC12 cells. n=4. Data are
presented as the mean ± SEM. *P<0.05, **P<0.01 vs. negative
control (no treatment or transfection); #P<0.05,
##P<0.01 vs. si-control + 20 µM Aβ25-35
group; &&P<0.01 vs. si-control + 20 µM
Aβ25-35 + 1,000 µg/ml NGR1group. Aβ25-35,
amyloid-β-protein fragment 25–35; NGR1, notoginsenoside R1; ns, not
significant; p-, phosphorylated; siRNA/si, small interfering RNA;
SphK1, sphingosine kinase 1. |
Discussion
The principal cellular characteristic of AD is
Aβ-related neurotoxicity that prompts apoptosis, which is
responsible for extensive neuronal degeneration and loss in the
brain (36). Although ongoing
clinical trials for AD therapies that target the degradation of Aβ
deposition have been unsatisfactory, widespread research has
indicated high toxicity of soluble Aβ oligomers (37). In general, Aβ1-40 and
Aβ1-42 are the most abundant species of Aβ peptides, of
which Aβ1-42 has been detected as the primary component
in amyloid plaque depositions in patients with AD (38). Among the Aβ fragments available at
present, the Aβ25-35 peptide consisting of 11 amino acid
residues has been found in the in the brain of patients with AD
(39). In addition, a comparison
study demonstrated that both Aβ1-42 and
Aβ25-35 induced similar cell toxicity in organotypic
hippocampal slices. Nevertheless, the authors highlighted that
synthetic Aβ25-35 peptide was convenient for clarifying
the neurotoxic mechanisms involved in the pathogenesis of AD
(40). Aβ25-35 peptide
is a cheap and convenient alternative in the pathological
investigations of AD, since this smaller peptide retains the
toxicity of the full-length peptide and induces a vast amount of
neuron death (41). Furthermore,
the Aβ25-35 peptide forms a series of β-barrel
aggregates and a dominant hexamer structure, the C terminus region
of which provides the most potent interactions between two
Aβ25-35 monomers (42).
Notably, aging aggravates the release of soluble Aβ1-40
from plaques and Aβ1-40 is converted by proteolysis to
toxic Aβ25-35/40, which causes detrimental damage in the
hippocampal CA1 neurons to promote neurotoxicity in AD (39). Collectively, the present study used
the Aβ25-35 peptide as a potent inducer to simulate cell
toxicity and apoptosis, effectively mimicking the pathological
conditions associated with AD in vitro.
PC12 cells are derived from the adrenal medulla of a
rat with pheochromocytoma and are widely utilized as a neuronal
precursor cell line in neuroscience research (43). PC12 cells exhibit morphological and
physiological traits resembling those of adrenal gland cells in the
undifferentiated state (43).
These cells are commonly employed in numerous studies to
investigate the neurotoxic effects of various substances (44,45).
For instance, PC12 cells are frequently used to study Aβ-induced
neurotoxicity, as an in vitro model simulating aspects of AD
(46). Our previous study revealed
that the Aβ25-35 peptide markedly contributed to severe
apoptosis and oxidative stress in PC12 cells (22). A recent study also revealed that
Aβ25-35 administration enhanced apoptosis and activated
the NF-κB signaling pathway in an APP/presenilin 1
double-transgenic mouse model of AD and BV2 microglia cell line
(47). Consistent with these
previous findings, the present study used Aβ25-35
peptide to establish AD-related phenotypes in PC12 cells, including
cellular apoptosis and neuroinflammation. In the present study, 20
µM Aβ25-35 treatment for 24 h was used and significantly
induced apoptosis, as demonstrated by the decrease in cell
viability, increased mRNA expression levels of pro-apoptotic
markers (Bax and Caspase3) and increased percentage
of TUNEL-labeled apoptotic cells. In addition, Aβ25-35
markedly activated the NF-κB inflammatory signaling pathway
associated with the increased levels of Sphk1 signaling in PC12
cells. Notably, the present study demonstrated that NGR1 blocked
the NF-κB inflammatory pathway by inhibiting SphK1 signaling,
thereby reversing Aβ25-35-induced apoptosis and
inflammatory damage in PC12 cells. However, a limitation of the
present study was the absence of protein detection of cleaved
caspase3, necessitating further investigations to conclusively
ascertain the impact of NGR1 on apoptosis.
Prospective studies have demonstrated the advantages
of P. notoginseng in alleviating inflammation, inhibiting
cell apoptosis and reducing thrombosis (48,49).
As the bioactive component of P. notoginseng, PNS exhibits
neuroprotective effects on the progression of AD by inhibiting Aβ
production and aggregation and alleviating inflammation (50). Notably, NGR1, a primary active
ingredient of PNSs, has also been reported to have
beneficial effects on neuronal activity repairment in AD
development (51,52). For instance, oral administration of
NGR1 effectively improved Aβ-induced synaptic plasticity deficits
in brain slices and reduced Aβ neurotoxicity by recovering
Aβ1-42 oligomer-induced long-term potentiation
impairment (51). Hu et al
(21) demonstrated that NGR1
treatment restored cell viability of primary cultured mouse neurons
after Aβ1-42 administration and reduced neuronal
hyperexcitability via the reallocation of Nav1.1α and prevention of
excessive cleavage of Navβ2. The present findings demonstrated that
NGR1 supplementation increased viability in a dose-dependent manner
after incubation of PC12 cells with Aβ25-35 and
decreased apoptosis. Another study also demonstrated that NGR1
counteracted the cell damage induced by Aβ25-35 by
increasing cell viability, alleviating cellular oxidative damage
and normalizing mitochondrial membrane potential via the
suppression of the MAPK signaling pathway in PC12 cells (52). Therefore, we hypothesized that NGR1
exerted neuroprotective effects against Aβ25-35-induced
cell injury by alleviating apoptosis in PC12 cells.
Extracellular Aβ deposition indicates neurotoxicity
resulting in neuronal death, microglia activation and excessive
production of pro-inflammatory cytokines, which are mediated
through binding to neurotrophic factors, such as
N-methyl-D-aspartate receptor and insulin receptor (53). The present study revealed
activation of the NF-κB inflammatory signaling pathway in
Aβ25-35-treated PC12 cells. In addition, in the present
study, Aβ25-35 treatment markedly increased the mRNA
expression levels of MMP-3 and MMP-9, which are located around
amyloid plaques and elevated in the brain of patients with AD
(54,55). In the Ra2 microglia cell line,
excessive Aβ accumulation elevated the activity of MMP-3 and
subsequently activated PI3K/Akt signaling inflammatory cascades
(56). In addition, the
simultaneous accumulation of cortical MMP-9 may be recognized as a
pathological marker of the early onset of AD (57), and MMP-3 has been demonstrated to
activate MMP-9 effectively and to indirectly facilitate AD
pathology (58). The present study
revealed inhibition of phosphorylation of NF-κB p65 and decreased
mRNA expression levels of MMP-3 and MMP-9 in NGR1-treated groups
compared with the group treated with only Aβ25-35.
Although the substantial role and mechanism of MMP-3 and MMP-9 in
the Aβ-induced inflammatory response remain elusive, the present
data suggested that MMP-3 and MMP-9 were involved in the
Aβ25-35-related neurotoxicity and inflammation.
Therefore, further investigation is required to examine the
potential role and possible mechanism of MMPs in the Aβ
neurotoxicity during AD progression.
In addition to the downregulation of NF-κB
activation, the present study also revealed a marked decrease in
SphK1 mRNA and protein expression in NGR1-treated groups. SphK1 is
one of the rate-limiting enzymes catalyzing S1P generation, which
subsequently binds to a series of intracellular S1P receptors and
is involved in various inflammatory diseases (11). One study reported that SphK1 and
its product S1P acted as viable therapeutic targets for mediating
neuroinflammation via the phosphorylation of p38 MAPK and ultimate
activation of NF-κB p65 (14).
Conversely, treatment with PF543, a validated inhibitor of SphK1,
reversed pro-inflammatory M1-type microglia-facilitated neuron
apoptosis and NF-κB p65 activation in PC12 cells (14). Consistently, the present data
demonstrated that NGR1 decreased the mRNA and protein levels of
SphK1 and inhibited the phosphorylation level of NF-κB p65. The
present study used 10 µM SKI–II as an effective SphK1 inhibitor
(23,24). It demonstrated that pharmacological
inhibition of SphK1 markedly decreased apoptosis and decreased the
ratio of p-p65/p65 in PC12 cells. A prior study of the
pharmacokinetics of SKI–II suggested a pro-inflammatory effect in
atherosclerosis-prone mice (59).
However, to the best of our knowledge, the role of the
SphK1-triggered inflammatory signaling pathway in Aβ toxicity has
not been fully elucidated. In the present study, pharmacological
inhibition by SKI–II administration and siRNA-mediated SphK1
knockdown were performed in PC12 cells. Notably, the present study
revealed significant alleviation of Aβ25-35-induced
apoptosis and the NF-κB inflammatory response when SphK1 was
inhibited or knocked down. However, the underlying mechanism of
SphK1 signaling appears to be complicated and controversial in Aβ
neurotoxicity (24). In
APP-transfected PC12 cells, the presence of endogenous Aβ peptides
led to a reduction in SphK1 activity, and treatment with SKI–II
decreased cell viability (24). In
addition, a reduction of SphK1 activity was observed in the neurons
of patients with AD and mice (60), while COX2 acetylated by the
activated SphK1 signaling contributed to the resolution of
neuroinflammation and Aβ phagocytosis by microglia in AD (15). Furthermore, glial cells, mainly
microglia, and astrocytes are involved in the pathogenesis of AD by
regulating the phagocytosis of Aβ proteins and the release of
pro-inflammatory cytokines (61).
Therefore, further investigations involving the concrete mechanism
regarding NGR1 and Aβ25-35-activated neuroinflammation
and subsequent glial cell activation via SphK1 signaling are
required. Notably, the increased mRNA expression levels of MMP-3
and MMP-9 following treatment with Aβ25-35 were
decreased when SphK1 was inhibited or knocked down. Although few
studies have focused on the role of SphK1 in
Aβ25-35-related neurotoxicity via the regulation of
MMP-3 and MMP-9, the activity of S1P, a downstream product of
SphK1, elevated the levels of MMP-3 and subsequently promoted the
progression of osteoarthritis (62). In addition, N,
N-dimethylsphingosine, a potent SphK1 inhibitor, also decreased the
levels of MMP-9 and other pro-inflammatory cytokines in the
cellular junctions between Jurkat-U937 cells and rheumatoid
arthritis human peripheral blood mononuclear cells (63). Therefore, SphK1-regualted MMP-3 and
MMP-9 may be a potential mechanism for investigating
Aβ25-35-related neurotoxicity and the inflammatory
response. Notably, the inhibition of SphK1 in PC12 cells did not
result in any alterations in the mRNA expression levels of MMP-3 or
MMP-9, nor did it affect the ratio of p-p65/p65 compared with those
of cells treated with NGR1 and Aβ25-35. These findings
suggested that the SphK1-mediated activity of MMP-3 and MMP-9 may
serve a role in the NGR1-related neuroprotective effect on
Aβ25-35-induced neurotoxicity and inflammation.
Taken together, the present study revealed that
NGR1 supplementation increased the viability and protected PC12
cells against Aβ25-35-induced apoptosis in a
dose-dependent manner. Furthermore, NGR1 treatment alleviated the
Aβ25-35-activated inflammatory response by inhibiting
SphK1-mediated NF-κB signaling pathway activation. These findings
highlighted the neuroprotective role of NGR1 in
Aβ25-35-related neurological impairment and provided a
theoretical basis for further understanding the mechanism of NGR1
in Aβ neurotoxicity.
However, a major limitation of the present study
was its focus on detecting changes of apoptosis markers at the gene
level without considering alterations at the protein level, which
could have been detected by western blotting. Therefore, further
investigations involving protein analyses are required to confirm
the validity of these results. Further studies using an in
vivo AD model and corresponding primary neuron cells are
required to identify the exact role and mechanism of the SphK1
signaling in the NGR1-mediated anti-inflammatory effects during the
progression of AD.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Science and Technology
Program of Zhejiang Traditional Chinese Medicine (grant no.
2014ZB077), Hangzhou Science and Technology development program
(grant no. 20150733Q13), Zhejiang Provincial Key Laboratory of
Traditional Chinese Medicine for the Prevention and Treatment of
Major Chronic Diseases of the Elderly, and the Construction Fund of
Key Medical Disciplines of Hangzhou (grant no. OO20200055).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
XW, BL, XY, YZ, KW and YG collected samples, and
acquired and analyzed data. XW wrote the manuscript. XW and YG
contributed to the study concept, and drafted and revised the
manuscript. XW and YG confirm the authenticity of all the raw data.
All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Weintraub S, Karpouzian-Rogers T, Peipert
JD, Nowinski C, Slotkin J, Wortman K, Ho E, Rogalski E, Carlsson C,
Giordani B, et al: ARMADA: Assessing reliable measurement in
Alzheimer's disease and cognitive aging project methods. Alzheimers
Dement. 18:1449–1460. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
GBD 2019 Dementia Forecasting
Collaborators, . Estimation of the global prevalence of dementia in
2019 and forecasted prevalence in 2050: An analysis for the global
burden of disease study 2019. Lancet Public Health. 7:e105–e125.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zetterberg H and Mattsson N: Understanding
the cause of sporadic Alzheimer's disease. Expert Rev Neurother.
14:621–630. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Leng F and Edison P: Neuroinflammation and
microglial activation in Alzheimer disease: Where do we go from
here? Nat Rev Neurol. 17:157–172. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Del Bo R, Angeretti N, Lucca E, De Simoni
MG and Forloni G: Reciprocal control of inflammatory cytokines,
IL-1 and IL-6, and beta-amyloid production in cultures. Neurosci
Lett. 188:70–74. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Akiyama H, Barger S, Barnum S, Bradt B,
Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich
BL, et al: Inflammation and Alzheimer's disease. Neurobiol Aging.
21:383–421. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Busche MA and Hyman BT: Synergy between
amyloid-β and tau in Alzheimer's disease. Nat Neurosci.
23:1183–1193. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Haass C and Selkoe D: If amyloid drives
Alzheimer disease, why have anti-amyloid therapies not yet slowed
cognitive decline? PLoS Biol. 20:e30016942022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thakur S, Dhapola R, Sarma P, Medhi B and
Reddy DH: Neuroinflammation in Alzheimer's disease: Current
progress in molecular signaling and therapeutics. Inflammation.
46:1–17. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hofmann LP, Ren S, Schwalm S,
Pfeilschifter J and Huwiler A: Sphingosine kinase 1 and 2 regulate
the capacity of mesangial cells to resist apoptotic stimuli in an
opposing manner. Biol Chem. 389:1399–1407. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kunkel GT, Maceyka M, Milstien S and
Spiegel S: Targeting the sphingosine-1-phosphate axis in cancer,
inflammation and beyond. Nat Rev Drug Discov. 12:688–702. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Spiegel S and Milstien S:
Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat Rev Mol
Cell Biol. 4:397–407. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Alvarez SE, Harikumar KB, Hait NC,
Allegood J, Strub GM, Kim EY, Maceyka M, Jiang H, Luo C, Kordula T,
et al: Sphingosine-1-phosphate is a missing cofactor for the E3
ubiquitin ligase TRAF2. Nature. 465:1084–1088. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang C, Xu T, Lachance BB, Zhong X, Shen
G, Xu T, Tang C and Jia X: Critical roles of sphingosine kinase 1
in the regulation of neuroinflammation and neuronal injury after
spinal cord injury. J Neuroinflammation. 18:502021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee JY, Han SH, Park MH, Song IS, Choi MK,
Yu E, Park CM, Kim HJ, Kim SH, Schuchman EH, et al: N-AS-triggered
SPMs are direct regulators of microglia in a model of Alzheimer's
disease. Nat Commun. 11:23582020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu H, Lu X, Hu Y and Fan X: Chemical
constituents of Panax ginseng and Panax notoginseng explain
why they differ in therapeutic efficacy. Pharmacol Res.
161:1052632020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chang D, Liu J, Bilinski K, Xu L, Steiner
GZ, Seto SW and Bensoussan A: Herbal medicine for the treatment of
vascular dementia: An overview of scientific evidence. Evid Based
Complement Alternat Med. 2016:72936262016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Feng L, Han F, Zhou L, Wu S, Du Y, Zhang
D, Zhang C and Gao Y: Efficacy and safety of Panax notoginseng
saponins (Xueshuantong) in patients with acute ischemic stroke
(EXPECT) trial: Rationale and design. Front Pharmacol.
12:6489212021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen M, Zhou SQ, Liu L, Wen YX and Chen
LB: Notoginsenoside R1 alleviates the inflammation of
osteoarthritis by activating the Nrf2/HO-1 signalling pathway in
vitro and in vivo. J Funct Foods. 85:1046662021. View Article : Google Scholar
|
|
20
|
Wang M, Liu H, Xu L, Li M and Zhao M: The
protective effect of notoginsenoside R1 on isoflurane-induced
neurological impairment in the rats via regulating miR-29a
expression and neuroinflammation. Neuroimmunomodulation. 29:70–76.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu T, Li S, Liang WQ, Li SS, Lu MN, Chen
B, Zhang L, Mao R, Ding WH, Gao WW, et al: Notoginsenoside
R1-induced neuronal repair in models of Alzheimer disease is
associated with an alteration in neuronal hyperexcitability, which
is regulated by Nav. Front Cell Neurosci. 14:2802020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang X, Li B, Yu X, Zhou Y and Gao Y: The
neuroprotective effect of GM-1 ganglioside on the
amyloid-beta-induced oxidative stress in PC-12 cells mediated by
Nrf-2/ARE signaling pathway. Neurochem Res. 47:2405–2415. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cieślik M, Czapski GA and Strosznajder JB:
The molecular mechanism of amyloid β42 peptide toxicity: The role
of sphingosine kinase-1 and mitochondrial sirtuins. PLoS One.
10:e01371932015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gassowska M, Cieslik M, Wilkaniec A and
Strosznajder JB: Sphingosine kinases/sphingosine-1-phosphate and
death signalling in APP-transfected cells. Neurochem Res.
39:645–652. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang X, Wei L, Zhu J, He B, Kong B, Jin Y
and Fu Z: Tetrabromoethylcyclohexane (TBECH) exhibits
immunotoxicity in murine macrophages. Environ Toxicol. 35:159–166.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ni L, Zhuge F, Yang S, Ma L, Zheng A, Zhao
Y, Hu L, Fu Z and Ni Y: Hydrolyzed chicken meat extract attenuates
neuroinflammation and cognitive impairment in middle-aged mouse by
regulating M1/M2 microglial polarization. J Agric Food Chem.
69:9800–9812. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ni Y, Ni L, Ma L, Wang Z, Zhao Y, Hu L,
Zheng L and Fu Z: Neuroprotective effects of ProBeptigen/CMI-168 on
aging-induced cognitive decline and neuroinflammation in mice: A
comparison with essence of chicken. Acta Biochim Biophys Sin
(Shanghai). 53:419–429. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ma L, Ni Y, Wang Z, Tu W, Ni L, Zhuge F,
Zheng A, Hu L, Zhao Y, Zheng L and Fu Z: Spermidine improves gut
barrier integrity and gut microbiota function in diet-induced obese
mice. Gut Microbes. 12:1–19. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qiao L, Chen Y, Dou X, Song X and Xu C:
Biogenic selenium nanoparticles attenuate Aβ25-35-induced toxicity
in PC12 cells via Akt/CREB/BDNF signaling pathway. Neurotox Res.
40:1869–1881. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Meng XL, Liu SY, Xue JS, Gou JM, Wang D,
Liu HS, Chen CL and Xu CB: Protective effects of liensinine,
isoliensinine, and neferine on PC12 cells injured by amyloid-β. J
Food Biochem. 46:e143032022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li LX, Liu MY, Jiang X, Xia ZH, Wang YX,
An D, Wang HG, Heng B and Liu YQ: Metformin inhibits
Aβ25-35-induced apoptotic cell death in SH-SY5Y cells.
Basic Clin Pharmacol Toxicol. 125:439–449. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Avni D, Harikumar KB, Sanyal AJ and
Spiegel S: Deletion or inhibition of SphK1 mitigates fulminant
hepatic failure by suppressing TNFα-dependent inflammation and
apoptosis. FASEB J. 35:e214152021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Etemadi N, Chopin M, Anderton H, Tanzer
MC, Rickard JA, Abeysekera W, Hall C, Spall SK, Wang B, Xiong Y, et
al: TRAF2 regulates TNF and NF-κB signalling to suppress apoptosis
and skin inflammation independently of Sphingosine kinase 1. Elife.
4:e105922015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang XX, Tan MS, Yu JT and Tan L: Matrix
metalloproteinases and their multiple roles in Alzheimer's disease.
Biomed Res Int. 2014:9086362014.PubMed/NCBI
|
|
36
|
LaFerla FM, Tinkle BT, Bieberich CJ,
Haudenschild CC and Jay G: The Alzheimer's A beta peptide induces
neurodegeneration and apoptotic cell death in transgenic mice. Nat
Genet. 9:21–30. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Viola KL and Klein WL: Amyloid β oligomers
in Alzheimer's disease pathogenesis, treatment, and diagnosis. Acta
Neuropathol. 129:183–206. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Masliah E, Alford M, Adame A, Rockenstein
E, Galasko D, Salmon D, Hansen LA and Thal LJ: Abeta1-42 promotes
cholinergic sprouting in patients with AD and Lewy body variant of
AD. Neurology. 61:206–211. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kubo T, Nishimura S, Kumagae Y and Kaneko
I: In vivo conversion of racemized beta-amyloid [(D-Ser 26)A beta
1–40] to truncated and toxic fragments [(D-Ser 26)A beta 25–35/40]
and fragment presence in the brains of Alzheimer's patients. J
Neurosci Res. 70:474–483. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Frozza RL, Horn AP, Hoppe JB, Simão F,
Gerhardt D, Comiran RA and Salbego CG: A comparative study of
beta-amyloid peptides Abeta1-42 and Abeta25-35 toxicity in
organotypic hippocampal slice cultures. Neurochem Res. 34:295–303.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Varadarajan S, Kanski J, Aksenova M,
Lauderback C and Butterfield DA: Different mechanisms of oxidative
stress and neurotoxicity for Alzheimer's A beta(1–42) and A
beta(25–35). J Am Chem Soc. 123:5625–5631. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu X, Ganguly P, Jin Y, Jhatro MJ, Shea
JE, Buratto SK and Bowers MT: Tachykinin neuropeptides and amyloid
β (25–35) assembly: Friend or foe? J Am Chem Soc. 144:14614–14626.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wiatrak B, Kubis-Kubiak A, Piwowar A and
Barg E: PC12 cell line: Cell types, coating of culture vessels,
differentiation and other culture conditions. Cells. 9:9582020.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gao X, Han Z, Huang C, Lei H, Li G, Chen
L, Feng D, Zhou Z, Shi Q, Cheng L and Zhou X: An anti-inflammatory
and neuroprotective biomimetic nanoplatform for repairing spinal
cord injury. Bioact Mater. 18:569–582. 2022.PubMed/NCBI
|
|
45
|
Changhong K, Peng Y, Yuan Z and Cai J:
Ginsenoside Rb1 protected PC12 cells from
Aβ25-35-induced cytotoxicity via PPARγ activation and
cholesterol reduction. Eur J Pharmacol. 893:1738352021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Geng XD, Wang WW, Feng Z, Liu R, Cheng XL,
Shen WJ, Dong ZY, Cai GY, Chen XM, Hong Q and Wu D: Identification
of key genes and pathways in diabetic nephropathy by bioinformatics
analysis. J Diabetes Investig. 10:972–984. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gao JM, Zhang X, Shu GT, Chen NN, Zhang
JY, Xu F, Li F, Liu YG, Wei Y, He YQ, et al: Trilobatin rescues
cognitive impairment of Alzheimer's disease by targeting HMGB1
through mediating SIRT3/SOD2 signaling pathway. Acta Pharmacol Sin.
43:2482–2494. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gao J, Yao M, Zhang W, Yang B, Yuan G, Liu
JX and Zhang Y: Panax notoginseng saponins alleviates
inflammation induced by microglial activation and protects against
ischemic brain injury via inhibiting HIF-1α/PKM2/STAT3 signaling.
Biomed Pharmacother. 155:1134792022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu Y, Liu T, Ding K, Liu Z, Li Y, He T,
Zhang W, Fan Y, Ma W, Cui L and Song X: Phospholipase Cγ2 signaling
cascade contribute to the antiplatelet effect of notoginsenoside
Fc. Front Pharmacol. 9:12932018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Du Y, Fu M, Wang YT and Dong Z:
Neuroprotective effects of ginsenoside Rf on amyloid-β-induced
neurotoxicity in vitro and in vivo. J Alzheimers Dis. 64:309–322.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yan S, Li Z, Li H, Arancio O and Zhang W:
Notoginsenoside R1 increases neuronal excitability and ameliorates
synaptic and memory dysfunction following amyloid elevation. Sci
Rep. 4:63522014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ma B, Meng X, Wang J, Sun J, Ren X, Qin M,
Sun J, Sun G and Sun X: Notoginsenoside R1 attenuates
amyloid-β-induced damage in neurons by inhibiting reactive oxygen
species and modulating MAPK activation. Int Immunopharmacol.
22:151–159. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fricker M, Tolkovsky AM, Borutaite V,
Coleman M and Brown GC: Neuronal cell death. Physiol Rev.
98:813–880. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yoshiyama Y, Asahina M and Hattori T:
Selective distribution of matrix metalloproteinase-3 (MMP-3) in
Alzheimer's disease brain. Acta Neuropathol. 99:91–95. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Backstrom JR, Lim GP, Cullen MJ and Tökés
ZA: Matrix metalloproteinase-9 (MMP-9) is synthesized in neurons of
the human hippocampus and is capable of degrading the amyloid-beta
peptide (1–40). J Neurosci. 16:7910–7919. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ito S, Kimura K, Haneda M, Ishida Y,
Sawada M and Isobe KI: Induction of matrix metalloproteinases
(MMP3, MMP12 and MMP13) expression in the microglia by amyloid-beta
stimulation via the PI3K/Akt pathway. Exp Gerontol. 42:532–537.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lorenzl S, Buerger K, Hampel H and Beal
MF: Profiles of matrix metalloproteinases and their inhibitors in
plasma of patients with dementia. Int Psychogeriatr. 20:67–76.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Nübling G, Levin J, Bader B, Israel L,
Bötzel K, Lorenzl S and Giese A: Limited cleavage of tau with
matrix-metalloproteinase MMP-9, but not MMP-3, enhances tau
oligomer formation. Exp Neurol. 237:470–476. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Poti F, Ceglarek U, Burkhardt R, Simoni M
and Nofer JR: SKI–II-a sphingosine kinase 1 inhibitor-exacerbates
atherosclerosis in low-density lipoprotein receptor-deficient
(LDL-R-/-) mice on high cholesterol diet. Atherosclerosis.
240:212–215. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lee JY, Han SH, Park MH, Baek B, Song IS,
Choi MK, Takuwa Y, Ryu H, Kim SH, He X, et al: Neuronal SphK1
acetylates COX2 and contributes to pathogenesis in a model of
Alzheimer's disease. Nat Commun. 9:14792018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Uddin MS and Lim LW: Glial cells in
Alzheimer's disease: From neuropathological changes to therapeutic
implications. Ageing Res Rev. 78:1016222022. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Cherifi C, Latourte A, Vettorazzi S,
Tuckermann J, Provot S, Ea HK, Ledoux A, Casas J, Cuvillier O,
Richette P, et al: Inhibition of sphingosine 1-phosphate protects
mice against chondrocyte catabolism and osteoarthritis.
Osteoarthritis Cartilage. 29:1335–1345. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lai WQ, Irwan AW, Goh HH, Howe HS, Yu DT,
Valle-Oñate R, McInnes IB, Melendez AJ and Leung BP:
Anti-inflammatory effects of sphingosine kinase modulation in
inflammatory arthritis. J Immunol. 181:8010–8017. 2008. View Article : Google Scholar : PubMed/NCBI
|