Introduction
Heart failure, which is characterized by increased
intracardiac pressures or decreased cardiac output, is a
life-threatening condition and is recognized as a global health
issue (1). In the early stages of
cardiovascular diseases like hypertension and aortic valve
stenosis, cardiac hypertrophy often occurs as a response to
increased pressure overload (2).
This hypertrophy initially boosts cardiac work efficiency. However,
if it persists under adverse conditions, it can lead to excessive
cell enlargement, cardiomyocyte death and impaired contraction
(3–5). These changes will ultimately result
in irreversible heart failure (6).
Despite significant progress in understanding pressure
overload-induced heart failure, its incidence is still increasing
(7,8). Currently, there are no effective
approved pharmacological treatments for this condition (9). Therefore, it is important to further
explore the mechanisms behind pressure overload-induced heart
failure, which will help identify new and promising therapeutic
targets.
The endoplasmic reticulum (ER) is a vital cellular
organelle with multiple functions, including protein synthesis,
folding and translocation, as well as the uptake and storage of
cellular calcium and the production of cellular lipids (10). There is mounting evidence that
pressure overload increases the protein synthesis and folding load
of the ER, resulting in the accumulation of misfolded and unfolded
proteins, which in turn disturbs ER homeostasis (11). These disturbances trigger ER
stress, also known as the unfolded protein response, which aims to
eliminate accumulated unfolded proteins and maintain ER homeostasis
(12,13). However, prolonged or severe ER
stress from continued pressure overload can escalate and lead to
increased levels of ER chaperones, like GRP78, and activation of
the pPERK/PERK-ATF4-CHOP signaling pathway (12). These changes can result in
myocardial apoptosis and potentially fatal heart failure (14,15).
Autophagy is a cellular process that helps maintain
intracellular homeostasis by removing damaged proteins and
organelles and recycling intracellular components through lysosomal
degradation (16–18). Autophagy also promotes protein
synthesis and energy production (19). Numerous studies have demonstrated
the role of autophagy in protecting against heart failure induced
by pressure overload (20–22). The mammalian target of rapamycin
(mTOR), a well-established negative regulator of autophagy, has
been increasingly recognized for its role in cardiac health.
Evidence suggests that inhibiting the mTOR signaling pathway can
enhance autophagy in cardiomyocytes. This enhancement in turn may
reduce ER stress-induced apoptosis in cardiomyocytes, offering
potential protection against heart failure (23–25).
Transient receptor potential cation (TRPC) channels,
comprising seven subfamilies (TRPC1-7), play a crucial role in
regulating the functions of cardiomyocytes, smooth muscle cells and
endothelial cells (26,27). Among these, TRPC6 is particularly
significant in the pathophysiology of cardiac hypertrophy and heart
failure (28,29). Larixyl acetate, extracted from
larch balsam, has been identified as a specific inhibitor of TRPC6.
It has been reported that larixyl acetate can effectively block the
Ca2+ entry and ionic current following the activation of
TRPC6 channels (30). Notably,
this compound demonstrates 12- and 5-fold selectivity over the
closely related TRPC3 and TRPC7 channels, respectively (30). Previous studies have shown that
larixyl acetate can prevent traumatic brain injury-induced systemic
endothelial dysfunction (31),
ozone or lipopolysaccharide-induced airway or lung injury (32,33)
and neuropathic pain resulting from spared nerve injury (34) through TPRC6-dependent mechanisms.
These findings underscore the potential of larixyl acetate as a
therapeutic TRPC6 inhibitor in various diseases. However, its
effects on cardiac hypertrophy and heart failure remain
unexplored.
Thus, the present study employed both in vivo
and in vitro experiments, along with pharmacological
interventions, to investigate this issue. While the calcineurin
A/NFAT pathway has been extensively studied (28,35),
the present research aimed to uncover other potential new
mechanisms. It is widely recognized that inhibiting the mTOR
signaling pathway can alleviate heart failure by promoting
autophagy (36). Furthermore,
recent research has demonstrated that the inhibition of TRPC6 can
reduce mTOR phosphorylation (37).
Therefore, it is reasonable to hypothesize that larixyl acetate, as
a TRPC6 inhibitor, might protect against pressure overload-induced
heart failure. Mechanistically, the protective role of larixyl
acetate may involve promoting autophagy through the inhibition of
the mTOR signaling pathway, which could subsequently reduce ER
stress-induced cardiomyocyte apoptosis.
Materials and methods
Reagents
Angiotensin II (Ang II) was purchased from
MilliporeSigma (Merck KGaA), and larixyl acetate and MHY1485 were
purchased from MedChemExpress.
Animals and treatments
A total of 70 male C57BL/6 mice (age, 8–10 weeks;
weight, 23–25 g) were purchased from GemPharmatech Co. Ltd. All
animal procedures were performed in accordance with the National
Institutes of Health (NIH) Guide for the Care and Use of Laboratory
Animals (38). All the
experimental procedures were approved by The Ethical Review Board
of Drum Tower Hospital of Nanjing University Medical School
(Nanjing, China; approval no. 2022AE01017). The mice were housed in
a controlled environment, with a temperature of 23°C (±2°C), a
relative humidity of 50–60%, and under a 12-h light/dark cycles.
Food and water were provided ad libitum. Throughout the
course of the experiment, eight mice reached the predetermined
humane endpoints and were subsequently euthanized. The remaining
mice were euthanized upon completion of the experiment, and their
heart tissues were harvested for further experimental analysis. The
method of euthanasia was rapid decapitation following anesthesia
induced with 5% isoflurane.
In the present study, specific humane endpoints were
established to ensure ethical treatment of the animals. These
endpoints were designed to minimize suffering and distress. They
included: i) Clinical signs: Animals showing severe signs of heart
failure, such as labored breathing, inability to remain upright or
decreased activity levels; ii) weight loss: A weight loss more than
20% of the baseline body weight of the animal; iii) failure to eat
or drink: Animals with a complete loss of appetite for 24 h or poor
appetite (<50% of the normal amount) for 3 days; and iv) disease
progression: Animals exhibiting rapid progression of disease
symptoms without any signs of recovery. These endpoints were
determined in consultation with veterinary staff and were in
compliance with the guidelines provided by The Experimental Animal
Ethics Committee of Drum Tower Hospital of Nanjing University
Medical School. Throughout the present study, animals were
monitored daily for these indicators, and any decisions for
euthanasia were made with the utmost consideration for the welfare
of the animals. Throughout the course of the experiment, eight mice
reached the predetermined humane endpoints and were subsequently
euthanized. The remaining mice were euthanized upon completion of
the experiment, and their heart tissues were harvested for further
experimental analysis.
Pressure overload model,
echocardiography and treatment
Transverse aortic constriction (TAC) or sham surgery
was performed according to previous studies with mild modifications
(39,40). Briefly, mice were anesthetized
using 5% isoflurane for induction, followed by maintenance with
1.5% isoflurane. The aortic arch was exposed through a median
incision at the upper sternal segment under a stereomicroscope. A
6–0 silk ligature was made between the right brachiocephalic and
left common carotid artery around a 27-gauge, and then the wire was
removed to generate a defined constriction of the aorta. As for the
sham group, an identical operation was performed except for the
ligature of the aorta. Doppler echocardiography was applied on mice
after TAC to assess the pressure gradient across the constriction.
A pressure gradient >45 mmHg was considered a successful
surgery. A total of four weeks after TAC, echocardiography was
performed and analyzed in a blinded manner to evaluate the cardiac
function using a Small Animal Ultrasound Imaging System (VEVO2100,
FUJIFILM VisualSonics, Inc.). The measurements were taken in M-mode
with a 30 MHz linear ultrasonic transducer.
Previous studies have reported that the
intraperitoneal (i.p.) injection of larixyl acetate at a dosage of
5 mg/kg/day for 1 week can safely and effectively inhibit TRPC6
(31,34). In the present study, the same
dosage of larixyl acetate was selected, with 5 mg/kg i.p.
administered daily after surgery to investigate its protective
effects against pressure overload-induced heart failure using a
4-week TAC animal model. It has been reported that i.p.
administration of 10 mg/kg MHY1485, an mTOR activator, is safe and
effective for activating the mTOR signaling pathway when
administered daily for 28 days (41). Therefore, this dosage was selected
to investigate the impact of the mTOR signaling pathway on the
efficacy of larixyl acetate.
Group and tissue harvest
A total mortality rate of 18.18% (8/44) was observed
in the TAC model. To investigate the effects of larixyl acetate on
pressure overload-induced heart failure, the number of mice in each
experimental group was adjusted as necessary. The goal was to
ensure that 10 mice/group successfully completed the 4-week TAC
procedure, providing robust data for analysis and sufficient tissue
samples for subsequent experiments. The final composition of each
group was as follows: i) Sham + Vehicle (n=10); ii) Sham + Larixyl
(n=10); iii) TAC + Vehicle (n=13; with 3 mice deceased); and iv)
TAC + Larixyl (n=12; with 2 mice deceased). Additionally, all spare
mice were euthanized at the conclusion of the experiment. For the
subsequent experiments, the mice were further divided in each
group. The hearts of 5 mice underwent apical perfusion with ice
saline and paraformaldehyde, followed by fixation, dehydration, OCT
embedding and preparation for tissue staining. In the remaining 5
mice, after blood removal, the ventricles were divided for western
blotting analysis and revese transcription quantitative polymerase
chain reaction (RT-qPCR).
In experiments testing the interaction of larixyl
acetate with the mTOR activator, MHY1485, it was aimed to have 8
mice/group complete the 4-week TAC procedure. The groups were: i)
TAC + Larixyl (n=9; with 1 mouse deceased); and ii) TAC + Larixyl +
MHY (n=10; with 2 mice deceased). Only one additional spare mouse
was euthanized post-experiment.
Hematoxylin-eosin staining
The heart tissues embedded in OCT were stored at
−80°C. Frozen heart sections (10 µm) were prepared in a cryostat at
−20°C. The sections were then recovered to room temperature before
staining and then rinsed 3×5 min with ddH2O, incubated
for 2 min with hematoxylin staining solution (Beijing Solarbio
Science & Technology Co., Ltd.; H8070) followed by 1%
hydrochloric acid alcohol for a few sec, and then rinsed with
ddH2O. After that, the sections were soaked in eosin
staining solution (Beijing Solarbio Science & Technology Co.,
Ltd., G1100) for 1 min and then rinsed with ddH2O, and then
dehydrated with gradient alcohol, and transparentized with dimethyl
benzene, and sealed the sections with sealing cement. All
incubations were carried out at room temperature. The images were
captured under an Olympus BX43 light microscope (Olympus
Corporation).
Sirius red staining
The heart tissues embedded in OCT were stored at
−80°C. Frozen heart sections (10 µm) were prepared in a cryostat at
−20°C. The sections were then recovered to room temperature before
staining and then rinsed 3×5 min with ddH2O, and the
staining was performed according to the manual of the modified
Sirius Red Stain Kit (Beijing Solarbio Science & Technology
Co., Ltd; cat. no. G1472) at room temperature. Briefly, heart
frozen sections were rinsed 3×5 min with ddH2O and then
incubated with iron hematoxylin staining solution for 2 min and
rinsed with tap water for 5 min. Next, sections were incubated with
sirius red staining solution for 30 min, rinsed with running water
and then dehydrated with gradient alcohol and transparentized with
dimethyl benzene. Finally, the sections were sealed with sealing
cement. The images were captured by Olympus BX43 light microscope
(Olympus Corporation). The collagen volume fraction of the left
ventricular (LV) was analyzed with Fiji software (Version 1.54f;
NIH) by using the interest grayscale threshold analysis and was
calculated as sirius red staining area divided by total area. A
total of three sections/heart and five mice/group were sampled. The
images were captured and analyzed by two individuals blinded to the
experimental group. Average data were used to represent the data
for each mouse.
TUNEL staining
The heart tissues embedded in OCT were stored at
−80°C. Frozen heart sections (10 µm) were prepared in a cryostat at
−20°C. The sections were then recovered to room temperature before
staining and then rinsed 3×5 min with ddH2O, and the
staining was performed with a One-step TUNEL cell apoptosis
detection kit (Beyotime Institute of Biotechnology; cat. no. C1090)
according to the manufacturer's instructions. Briefly, heart frozen
sections were rinsed 2×10 min with phosphate buffer saline (PBS),
then permeabilized by 0.3% Triton X-100 in PBS for 5 min at room
temperature, and again rinsed 2×10 min with PBS. Next, sections
were incubated with TUNEL solution at 37°C for 60 min, rinsed 3×10
min with PBS and then sealed with anti-fluorescence quenching seal
liquid. The images were captured by the Olympus FV3000 confocal
microscope (Olympus Corporation) with 550 nm excitation light and
analyzed with Fiji software (Version 1.54f; NIH). A total of three
random fields from the LV-free wall/section, three sections/mouse
and five mice/group were sampled. The images were captured and
analyzed blindly. Average data were used to represent the data for
each mouse.
Cell culture
The rat cardiomyocyte line, H9c2 was cultured in
DMEM (cat. no. 319-015; Wisent, Inc.) supplemented with 10% fetal
bovine serum (cat. no. 085-150; Wisent, Inc.) and 1%
penicillin/streptomycin (cat. no. 15140-122; Gibco) at 37°C.
Previous studies have demonstrated that Ang II is capable of
activating TRPC6 and the mTOR signaling pathway, leading to
cardiomyocyte hypertrophy, ER stress and apoptosis (23,28,42–44).
Therefore, Ang II (100 nM) was administered to H9c2 cells for 72 h
at 37°C to establish a cell model and to confirm whether larixyl
acetate could provide protective effects by upregulating autophagy
via inhibiting the mTOR signaling pathway and reducing ER stress.
Groups were incubated with larixyl acetate (5 µM) with or without
MHY1485 (10 µM) at 37°C to block TRPC6 only or to activate mTOR
signaling simultaneously, 30 min before Ang II administration.
Cellular size, and the expression of cardiac hypertrophy-related
genes, ER stress markers, autophagy-related proteins and
apoptosis-related proteins were then assessed. The specified dosage
for administering larixyl acetate (30,45)
and MHY1485 (46) were determined
based on previous research.
Wheat germ agglutinin (WGA)
staining
WGA staining was used to measure the cardiomyocyte
crossectional area. Briefly, heart tissues embedded in OCT were
stored at −80°C. Frozen heart sections (10 µm) were prepared in a
cryostat at −20°C. The sections were then recovered to room
temperature before staining and the sections were then rinsed for
3×5 min with PBS. Next, sections were incubated with 1 ug/ml WGA
(cat. no. W6748; Invitrogen; Thermo Fisher Scientific, Inc.) at
37°C for 10 min, rinsed 3×5 min with PBS and then sealed with
anti-fluorescence quenching seal liquid. The images were taken by
an Olympus FV3000 confocal microscope (Olympus Corporation) with
488 nm excitation light and analyzed with Fiji software (Version
1.54f; NIH). A total of three random fields from the LV-free
wall/section, three sections/mouse and five mice/group were
sampled. The images were captured and analyzed blindly. Average
data was used to represent the data for each mouse.
Phalloidin staining
Phalloidin staining was used to measure the cellular
area of H9c2 cells. Briefly, cells were fixed with 4%
paraformaldehyde for 10 min at room temperature, rinsed 3×5 min
with PBS and further permeabilized by 0.3% Triton X-100 in PBS for
5 min. Next, cells were rinsed 3×5 min with PBS and then incubated
with 0.005 unit/µl phalloidin (A12381, Invitrogen; Thermo Fisher
Scientific, Inc.) for 60 min at room temperature, rinsed 3×5 min
with PBS and then sealed with anti-fluorescence quenching seal
liquid. The images were taken by an Olympus FV3000 confocal
microscope (Olympus Corporation) with 594 nm excitation light and
analyzed with Fiji software (Version 1.54f; NIH). A total of five
random fields from each coverslip and five coverslips/group were
sampled. The images were captured and analyzed in a blind manner.
Average data were used to represent the data for each
coverslip.
Western blotting
Proteins in LV tissues or H9c2 cells were extracted
using RIPA lysis buffer (cat. no. P0013B; Beyotime Institute of
Biotechnology) containing proteinase and phosphatase inhibitors.
BCA Protein Assay Kit (cat. no. 23225; Thermo Fisher Scientific,
Inc.) was used. A total of 20 µg protein samples were separated by
SDS/PAGE on 4–20% gels and then transferred to PVDF membranes. The
membranes were blocked in 5% defatted milk for 1 h at room
temperature and then incubated with primary antibodies including
TRPC6 (1:500; cat. no. 18236-1AP; Proteintech Group, Inc.), GRP78
(1:1,000; cat. no. sc-13539; Santa Cruz Biotechnology, Inc.), pPERK
(1:500; cat. no. sc-32577; Santa Cruz Biotechnology, Inc.), PERK
(1:1,000; cat. no. sc-377400; Santa Cruz Biotechnology, Inc.), ATF4
(1:1,000; cat. no. sc-390063; Santa Cruz Biotechnology, Inc.), CHOP
(1:1,000; cat. no. sc-7351; Santa Cruz Biotechnology, Inc.), pmTOR
(1:500; cat. no. 2971; CST Biological Reagents Co., Ltd.), mTOR
(1:500; cat. no. 2972; CST Biological Reagents Co., Ltd.), P62
(1:500; cat. no. ab155686; Abcam), LC3B (1:1,000; cat. no. ab51520;
Abcam), Bcl-2 (1:500; cat. no. 3498; CST Biological Reagents Co.,
Ltd.), Bax (1:1,000; cat. no. ab32503; Abcam), cleaved caspase-3
(1:100; cat. no. 9664; CST Biological Reagents Co., Ltd.) and GAPDH
(1:2,000; cat. no. ab8245; Abcam) at 4°C overnight. The membranes
were rinsed 3×5 min with TBST (0.1% Tween; cat. no. ST825; Beyotime
Institute of Biotechnology) and then incubated with goat anti-mouse
(1:2,000; cat. no. A0216), goat anti-rat (1:2,000; cat. no. A0192),
or goat anti-rabbit (1:2,000; cat. no. A0208) HRP-conjugated
secondary antibodies (all from Beyotime Institute of Biotechnology)
according to primary antibodies at room temperature for 1 h, rinsed
3×5 min with TBST and detected by enhanced chemiluminescence
solutions (cat. no. KGC4602; Nanjing KeyGen Biotech Co., Ltd.). The
protein expression level was quantified using Fiji software
(Version 1.54f; NIH) with GAPDH as the loading control.
RNA isolation and RT-qPCR
Total mRNA in LV tissues or H9c2 cells was extracted
using Trizol reagent (cat. no. 15596026; Invitrogen; Thermo Fisher
Scientific, Inc.). A total of 1 µg total RNA was used to reverse
transcribe into cDNA using HiScript III RT SuperMix for qPCR (cat.
no. R323-01; Vazyme Biotech Co., Ltd.). The obtained cDNA was mixed
with gene-specific primers (Table
I) and ChamQ Universal SYBR qPCR Master Mix (cat. no. Q711-02;
Vazyme Biotech Co., Ltd.) was used for RT-qPCR in light cycler 480
instruments (Roche Diagnostics). The thermocycling conditions were
as follows: Initial denaturation at 95°C for 10 m, followed by 40
cycles of denaturation at 95°C for 10 sec and annealing/extension
at 60°C for 30 sec. The cycle time values were standardized to
GAPDH of the same sample and 2−ΔΔCq was used to
represent relative quantity (47).
 | Table I.Primers for qPCR. |
Table I.
Primers for qPCR.
| Genes | Forward sequence
(5′-3′) | Reverse sequence
(5′-3′) |
|---|
| Mouse
Anp |
GCTTCCAGGCCATATTGGAG |
GGGGGCATGACCTCATCTT |
| Mouse
Bnp |
GAGGTCACTCCTATCCTCTGG |
GCCATTTCCTCCGACTTTTCTC |
| Mouse
Myh7 |
CAACCTGTCCAAGTTCCGCA |
TACTCCTCATTCAGGCCCTTG |
| Mouse
Col1a1 |
TGCTAACGTGGTTCGTGACCGT |
ACATCTTGAGGTCGCGGCATGT |
| Mouse
Col3a1 |
ACGTAAGCACTGGTGGACAG |
CCGGCTGGAAAGAAGTCTGA |
| Mouse
Gapdh |
ATGTGTCCGTCGTGGATCTG |
AGTTGGGATAGGGCCTCTCTT |
| Rat Anp |
AAAGCAAACTGAGGGCTCTGCTCG |
TTCGGTACCGGAAGCTGTTGCA |
| Rat Bnp |
TGCCCCAGATGATTCTGCTC |
TGTAGGGCCTTGGTCCTTTG |
| Rat
Myh7 |
AGTTCGGGCGAGTCAAAGATG |
CAGGTTGTCTTGTTCCGCCT |
| Rat
Gapdh |
ACTCTACCCACGGCAAGTTC |
TGGGTTTCCCGTTGATGACC |
Statistical analysis
All data were presented as means ± SD. An unpaired
t-test was utilized to evaluate the statistical significance
between two groups. For multiple comparisons, one-way ANOVA
followed by Tukey's or Welch ANOVA followed by Dunnett's post hoc
test was employed, contingent on the results of the homogeneity
test of variance. The analysis of data and the creation of figures
were carried out with GraphPad Prism 9.0 (Dotmatics). P<0.05 was
considered to indicate a statistically significant difference.
Results
Larixyl acetate attenuates pressure
overload-induced heart failure
Compared with the Sham + Vehicle group, the TAC +
Vehicle group exhibited significant increases in heart weight
(HW)/body weight (BW; P<0.001; t=12.45, df=36), HW/tibia length
(TL; P<0.001; t=13.50, df=36; Fig.
1B), LV end-systolic diameter (LVESd; P<0.001; t=7.11,
df=36) and LV end-diastolic diameter (LVEDd; P<0.001; t=10.16,
df=36) (Fig. 1D). Whereas the
fractional shortening (FS; P<0.001; t=6.99, df=36) and ejection
fraction (EF) were significantly decreased (P<0.001; t=7.57,
df=36; Fig. 1D). In addition, TAC
induced upregulation of cardiac hypertrophy-related genes, such as
atrial natriuretic peptide (Anp; P=0.0016; t=11.14,
df=4.141), B-type natriuretic peptide (Bnp; P<0.001;
t=27.13, df=16) and β-myosin heavy chain (Mh7; P<0.001;
t=13.77, df=16; Fig. 1E). However,
treatment with larixyl acetate for 4 weeks reversed the above
alterations in the TAC + Larixyl group compared with the TAC +
Vehicle group.
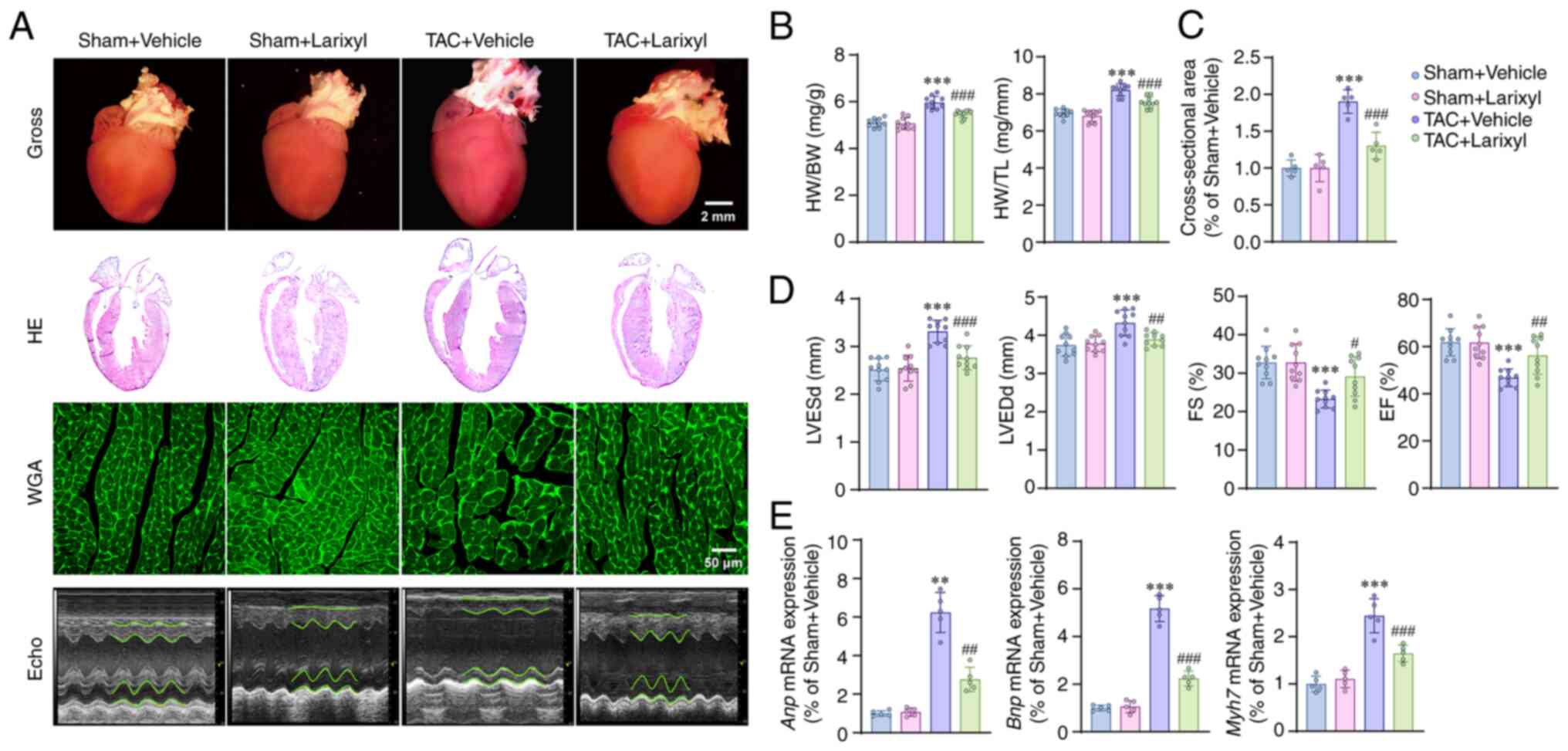 | Figure 1.Larixyl acetate attenuates pressure
overload-induced heart failure. (A) Representative images of gross
morphology of hearts, HE staining, WGA staining and transthoracic
echocardiography of the hearts in the Sham + Vehicle, Sham +
Larixyl, TAC + Vehicle and TAC + Larixyl groups at 4 weeks after
TAC (n=10/group). (B) HW/BW, HW/TL ratios in the Sham + Vehicle,
Sham + Larixyl, TAC + Vehicle and TAC + Larixyl groups at 4 weeks
after TAC (n=10/group). (C and D) Quantitative results of the
cross-sectional area of cardiomyocyte (n=5/group), LVESd, LVEDd, FS
and EF of the hearts (n=10/group) from indicated groups. (E)
Relative mRNA levels of hypertrophy marker genes, Anp, Bnp
and Myh7 in the hearts from the indicated groups
(n=5/group). Data are presented as the mean ± SD. For Anp
mRNA expression, the homogeneity of variance test showed
significant differences, therefore, these data were analyzed using
Welch's ANOVA. For the rest of the datasets, where the homogeneity
of variance test did not reveal significant differences, one-way
ANOVA was employed for analysis. **P<0.01, ***P<0.001 vs. the
Sham + Vehicle group; #P<0.05, ##P<0.01
and ###P<0.001 vs. the TAC + Vehicle group. HE,
hematoxylin-eosin; WGA, wheat germ agglutinin; TAC, transverse
aortic constriction; HW, heart weigh; BW, body weight; TL, tibia
length; LVESd, left ventricular end-systolic diameter; LVEDd, left
ventricular end-diastolic diameter; FS, fractional shortening; EF,
ejection fraction; Anp, atrial natriuretic peptide;
Bnp, B-type natriuretic peptide; Myh7, β-myosin heavy
chain. |
The expression of TRPC6 among groups was also
detected and the results showed that 4 weeks of TAC induced a
significant upregulation of TRPC6 expression when compared with the
Sham + Vehicle group (P<0.001; t=9.94, df=8; Fig. S1A). Whereas the administration of
larixyl acetate reversed the aforementioned alteration.
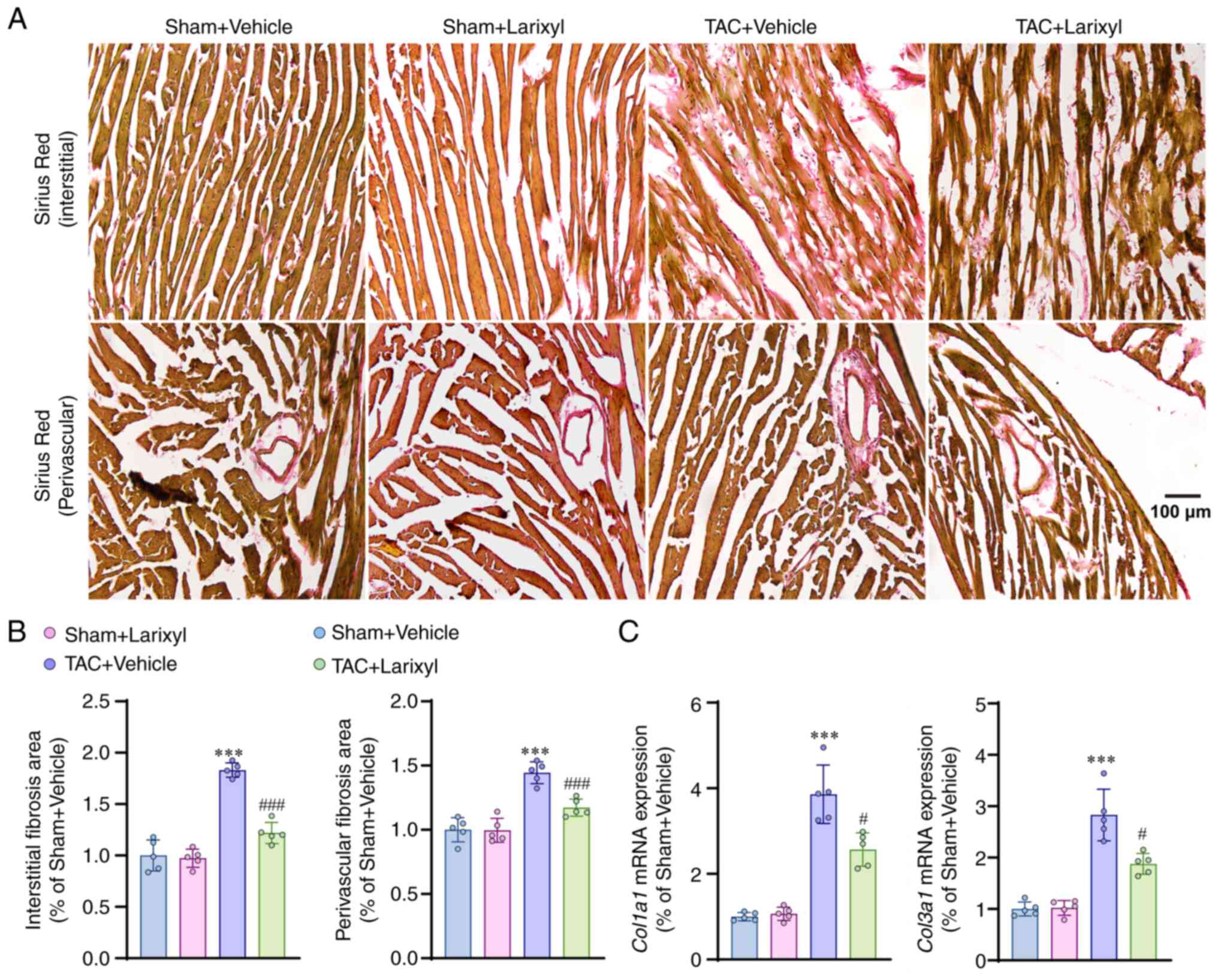
Larixyl acetate ameliorates pressure
overload-induced cardiac fibrosis
Next, the effect of larixyl acetate on cardiac
fibrosis, an important pathophysiological mechanism of heart
failure, was evaluated. The findings revealed that TAC induced a
substantial interstitial (P<0.001; t=17.30, df=16) and
perivascular (P<0.001; t=11.63, df=16) fibrosis of the heart, as
observed through sirus red staining when compared with the Sham +
Vehicle group (Fig. 2A and B).
Additionally, a significant upregulation of fibrosis-related genes,
such as collagen type I α1 (Col1a1; P=0.0033; t=9.27,
df=4.155) and collagen type III α1 (Col3a1; P=0.0025;
t=7.88, df=4.596), in the TAC + Vehicle group (Fig. 2C) was observed. The results also
demonstrated that the effects of larixyl acetate on fibrosis were
consistent with the observations made during cardiac hypertrophy
and heart failure. Specifically, the administration of larixyl
acetate effectively inhibited the development of cardiac fibrosis
after TAC.
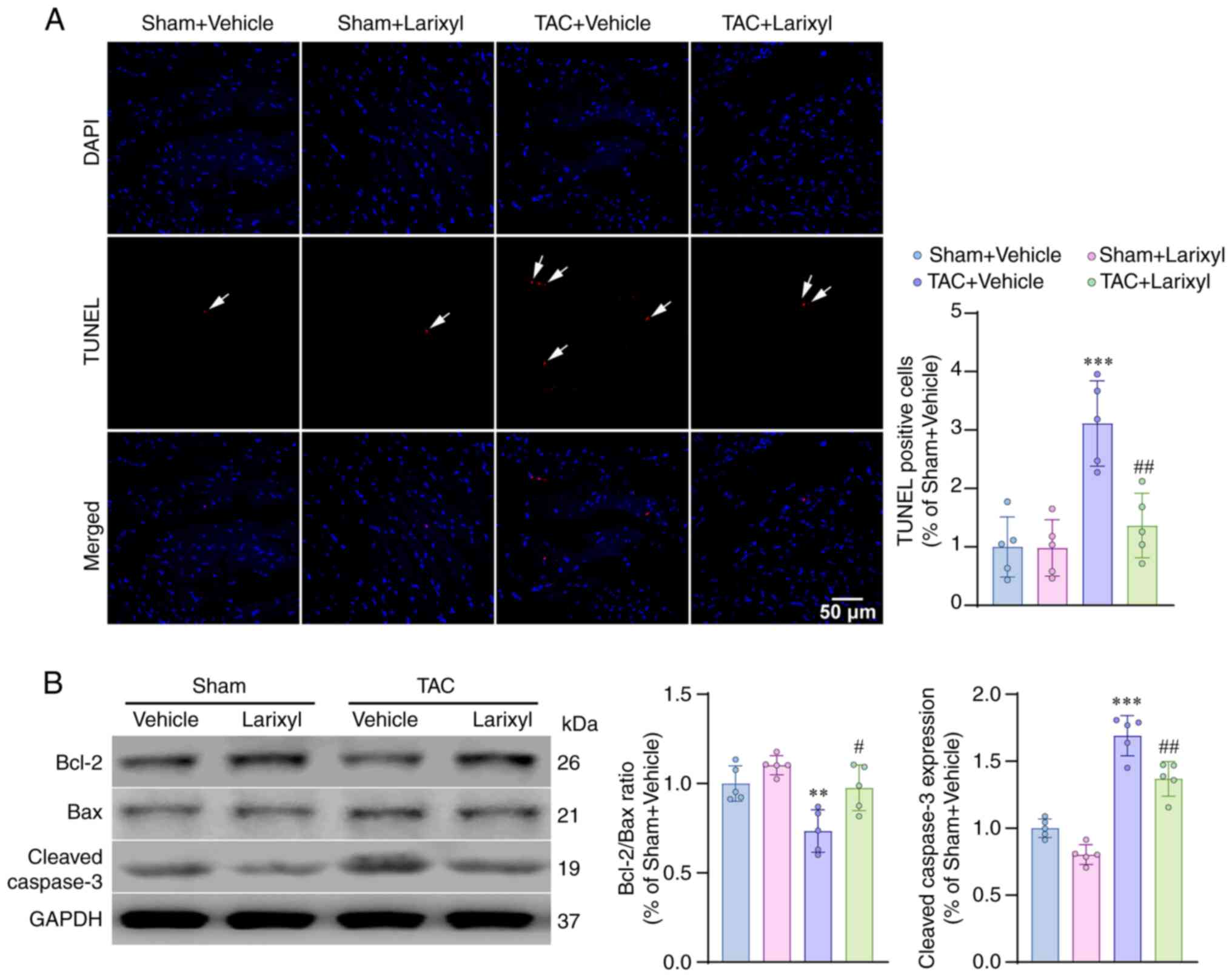
Larixyl acetate decreases pressure
overload-induced cardiomyocyte apoptosis
TUNEL staining revealed a significant increase in
apoptotic cells in the TAC + Vehicle group compared with the Sham +
Vehicle group (P<0.001; t=8.18, df=16; Fig. 3A). Western blotting analysis
demonstrated that TAC downregulated the anti-apoptotic Bcl2/Bax
pathway (P=0.0048; t=5.70, df=16) and upregulated the pro-apoptotic
cleaved caspase-3 pathway (P<0.001; t=13.89, df=16; Fig. 3B). These results indicated that TAC
induced cardiomyocyte apoptosis by disrupting the balance between
anti- and pro-apoptotic pathways. Treatment with larixyl acetate
via i.p. injection for 4 weeks effectively reversed these
impairments.
Larixyl acetate inhibits ER stress and
the mTOR signaling pathway and promotes autophagy
To elucidate the mechanisms underlying the
protective effect of larixyl acetate against cardiomyocyte
apoptosis and heart failure, the involvement of ER stress, the mTOR
signaling pathway and autophagy was investited. Firstly, it was
confirmed that 4 weeks of TAC significantly upregulated the
expression of ER stress-related proteins, such as GRP78
(P<0.001; t=7.30, df=16), pPERK/PERP (P<0.001; t=11.29,
df=16), ATF4 (P<0.001; t=12.49, df=16) and CHOP (P<0.001;
t=12.39, df=16), which are closely related to promoting
cardiomyocyte apoptosis, in the TAC + Vehicle group compared with
the Sham + Vehicle group (Fig.
4A). Secondly, it was found that TAC increased the level of
pmTOR/mTOR (P 0.001; t=9.61, df=16) in the TAC + Vehicle group,
indicating the activation of the mTOR signaling pathway (Fig. 4B). Thirdly, whether mTOR activation
was accompanied by inhibition of autophagy was investigated by
assessing the autophagy markers P62 and the ratio of light chain 3B
II to light chain 3B I (LC3B II/I). The results showed that the
expression level of P62 (P=0.0047; t=5.71, df=16) was increased,
while LC3B II/I (P<0.001; t=8.40, df=16) was decreased in the
TAC + Vehicle group compared with the Sham + Vehicle group
(Fig. 4B), suggesting that
autophagy was inhibited. Treatment with larixyl acetate effectively
reversed these alterations.
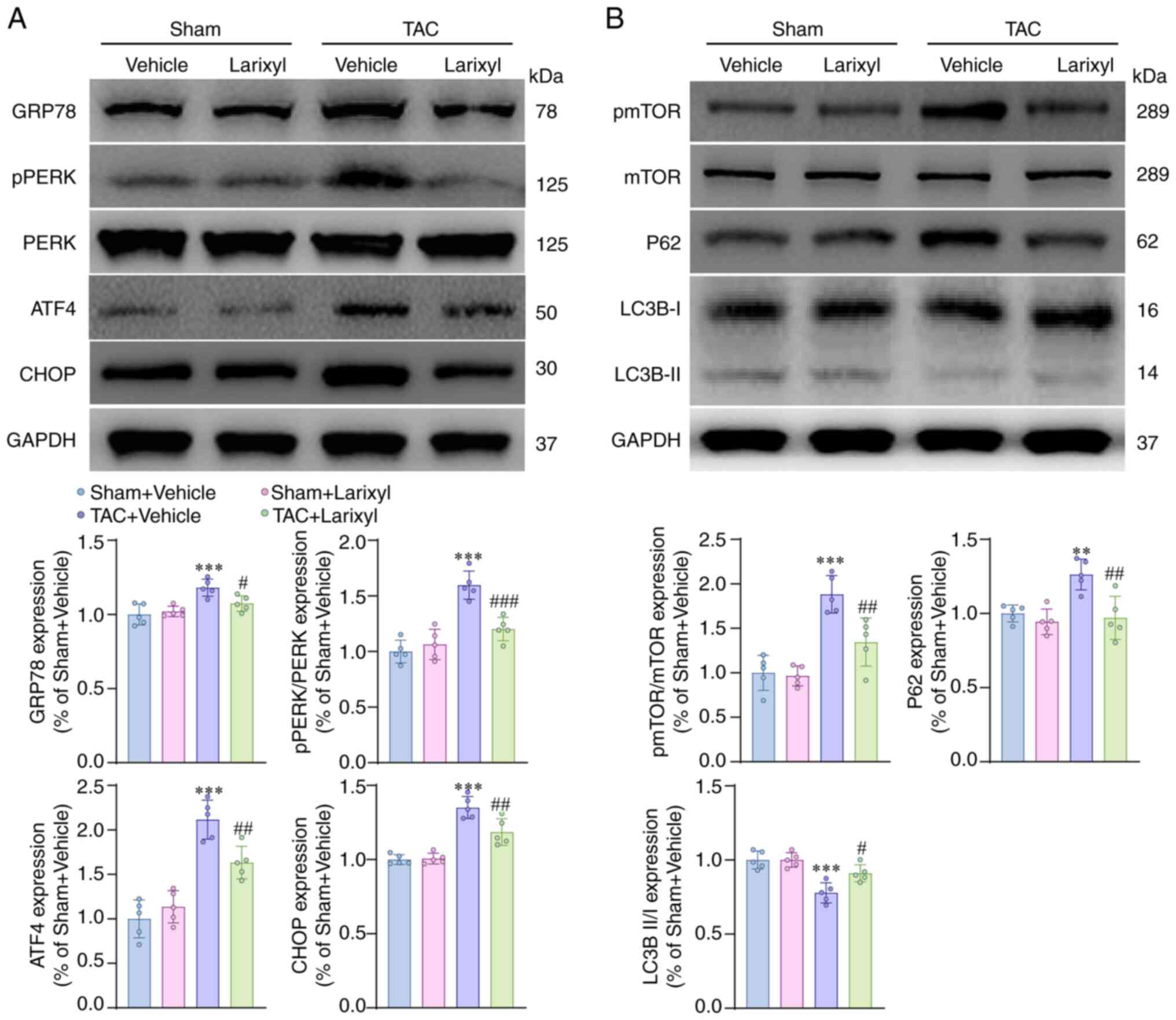 | Figure 4.Larixyl acetate inhibits ER stress
and mTOR signal pathway and promotes autophagy. (A) Relative
protein levels of ER stress markers, GRP78, PERK, ATF4 and CHOP in
the heart from the Sham + Vehicle, Sham + Larixyl, TAC + Vehicle
and TAC + Larixyl groups at 4 weeks after TAC (n=5/group). (B)
Relative protein levels of pmTOR/mTOR and autophagy markers, P62
and LC3BII/I in the heart from indicated groups (n=5/group). The
data are presented as the mean ± SD. The homogeneity of variance
test for the data did not reveal significant differences,
therefore, the data were analyzed using one-way ANOVA. **P<0.01
and ***P<0.001 vs. the Sham + Vehicle group;
#P<0.05, ##P<0.01 and
###P<0.001 vs. the TAC + Vehicle group. ER,
endoplasmic reticulum; mTOR, mammalian target of rapamycin; TAC,
transverse aortic constriction. |
Larixyl acetate alleviates Ang
II-induced H9c2 cell hypertrophy, ER stress and apoptosis while
promoting autophagy; mTOR activation reverses these alterations,
except for cellular hypertrophy
Compared with the Con + Vehicle group, the Ang II +
Vehicle group cells showed significant hypertrophy, as evidenced by
an enlarged cellular area (P<0.001; t=17.91, df=16),
upregulation of Anp (P<0.001; t=13.58, df=16), Bnp
(P<0.001; t=14.28, df=16) and Myh7 (P<0.001; t=16.75,
df=16) genes (Fig. 5A).
Additionally, Ang II significantly increased the expression levels
of GRP78 (P<0.001; t=10.36, df=16), pPERK/PERK (P<0.001;
t=13.08, df=16), ATF4 (P<0.001; t=13.31, df=16), CHOP
(P<0.001; t=7.71, df=16), pmTOR/mTOR (P=0.0056; t=5.84,
df=5.569), P62 (P=0.0033; t=5.97, df=16) and cleaved caspase-3
(P<0.001; t=7.80, df=16), while decreasing the ratio of
LC3B-II/I (P<0.001; t=8.87, df=16) and Bcl-2/Bax (P<0.001;
t=10.14, df=16; Fig. 5B-D). These
results suggested that Ang II increases ER stress, mTOR signaling
pathway and apoptosis, while inhibiting autophagy. Preincubation
with larixyl acetate alleviated the above impairments, whereas the
co-incubation with MHY1485 abolished the effects of larixyl
acetate, except for cellular hypertrophy. These findings indicated
that larixyl acetate can enhance autophagy and reduce ER stress and
apoptosis by inhibiting the mTOR signal pathway, thereby playing a
protective role in cardiac dysfunction.
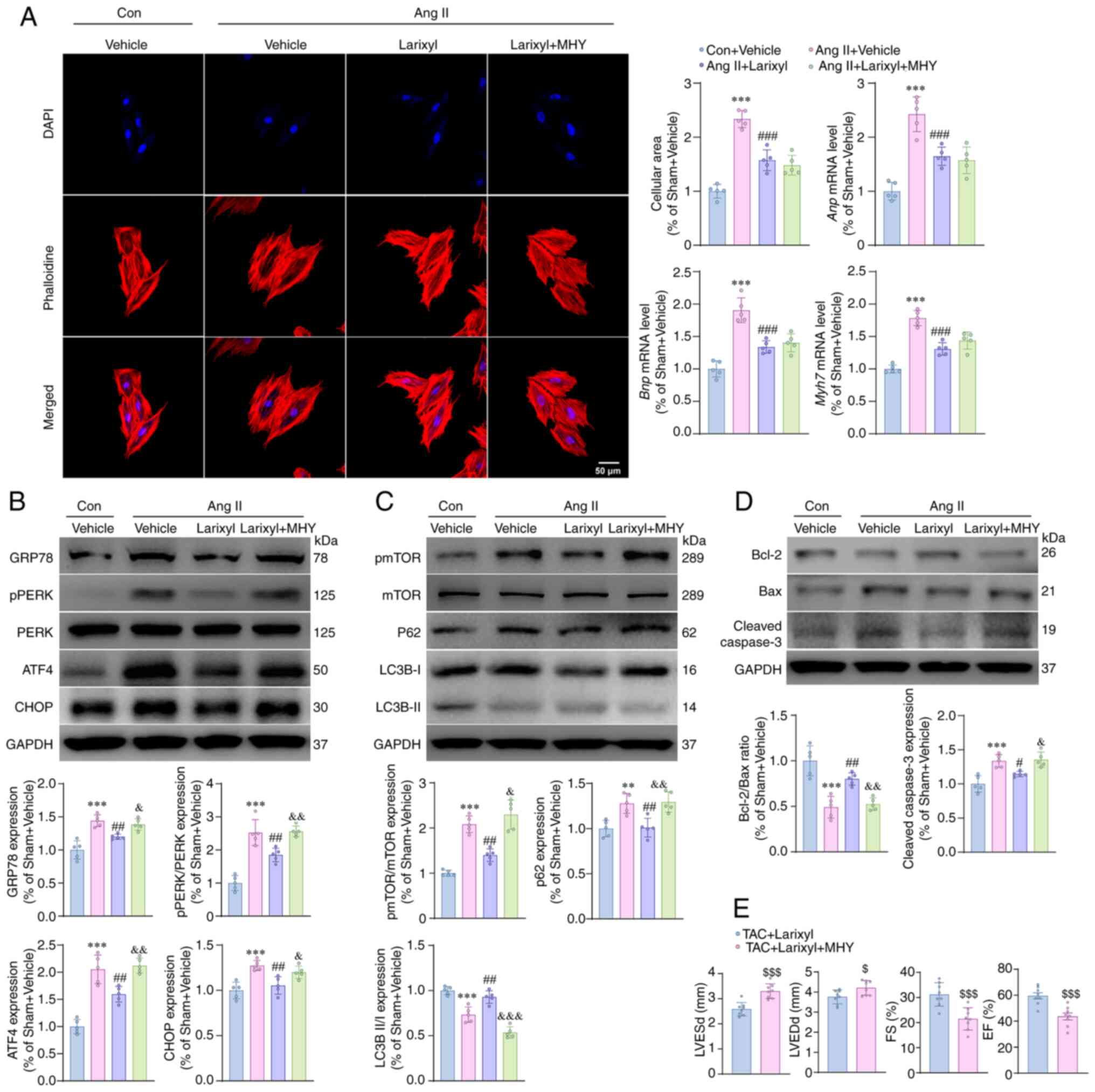 | Figure 5.Activation of the mTOR signal
reverses the protective effects of larixyl acetate in vitro
and in vivo. (A) Representative images of phalloidine
staining (left) of H9c2 cells from the Con + vehicle, Ang II +
Vehicle, Ang II + Larixyl and Ang II + Larixyl + MYH groups at 72 h
after drugs treatment. Quantitative results (right) of cellular
area (n=5/group) and relative mRNA levels of hypertrophy markers of
cells (n=5/group) from the indicated groups. (B) Relative protein
levels of ER stress markers in cells from the indicated groups
(n=5/group). (C) Relative protein levels of pmTOR/mTOR and
autophagy markers in the heart from indicated groups (n=5/group).
(D) Relative protein levels of apoptosis markers in cells from the
indicated groups (n=5/group). (E) EF of the hearts from indicated
groups evaluated by transthoracic echocardiograph (n=8/group). Data
are presented as the mean ± SD. For pmTOR/mTOR expression, the
homogeneity of variance test showed significant differences,
therefoere, the data were analyzed using Welch ANOVA. The
homogeneity of variance test for the other data did not reveal
significant differences, thus, the data were analyzed using one-way
ANOVA followed. **P<0.01 and ***P<0.001 vs. the Con + Vehicle
group; #P<0.05, ##P<0.01 and
###P<0.001 vs. the Ang II + Vehicle group;
&P<0.05, &&P<0.01 and
&&&P<0.001 vs. the Ang II + Larixyl
group; $P<0.05 and $$$P<0.001 vs. the
TAC + Larixyl group. mTOR, mammalian target of rapamycin; Con,
control; Ang II, angiotensin II; MHY, MYH1485; ER, endoplasmic
reticulum. |
Additionally, the expression of TRPC6 across
different groups was assessed. The results showed that Ang II
significantly upregulated TRPC6 expression compared with the Con +
Vehicle group (P<0.001; t=9.41, df=8; Fig. S1B). The administration of larixyl
acetate, with or without MHY1485, reversed this alteration. This
finding aligns with the in vitro experiments, demonstrating
that not only mechanical but also chemical activation of TRPC6 can
further enhance its expression. Since MHY1485 did not have an
additional influence, it suggests that the mTOR pathway is not
involved in this feedback loop.
Activation of the mTOR signal
abrogates the protective effects of larixyl acetate in vitro
Based on the evidence presented, it was hypothesized
that mTOR activation may influence the protective effects of
larixyl acetate in TAC-induced heart failure. Accordingly, larixyl
acetate was injected i.p. at a dose of 5 mg/kg, once/day, with or
without the mTOR activator MHY1485 at 10 mg/kg, also once/day. This
treatment was continued for 4 weeks following TAC to test the
hypothesis. At the end of the TAC model, heart function was
evaluated using transthoracic echocardiography. The results showed
that LVESd (P<0.001; t=5.15, df=14) and LVEDd (P=0.0236; t=2.54,
df=14) were significantly increased, whereas FS (P<0.001;
t=4.27, df=14) and EF (P<0.001; t=4.40, df=14) were
significantly decreased in the TAC + Larixyl + MHY group when
compared with the TAC + Larixyl group (Fig. 5E). These findings suggested that
inhibition of the mTOR signal pathway is essential for the
protective effects of larixyl acetate in maintaining cardiac
function under pressure overload stress.
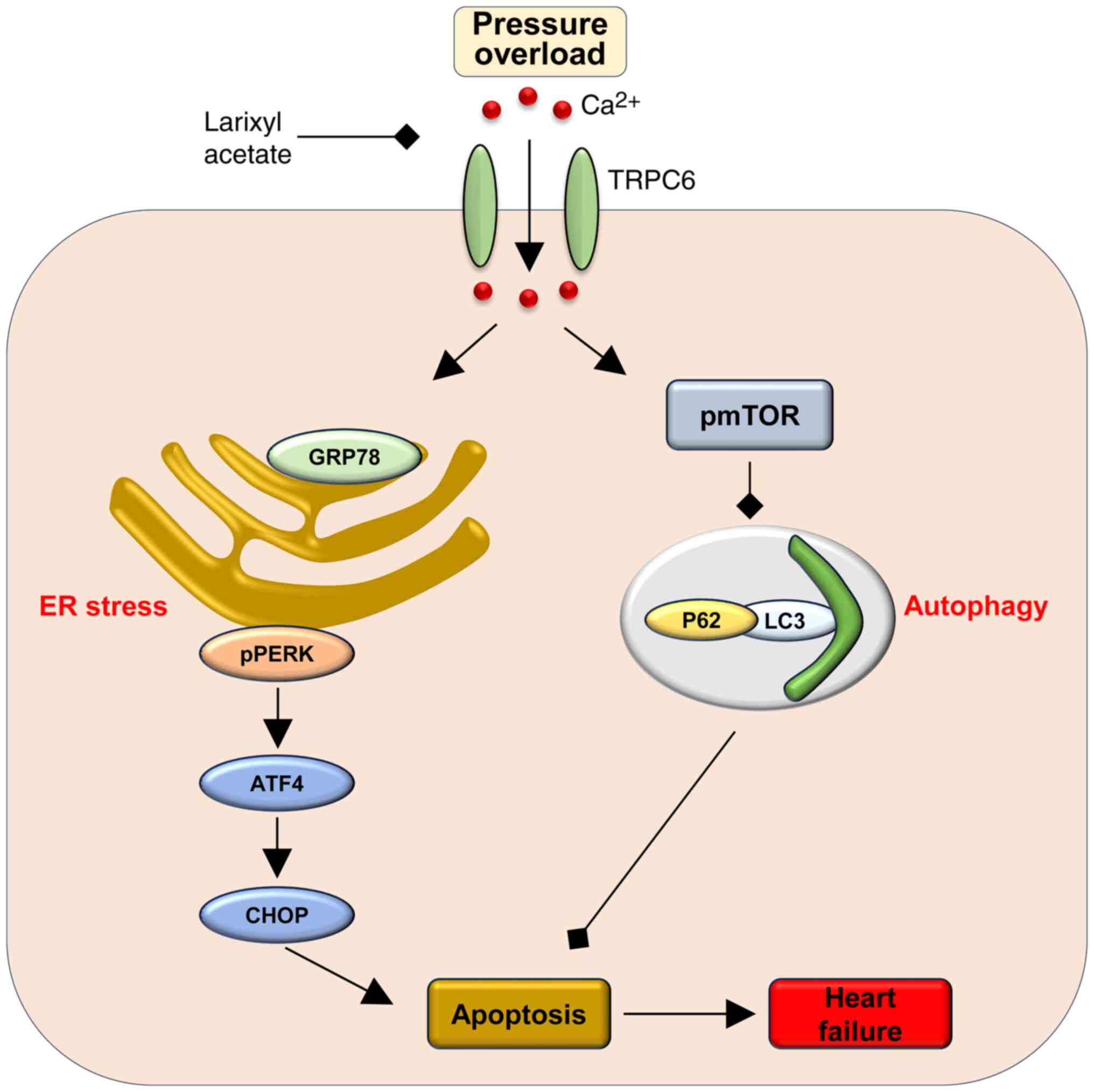
Discussion
It is widely recognized that persistent pressure
overload resulting from pathological conditions can induce heart
failure, which is a leading cause of morbidity and mortality
(2,8). To improve the understanding of the
pathophysiologic mechanisms underlying heart failure and identify
promising therapeutic targets, it is necessary to conduct further
research. The present study has demonstrated that larixyl acetate,
a TRPC6 inhibitor, can effectively reverse cardiac dysfunction
induced by pressure overload. Notably, it was observed that the
protecve role of larixyl acetate involves the inhibition of
autophagy through decreased phosphorylation of mTOR, which
consequently diminishes ER stress and apoptosis in cardiomyocytes.
These findings provided valuable insights into the pathogenesis of
heart failure and highlight the potential utility of larixyl
acetate as a therapeutic intervention (Fig. 6).
The ER is a multifunctional organelle that plays an
important role in the folding of secretory and membrane proteins
(12,48). In cases of pressure overload, there
is an increase in protein synthesis, which can lead to ER stress
and subsequent cardiomyocyte apoptosis (49,50).
Autophagy is a crucial lysosome-dependent catabolic process that
plays a vital role in the degradation and recycling of cytoplasmic
components to maintain intracellular homeostasis (51,52).
Numerous studies have demonstrated that autophagy can protect
against ER stress-induced apoptosis of myocardiocytes by limiting
the accumulation of misfolded proteins, thereby preventing the
deterioration of cardiac function (23,53).
This suggests that autophagy serves as a critical mechanism to
maintain the proper functioning of cardiac cells under stressful
conditions (54,55). The present study revealed that
either 4 weeks of TAC or 72 h of incubation with Ang II
significantly upregulated ER stress while downregulating autophagy,
which are key factors for the malfunction of cardiomyocytes
(25). However, it was found that
the administration of larixyl acetate can mitigate this damage by
promoting autophagy and limiting ER stress. This highlights the
potential therapeutic value of targeting TRPC6 to alleviate cardiac
dysfunction associated with ER stress and impaired autophagy.
Previous studies have indicated that pressure
overload or Ang II-induced activation of TRPC6 can initiate a
positive feedback loop, resulting in increased TRPC6 expression.
This activation and upregulation contribute to cardiac hypertrophy
through the calcineurin/NFAT signaling pathway (35,56).
However, it is unclear whether TRPC6 inhibition can modulate ER
stress and autophagy, which are key processes involved in
cardiomyocyte function and survival. It has been reported that
TRPC1 can regulate ER stress in dopaminergic neurons (57), and TRPC6 has been shown to regulate
the mTOR signaling pathway, a well-established mediator of
autophagy, in human pulmonary arterial smooth muscle cells
(37). Therefore, it is reasonable
to hypothesize that TRPC6 may also modulate ER stress and autophagy
in the context of cardiac function. Additioinally, it has been well
established that pressure overload-induced heart failure is partly
driven by excessive cardiomyocyte apoptosis following TAC (46). Furthermore, recent studies have
highlighted that downregulation or inhibition of TRPC6 can exert
anti-apoptotic effects (47,48).
Consequently, it can be hypothesized that larixyl acetate may
preserve cardiac function post-TAC, potentially by mitigating
cardiomyocyte apoptosis. Consistent with the aforementioned
hypothesis, the present study found that blocking TRPC6 by larixyl
acetate significantly decreased ER stress and phosphorylation of
mTOR, while promoting autophagy and decreased apoptosis.
The aforementioned results have established that
TRPC6 inhibition can mitigate ER stress and apoptosis; however,
these effects might be attributed mainly to the inhibition of the
calcineurin A/NFAT pathway (12).
It remains unclear whether the reduced phosphorylation of the mTOR
signaling pathway and the resulting enhancement of autophagy also
contribute to the reduction of ER stress and the protection against
apoptosis. To investigate this, larixyl acetate was administered to
block TRPC6, while concurrently using the mTOR activator MHY1485 to
specifically activate the mTOR pathway and suppress autophagy in an
Ang II-induced H9C2 cellular hypertrophy model. The findings of the
present study revealed a notable phenomenon that although MHY1485
does not affect the anti-hypertrophic effects of larixyl acetate,
it still abolished the protective role of larixyl acetate by
pronounced significant ER stress and apoptosis through increasing
mTOR phosphorylation and inhibiting autophagy.
A reasonable explanation is that administering
larixyl acetate, both in vivo and in vitro, does not
completely diminish cardiac hypertrophy. This partial effect is
likely because there are multiple mechanisms (2), apart from TRPC6, that mediate
pressure overload or Ang II-induced cardiac hypertrophy and heart
failure. The results from the present study lend strong support to
this hypothesis. Although some hypertrophy persists, the blockade
of larixyl acetate of the mTOR signaling pathway simultaneously
promotes the protective effects of autophagy. This action
effectively counters the adverse effects of residual hypertrophy
and prevents myocardial damage. Conversely, the activation of mTOR
using MHY1485 can negate this protective effect. This evidence
indicated that TRPC6 inhibitors may exert a cardioprotective role
through two mechanisms. The first is the traditional inhibition of
the calcineurin A/NFAT pathway and the second is via the newly
identified mTOR signaling pathway. This dual-mechanism action
potentially makes TRPC6 inhibitors more effective than drugs
operating through a single mechanism.
In addition to cardiomyocyte apoptosis, myocardial
fibrosis represents a key pathological alteration in cardiac
hypertrophy and heart failure (58). Studies have shown a strong
association between TRPC6 activation and fibrosis. For example,
cardiac-specific overexpression of TRPC6 in transgenic mice
triggers cardiac fibrosis, hypertrophy and heart failure (35). In cases of renal injury induced by
unilateral ureteral obstruction in mice, an increase in
TRPC6 gene expression is observed, leading to significant
interstitial fibrosis. This effect is notably reduced in
TRPC6-deficient mice (59).
Similarly, TRPC6 knockout mice exhibit less lung fibrosis in the
bleomycin-treated mice model (60). In the present research, it was also
discovered that the administration of larixyl acetate significantly
reduces myocardial fibrosis. This anti-fibrotic effect is two-fold,
firstly, through the inhibition of cardiomyocyte apoptosis, as
aformentioned, and secondly, it may directly inhibit the
transdifferentiation of fibroblasts into myofibroblasts. A previous
study indicated that overexpression of TRPC6 in fibroblasts
markedly facilitates their conversion to myofibroblasts, whereas
the absence of TRPC6 prevents TGF-β-mediated myofibroblast
conversion (61).
In the present study exploring cardiac hypertrophy,
heart failure and the effects of larixyl acetate, some limitations
were acknowledged and future research directions identied. Firstly,
the use of the H9C2 rat cardiomyocyte line, instead of primary
cultured cardiomyocytes, may not fully replicate the complex in
vivo environment or the exact behavior of primary
cardiomyocytes, potentially biasing the conclusions of the present
study. Secondly, while larixyl acetate significantly reduced
myocardial fibrosis in vivo, there was a lack of in
vitro evidence to confirm its direct impact on fibrosis.
Additionally, its high selectivity in inhibiting TRPC6 may not rule
out off-target effects. Future research should focus on validating
the findings of the present study with primary cultured
cardiomyocytes for more accurate insights, exploring the direct
effects of larixyl acetate on fibroblasts, and using more specific
molecular or genetic interventions to clarify the role of TRPC6 in
cardiac fibrosis and hypertrophy.
In summary, the present study demonstrated that
larixyl acetate, a TRPC6 inhibitor, plays a significant
cardioprotective role against cardiac hypertrophy and heart
failure. The mTOR signaling pathway was newly identifed as a key
component in this cardioprotective effect. This novel mechanism
involves the enhancement of autophagy through the decrease in
phosphorylation of mTOR, which subsequently reduces ER stress and
cardiomyocyte apoptosis. These findings not only enhance
understanding of the pathophysiology of heart failure but also
underscore the therapeutic potential of larixyl acetate. It offers
a dual-mechanism approach that could potentially surpass the
efficacy of single-mechanism drugs in heart failure treatment.
Additionally, this research provides a new perspective for
developing novel anti-heart failure drugs targeting the mTOR
signaling pathway.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The study was supported by a grant from The National Natural
Science Foundation of China (grant nos. 82301368, 82370482 and
82270346).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
MJ and WXL conducted the experiments and contributed
to data acquisition; KYZ, RSL and ZGW performed data analysis; MJ
and WXL wrote the manuscript; JP, JJY and DJW made significant
contributions to the conception and design of the study. JJY and
DJW reviewed the manuscript and gave critical suggestions for
revision. MJ, WXL, KYZ, ZGW, RSL, JP, JJY and DJW confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
All the experimental procedures were approved by The
Experimental Animal Ethics Committee of Drum Tower Hospital of
Nanjing University Medical School (Nanjing, China; approval no.
2022AE01017).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Njoroge JN and Teerlink JR:
Pathophysiology and therapeutic approaches to acute decompensated
heart failure. Circ Res. 128:1468–1486. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nakamura M and Sadoshima J: Mechanisms of
physiological and pathological cardiac hypertrophy. Nat Rev
Cardiol. 15:387–407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guo J, Mihic A, Wu J, Zhang Y, Singh K,
Dhingra S, Weisel RD and Li RK: Canopy 2 attenuates the transition
from compensatory hypertrophy to dilated heart failure in
hypertrophic cardiomyopathy. Eur Heart J. 36:2530–2540. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gao M, Cai Q, Si H, Shi S, Wei H, Lv M,
Wang X and Dong T: Isoliquiritigenin attenuates pathological
cardiac hypertrophy via regulating AMPKα in vivo and in vitro. J
Mol Histol. 53:679–689. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kou T, Luo H, Shen Y, Su Y and Yin L:
Effects of berberine hydrochloride on left ventricular structure
and function in rats with myocardial hypertrophy. Acta Cardiol.
78:433–441. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dickhout JG, Carlisle RE and Austin RC:
Interrelationship between cardiac hypertrophy, heart failure, and
chronic kidney disease: Endoplasmic reticulum stress as a mediator
of pathogenesis. Circ Res. 108:629–642. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ziaeian B and Fonarow GC: Epidemiology and
aetiology of heart failure. Nat Rev Cardiol. 13:368–378. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tham YK, Bernardo BC, Ooi JY, Weeks KL and
McMullen JR: Pathophysiology of cardiac hypertrophy and heart
failure: Signaling pathways and novel therapeutic targets. Arch
Toxicol. 89:1401–1438. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sherrid MV: Drug therapy for hypertrophic
cardiomypathy: Physiology and practice. Curr Cardiol Rev. 12:52–65.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hetz C, Zhang K and Kaufman RJ:
Mechanisms, regulation and functions of the unfolded protein
response. Nat Rev Mol Cell Biol. 21:421–438. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang S, Binder P, Fang Q, Wang Z, Xiao W,
Liu W and Wang X: Endoplasmic reticulum stress in the heart:
insights into mechanisms and drug targets. Br J Pharmacol.
175:1293–1304. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ren J, Bi Y, Sowers JR, Hetz C and Zhang
Y: Endoplasmic reticulum stress and unfolded protein response in
cardiovascular diseases. Nat Rev Cardiol. 18:499–521. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim I, Xu W and Reed JC: Cell death and
endoplasmic reticulum stress: Disease relevance and therapeutic
opportunities. Nat Rev Drug Discov. 7:1013–1030. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yao Y, Lu Q, Hu Z, Yu Y, Chen Q and Wang
QK: A non-canonical pathway regulates ER stress signaling and
blocks ER stress-induced apoptosis and heart failure. Nat Commun.
8:1332017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang J, Hu X and Jiang H: ER
stress-induced apoptosis: A novel therapeutic target in heart
failure. Int J Cardiol. 177:564–565. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gatica D, Chiong M, Lavandero S and
Klionsky DJ: Molecular mechanisms of autophagy in the
cardiovascular system. Circ Res. 116:456–467. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tagashira H, Bhuiyan MS, Shinoda Y,
Kawahata I, Numata T and Fukunaga K: Sigma-1 receptor is involved
in modification of ER-mitochondria proximity and Ca(2+) homeostasis
in cardiomyocytes. J Pharmacol Sci. 151:128–133. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun X, Zhou L, Han Y, Yang Q, Li X, Xin B,
Chi M, Wang Y and Guo C: Scutellarin attenuates doxorubicin-induced
cardiotoxicity by inhibiting myocardial fibrosis, apoptosis and
autophagy in rats. Chem Biodivers. 20:e2022004502023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lindqvist LM, Tandoc K, Topisirovic I and
Furic L: Cross-talk between protein synthesis, energy metabolism
and autophagy in cancer. Curr Opin Genet Dev. 48:104–111. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shirakabe A, Zhai P, Ikeda Y, Saito T,
Maejima Y, Hsu CP, Nomura M, Egashira K, Levine B and Sadoshima J:
Drp1-dependent mitochondrial autophagy plays a protective role
against pressure overload-induced mitochondrial dysfunction and
heart failure. Circulation. 133:1249–1263. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang Z, Su W, Zhang Y, Zhou L, Xia ZY and
Lei S: Selective inhibition of PKCβ2 improves Caveolin-3/eNOS
signaling and attenuates lipopolysaccharide-induced injury by
inhibiting autophagy in H9C2 cardiomyocytes. J Mol Histol.
52:705–715. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guo X, Zhang Y, Lu C, Qu F and Jiang X:
Protective effect of hyperoside on heart failure rats via
attenuating myocardial apoptosis and inducing autophagy. Biosci
Biotechnol Biochem. 84:714–724. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gao G, Chen W, Yan M, Liu J, Luo H, Wang C
and Yang P: Rapamycin regulates the balance between cardiomyocyte
apoptosis and autophagy in chronic heart failure by inhibiting mTOR
signaling. Int J Mol Med. 45:195–209. 2020.PubMed/NCBI
|
|
24
|
Buss SJ, Muenz S, Riffel JH, Malekar P,
Hagenmueller M, Weiss CS, Bea F, Bekeredjian R, Schinke-Braun M,
Izumo S, et al: Beneficial effects of Mammalian target of rapamycin
inhibition on left ventricular remodeling after myocardial
infarction. J Am Coll Cardiol. 54:2435–2446. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pires Da Silva J, Monceaux K, Guilbert A,
Gressette M, Piquereau J, Novotova M, Ventura-Clapier R, Garnier A
and Lemaire C: SIRT1 protects the heart from ER stress-induced
injury by promoting eEF2K/eEF2-dependent autophagy. Cells.
9:4262020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Martín-Bórnez M, Galeano-Otero I, Del Toro
R and Smani T: TRPC and TRPV channels' role in vascular remodeling
and disease. Int J Mol Sci. 21:61252020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Eder P and Molkentin JD: TRPC channels as
effectors of cardiac hypertrophy. Circ Res. 108:265–272. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kinoshita H, Kuwahara K, Nishida M, Jian
Z, Rong X, Kiyonaka S, Kuwabara Y, Kurose H, Inoue R, Mori Y, et
al: Inhibition of TRPC6 channel activity contributes to the
antihypertrophic effects of natriuretic peptides-guanylyl cyclase-A
signaling in the heart. Circ Res. 106:1849–1860. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tang N, Tian W, Ma GY, Xiao X, Zhou L, Li
ZZ, Liu XX, Li CY, Wu KH, Liu W, et al: TRPC channels blockade
abolishes endotoxemic cardiac dysfunction by hampering
intracellular inflammation and Ca(2+) leakage. Nat Commun.
13:74552022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Urban N, Wang L, Kwiek S, Rademann J,
Kuebler WM and Schaefer M: Identification and validation of Larixyl
acetate as a potent TRPC6 inhibitor. Mol Pharmacol. 89:197–213.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen X, Taylor-Nguyen NN, Riley AM,
Herring BP, White FA and Obukhov AG: The TRPC6 inhibitor, larixyl
acetate, is effective in protecting against traumatic brain
injury-induced systemic endothelial dysfunction. J
Neuroinflammation. 16:212019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen QZ, Zhou YB, Zhou LF, Fu ZD, Wu YS,
Chen Y, Li SN, Huang JR and Li JH: TRPC6 modulates adhesion of
neutrophils to airway epithelial cells via NF-kappaB activation and
ICAM-1 expression with ozone exposure. Exp Cell Res. 377:56–66.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang M, Zhang X, Guo J, Yang S, Yang F and
Chen X: TRPC6 deletion enhances eNOS expression and reduces
LPS-induced acute lung injury. Int J Mol Sci. 24:167562023.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang J, Zhao M, Jia P, Liu FF, Chen K,
Meng FY, Hong JH, Zhang T, Jin XH and Shi J: The analgesic action
of larixyl acetate, a potent TRPC6 inhibitor, in rat neuropathic
pain model induced by spared nerve injury. J Neuroinflammation.
17:1182020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kuwahara K, Wang Y, McAnally J, Richardson
JA, Bassel-Duby R, Hill JA and Olson EN: TRPC6 fulfills a
calcineurin signaling circuit during pathologic cardiac remodeling.
J Clin Invest. 116:3114–3126. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sciarretta S, Forte M, Frati G and
Sadoshima J: New insights into the role of mTOR signaling in the
cardiovascular system. Circ Res. 122:489–505. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jain PP, Lai N, Xiong M, Chen J, Babicheva
A, Zhao T, Parmisano S, Zhao M, Paquin C, Matti M, et al: TRPC6, a
therapeutic target for pulmonary hypertension. Am J Physiol Lung
Cell Mol Physiol. 321:L1161–L1182. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals, . Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press (US); Washington, DC: 2011
|
|
39
|
Wilkins BJ, Dai YS, Bueno OF, Parsons SA,
Xu J, Plank DM, Jones F, Kimball TR and Molkentin JD:
Calcineurin/NFAT coupling participates in pathological, but not
physiological, cardiac hypertrophy. Circ Res. 94:110–118. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vanhoutte D, Schips TG, Vo A, Grimes KM,
Baldwin TA, Brody MJ, Accornero F, Sargent MA and Molkentin JD:
Thbs1 induces lethal cardiac atrophy through PERK-ATF4 regulated
autophagy. Nat Commun. 12:39282021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ye T, Yan Z, Chen C, Wang D, Wang A, Li T,
Yang B, Ding X and Shen C: Lactoferrin attenuates cardiac fibrosis
and cardiac remodeling after myocardial infarction via inhibiting
mTORC1/S6K signaling pathway. Theranostics. 13:3419–3433. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Okada K, Minamino T, Tsukamoto Y, Liao Y,
Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani
T, et al: Prolonged endoplasmic reticulum stress in hypertrophic
and failing heart after aortic constriction: Possible contribution
of endoplasmic reticulum stress to cardiac myocyte apoptosis.
Circulation. 110:705–712. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Klaiber M, Kruse M, Völker K, Schröter J,
Feil R, Freichel M, Gerling A, Feil S, Dietrich A, Londoño JE, et
al: Novel insights into the mechanisms mediating the local
antihypertrophic effects of cardiac atrial natriuretic peptide:
Role of cGMP-dependent protein kinase and RGS2. Basic Res Cardiol.
105:583–595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sadoshima J and Izumo S: Rapamycin
selectively inhibits angiotensin II-induced increase in protein
synthesis in cardiac myocytes in vitro. Potential role of 70-kD S6
kinase in angiotensin II-induced cardiac hypertrophy. Circ Res.
77:1040–1052. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Scheuble J, Rössler OG, Ulrich M and Thiel
G: Pharmacological and genetic inhibition of TRPC6-induced gene
transcription. Eur J Pharmacol. 886:1733572020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gao L, Lv G, Li R, Liu WT, Zong C, Ye F,
Li XY, Yang X, Jiang JH, Hou XJ, et al: Glycochenodeoxycholate
promotes hepatocellular carcinoma invasion and migration by
AMPK/mTOR dependent autophagy activation. Cancer Lett. 454:215–223.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nagalingam RS, Chattopadhyaya S, Al-Hattab
DS, Cheung DYC, Schwartz LY, Jana S, Aroutiounova N, Ledingham DA,
Moffatt TL, Landry NM, et al: Scleraxis and fibrosis in the
pressure-overloaded heart. Eur Heart J. 43:4739–4750. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Travers KJ, Patil CK, Wodicka L, Lockhart
DJ, Weissman JS and Walter P: Functional and genomic analyses
reveal an essential coordination between the unfolded protein
response and ER-associated degradation. Cell. 101:249–258. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Minamino T, Komuro I and Kitakaze M:
Endoplasmic reticulum stress as a therapeutic target in
cardiovascular disease. Circ Res. 107:1071–1082. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ghosh R and Pattison JS: Macroautophagy
and chaperone-mediated autophagy in heart failure: The known and
the unknown. Oxid Med Cell Longev. 2018:86020412018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Nemchenko A, Chiong M, Turer A, Lavandero
S and Hill JA: Autophagy as a therapeutic target in cardiovascular
disease. J Mol Cell Cardiol. 51:584–593. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wang ZV, Ferdous A and Hill JA:
Cardiomyocyte autophagy: Metabolic profit and loss. Heart Fail Rev.
18:585–594. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chen J, Li L, Bai X, Xiao L, Shangguan J,
Zhang W, Zhang X, Wang S and Liu G: Inhibition of autophagy
prevents panax notoginseng saponins (PNS) protection on cardiac
myocytes against endoplasmic reticulum (ER) stress-induced
mitochondrial injury, Ca(2+) homeostasis and associated apoptosis.
Front Pharmacol. 12:6208122021. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hariharan N, Ikeda Y, Hong C, Alcendor RR,
Usui S, Gao S, Maejima Y and Sadoshima J: Autophagy plays an
essential role in mediating regression of hypertrophy during
unloading of the heart. PLoS One. 8:e516322013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Shi S and Jiang P: Therapeutic potentials
of modulating autophagy in pathological cardiac hypertrophy. Biomed
Pharmacother. 156:1139672022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Onohara N, Nishida M, Inoue R, Kobayashi
H, Sumimoto H, Sato Y, Mori Y, Nagao T and Kurose H: TRPC3 and
TRPC6 are essential for angiotensin II-induced cardiac hypertrophy.
EMBO J. 25:5305–5316. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Selvaraj S, Sun Y, Watt JA, Wang S, Lei S,
Birnbaumer L and Singh BB: Neurotoxin-induced ER stress in mouse
dopaminergic neurons involves downregulation of TRPC1 and
inhibition of AKT/mTOR signaling. J Clin Invest. 122:1354–1367.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
González A, Schelbert EB, Díez J and
Butler J: Myocardial interstitial fibrosis in heart failure:
Biological and translational perspectives. J Am Coll Cardiol.
71:1696–1706. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Wu YL, Xie J, An SW, Oliver N, Barrezueta
NX, Lin MH, Birnbaumer L and Huang CL: Inhibition of TRPC6 channels
ameliorates renal fibrosis and contributes to renal protection by
soluble klotho. Kidney Int. 91:830–841. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hofmann K, Fiedler S, Vierkotten S, Weber
J, Klee S, Jia J, Zwickenpflug W, Flockerzi V, Storch U, Yildirim
AÖ, et al: Classical transient receptor potential 6 (TRPC6)
channels support myofibroblast differentiation and development of
experimental pulmonary fibrosis. Biochim Biophys Acta Mol Basis
Dis. 1863:560–568. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Davis J, Burr AR, Davis GF, Birnbaumer L
and Molkentin JD: A TRPC6-dependent pathway for myofibroblast
transdifferentiation and wound healing in vivo. Dev Cell.
23:705–715. 2012. View Article : Google Scholar : PubMed/NCBI
|