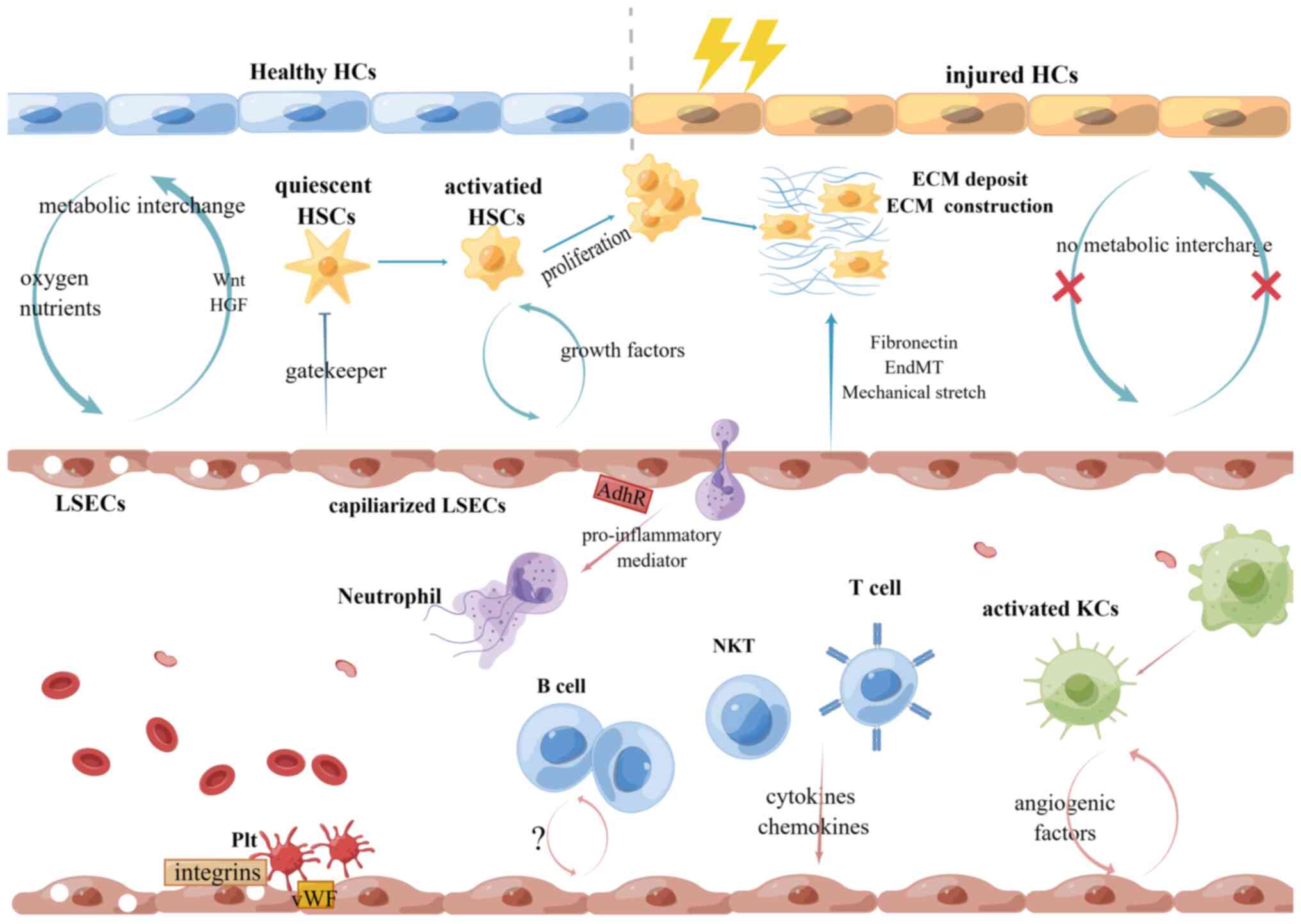
As it has long been linked to severe morbidity and
mortality, liver fibrosis is one of the most significant
contributors to the worldwide disease burden (1). The liver, the most important
detoxification organ, maintains a stable metabolic balance within
the body. After exposure to toxic metabolites, viruses and alcohol
that interfere with the normal physiological functions, the liver
develops drug-induced liver injury, viral hepatitis and alcoholic
liver disease (2), eventually
progressing to fibrosis. In addition, nonalcoholic fatty liver
disease (NAFLD) is a continuum of liver diseases, starting from
simple steatosis (NAFL) and gradually progressing to nonalcoholic
steatohepatitis (NASH), which can progress to liver fibrosis and
cirrhosis. As NAFLD is a disease caused by excessive fat
accumulation in the liver closely related to metabolic dysfunction,
which excludes alcohol and other diseases, a recent consensus among
international experts has proposed NAFLD as a change of
nomenclature for metabolic dysfunction-associated fatty liver
disease (3). Fibrosis is a risk
factor for cirrhosis and hepatocellular carcinoma (4). In addition, liver fibrosis can result
in chronic portal hypertension, a common clinical complication
(5).
Liver fibrosis is characterized by excessive
deposition of extracellular matrix (ECM) and its reduced
degradation. The activation of hepatic stellate cells (HSCs) is a
well-known event in liver fibrosis. HSCs are primarily activated by
stimulated Kupffer cells (KCs) and liver sinusoidal endothelial
cells (LSECs) via paracrine production of pro-inflammatory and
pro-fibrotic cytokines such as TGF-β, connective tissue growth
factor and platelet-derived growth factor (PDGF) and then transform
into resilient myofibroblast which produces ECM to form fibrous
scars around the damaged area (6).
If liver injury is acute or self-limiting, the liver returns to
normal due to the specific destruction of collagen and ECM by
matrix metalloproteinases (MMPs). Otherwise, persistent injury can
cause chronic inflammation and ECM deposition, eventually leading
to replacement of parenchymal cells by scar tissues and ultimately
cirrhosis. How to balance the synthesis and degradation of ECM is
still challenging. Although regression of liver fibrosis in
patients with chronic liver disease has been recognized for
decades, there are still no FDA-approved anti-fibrosis drugs.
Although the exact definition of fibrotic regression has not been
properly established (7), the
success of antiviral therapy has given us hope to elucidate the
cellular and molecular mechanisms and develop therapeutic agents
that mimic the endogenous capabilities of the liver (8).
Dysfunction of LSECs allows initiation and
progression of liver fibrosis and is responsible for its clinical
complication, portal hypertension. LSECs create a bridge between
hepatocytes and hepatic sinusoids, acting as protective gatekeepers
of the microvascular environment (9). Although HSC activation is the primary
event in liver fibrosis, it is still important and necessary to
seek other new directions to reverse the condition.
The present review summarized the structural and
functional alterations of LSECs and their crosstalk with hepatic
microenvironment during liver fibrosis and highlighted the role of
LSECs in portal hypertension. Several novel therapeutic strategies
targeting LSECs are also reviewed.
Compared to other endothelial cells, LSECs are
morphologically and functionally specialized. LSECs are the most
abundant non-parenchymal cells in the liver and form the sinusoidal
wall (10). LSECs represent a
gatekeeper between hepatocytes and hepatic sinusoids during liver
injury. They are highly specialized as nondiaphragmatic fenestrae
(cytoplasmic pores) and lack a basal membrane, facilitating
high-efficient material exchange from the bloodstream into the
space of Disse and regulate hepatic stellate cell activation status
(11). The fenestrae are 100–150
nm in size and are organized in clusters termed sieve plates
(12). Their size and number
varies across liver lobules with larger but fewer fenestrae per
sieve plate in the periportal region and smaller but more numerous
fenestrae per sieve plate in the centrilobular region (13,14),
which may be related to the need for oxygen exchange along the
lobules. Notably, heterogeneities between periportal and
centrilobular LSECs exist in morphology (15), antigen expression (16) and lectin affinity (17). Di Martino et al (18) established an approach of stimulated
emission depletion (STED) microscopy to visualize and analyze LSEC
fenestrae and revealed that cytoskeleton, especially actin, is
closely associated with fenestrae formation. Herrnberger et
al (19) found that
plasmalemma vesicle-associated protein (PLVAP) is required for the
formation of endothelial diaphragms and endothelial fenestrae and
opening fenestrae in LSECs are critical for the passage of
lipoproteins.
In addition to their unique structure, LSECs contain
various receptors to perform multiple functions. A total of three
endocytosis receptors have been identified on LSECs,
collagen-α-chain/mannose receptor, hyaluronan/scavenger receptor
and FcγIIb2 receptor (20). Among
them, mannose receptor is the main candidate for endocytosis of
denatured collagen on LSECs and functions as MMPs, making it a
promising target in the study of hepatic fibrosis (21). Through the combined effect of
endocytic vesicles and receptor-mediated endocytosis, LSECs can
eliminate antigens, cell debris and immune complexes.
SR-H/stabillin-1 and SR-H/stabillin2, the main scavenger receptors
of LSECs, mediate endocytosis of polyanionic molecules, including
oxidized low-density lipoproteins, hyaluronan, chondroitin sulfate,
formaldehyde treated serum albumin, procollagen type I and III
N-terminal peptides and advanced glycation end products (22). FcγIIb2 receptor on LSECs mediates
vascular immunity mainly through cleaning circulating IgG immune
complexes (23). Under homeostatic
conditions, LSECs are not merely endocytic fenestrated endothelial
cells, they also exhibit vasodilatory, anti-inflammatory,
anti-thrombotic and anti-fibrotic phenotypes, which are important
for orchestrating liver microenvironment response and regulating
intrahepatic vascular tone and immune cell function (11).
Maintenance of the highly specialized phenotype of
LSECs is crucial for liver homeostasis. During liver fibrosis and
cirrhosis, loss of LSEC phenotype and functions amplify liver
damage by undergoing a process called ‘capillarization’, which is
clarified by the loss of fenestrae (cytoplasmic pores) and the
manifestation of a basement membrane, leading to LSEC dysfunction
(20).
The exact mechanisms regulating the loss of
fenestrae have not been fully elucidated, but several molecules and
pathways have been identified. Maintenance of the fenestrated LSEC
phenotype requires VEGF, which is derived from hepatocytes and
HSCs. VEGF is a key regulator for the LSEC phenotype (27). It has been established from various
in vitro and in vivo studies that VEGF operates in
two pathways, the NO-dependent and NO-independent (28). In the NO-dependent pathway,
endothelial nitric oxide synthase (eNOS)-NO-cyclic guanosine
monophosphate signaling is critical for regulating the formation of
fenestrae in LSECs (29).
Endothelial Notch activation leads to LSEC dedifferentiation and
promotes liver fibrogenesis through eNOS-soluble guanylate cyclase
(sGC) signaling. Regarding Notch ligands, Delta-like ligand 4
(DLL4) is predominantly expressed in endothelial cells under
hypoxic conditions during liver fibrosis (30). It is reported that DLL4
overexpression accelerated defenestration of LSECs and production
of basement membrane in both human fibrotic livers and carbon
tetrachloride (CCl4)-induced mouse livers (31). In addition to Notch signaling, the
Hedgehog (Hh) pathway also induces LSEC capillarization and
formation of vascular tubes in vitro by Hh ligands binding
to their transmembrane receptors [Patched (Ptc)] to activate
Smoothened (32). Besides,
miR-511-3p was found to positively regulate Hh signaling by
targeting Ptch1 in hepatic sinusoidal obstruction syndrome
(33). Long noncoding RNA (lncRNA)
Airn was identified to interact with enhancer of zeste homolog 2 to
maintain LSEC differentiation through Krüppel-like factor 2
(KLF2)-eNOS-sGC pathway, thus indirectly inhibiting HSC activation
(34). Studies have found that
several transcription factors in liver endothelial cells also play
key roles in regulating the characteristics and function of LSECs.
Genetic endothelial GATA4 deletion leads to liver fibrosis and
hepatopathy by GATA4-MYC-PDGFB axis (35). In 2015, transcriptional factor KLF2
was found to upregulate eNOS expression, which is essential for
maintaining a functional endothelial phenotype and also explains
the molecular mechanisms of statins (36). In 2022, de Haan et al
(37) found that LSEC-enriched
zinc-finger E-box-binding homeobox2 (Zeb2) can preserve the liver
angioarchitecture and prevent liver fibrosis.
On the other hand, capillarization is thought to be
the repair of damaged LSECs by bone marrow endothelial progenitors
that engraft but fail to fully mature through cell autonomous
pathways that inhibit the NO-dependent pathway. Maretti-Mira et
al (38) identified
heparin-binding EGF-like growth factor (HB-EGF) as a signal to
maintain HSCs quiescence while the immature LSECs from bone marrow
were unable to shed HB-EGF from the cytosolic membrane. These data
demonstrate that capillarization is an early event in the fibrotic
process and is a perfect target for the treatment of liver
fibrosis. The number of fenestrae was shown to be significantly
decreased in LSECs from BMP9-knockout mice and addition of BMP9 to
the LSECs culture recovers fenestrae and elevates the expression of
GATA4 and PLVAP, suggesting that BMP9 is a key paracrine regulator
of LSEC fenestration (39).
Conversely, Haristi et al found that high BMP9 in
combination with LPS stimulation induced the expression of certain
capillarization markers in LSEC (40). Desroches-Castan et al
(41) found that the role of BMP9
in LSEC differentiation depends on genetic background of C57BL/6,
BALB/c and 129/Ola mice. Further studies may warrant the exact
roles of BMP9 in LSEC capillarization.
In addition, cell-specific junctional adhesion
molecule A (JAM-A) is reported to exert crucial functions in
hepatic fibrogenesis (10). Loss
of endothelial JAM-A induces LSEC capillarization and HSC
activation by triggering Hh signaling and loss of VEGFR1/2 with no
alterations in myeloid recruitment (10). Elevated portal blood
lipopolysaccharide (LPS) due to leaky gut induces LSEC
capillarization in mice and in vitro, while antimicrobial
treatment lowers portal blood LPS concentration and inhibits LSEC
capillarization (42). Overall,
the signaling pathways that regulate LSEC fenestration varies under
physiological and pathological conditions and the mechanisms
involved by various factors are summarized in Table I.
Increasing evidence has indicated that
fibroblast-like cells in liver fibrosis are mainly derived from
activated hepatic stellate cells (aHSCs). Therefore, HSC activation
is considered a pivotal event leading to fibrosis. HSC activation
is fine-tuned by the angiocrine signaling from LSECs and is also
tightly modulated by dynamic interactions between LSECs and other
cell types in the liver. Understanding these crosstalk is expected
to improve liver disease therapy (Fig.
1).
HSCs are the main source of ECM and thus have been
the central to the development of liver fibrosis. The interaction
between LSECs and HSCs forms a positive feedback loop. The starting
point of the loop is VEGF. As described above, VEGF from
hepatocytes and HSCs maintains the phenotype of fenestrated LSECs
and, in turn, NO from VEGF-stimulated LSECs prevents HSC
activation. Therefore, VEGF may have a dual role in fibrosis. It
induces fibrosis-associated angiogenesis while recruiting monocytes
mainly through the CXC chemokine (CXCL)9-MMP13 axis to control the
secretion of multiple MMPs and tissue inhibitor of MMPs during
fibrosis resolution (43).
Therefore, as a key mediator of angiocrine signaling, the
regulatory role of VEGF in liver diseases remains to be elucidated.
Endothelin-1 from HSCs also plays a key role in the regulation of
eNOS activation in LSECs (44). In
addition, capillarized LSECs activate HSCs through secreting PDGF,
TGF-β and lowering a transcription factor KLF2, which acts as a
vasoprotective molecule of the liver endothelium, subsequently
activating HSCs (45). Activated
HSCs further act on quiescent HSCs and LSECs by autocrine TGF-β1
(46). Thrombospondin-1 can be
induced by HSC activation, enhancing LSEC capillarization by
blocking the NO-dependent pathway (47). In addition, LSECs stimulate HSC
migration by producing CXCL12/stromal-derived factor-1. CXCL12
binds to CXC chemokine receptor (CXCR)4 on HSCs, thereby inducing
HSC proliferation and collagen production perpetuating fibrosis,
while CXCR7-expressing LSECs activated by CXCL12 initiate liver
regeneration (48). Enhancement of
intracellular adipocyte fatty acid-binding protein (A-FABP)
potentiates the LSEC capillarization by inducing Hh signaling,
leading to impairment of the gatekeeper function of LSECs on HSC
activation. On the other hand, LSEC derived A-FABP releases and
acts on HSCs in a paracrine manner to potentiate the
transactivation of TGF-β1 by activating JNK/c-Jun signaling.
Elevated TGF-β1 subsequently promotes the expression of ECM in both
paracrine and autocrine manners. These findings define that A-FABP
is a novel therapeutic target of liver fibrosis (49). The contribution of exosomes and
their cargoes has been proposed as a new model of intercellular
communication between LSECs and HSCs. LSEC-derived sphingosine
kinase 1-containing exosomes regulate HSC signaling and migration
through fibronectin-integrin-dependent exosome adherence and
dynamin-dependent exosome internalization (50).
Loss of fenestration of LSECS protects the liver
from ongoing damage by limiting toxins to specific areas (19). However, the formation of a
continuous basement membrane lining the sinusoids disrupts the
bidirectional exchange of oxygen and nutrients between hepatocytes
and sinusoids (51), as well as
the lipoprotein secretion of hepatocytes and the de novo
lipogenesis in hepatocytes which aggravates hepatic steatosis in
NASH (52). LSECs can also secrete
Wnt and hepatocyte growth factor (HGF), which together act as key
hepatocyte mitogens with a potent liver regenerative capacity that
is readily switched to a pro-fibrotic phenotype, in which the
Erk1/2/Akt axis in LSECs acts as a switch (53). Growth factors from LSECs, such as
bone morphogenetic protein (BMP)2, BMP6 and TGFβ1 act on
hepatocytes and HSCs to control systemic iron homeostasis and
fibrotic processes in a paracrine manner, respectively (54). Notch activation can alter the
angiocrine profile of LSECs to compromise hepatocyte proliferation
and liver regeneration through downregulating critical hepatocyte
mitogens, including Wnt2a, Wnt9b and HGF (31). A recent study found that lncRNA
Airn, a nuclear localized and highly unstable in the form of
non-splicing 108 kb ncRNA from LSECs, promoted hepatocyte
proliferation by increasing paracrine secretion of Wnt2a and HGF
from LSECs (34). lncRNA Airn is
inherent and might be a serum biomarker for liver fibrogenesis.
Indeed, after acute injury, endothelial cells determine if the
liver undergoes regeneration or fibrosis by angiocrine signaling
(55). LSECs and hepatocytes also
communicate with each other through the VEGF-A/VEGFR-2 signaling
which is regarding to angiogenesis. Yan et al (56) investigated the biological roles of
cluster of differentiation (CD)147 in a CCl4-induced
liver fibrosis mouse model and found that CD147 promoted liver
fibrosis progression via VEGF-A/VRGFR-2 signaling, mediating
crosstalk between hepatocytes and LSECs. A combination of leukocyte
derived chemokine2 expressed by hepatocytes and Tie1 produced by
LSECs can promote LSECs capillarization and reduce portal
angiogenesis (57).
Hepatic macrophages are important to the
pathogenesis of chronic liver injury. Activated KCs can secrete a
wide variety of proinflammatory activating HSCs. KCs and
infiltrating macrophages can usually be divided into two subtypes,
including M1 macrophages, which produce proinflammatory cytokines
such as TNF-α, IL-1b, CCL2 and CCL5, and M2 macrophages which
secrete a distinct set of mediators including IL-13, IL-10, IL-4
and TGF-b (58,59). Imbalance of M1/M2 ratio may be the
key to the progression of NASH to liver fibrosis (52). Pro-inflammatory KCs may also
produce cytokines and chemokines which increase the expression of
adhesion molecules by LSECs, leading to leukocyte infiltration and
activation (60). In a murine
model of acute liver injury caused by overdose of acetaminophen,
You et al (61) found that
liver resident macrophages and infiltrating macrophages express an
array of angiogenic factors and induce LSEC proliferation and
migration to repair liver blood vessels, while in NASH, the
capillarization of LSECs is necessary for activating KCs in that
LSECs injury appeared during the simple steatosis phase and
preceded the appearance of activate KCs and HSCs (62). Ford et al (63) investigated the effects of KCs on
human liver sinusoidal endothelial cells (hLSECs) phenotype on soft
materials, which indicate that TNF-α secreted by KCs undermines the
effects of a soft matrix that is representative of healthy tissues
and leads to loss of LSEC fenestrae. The relationship between LSECs
and macrophages in liver fibrosis needs further investigation.
LSECs belong to nonprofessional antigen presenting
cells, plasmacytoid DCs and are primary immunologic mediators
underlying hepatic tolerance. They express major histocompatibility
complex (MHC) class I, MHC class II and the co-stimulatory
molecules CD80, CD86 and CD40 (11). The LSECs mediate hepatic immune
tolerance toward self or foreign antigens by expression of
anti-inflammatory mediators under physiological conditions, but
they gain pro-inflammatory functions upon viral infection (64). LSECs release cytokines via
toll-like receptors and contribute to viral infection of the liver,
such as hepatitis B or hepatitis C virus (65). The physiologic consequences of
higher antigen-presenting cell-related immunogenicity is associated
with elevated intrahepatic inflammatory milieu in fibrosis. These
LSECs gain enhanced capacity to capture antigens and induce the
immunogenic T cell to sensitize endogenous cytotoxic T lymphocytes
(66). During liver fibrosis, Th1
and Th2 cells recruit and adhere to liver sinusoids using
α4β1-integrin and vascular adhesion protein-1 (VAP-1), respectively
(67). However, the specific
mechanisms by which T cells and LSECs crosstalk regulate sinusoidal
capillarization remain largely unknown. Emerging evidences
suggested B-cell activation is involved in NAFLD (68), while crosstalk between LSECs and
B-cell remains to be elucidated. In addition, chemokine receptor
CXCR6 on LSECs and its ligand CXCL16 on natural killer T cells
(NKTs) control NKT cell migration (69).
Inflammation is an essential part involved in the
process of liver fibrosis. Hepatitis and NAFLD, especially NASH,
have recently received increasing attention (70). LSECs interact with portal
circulation via expression of surface receptors and release of
paracrine factors, such as intercellular adhesion molecule,
vascular cell adhesion molecule-1, VAP-1, chemokines and cytokines
(71), recruiting the circulating
leucocytes to enter the liver parenchyma. Exploring these
inflammatory mediators may help to discover new therapeutic targets
for the treatment of liver fibrosis. Gola et al (72) considered LSECs as a microbiome
sensor, sustaining commensal-induced MyD88-dependent signaling to
regulate the composition of the peri-cellular matrix involved in
chemokine gradient formation. In vitro and in vivo
findings demonstrate that specific focal adhesion proteins
recruited by phosphofructokinase can parlay LSEC
mechanotransduction into stiffness-induced angiocrine signaling and
mediate neutrophil infiltration and promote liver fibrosis
(73). By contrast, Brozat et
al found that LSEC-derived JAM-A had no effect on cell
migration from sinusoids or adhesion to LSECs (10), suggesting a complex role of
leukocyte-endothelial cell interactions in promoting liver
fibrosis.
At present, an increasing number of individuals
realize that the platelets are no longer bystanders in liver
diseases and anti-platelet therapy has been proven effective in
treating chronic liver injury (74,75).
In vitro studies show that platelet adhesion is partly
mediated by integrin (glycoprotein IIb/IIIa and αvβ3) with human
hepatic sinusoidal endothelial cells (76), leading to platelet and endothelial
activation and leukocyte recruitment. On the other hand, in
wild-type mice after chronic CCl4 challenge plasma Von
Willebrand factor levels are increased (77), which could be associated with the
interaction between platelets and LSECs. However, it is not clear
whether platelet adhesion is associated with capillarization.
Platelets have well appreciated roles in physiological and
pathological processes including inflammation, tissue repair,
angiogenesis and tumor growth (78). A few studies have focused on the
release of platelet granule content or platelet RNA transfer to
activated HSCs (79,80), but the influence of platelets on
LSECs needs more research. Although platelets have been shown to
simulate proliferation of cultured hepatocytes (81), this is a long way to elucidating
the role of modulators of platelets in chronic liver diseases.
While most researchers focused on intercellular
crosstalk of LSECs, Brougham-Cook et al (82) redirected attention to identify the
unique phenotypes that LSECs exhibit due to ECM composition,
stiffness and soluble factor alterations in fibrotic models by a
high-throughput cellular microarray. LSECs play a crucial role in
hepatic fibrogenesis by participating in ECM deposition in the form
of collagen and fibronectin synthesis and regulating ECM
metabolism. Extra domain A from injured LSECs promotes stellate
cell motility and parenchymal liver fibrosis (83). Liu et al (84) used the principles of mechanical
mechanics to explain capillarization of LSECs, which might
contribute to early-stage collagen contraction, then collagen
fibers function as mechanical transducer to activate HSCs. Notably,
researchers found that capillarized LSECs undergo a partial
endothelial-mesenchymal transition characterized by increased ECM
production without activating cell mobility, leading to
perisinusoidal ECM deposition preferentially in liver sinusoids but
not septal/portal scars (85)
(Fig. 1)
Portal hypertension, defined as increased pressure
in the portal vein, develops as a consequence of increased
intrahepatic vascular resistance and is the initial step towards
complications of chronic liver disease (86). It can cause ascites and
gastroesophageal varices, as well as hepatic encephalopathy due to
portosystemic shunting, hepatorenal syndrome and hypersplenism
(87). To date, a number of
researchers have considered that the effect of increased
intrahepatic vascular resistance in liver fibrosis is closely
related to LSECs, ultimately leading to portal hypertension and its
complications (88–90). Decreased NO levels and
capillarization can increase vascular resistance (91). In addition, endothelial autophagy
(92) and aging (93) are also involved in the occurrence
of portal hypertension and liver fibrosis.
Recently, an increasing number of studies have
suggested that thrombosis is one of the key pathological factors
mediating portal hypertension and anticoagulation therapy seems to
be beneficial in the treatment of liver fibrosis and portal
hypertension in rats, although it depends on the stage of cirrhosis
(86,94). The current consensus is that most
portosystemic collateral vessels are formed through angiogenesis,
the formation of new blood vessels from pre-existing vasculature
especially within fibrous scar (95). In addition, mechanical stretch
increases expression of CXCL1 in LSECs to recruit neutrophils,
generate sinusoidal microthrombi and promote portal hypertension
(96). Therefore, reversing LSEC
dysregulation maybe an attractive strategy for portal
hypertension.
It is well-known that statins have beneficial
effects on dysfunctional sinusoidal cells by selectively NO
bioavailability and inhibiting RhoA/Rho-kinase in the liver. This
was confirmed in portal hypertension of rats with NASH (88). In addition, simvastatin
administration for 15 days in aged cirrhotic rats improves the
hepatic sinusoidal milieu, demonstrating its therapeutic potential
in advanced chronic liver disease (93). Simvastatin restores the quiescence
of aHSCs via stimulation of KLF2-NO signaling in LSECs (97). Treatment with the pan-peroxisome
proliferator-activated receptor (pan-PPAR) agonist lanifibranor
demonstrates phenotypic improvement in a rat model of cBDL as well
as in human hepatocytes from patients with cirrhosis (98). Although further validation is
needed in human trials, it paves a way to develop novel drugs that
target both HSCs and LSECs to treat liver fibrosis.
The property of containing numerous receptors to
endocytose soluble macromolecules and small particles (20) makes LSECs suitable for specific
drug delivery. To date, several modified polymers targeting LSECs
have been developed to delivery drugs. Hide et al (99) designed simvastatin-loaded polymeric
micelles to treat chronic liver disease and showed promising
results, thus suggesting that nanoparticles are a promising
therapeutic approach. On the other hand, liver sinusoidal
capillarization and ECM deposition are dual pathological barriers
to drug delivery (20), Zhang
et al (100) constructed
an efficient nanodrug delivery system with LSEC-targeting and
fenestrae-repairing nanoparticles (named HA-NPs/SMV) on the basis
of the modification with hyaluronic acid.
Based on a specific route of drug delivery targeting
LSECs, strategies that reverse endothelial dysfunction by
inhibiting Notch signaling (30,101) and Hh signaling (102,103), downregulating VEGF-R2 (104), or suppressing KLF5-mediated LSEC
angiogenesis (105), inhibiting
hypoxia-inducible factor-1α (HIF-1α) (106) or enhancing endothelial barrier
function through the cyclic adenosine monophosphate/Rac/cortactin
pathway (107), can be applied to
improve liver fibrosis. Besides special targeting of LSECs, there
are other methods of liver fibrosis treatment that act via an
indirect effect on LSECs, such as simultaneous consumption of LSECs
and aHSCs (108), artificial
control of the switch of the Erk1/2-Akt axis in LSECs to reverse
LSECs from a proregenerative to a profibrotic phenotype (53), prevention of interstellar
interaction of LSECs (109,110), selective enhancement of autophagy
in LSEC in the early stages (111) and treatment of liver fibrosis
with anti-angiogenic agents (112). Finally, a more detailed
understanding of cytoskeletal structure and function of LSECs may
help with the design of drugs that control fenestrae opening, which
seems to be a simple and straightforward anti-liver fibrosis
approach.
The present study reviewed the capillarization of
LSECs and involved mechanisms and sinusoidal communication mediated
by LSECs in liver fibrosis. LSECs play a central role in the
maintenance of HSC quiescence and modulating the crosstalk among
various types of liver cells and dysfunction of LSEC characterized
by loss of fenestration is widely considered to be an early event
in the development of liver fibrosis. In recent years, a number of
anti-fibrotic drug candidates have shown potent effects in
experimental animal models; however, an urgent question remains
that they have limited or no anti-fibrotic effects in clinical
trials. At present, there are no approved drugs for liver fibrosis.
The studies on LSECs suggest that protection of LSECs may be an
effective strategy to prevent fibrosis initiation and progression.
LSEC-targeted drugs or technologies will provide some novel
strategies for the treatment of liver fibrosis in the future.
Meanwhile, exploring the regulatory mechanisms of LSEC development
and crosstalk with other liver cells will help understand the
initiation progress of liver fibrosis and develop improved
therapeutic targets.
Not applicable.
Funding: No funding was received.
Not applicable.
JG prepared and wrote the original draft of the
manuscript. BZ and YH wrote, reviewed and edited the manuscript.
All authors read and approved the final manuscript. Data
authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Parola M and Pinzani M: Liver fibrosis:
Pathophysiology, pathogenetic targets and clinical issues. Mol
Aspects Med. 65:37–55. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mallat A and Lotersztajn S: Cellular
mechanisms of tissue fibrosis. 5. Novel insights into liver
fibrosis. Am J Physiol Cell Physiol. 305:C789–C799. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gofton C, Upendran Y, Zheng MH and George
J: MAFLD: How is it different from NAFLD? Clin Mol Hepatol. 29
(Suppl):S17–S31. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Asrani SK, Devarbhavi H, Eaton J and
Kamath PS: Burden of liver diseases in the world. J Hepatol.
70:151–171. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
D'Amico G, Morabito A, D'Amico M, Pasta L,
Malizia G, Rebora P and Valsecchi MG: New concepts on the clinical
course and stratification of compensated and decompensated
cirrhosis. Hepatol Int. 12 (Suppl 1):S34–S43. 2018. View Article : Google Scholar
|
|
6
|
Dewidar B, Meyer C, Dooley S and
Meindl-Beinker AN: TGF-β in hepatic stellate cell activation and
liver fibrogenesis-updated 2019. Cells. 8:14192019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rockey DC and Friedman SL: Fibrosis
regression after eradication of hepatitis C virus: From bench to
bedside. Gastroenterology. 160:1502–1520.e1. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Friedman SL and Pinzani M: Hepatic
fibrosis 2022: Unmet needs and a blueprint for the future.
Hepatology. 75:473–488. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu M, Wang X, Zou Y and Zhong Y: Key role
of liver sinusoidal endothelial cells in liver fibrosis. Biosci
Trends. 11:163–168. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brozat JF, Brandt EF, Stark M, Fischer P,
Wirtz TH, Flaßhove A, Rodenhausen AN, Vajen T, Heinzmann ACA,
Schmitz SM, et al: JAM-A is a multifaceted regulator in hepatic
fibrogenesis, supporting LSEC integrity and stellate cell
quiescence. Liver Int. 42:1185–1203. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lafoz E, Ruart M, Anton A, Oncins A and
Hernández-Gea V: The endothelium as a driver of liver fibrosis and
regeneration. Cells. 9:9292020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Poisson J, Lemoinne S, Boulanger C, Durand
F, Moreau R, Valla D and Rautou PE: Liver sinusoidal endothelial
cells: Physiology and role in liver diseases. J Hepatol.
66:212–227. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xie G, Wang L, Wang X, Wang L and DeLeve
LD: Isolation of periportal, midlobular, and centrilobular rat
liver sinusoidal endothelial cells enables study of zonated drug
toxicity. Am J Physiol Gastrointest Liver Physiol. 299:G1204–G1210.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mönkemöller V, Øie C, Hübner W, Huser T
and McCourt P: Multimodal super-resolution optical microscopy
visualizes the close connection between membrane and the
cytoskeleton in liver sinusoidal endothelial cell fenestrations.
Sci Rep. 5:162792015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wack KE, Ross MA, Zegarra V, Sysko LR,
Watkins SC and Stolz DB: Sinusoidal ultrastructure evaluated during
the revascularization of regenerating rat liver. Hepatology.
33:363–378. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Scoazec JY, Racine L, Couvelard A, Flejou
JF and Feldmann G: Endothelial cell heterogeneity in the normal
human liver acinus: In situ immunohistochemical demonstration.
Liver. 14:113–123. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Barberá-Guillem E, Rocha M, Alvarez A and
Vidal-Vanaclocha F: Differences in the lectin-binding patterns of
the periportal and perivenous endothelial domains in the liver
sinusoids. Hepatology. 14:131–139. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Di Martino J, Mascalchi P, Legros P,
Lacomme S, Gontier E, Bioulac-Sage P, Balabaud C, Moreau V and
Saltel F: Actin depolymerization in dedifferentiated liver
sinusoidal endothelial cells promotes fenestrae re-formation.
Hepatol Commun. 3:213–219. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Herrnberger L, Hennig R, Kremer W,
Hellerbrand C, Goepferich A, Kalbitzer HR and Tamm ER: Formation of
fenestrae in murine liver sinusoids depends on plasmalemma
vesicle-associated protein and is required for lipoprotein passage.
PLoS One. 9:e1150052014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
DeLeve LD: Liver sinusoidal endothelial
cells in hepatic fibrosis. Hepatology. 61:1740–1746. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Malovic I, Sørensen KK, Elvevold KH,
Nedredal GI, Paulsen S, Erofeev AV, Smedsrød BH and McCourt PA: The
mannose receptor on murine liver sinusoidal endothelial cells is
the main denatured collagen clearance receptor. Hepatology.
45:1454–1461. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Verwilligen RAF, Mulder L, Rodenburg FJ,
Van Dijke A, Hoekstra M, Bussmann J and Van Eck M: Stabilin 1 and 2
are important regulators for cellular uptake of apolipoprotein
B-containing lipoproteins in zebrafish. Atherosclerosis. 346:18–25.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sørensen KK, McCourt P, Berg T, Crossley
C, Le Couteur D, Wake K and Smedsrød B: The scavenger endothelial
cell: A new player in homeostasis and immunity. Am J Physiol Regul
Integr Comp Physiol. 303:R1217–R1230. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Su T, Yang Y, Lai S, Jeong J, Jung Y,
McConnell M, Utsumi T and Iwakiri Y: Single-cell transcriptomics
reveals zone-specific alterations of liver sinusoidal endothelial
cells in cirrhosis. Cell Mol Gastroenterol Hepatol. 11:1139–1161.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Deleve LD, Wang X and Guo Y: Sinusoidal
endothelial cells prevent rat stellate cell activation and promote
reversion to quiescence. Hepatology. 48:920–930. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Horn T, Christoffersen P and Henriksen JH:
Alcoholic liver injury: Defenestration in noncirrhotic livers-a
scanning electron microscopic study. Hepatology. 7:77–82. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jin Y, Guo YH, Li JC, Li Q, Ye D, Zhang XX
and Li JT: Vascular endothelial growth factor protein and gene
delivery by novel nanomaterials for promoting liver regeneration
after partial hepatectomy. World J Gastroenterol. 29:3748–3757.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xie G, Wang X, Wang L, Wang L, Atkinson
RD, Kanel GC, Gaarde WA and Deleve LD: Role of differentiation of
liver sinusoidal endothelial cells in progression and regression of
hepatic fibrosis in rats. Gastroenterology. 142:918–927.e6. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Carpenter B, Lin Y, Stoll S, Raffai RL,
McCuskey R and Wang R: VEGF is crucial for the hepatic vascular
development required for lipoprotein uptake. Development.
132:3293–3303. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen L, Gu T, Li B, Li F, Ma Z, Zhang Q,
Cai X and Lu L: Delta-like ligand 4/DLL4 regulates the
capillarization of liver sinusoidal endothelial cell and liver
fibrogenesis. Biochim Biophys Acta Mol Cell Res. 1866:1663–1675.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Duan JL, Ruan B, Yan XC, Liang L, Song P,
Yang ZY, Liu Y, Dou KF, Han H and Wang L: Endothelial Notch
activation reshapes the angiocrine of sinusoidal endothelia to
aggravate liver fibrosis and blunt regeneration in mice.
Hepatology. 68:677–690. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Francis H, Bohanan J and Alpini G:
Hedgehog signalling and LSEC capillarisation: Stopping this one in
its tracks. Gut. 61:1243–1244. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang L, Xu X, Chen Z, Zhang Y, Chen H and
Wang X: miR-511-3p promotes hepatic sinusoidal obstruction syndrome
by activating hedgehog pathway via targeting Ptch1. Am J Physiol
Gastrointest Liver Physiol. 321:G344–G354. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen T, Shi Z, Zhao Y, Meng X, Zhao S,
Zheng L, Han X, Hu Z, Yao Q, Lin H, et al: LncRNA Airn maintains
LSEC differentiation to alleviate liver fibrosis via the
KLF2-eNOS-sGC pathway. BMC Med. 20:3352022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Winkler M, Staniczek T, Kürschner SW,
Schmid CD, Schönhaber H, Cordero J, Kessler L, Mathes A, Sticht C,
Neßling M, et al: Endothelial GATA4 controls liver fibrosis and
regeneration by preventing a pathogenic switch in angiocrine
signaling. J Hepatol. 74:380–393. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Marrone G, Maeso-Díaz R, García-Cardena G,
Abraldes JG, García-Pagán JC, Bosch J and Gracia-Sancho J: KLF2
exerts antifibrotic and vasoprotective effects in cirrhotic rat
livers: Behind the molecular mechanisms of statins. Gut.
64:1434–1443. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
de Haan W, Dheedene W, Apelt K,
Décombas-Deschamps S, Vinckier S, Verhulst S, Conidi A, Deffieux T,
Staring MW, Vandervoort P, et al: Endothelial Zeb2 preserves the
hepatic angioarchitecture and protects against liver fibrosis.
Cardiovasc Res. 118:1262–1275. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Maretti-Mira AC, Wang X, Wang L and DeLeve
LD: Incomplete differentiation of engrafted bone marrow endothelial
progenitor cells initiates hepatic fibrosis in the rat. Hepatology.
69:1259–1272. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Desroches-Castan A, Tillet E, Ricard N,
Ouarné M, Mallet C, Belmudes L, Couté Y, Boillot O, Scoazec JY,
Bailly S and Feige JJ: Bone morphogenetic protein 9 is a paracrine
factor controlling liver sinusoidal endothelial cell fenestration
and protecting against hepatic fibrosis. Hepatology. 70:1392–1408.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gaitantzi H, Karch J, Germann L, Cai C,
Rausch V, Birgin E, Rahbari N, Seitz T, Hellerbrand C, König C, et
al: BMP-9 modulates the hepatic responses to LPS. Cells. 9:6172020.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Desroches-Castan A, Tillet E, Ricard N,
Ouarné M, Mallet C, Feige JJ and Bailly S: Differential
Consequences of Bmp9 deletion on sinusoidal endothelial cell
differentiation and liver fibrosis in 129/Ola and C57BL/6 Mice.
Cells. 8:10792019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kawai H, Osawa Y, Matsuda M, Tsunoda T,
Yanagida K, Hishikawa D, Okawara M, Sakamoto Y, Shimagaki T,
Tsutsui Y, et al: Sphingosine-1-phosphate promotes tumor
development and liver fibrosis in mouse model of congestive
hepatopathy. Hepatology. 76:112–125. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kantari-Mimoun C, Krzywinska E, Castells
M, Milien C, Klose R, Meinecke AK, Lemberger U, Mathivet T,
Gojkovic M, Schrödter K, et al: Boosting the hypoxic response in
myeloid cells accelerates resolution of fibrosis and regeneration
of the liver in mice. Oncotarget. 8:15085–15100. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kwok W, Lee SH, Culberson C, Korneszczuk K
and Clemens MG: Caveolin-1 mediates endotoxin inhibition of
endothelin-1-induced endothelial nitric oxide synthase activity in
liver sinusoidal endothelial cells. Am J Physiol Gastrointest Liver
Physiol. 297:G930–G939. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li H: Intercellular crosstalk of liver
sinusoidal endothelial cells in liver fibrosis, cirrhosis and
hepatocellular carcinoma. Dig Liver Dis. 54:598–613. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Trautwein C, Friedman SL, Schuppan D and
Pinzani M: Hepatic fibrosis: Concept to treatment. J Hepatol. 62 (1
Suppl):S15–S24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Venkatraman L and Tucker-Kellogg L: The
CD47-binding peptide of thrombospondin-1 induces defenestration of
liver sinusoidal endothelial cells. Liver Int. 33:1386–1397. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liepelt A and Tacke F: Stromal
cell-derived factor-1 (SDF-1) as a target in liver diseases. Am J
Physiol Gastrointest Liver Physiol. 311:G203–G209. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wu X, Shu L, Zhang Z, Li J, Zong J, Cheong
LY, Ye D, Lam KSL, Song E, Wang C, Xu A and Hoo RLC: Adipocyte
fatty acid binding protein promotes the onset and progression of
liver fibrosis via mediating the crosstalk between liver sinusoidal
endothelial cells and hepatic stellate cells. Adv Sci (Weinh).
8:e20037212021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang R, Ding Q, Yaqoob U, de Assuncao TM,
Verma VK, Hirsova P, Cao S, Mukhopadhyay D, Huebert RC and Shah VH:
Exosome adherence and internalization by hepatic stellate cells
triggers Sphingosine 1-Phosphate-dependent Migration. J Biol Chem.
290:30684–30696. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zapotoczny B, Szafranska K, Kus E, Braet
F, Wisse E, Chlopicki S and Szymonski M: Tracking fenestrae
dynamics in live murine liver sinusoidal endothelial cells.
Hepatology. 69:876–888. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Du W and Wang L: The crosstalk between
liver sinusoidal endothelial cells and hepatic microenvironment in
NASH related liver fibrosis. Front Immunol. 13:9361962022.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lao Y, Li Y, Zhang P, Shao Q, Lin W, Qiu
B, Lv Y, Tang L, Su S, Zhang H, et al: Targeting Endothelial
Erk1/2-Akt axis as a regeneration strategy to bypass fibrosis
during chronic liver injury in mice. Mol Ther. 26:2779–2797. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Colucci S, Altamura S, Marques O, Dropmann
A, Horvat NK, Müdder K, Hammad S, Dooley S and Muckenthaler MU:
Liver sinusoidal endothelial cells suppress bone morphogenetic
protein 2 production in response to TGFβ pathway activation.
Hepatology. 74:2186–2200. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ding BS, Cao Z, Lis R, Nolan DJ, Guo P,
Simons M, Penfold ME, Shido K, Rabbany SY and Rafii S: Divergent
angiocrine signals from vascular niche balance liver regeneration
and fibrosis. Nature. 505:97–102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yan Z, Qu K, Zhang J, Huang Q, Qu P, Xu X,
Yuan P, Huang X, Shao Y, Liu C, et al: CD147 promotes liver
fibrosis progression via VEGF-A/VEGFR2 signalling-mediated
cross-talk between hepatocytes and sinusoidal endothelial cells.
Clin Sci (Lond). 129:699–710. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Su T and Iwakiri Y: Endothelial Leukocyte
Cell-Derived Chemotaxin 2/Tyrosine Kinase with immunoglobulin-like
and epidermal growth factor-like domains 1 signaling in liver
fibrosis. Hepatology. 72:347–349. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Murray PJ, Allen JE, Biswas SK, Fisher EA,
Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence
T, et al: Macrophage activation and polarization: Nomenclature and
experimental guidelines. Immunity. 41:14–20. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Arrese M, Cabrera D, Kalergis AM and
Feldstein AE: Innate Immunity and Inflammation in NAFLD/NASH. Dig
Dis Sci. 61:1294–1303. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Shetty S, Lalor PF and Adams DH: Liver
sinusoidal endothelial cells-gatekeepers of hepatic immunity. Nat
Rev Gastroenterol Hepatol. 15:555–567. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
You Q, Holt M, Yin H, Li G, Hu CJ and Ju
C: Role of hepatic resident and infiltrating macrophages in liver
repair after acute injury. Biochem Pharmacol. 86:836–843. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Miyao M, Kotani H, Ishida T, Kawai C,
Manabe S, Abiru H and Tamaki K: Pivotal role of liver sinusoidal
endothelial cells in NAFLD/NASH progression. Lab Invest.
95:1130–1144. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ford AJ, Jain G and Rajagopalan P:
Designing a fibrotic microenvironment to investigate changes in
human liver sinusoidal endothelial cell function. Acta Biomater.
24:220–227. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Kamada Y, Sato M, Kida S, Akita M,
Mizutani K, Fujii H, Sobajima T, Yoshida Y, Shinzaki S, Takamatsu
S, et al: N-acetylglucosaminyltransferase V exacerbates
concanavalin A-induced hepatitis in mice. Mol Med Rep.
11:3573–3584. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ni Y, Li JM, Liu MK, Zhang TT, Wang DP,
Zhou WH, Hu LZ and Lv WL: Pathological process of liver sinusoidal
endothelial cells in liver diseases. World J Gastroenterol.
23:7666–7677. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Connolly MK, Bedrosian AS, Malhotra A,
Henning JR, Ibrahim J, Vera V, Cieza-Rubio NE, Hassan BU, Pachter
HL, Cohen S, et al: In hepatic fibrosis, liver sinusoidal
endothelial cells acquire enhanced immunogenicity. J Immunol.
185:2200–2208. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bonder CS, Norman MU, Swain MG, Zbytnuik
LD, Yamanouchi J, Santamaria P, Ajuebor M, Salmi M, Jalkanen S and
Kubes P: Rules of recruitment for Th1 and Th2 lymphocytes in
inflamed liver: A role for alpha-4 integrin and vascular adhesion
protein-1. Immunity. 23:153–163. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Barrow F, Khan S, Wang H and Revelo XS:
The Emerging Role of B Cells in the Pathogenesis of NAFLD.
Hepatology. 74:2277–2286. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wehr A, Baeck C, Heymann F, Niemietz PM,
Hammerich L, Martin C, Zimmermann HW, Pack O, Gassler N, Hittatiya
K, et al: Chemokine receptor CXCR6-dependent hepatic NK T Cell
accumulation promotes inflammation and liver fibrosis. J Immunol.
190:5226–5236. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Neumann K, Rudolph C, Neumann C, Janke M,
Amsen D and Scheffold A: Liver sinusoidal endothelial cells induce
immunosuppressive IL-10-producing Th1 cells via the Notch pathway.
Eur J Immunol. 45:2008–2016. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Dauphinee SM and Karsan A:
Lipopolysaccharide signaling in endothelial cells. Lab Invest.
86:9–22. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Gola A, Dorrington MG, Speranza E, Sala C,
Shih RM, Radtke AJ, Wong HS, Baptista AP, Hernandez JM, Castellani
G, et al: Commensal-driven immune zonation of the liver promotes
host defence. Nature. 589:131–136. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Greuter T, Yaqoob U, Gan C, Jalan-Sakrikar
N, Kostallari E, Lu J, Gao J, Sun L, Liu M, Sehrawat TS, et al:
Mechanotransduction-induced glycolysis epigenetically regulates a
CXCL1-dominant angiocrine signaling program in liver sinusoidal
endothelial cells in vitro and in vivo. J Hepatol. 77:723–734.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Fujita K, Nozaki Y, Wada K, Yoneda M, Endo
H, Takahashi H, Iwasaki T, Inamori M, Abe Y, Kobayashi N, et al:
Effectiveness of antiplatelet drugs against experimental
non-alcoholic fatty liver disease. Gut. 57:1583–1591. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Malehmir M, Pfister D, Gallage S,
Szydlowska M, Inverso D, Kotsiliti E, Leone V, Peiseler M,
Surewaard BGJ, Rath D, et al: Platelet GPIbα is a mediator and
potential interventional target for NASH and subsequent liver
cancer. Nat Med. 25:641–655. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Lalor PF, Herbert J, Bicknell R and Adams
DH: Hepatic sinusoidal endothelium avidly binds platelets in an
integrin-dependent manner, leading to platelet and endothelial
activation and leukocyte recruitment. Am J Physiol Gastrointest
Liver Physiol. 304:G469–G478. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Joshi N, Kopec AK, Ray JL, Cline-Fedewa H,
Groeneveld DJ, Lisman T and Luyendyk JP: Von Willebrand factor
deficiency reduces liver fibrosis in mice. Toxicol Appl Pharmacol.
328:540592017. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Weyrich AS: Platelets: More than a sack of
glue. Hematology Am Soc Hematol Educ Program. 2014:400–403. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Chauhan A, Adams DH, Watson SP and Lalor
PF: Platelets: No longer bystanders in liver disease. Hepatology.
64:1774–1784. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ripoche J: Blood platelets and
inflammation: Their relationship with liver and digestive diseases.
Clin Res Hepatol Gastroenterol. 35:353–357. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Lisman T and Luyendyk JP: Platelets as
modulators of liver diseases. Semin Thromb Hemost. 44:114–125.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Brougham-Cook A, Kimmel HRC, Monckton CP,
Owen D, Khetani SR and Underhill GH: Engineered matrix
microenvironments reveal the heterogeneity of liver sinusoidal
endothelial cell phenotypic responses. APL Bioeng. 6:0461022022.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Olsen AL, Sackey BK, Marcinkiewicz C,
Boettiger D and Wells RG: Fibronectin extra domain-A promotes
hepatic stellate cell motility but not differentiation into
myofibroblasts. Gastroenterology. 142:928–937.e3. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Liu L, You Z, Yu H, Zhou L, Zhao H, Yan X,
Li D, Wang B, Zhu L, Xu Y, et al: Mechanotransduction-modulated
fibrotic microniches reveal the contribution of angiogenesis in
liver fibrosis. Nat Mater. 16:1252–1261. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Ruan B, Duan JL, Xu H, Tao KS, Han H, Dou
GR and Wang L: Capillarized Liver sinusoidal endothelial cells
undergo partial endothelial-mesenchymal transition to actively
deposit sinusoidal ECM in liver fibrosis. Front Cell Dev Biol.
9:6710812021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Iwakiri Y and Trebicka J: Portal
hypertension in cirrhosis: Pathophysiological mechanisms and
therapy. JHEP Rep. 3:1003162021. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Trebicka J, Reiberger T and Laleman W:
Gut-Liver Axis Links Portal Hypertension to Acute-on-Chronic Liver
Failure. Visc Med. 34:270–275. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Bravo M, Raurell I, Hide D,
Fernández-Iglesias A, Gil M, Barberá A, Salcedo MT, Augustin S,
Genescà J and Martell M: Restoration of liver sinusoidal cell
phenotypes by statins improves portal hypertension and histology in
rats with NASH. Sci Rep. 9:201832019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Iwakiri Y: Endothelial dysfunction in the
regulation of portal hypertension. Liver Int. 32:199–213. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Rockey DC and Chung JJ: Reduced nitric
oxide production by endothelial cells in cirrhotic rat liver:
Endothelial dysfunction in portal hypertension. Gastroenterology.
114:344–351. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Gracia-Sancho J, Marrone G and
Fernández-Iglesias A: Hepatic microcirculation and mechanisms of
portal hypertension. Nat Rev Gastroenterol Hepatol. 16:221–234.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Hammoutene A, Biquard L, Lasselin J,
Kheloufi M, Tanguy M, Vion AC, Mérian J, Colnot N, Loyer X, Tedgui
A, et al: A defect in endothelial autophagy occurs in patients with
non-alcoholic steatohepatitis and promotes inflammation and
fibrosis. J Hepatol. 72:528–538. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Maeso-Díaz R, Ortega-Ribera M, Lafoz E,
Lozano JJ, Baiges A, Francés R, Albillos A, Peralta C, García-Pagán
JC, Bosch J, et al: Aging influences hepatic microvascular biology
and liver fibrosis in advanced chronic liver disease. Aging Dis.
10:684–698. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Bosch J, Groszmann RJ and Shah VH:
Evolution in the understanding of the pathophysiological basis of
portal hypertension: How changes in paradigm are leading to
successful new treatments. J Hepatol. 62 (1 Suppl):S121–S130. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Medina J, Arroyo AG, Sánchez-Madrid F and
Moreno-Otero R: Angiogenesis in chronic inflammatory liver disease.
Hepatology. 39:1185–1195. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Hilscher MB, Sehrawat T, Arab JP, Zeng Z,
Gao J, Liu M, Kostallari E, Gao Y, Simonetto DA, Yaqoob U, et al:
Mechanical stretch increases expression of CXCL1 in liver
sinusoidal endothelial cells to recruit neutrophils, generate
sinusoidal microthombi, and promote portal hypertension.
Gastroenterology. 157:193–209.e9. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Yu Z, Guo J, Liu Y, Wang M, Liu Z, Gao Y
and Huang L: Nano delivery of simvastatin targets liver sinusoidal
endothelial cells to remodel tumor microenvironment for
hepatocellular carcinoma. J Nanobiotechnology. 20:92022. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Boyer-Diaz Z, Aristu-Zabalza P,
Andrés-Rozas M, Robert C, Ortega-Ribera M, Fernández-Iglesias A,
Broqua P, Junien JL, Wettstein G, Bosch J and Gracia-Sancho J:
Pan-PPAR agonist lanifibranor improves portal hypertension and
hepatic fibrosis in experimental advanced chronic liver disease. J
Hepatol. 74:1188–1199. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Hide D, Gil M, Andrade F, Rafael D,
Raurell I, Bravo M, Barberá A, Gracia-Sancho J, Vargas V, Augustin
S, et al: Simvastatin-loaded polymeric micelles are more effective
and less toxic than conventional statins in a pre-clinical model of
advanced chronic liver disease. Nanomedicine. 29:1022672020.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Zhang LF, Wang XH, Zhang CL, Lee J, Duan
BW, Xing L, Li L, Oh YK and Jiang HL: Sequential nano-penetrators
of capillarized liver sinusoids and extracellular matrix barriers
for liver fibrosis therapy. ACS Nano. 16:14029–14042. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Gu T, Shen B, Li B, Guo Y, Li F, Ma Z,
Chen L, Zhang Q, Qu Y, Dong H, et al: miR-30c inhibits angiogenesis
by targeting delta-like ligand 4 in liver sinusoidal endothelial
cell to attenuate liver fibrosis. FASEB J. 35:e215712021.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Mookerjee RP, Mehta G, Balasubramaniyan V,
Mohamed Fel Z, Davies N, Sharma V, Iwakiri Y and Jalan R: Hepatic
dimethylarginine-dimethylaminohydrolase1 is reduced in cirrhosis
and is a target for therapy in portal hypertension. J Hepatol.
62:325–331. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Kumar K, Dong Y, Kumar V, Almawash S and
Mahato RI: The use of micelles to deliver potential hedgehog
pathway inhibitor for the treatment of liver fibrosis.
Theranostics. 9:7537–7555. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhao S, Zhang Z, Qian L, Lin Q, Zhang C,
Shao J, Zhang F and Zheng S: Tetramethylpyrazine attenuates carbon
tetrachloride-caused liver injury and fibrogenesis and reduces
hepatic angiogenesis in rats. Biomed Pharmacother. 86:521–530.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Gao L, Yang X, Li Y, Wang Z, Wang S, Tan
S, Chen A, Cao P, Shao J, Zhang Z, et al: Curcumol inhibits
KLF5-dependent angiogenesis by blocking the ROS/ERK signaling in
liver sinusoidal endothelial cells. Life Sci. 264:1186962021.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Yang X, Wang Z, Kai J, Wang F, Jia Y, Wang
S, Tan S, Shen X, Chen A, Shao J, et al: Curcumol attenuates liver
sinusoidal endothelial cell angiogenesis via regulating
Glis-PROX1-HIF-1α in liver fibrosis. Cell Prolif. 53:e127622020.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Bae CR, Zhang H and Kwon YG: The
endothelial dysfunction blocker CU06-1004 ameliorates
choline-deficient L-amino acid diet-induced non-alcoholic
steatohepatitis in mice. PLoS One. 15:e02434972020. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Turaga RC, Satyanarayana G, Sharma M, Yang
JJ, Wang S, Liu C, Li S, Yang H, Grossniklaus H, Farris AB, et al:
Targeting integrin αvβ3 by a rationally designed protein for
chronic liver disease treatment. Commun Biol. 4:10872021.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Ye Q, Zhou Y, Zhao C, Xu L and Ping J:
Salidroside Inhibits CCl4-Induced Liver fibrosis in mice by
reducing activation and migration of HSC induced by liver
sinusoidal endothelial cell-derived exosomal SphK1. Front
Pharmacol. 12:6778102021. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Guo Q, Furuta K, Islam S, Caporarello N,
Kostallari E, Dielis K, Tschumperlin DJ, Hirsova P and Ibrahim SH:
Liver sinusoidal endothelial cell expressed vascular cell adhesion
molecule 1 promotes liver fibrosis. Front Immunol. 13:9832552022.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Ruart M, Chavarria L, Campreciós G,
Suárez-Herrera N, Montironi C, Guixé-Muntet S, Bosch J, Friedman
SL, Garcia-Pagán JC and Hernández-Gea V: Impaired endothelial
autophagy promotes liver fibrosis by aggravating the oxidative
stress response during acute liver injury. J Hepatol. 70:458–469.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Lin Y, Dong MQ, Liu ZM, Xu M, Huang ZH,
Liu HJ, Gao Y and Zhou WJ: A strategy of vascular-targeted therapy
for liver fibrosis. Hepatology. 76:660–675. 2022. View Article : Google Scholar : PubMed/NCBI
|