Introduction
Chronic obstructive pulmonary disorder (COPD) is a
common, preventable, and treatable chronic disease characterized by
persistent respiratory symptoms and airflow limitation. It is
associated with an enhanced chronic inflammatory response of the
airways and the lungs to a variety of noxious particles or gases
(1,2) According to the 2019 Global Burden of
Disease Study report, 3.3 million people died from COPD and there
were 74.4 million disability-adjusted life years (DALYs) lost to
COPD, and it is thus considered a global major public health
problem (3). The etiology of COPD
is well established to be influenced by environmental factors,
especially smoking, and genetics (4–6). In
China, given it has the world's largest number of smokers, combined
with an ever-aging population, the COPD cases in China account for
25% of global COPD cases and it has thus become a significant
economic burden (7). COPD is
associated with chronic inflammation that predominantly affects the
lung parenchyma and peripheral airways (8). However, the exact pathogenesis of
COPD remains unclear. Therefore, an improved understanding of the
inflammatory responses is essential for the development of COPD
therapeutics and improved clinical treatment.
MicroRNAs (miRNAs/miRs) are endogenous non-coding
RNA molecules that regulate gene expression by binding to the
3′-untranslated region (3′-UTR) of multiple target mRNAs for
degradation or translational repression (9). miRNAs are involved in a wide variety
of biological processes including inflammation, cell
differentiation, cell proliferation, and cell apoptosis (10). Recent studies have demonstrated
that several miRNAs play an important role in positive and negative
regulation of the inflammatory response and participate in various
regulatory network motifs in respiratory disease (11–13).
Numerous studies have revealed that miRNAs have a role in the
pathogenesis of COPD via critical molecular pathways and have
become valuable biomarkers for the diagnosis and prognosis of COPD
(14–17). In a previous study, a significant
difference in the expression of miR-186-5p was found between
healthy controls and COPD patients (18). miR-186-5p has been shown to be
involved in the regulation of the inflammatory responses of various
diseases. For example, miR-186-5p knockdown repressed
oxygen-glucose deprivation/reperfusion (OGD/R)-induced pyroptosis
and suppressed lactate dehydrogenase and inflammatory cytokine
release (19). iR-186-5p
inhibition abolished the effects of SOX2-OT blocking on the
inflammatory responses, proliferation, and apoptosis of
OGD/R-challenged H2C9 cells (20).
Moreover, miR-186-5p inhibitor reduced the inflammatory factors and
oxidative stress in BV2 treated with lipopolysaccharide (LPS) and
reduced apoptosis (21). Li et
al (22) reported that
miR-186-5p may regulate COPD dysfunction. However, the relevance of
miR-186-5p in COPD and the underlying molecular mechanisms are
unclear. Therefore, it is necessary to develop novel therapeutic
methods and targets by understanding the mechanism of COPD
pathogenesis mediated by miR-186-5p.
HIF-1α is an important activator of inflammatory
responses. Increased serum levels of HIF-1α are associated
with the progression of COPD (23). The role of HIF-1α in the
development and progression of COPD has also been demonstrated
(24). Furthermore, NF-κB, the
downstream target gene of HIF, is a central regulator of immunity
and inflammation and is a key target in COPD therapy (25). In an osteosarcoma study,
HIF-1α was identified as a downstream target of miR-186-5p,
where it regulated osteosarcoma progression (26). However, the involvement of
miR-186-5p in relation to HIF-1α in controlling inflammation
of COPD remains unclear. Therefore, this investigation attempted to
evaluate the association between miR-186-5p and HIF-1α and
their roles in inflammation during COPD. It was found that
interfering with miR-186-5p reduced LPS-induced BEAS-2B cell
proliferation and promoted cell apoptosis, and miR-186-5p regulated
the inflammatory response of COPD by targeting HIF-1α. These
findings provide novel insights for further investigation of the
pathogenesis of COPD and may eventually contribute to novel
treatments for COPD.
Materials and methods
Cell culture and establishment of the
COPD inflammation model
Human bronchial epithelial cells (BEAS-2B),
purchased from Procell Life Science & Technology Co., Ltd.,
were incubated in DMEM (HyClone; Cytiva) with 10% FBS (HyClone;
Cytiva), 100 IU/ml penicillin (Gibco; Thermo Fisher Scientific,
Inc.), and 100 µg/ml streptomycin (Gibco; Thermo Fisher Scientific,
Inc.). The cells were cultured in an incubator (Likang Biomedical
Technology Co., Ltd.) at 37°C, with 5% CO2. BEAS-2B
cells were induced with LPS (MilliporeSigma) (0, 1, 2, 5, 10, 20,
or 40 mg/l) for 24 h to establish an in vitro inflammation
model of COPD, and cells treated with 0.1% DMSO were used as a
control. The cell inhibition rate and half maximal inhibitory
concentration (IC50) of LPS were detected and calculated
using a Cell Counting Kit 8 (CCK-8) assay to evaluate the COPD
inflammation model. Moreover, ELISA was used to detect the
expression levels of inflammatory factors IL-6 and
TNF-α in the supernatant of cells.
Cell transfection
The miR-186-5p mimics, miR-186-5p inhibitor, and
corresponding negative control (NCs) were synthesized by Suzhou
Jima Gene Co. Ltd. Cells were plated into a 6-well plate and
allowed to adhere. When the cell confluency reached 70–80%,
Lipofectamine® 2000 (Procell Life Science & Technology Co.,
Ltd.) was used for transfection according to the manufacturer's
protocol. The sequences of the miR-186-5p mimics, negative control
of mimics (mimics-NC), miR-186-5p inhibitor and negative control of
inhibitor (inhibitor-NC) are as follows: miR-186-5p mimics,
5′-CAAAGAAUUCUCCUUUUGGGCU-3′; mimics-NC,
5′-CGAUCGCAUCAGCAUCGAUUGC-3′; miR-186-5p inhibitor,
5′-AGCCCAAAAGGAGAAUUCUUUG-3′; and inhibitor-NC:
5′-CAGUACUUUUGUGUAGUACAA-3′.
Reverse transcription-quantitative PCR
(RT-qPCR)
RT-qPCR was used to measure the expression of
miR-186-5p, HIF-1α, IL-6, and TNF-α. Total RNA was
extracted from cells using TRIzol® according to the manufacturer's
instructions. RNA was reverse transcribed into cDNA using a
PrimeScript RT kit (Takara Bio, Inc.) according to the
manufacturer's protocol. qPCR was performed using a SYBR Premix Ex
Taq II kit (Takara Bio, Inc.). The expression levels of related
genes were detected on the ABI 7500 (Applied Biosystems; Thermo
Fisher Scientific, Inc.). Relative quantitative values were
calculated using the 2−ΔΔCq method (27). The relative expression was
normalized to that of U6 or GAPDH. The primers used in this study
were provided by Beijing Aogukang Biotechnology Co., Ltd. The
sequences of the PCR primers were: HIF-1α forward,
5′-GCCTCTGTGATGAGGCTTACC-3′ and reverse,
5′-CAGTGCAATACCTTCCATGTTGC-3′; IL-6 forward,
5′-CTCCTTCTCCACAAGCGCC-3′ and reverse, 5′-GATGCCGTCGAGGATGTACC-3′;
TNF-α forward, 5′-TGTAGCCCATGTTGTAGCAAACC-3′ and reverse,
5′-TGAGGTACAGGCCCTCTGAT; miR-186-5p forward,
5′-CGCCAAAGAATTCTCCTTTTGGGCT-3′ and reverse,
5′-AGCCCAAAAGGAGAATTCTTTGGCG-3′; and U6 forward,
5′-TGGAACGCTTCACGAATTTGCG-3′ and reverse,
5′-GGAACGATACAGAGAAGATTAGC-3′; GAPDH forward,
5′-ATCACTGCCACCCAGAAGAC3′ and reverse,
5′-TTTCTAGACGGCAGGTCAGG-3′.
CCK-8 assay
Cell proliferation was evaluated using a CCK-8 assay
(Biyuntian Co., Ltd.) according to the manufacturer's protocol. A
total of 5×103 cells per well were resuspended and
seeded in a 96-well plate and incubated for 24 h. After culture for
24, 48, 72, and 96 h, respectively, 10 µl CCK-8 solution was added
to each well and incubated for 4 h in the humidified incubator. An
Epoch microplate reader was used to detect the absorbance at 450 nm
(BioTek Instruments, Inc.).
Cell apoptosis assay
Cell apoptosis was detected using a Muse Annexin V
& Dead Cell Kit (MilliporeSigma). Stably transfected cells and
empty vector-transfected control cells were collected. Cells were
washed in PBS and resuspended in media supplemented with 1% FBS.
Next, 100 µl Muse Annexin V & Dead Cell Reagent was added to
the cell suspension, gently mixed, and incubated in the dark for 20
min at room temperature. The percentage of apoptotic cells was
determined using the Muse Cell Analyzer (Luminex Corp), according
to the manufacturer's instructions.
Cell cycle assay
Stably transfected cells and empty
vector-transfected control cells were collected. The adherent cells
were digested with pancreatin, and the cells were re-suspended in
DMEM. Next, cells were centrifuged at 2,000 × g for 5 min, and the
supernatant was discarded. The cells were washed twice with PBS for
5 min and fixed with pre-cooled 70% ethanol at 4°C overnight. The
following day, cells were washed with PBS twice, and then incubated
with propidium iodide (PI) in the dark for 30 min. PI staining was
detected using a flow cytometer (Gallios, Beckman Coulter, Inc.)
and analyzed using FlowJo (version 10.7.1; FlowJo LLC).
Western blotting
Total proteins from the cells were extracted using
RIPA lysis buffer (CWBIO), and the protein concentrations were
measured using a BCA Protein Assay Kit (Biyuntian Co., Ltd.).
Protein samples were separated using a 10% SDS gel by SDS-PAGE and
transferred to PVDF membranes. Subsequently, the membranes were
blocked using 5% non-fat milk and incubated with primary antibodies
against HIF-1α (1:500; Wanleibio Co., Ltd.; cat. no. WL01607),
p-p65 (1:1,000; Affinity Bioscience; cat. no. AF2006), p65
(1:1,000; ProteinTech Group, Inc.; cat. no. 66535-1-1g), and
β-actin (1:5,000; ProteinTech Group, Inc.; cat. no. 66009-1-lg)
overnight at 4°C. Subsequently, membranes were incubated with a
goat anti-rabbit horseradish peroxidase-conjugated IgG secondary
antibody (1:10,000; Zen Bio; cat. no. 511203) for 1 h at room
temperature. Signals were visualized using enhanced
chemiluminescence.
Cytokine quantification
ELISA kits (cat nos. JL10208 and JL14113; Shanghai
Jianglai Biotechnology Co., Ltd.) were used to measure the levels
of TNF-α and IL-6 in the supernatant of LPS-induced BEAS-2B cell
culture medium, according to the manufacturer's protocol.
Dual-luciferase reporter gene
assay
The potential target of miR-186-5p and HIF-1α
was predicted using TargetScan (http://www.targetscan.org). A dual-luciferase reporter
assay was performed to confirm the predicted interactions. The
region that contained the miR-186-5p binding site on HIF-1α
was inserted into the luciferase pGL3 reporter vector. This was
followed by co-transfection with luciferase plasmids and with
miR-186b-5p mimics, inhibitor, mimics-NC, or inhibitor-NC using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.), to confirm
binding between miR-186b-5p and HIF-1α. After 48 h, the
luciferase activity was measured using a Dual-Luciferase Reporter
Assay System (Promega Corporation).
Statistical analysis
All statistical tests were performed using GraphPad
Prism version 8.0 (GraphPad Software Inc.). Data are presented as
the mean ± SD of three repeats. A Student's t-test (unpaired) was
used to compare differences between two groups. A one-way ANOVA was
used to evaluate the statistical significance between multiple
groups, and a Tukey's Honey Significant Difference post hoc test
was used to identify which specific groups exhibited significant
differences. A two-sided P<0.05 was considered to indicate a
statistically significant difference.
Results
COPD inflammation model
To establish an in vitro model of COPD,
BEAS-2B cells were exposed to increasing concentrations of LPS to
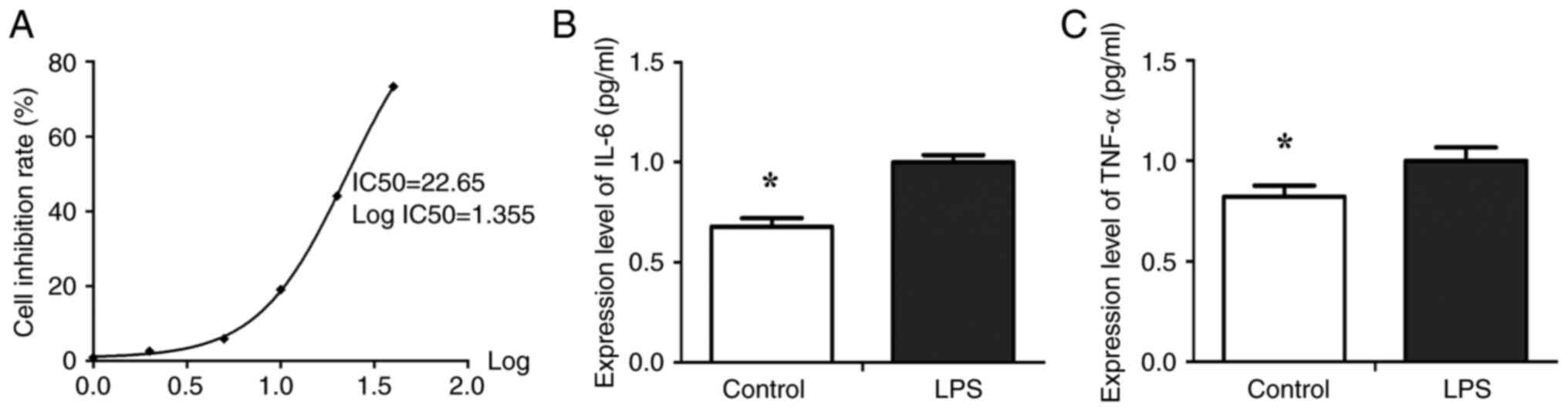
determine the optimum concentration. The results showed that
exposure to varying concentrations of LPS, namely 5 mg/l (5.87%),
10 mg/l (19.03%), 20 mg/l (44.06%), and 40 mg/l (73.29%), resulted
in significant inhibition of cell proliferation when compared to
the control group treated with DMSO (P<0.05, Table I). The IC50 value for
LPS was 22.65±3.03 mg/l (Fig. 1A,
P<0.05). LPS treatment resulted in a significant
concentration-dependent decrease in the number of viable cells
starting at a concentration of 10 mg/l with an inhibition rate of
19% (P<0.01). To verify whether 10 mg/l LPS induced cellular
inflammation, the expression levels of inflammatory factors IL-6
and TNF-α in the supernatant of cells were determined using ELISA.
The results indicated that the expression of IL-6 (Fig. 1B) and TNF-α (Fig. 1B) in the LPS-induced BEAS-2B cells
were significantly higher than that in the control group
(P<0.05). Therefore, in the subsequent experiments, cells were
pretreated with 10 mg/l LPS for 24 h.
 | Table I.Proliferation inhibition rate of
BEAS-2B cells following treatment with different concentrations of
LPS for 24 h |
Table I.
Proliferation inhibition rate of
BEAS-2B cells following treatment with different concentrations of
LPS for 24 h
| Group | Inhibitory rate of
proliferation (%, Mean ± SD) |
|---|
| 0.1% DMSO | 1.55±0.76 |
| LPS, mg/l |
|
| 1 | 0.84±0.08 |
| 2 | 2.58±1.25 |
| 5 |
5.87±2.14a |
| 10 |
19.03±2.36b |
| 20 |
44.06±6.43b |
| 40 |
73.29±4.77b |
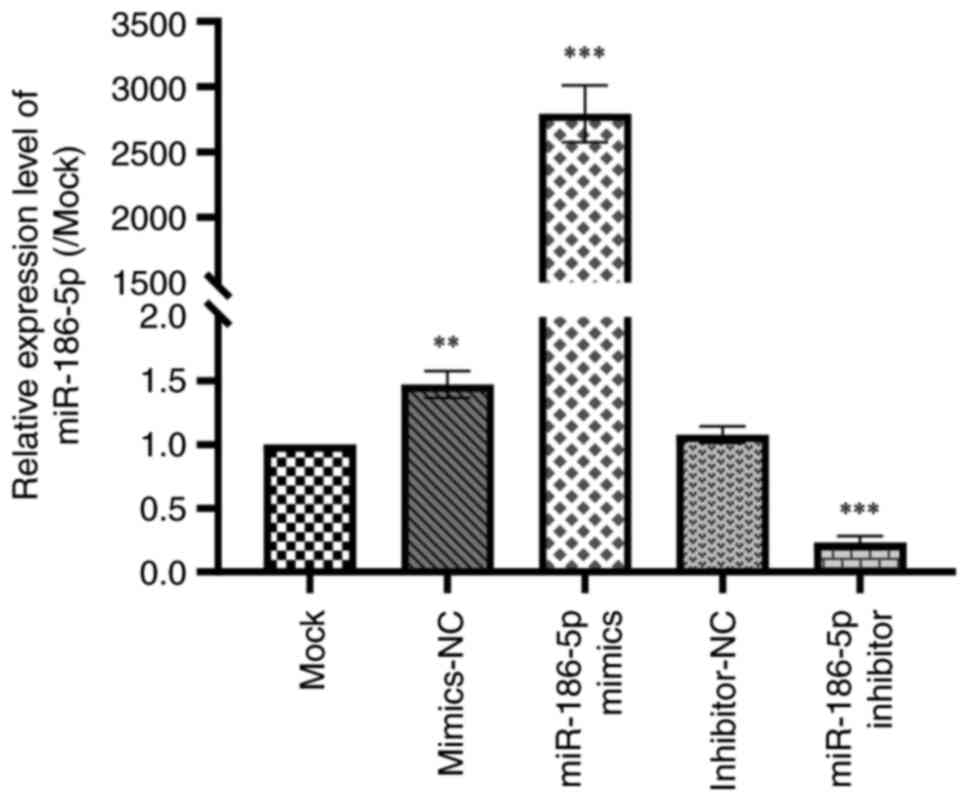
Transfection efficacy
To evaluate the impact of miRNA-186-5p on COPD
progression, miRNA-186-5p mimic or miRNA-186-5p inhibitor were
transfected into the LPS-induced COPD cells. The transfection
efficacies of miRNA-186-5p mimic and miRNA-186-5p inhibitor were
examined in LPS-induced BEAS-2B cells (Fig. 2). The results showed that
transfection of miRNA-186-5p mimic significantly increased the
expression of miRNA-186-5p in LPS-induced BEAS-2B cells, while
transfection of miRNA-186-5p inhibitor significantly inhibited the
levels of miRNA-186-5p in LPS-induced BEAS-2B cells
(P<0.001).
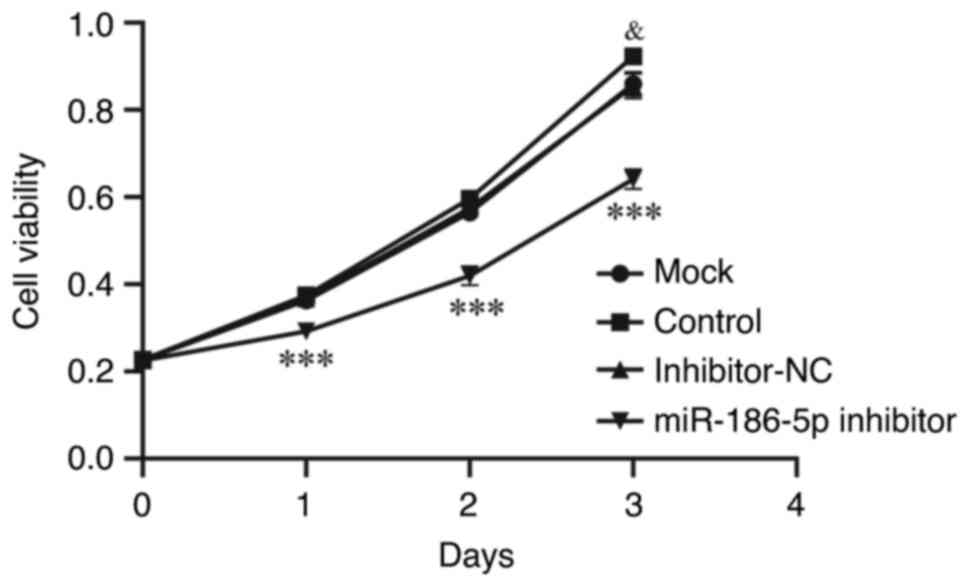
Inhibition of miR-186-5p reduces
proliferation and induces apoptosis in LPS-induced BEAS-2B
cells
To investigate the effect of miR-186-5p on the
proliferative activity of LPS-induced BEAS-2B cells, CCK-8 assays
were performed (Fig. 3). The
results showed that the miRNA-186-5p inhibitor significantly
decreased the viability of LPS-induced BEAS-2B cells between days 0
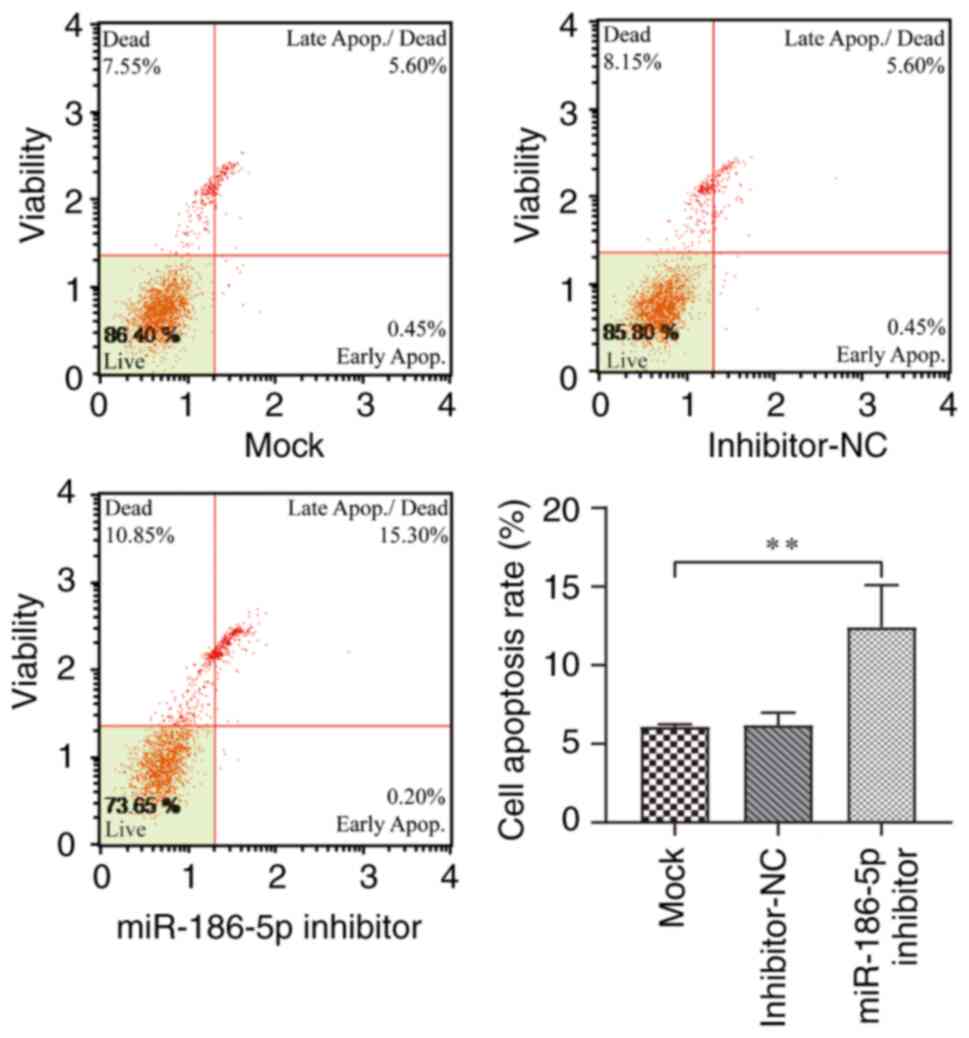
and 3 compared with the mock group (P<0.001). Apoptosis of
LPS-induced BEAS-2B cells was detected by flow cytometry after
transfection for 48 h (Fig. 4).
The findings revealed that the apoptotic rate of LPS-induced
BEAS-2B cells in the miR-186-5p inhibitor group was significantly
higher than that in the mock group (P<0.01). Additionally, cell
cycle distribution was also analyzed. The results of cell cycle
distribution indicated no significant difference between the mock
and miR-186-5p inhibitor groups in the G0/G1, S, and G2/M phase,
respectively (Fig. S1,
P>0.05). These findings suggest that inhibition of miR-186-5p
decreased the proliferation of and induced apoptosis in LPS-induced
BEAS-2B cells but had no effect on the cell cycle.
Role of miR-186-5p in
inflammation
To further evaluate the inflammatory effects of
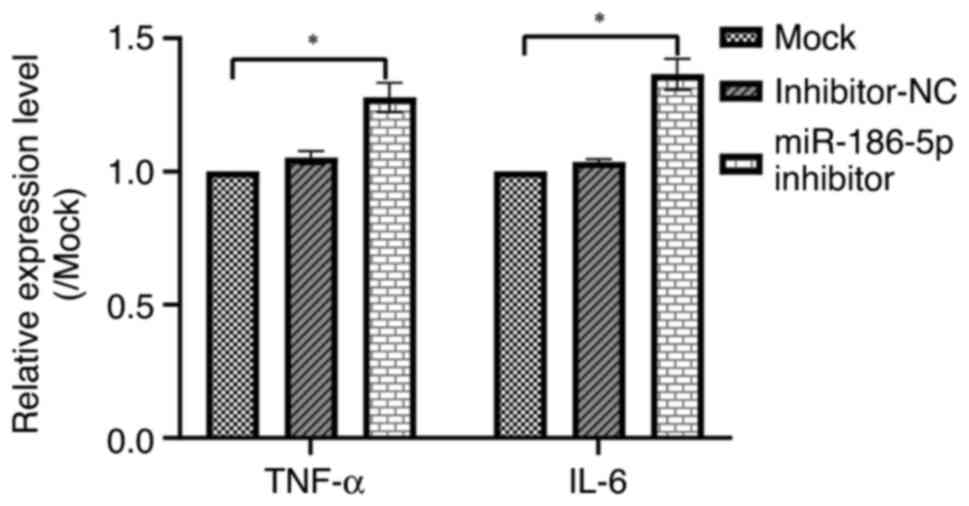
miR-186-5p, the levels of inflammatory cytokines (TNF-α and
IL-6) in LPS-induced BEAS-2B cells. The results indicated
that miR-186-5p inhibitor significantly increased the levels of
TNF-α and IL-6 compared with that in the control
group (Fig. 5, P<0.05).
miR-186-5p targets and regulates
HIF-1α
Predictions from the TargetScan database (http://www.targetscan.org) showed that miR-186-5p
bound to the 3′-UTR of HIF-1α (Fig. 6A). Dual-luciferase reporter gene
assays showed that the relative luciferase activity of the 3′ UTR +
miRNA group was significantly reduced compared with the 3′ UTR-NC +
miRNA group (Fig. 6B, P<0.01),
indicating that HIF-1α was the downstream target of
miRNA-186-5p. Moreover, RT-qPCR and western blotting further
confirmed that miR-186-5p could regulate the expression of
HIF-1α. The results revealed that the relative expression
levels of HIF-1α mRNA were significantly lower in the
control (LPS-induced COPD model) group (P<0.01) and miR-186-5p
mimics group (P<0.05), while the relative expression levels of
HIF-1α mRNA were significantly increased in the miR-186-5p
inhibitor group (P<0.001) compared with the mock group (Fig. 6C). Western blotting results showed
that the relative expression levels of HIF-1α were significantly
decreased in the control group (P<0.01), while the relative
expression levels of HIF-1α were significantly increased in the
miR-186-5p inhibitor group (P<0.001) compared with the mock
group (Fig. 6D). These findings
demonstrate that miR-186-5p could target and regulate the
expression of HIF-1α.
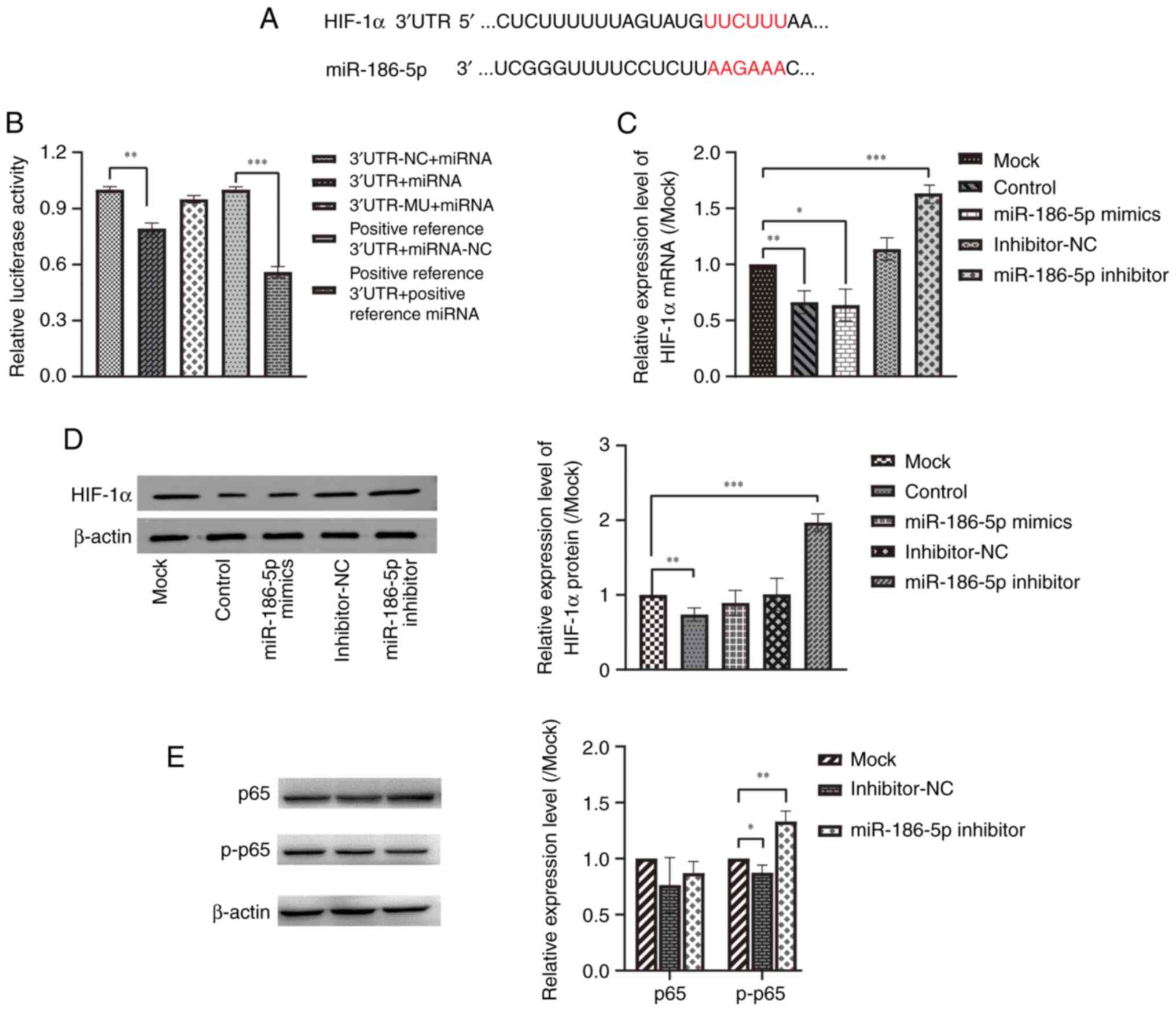 | Figure 6.Effect of miR-186-5p on the
expression of HIF-1α, inflammatory cytokines, and p-p65. (A)
Predicted miR-186-5p binding sites on HIF-1α 3′UTR. The binding
sites are labeled in red letters. (B) Dual-luciferase reporter
assays demonstrated that miR-186-5p bound to HIF-1α. (C) mRNA and
(D) protein expression levels of HIF-1α in BEAS-2B cells treated
with 10 mg/l LPS. (E) p65 and p-p65 protein expression in BEAS-2B
cells treated with 10 mg/l LPS after transfection with miR-486-5p
inhibitor. *P<0.05, **P<0.01, ***P<0.001. Data are
presented as the mean ± SD. Mock, LPS + Lip2000; Control, without
LPS; LPS, lipopolysaccharide; miR, microRNA; UTR, untranslated
region; NC, negative control. |
To investigate whether the downregulation of
miR-186-5p affected the NF-κB inflammatory pathway, the protein
expression levels of p-65, a crucial factor in the NF-κB pathway,
and its activated form, p-p65, were determined. The results
indicated that the miR-186-5p inhibitor group exhibited a
significant increase in the relative expression levels of p-p65
protein (P<0.05) compared to the mock group, while the relative
expression levels of p65 protein remained unchanged (Fig. 6E). These results suggest that the
downregulation of miR-186-5p may enhance the expression of
p-p65.
Discussion
COPD is a globally recognized and prevalent disease,
and although advances in clinical treatment have seen notable
progress, the pathogenesis of COPD remains poorly understood. In
this study, the potential role of miR-186-5p in COPD inflammation
was investigated by inducing BEAS-2B cells with LPS to establish an
in vitro COPD model. The results suggested that miR-186-5p
regulated the inflammatory response of lung epithelial cells
through targeted interactions with HIF-1α in COPD. It was
also shown that interfering with miR-186-5p was associated with
reduced LPS-induced BEAS-2B proliferation and enhanced LPS-induced
BEAS-2B apoptosis.
miR-186-5p has been shown to play an important role
in various diseases. Recent studies have suggested that miR-186 can
regulate cancer cell growth, proliferation, migration, apoptosis,
and other processes, and is associated with a variety of
physiological and pathological processes (28,29).
In our previous study, it was found that the expression of
miR-186-5p was upregulated in COPD compared to the control group
(19). Li et al (22) found, using bioinformatics analysis,
that miR-186-5p may be involved in regulating COPD dysfunction
blocks. The results of the present study found that downregulation
of miR-186-5p inhibited the proliferation of LPS-induced BEAS-2B
cells, promoted apoptosis in these cells, and significantly
increased the levels of inflammatory factors (TNF-α and
IL-6). However, further research is needed to confirm the
impact of miR-186-5p on the inflammatory response in COPD. For
example, in future studies, western blotting will be used to detect
the expression of proliferation-related genes and determine whether
the use of apoptosis inhibitors and necrosis inhibitors will rescue
cell death promoted by miR-186-5p knockdown.
Furthermore, the findings of the present study
suggested that miR-186-5p could target and regulate the expression
of HIF-1α in COPD. Our previous study showed that miRNA-186
was associated with the expression of HIF-1α in COPD
(30). HIF-1α serves as a
key regulator of cellular oxygen homeostasis during the development
of inflammation and various disorders. It activates a wide range of
genes involved in multiple processes, including glycolysis,
angiogenesis, proliferation, migration, autophagy, and apoptosis,
amongst other processes (31).
Several studies have investigated the relationship between
HIF-1α and COPD and found that HIF-1α expression is
increased in COPD patients, resulting in upregulated expression of
inflammatory factors, which is associated with disease severity
(23,24,32).
Furthermore, the HIF-1α signaling pathway has been shown to
be an important signaling pathway that drives COPD progression to
lung cancer (33). The present
study investigated the role of miR-186-5p in regulating
HIF-1α expression and its impact on COPD inflammation.
However, the direct effect of HIF-1α on COPD inflammation
was not confirmed. In addition, HIF inhibitors were not used to
ascertain whether the inflammatory response induced by miR-186-5p
could be rescued. Therefore, in future studies, it is necessary to
investigate the direct impact of HIF-1α on COPD inflammation
and observe whether the inflammatory response induced by miR-186-5p
can be suppressed using HIF inhibitors.
IL-6 can trigger a number of pro-inflammatory
cytokines and chemokines, and has been reported to exhibit
extensive crosstalk with NF-κB at multiple mechanistic levels to
regulate immune processes, as well as promote the development of
COPD by activating the NF-κB signaling pathway (34). The NF-κB signaling pathway is a
vital pro-inflammatory pathway that regulates the levels of
inflammatory factors in COPD patients' bronchial epithelial cells
(25,35). In the present study, the results
showed that miR-186-5p interference increased HIF-1α
expression whilst also upregulating the production of
pro-inflammatory cytokines TNF-α and IL-6 and
considerably increasing the phosphorylation of p65, a key regulator
of the NF-κB signaling pathway. Thus, the findings indicate that
miR-186-5p inhibits HIF-1α, which in turn contributes to the
inflammatory response in COPD. The underlying mechanism may be
associated with the NF-κB signaling pathway in epithelial cells.
However, this study also has the limitation of using a single cell
line to construct the COPD model. Therefore, further studies using
multiple cell lines are needed to validate the findings and assess
the generalizability of the results.
In conclusion, the results of the present study
suggested that miR-186-5p may regulate the inflammatory response of
COPD by targeting HIF-1α through regulating NF-κB signaling,
which could potentially impact the development and progression of
COPD. This discovery provides novel insights for the treatment of
COPD, and future research should further investigate the underlying
mechanism of the interaction between miR-186-5p and HIF-1α,
as well as the specific role of this interaction in COPD
inflammation.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural Science
Foundation of China (No. 81860015 and No. 82160011), and the
National Key Research and Development Program of China
(2018YFC2002304).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YF, JZ and JC wrote the original draft of the
manuscript, and created figures, tables and visual representations
of the data. YZ, RM, BZ and LZ analyzed and interpreted the data.
YF, JZ, JC, YD and TX conceived and designed the study. QL, CH, SL
and LL performed the experiments. YF, YD and TX confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors have declared that they have no conflict
of interest.
References
|
1
|
Christenson SA, Smith BM, Bafadhel M and
Putcha N: Chronic obstructive pulmonary disease. Lancet.
399:2227–2242. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Celli B, Fabbri L, Criner G, Martinez FJ,
Mannino D, Vogelmeier C, Montes de Oca M, Papi A, Sin DD, Han MK
and Agusti A: Definition and nomenclature of chronic obstructive
pulmonary disease: Time for its revision. Am J Respir Crit Care
Med. 206:1317–1325. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Safiri S, Carson-Chahhoud K, Noori M,
Nejadghaderi SA, Sullman MJM, Ahmadian Heris J, Ansarin K,
Mansournia MA, Collins GS, Kolahi AA and Kaufman JS: Burden of
chronic obstructive pulmonary disease and its attributable risk
factors in 204 countries and territories, 1990–2019: results from
the Global Burden of Disease Study 2019. BMJ. 378:e0696792022.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Silverman EK: Genetics of COPD. Annu Rev
Physiol. 82:413–431. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhang PD, Zhang XR, Zhang A, Li ZH, Liu D,
Zhang YJ and Mao C: Associations of genetic risk and smoking with
incident COPD. Eur Respir J. 59:21013202022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Adeloye D, Song P, Zhu Y, Campbell H,
Sheikh A and Rudan I: Global, regional, and national prevalence of,
and risk factors for, chronic obstructive pulmonary disease (COPD)
in 2019: A systematic review and modelling analysis. Lancet Respir
Med. 10:447–458. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yin P, Wu J, Wang L, Luo C, Ouyang L, Tang
X, Liu J, Liu Y, Qi J, Zhou M and Lai T: The Burden of COPD in
China and Its Provinces: Findings From the Global Burden of Disease
Study 2019. Front Public Health. 10:8594992022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Barnes PJ: Inflammatory mechanisms in
patients with chronic obstructive pulmonary disease. J Allergy Clin
Immunol. 138:16–27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ghanbarian H, Yıldız MT and Tutar Y:
MicroRNA Targeting. Methods Mol Biol. 2257:105–130. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vishnoi A and Rani S: miRNA Biogenesis and
Regulation of Diseases: An Updated Overview. Methods Mol Biol.
2595:1–12. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Specjalski K and Jassem E: MicroRNAs:
Potential Biomarkers and Targets of Therapy in Allergic Diseases?
Arch Immunol Ther Exp (Warsz). 67:213–223. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liang Q, He J, Yang Q, Zhang Q and Xu Y:
MicroRNA-335-5p alleviates inflammatory response, airway fibrosis,
and autophagy in childhood asthma through targeted regulation of
autophagy related 5. Bioengineered. 13:1791–1801. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li X, Gong Y, Lin X, Lin Q, Luo J, Yu T,
Xu J, Chen L, Xu L and Hu Y: Down-regulation of microRNA-155
suppressed Candida albicans induced acute lung injury by activating
SOCS1 and inhibiting inflammation response. J Microbiol.
60:402–410. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang X, Zhu Z, Guo X and Kong X: The
roles of microRNAs in the pathogenesis of chronic obstructive
pulmonary disease. Int Immunopharmacol. 67:335–347. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Roffel MP, Bracke KR, Heijink IH and Maes
T: miR-223: A key regulator in the innate immune response in asthma
and COPD. Front Med (Lausanne). 7:1962020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang J, Xu Z, Kong L, Gao H, Zhang Y,
Zheng Y and Wan Y: miRNA-486-5p promotes COPD progression by
targeting HAT1 to regulate the TLR4-Triggered inflammatory response
of alveolar macrophages. Int J Chron Obstruct Pulmon Dis.
15:2991–3001. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim RY, Sunkara KP, Bracke KR, Jarnicki
AG, Donovan C, Hsu AC, Ieni A, Beckett EL, Galvão I, Wijnant S, et
al: A microRNA-21-mediated SATB1/S100A9/NF-κB axis promotes chronic
obstructive pulmonary disease pathogenesis. Sci Transl Med.
13:eaav72232021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ding Y, Tian Z, Yang H, Yao H, He P,
Ouyang Y, Yao J, Li M and Jin T: MicroRNA expression profiles of
whole blood in chronic obstructive pulmonary disease. Int J Clin
Experiment Pathol. 10:4860–4865. 2017.
|
|
19
|
Cai SC, Li XP, Li X, Tang GY, Yi LM and Hu
XS: Oleanolic Acid Inhibits Neuronal Pyroptosis in Ischaemic Stroke
by Inhibiting miR-186-5p Expression. Exp Neurobiol. 30:401–414.
2021. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang P, Liang K, Wang W, Zhou D, Chen Y,
Jiang X, Fu R, Zhu B and Lin X: LncRNA SOX2-OTinhibitionprotects
against myocardialischemia/reperfusion-inducedinjury via
themicroRNA-186-5p (miR-186-5p)/Yin Yang 1 (YY1) pathway.
Bioengineered. 13:280–290. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nie Y and Wang F: Inhibiting miR-186-5p
relieves traumatic brain injury by regulating insulin-like growth
factor-I-NLRP3/ASC/caspase-1 signaling pathway. Neuroreport.
34:156–164. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li R, Xu F, Wu X, Ji S and Xia R:
CUL1-Mediated organelle fission pathway inhibits the development of
chronic obstructive pulmonary disease. Comput Math Methods Med.
2020:53901072020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rong B, Liu Y, Li M, Fu T, Gao W and Liu
H: Correlation of serum levels of HIF-1α and IL-19 with the disease
progression of COPD: A retrospective study. Int J Chron Obstruct
Pulmon Dis. 13:3791–3803. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fu X and Zhang F: Role of the HIF-1
signaling pathway in chronic obstructive pulmonary disease. Exp
Ther Med. 16:4553–4561. 2018.PubMed/NCBI
|
|
25
|
Alharbi KS, Fuloria NK, Fuloria S, Rahman
SB, Al-Malki WH, Javed Shaikh MA, Thangavelu L, Singh SK, Rama Raju
Allam VS, Jha NK, et al: Nuclear factor-kappa B and its role in
inflammatory lung disease. Chem Biol Interact. 345:1095682021.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tan H and Zhao L: lncRNA nuclear-enriched
abundant transcript 1 promotes cell proliferation and invasion by
targeting miR-186-5p/HIF-1α in osteosarcoma. J Cell Biochem.
120:6502–6514. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Z, Sha HH and Li HJ: Functions and
mechanisms of miR-186 in human cancer. Biomed Pharmacother.
119:1094282019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Becker V, Yuan X, Boewe AS, Ampofo E,
Ebert E, Hohneck J, Bohle RM, Meese E, Zhao Y, Menger MD, et al:
Hypoxia-induced downregulation of microRNA-186-5p in endothelial
cells promotes non-small cell lung cancer angiogenesis by
upregulating protein kinase C alpha. Molecular therapy. Mol Ther
Nucleic Acids. 31:421–436. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin L, Sun J, Wu D, Lin D, Sun D, Li Q,
Chen J, Niu H, He P and Ding Y: MicroRNA-186 is associated with
hypoxia-inducible factor-1α expression in chronic obstructive
pulmonary disease. Mol Genet Genomic Med. 7:e5312019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
De la Garza MM, Cumpian AM, Daliri S,
Castro-Pando S, Umer M, Gong L, Khosravi N, Caetano MS,
Ramos-Castañeda M, Flores AG, et al: COPD-Type lung inflammation
promotes K-ras mutant lung cancer through epithelial HIF-1α
mediated tumor angiogenesis and proliferation. Oncotarget.
9:32972–32983. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang HX, Yang JJ, Zhang SA, Zhang SM,
Wang JX, Xu ZY and Lin RY: HIF-1α promotes inflammatory response of
chronic obstructive pulmonary disease by activating EGFR/PI3K/AKT
pathway. Eur Rev Med Pharmacol Sci. 22:6077–6084. 2018.PubMed/NCBI
|
|
33
|
Xu YR, Wang AL and Li YQ:
Hypoxia-inducible factor 1-alpha is a driving mechanism linking
chronic obstructive pulmonary disease to lung cancer. Front Oncol.
12:9845252022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jiang H, Zhu Y, Xu H, Sun Y and Li Q:
Activation of hypoxia-inducible factor-1α via nuclear factor-κB in
rats with chronic obstructive pulmonary disease. Acta Biochim
Biophys Sin (Shanghai). 42:483–488. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schuliga M: NF-kappaB signaling in chronic
inflammatory airway disease. Biomolecules. 5:1266–1283. 2015.
View Article : Google Scholar : PubMed/NCBI
|