Introduction
Cardiac hypertrophy is an early pathological
structural feature of heart failure, and is a risk factor for
cardiac dysfunction, arrhythmia and myocardial infarction (1). It is characterized by an increase in
heart mass and individual cardiomyocyte volume, which is
accompanied by reactivated fetal genes, increased protein synthesis
and altered metabolism (1,2). Research into the molecular mechanisms
of cardiac hypertrophy serve a role in preventing and treating
cardiomyopathy and heart failure (1,2).
However, there are few feasible targets, such as Rho and
Rho-associated kinase (ROCK), for preventing or reversing cardiac
hypertrophy (3).
It has been reported that small GTPases are involved
in the development of cardiac hypertrophy (3–6).
Small GTPases are molecular switches that either exist in the
active GTP-bound or inactive GDP-bound states (7–9).
Small GTPases are divided into subfamilies, including Ras, Rho,
Rab, Ran and Arf/Sar (10).
Activated Ras and the upregulation of Rab1 are causes of cardiac
hypertrophy (5,6). Conversely, inactivation of RhoA can
suppress cardiac hypertrophy (11,12).
GTPase-activating proteins (GAPs) can induce inactivation of small
GTPases by inducing GTP hydrolysis (13,14).
Notably, the inhibitory roles of RabGAPs and RasGAPs in cardiac
hypertrophy have previously been reported (15–18).
The ablation of the RabGAP TBC1 domain family member 1 results in
significant cardiac hypertrophy in male rats and increased
myocardial damage after ischemia/reperfusion (15,16).
Similarly, depletion of Carabin, another RabGAP, can exacerbate
cardiac hypertrophy and significantly reduce fractional shortening
in mice (17,18). Furthermore, the RasGAP
neurofibromatosis type 1 (NF1) is involved in cardiac hypertrophy.
It has been reported that the cardiomyocyte-specific NF1 knockout
promotes cardiac hypertrophy and dysfunction in mice (19). However, compared with RabGAPs and
RasGAPs, the role of RhoGAPs in cardiac hypertrophy is unclear.
RhoGAP interacting with CIP4 homologs protein 1
(RICH1) is a RhoGAP that is also known as ARHGAP17. RICH1 contains
three functional domains: An N-terminal BAR domain; a RhoGAP
domain; and a C-terminal tail consisting of multiple proline-rich
motifs (20,21). RICH1 is a selective GAP of Cdc42
and Rac1, and associates with tight junctions in epithelial cells
(21,22). The GAP activity of RICH1 and
regulation of Cdc42 by RICH1 is required for maintaining proper
tight junctions in epithelial cells (21). RICH1 is widely distributed and
highly expressed in human heart and placenta tissues (23), and it has been reported that RICH1
has multiple functions in vivo (24–26).
RICH1 can inhibit the activity of Rac1 by catalyzing the cleavage
of Rac1-GTP to Rac1-GDP, which then inhibits the MAPK signaling
pathway, which in turn reduces cellular proliferation and
tumorigenesis (27,28). It has been reported that the
overexpression of RICH1 inhibits the invasion and metastasis of
breast cancer through downregulating Hippo signaling (29). However, the role of RICH1 in
cardiac hypertrophy remains unknown. The present study aimed to
clarify whether RICH1 is associated with myocardial cell
hypertrophy and how it serves a role in myocardial cell
hypertrophy.
Materials and methods
Cell culture and treatments
The H9c2 cardiomyocyte cell line was purchased from
Procell Life Science & Technology Co., Ltd. Cells were
maintained in high-glucose Dulbecco's modified Eagle's medium
(DMEM; Procell Life Science & Technology Co., Ltd.) containing
10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and
1% penicillin-streptomycin (Biosharp Life Sciences) in a 5%
CO2 incubator at 37°C. To induce the cardiomyocyte
hypertrophy model, H9c2 cells were treated with either 60 µM ISO
(MilliporeSigma) dissolved in phosphate-buffered saline (PBS) for
48 h, or 3 µM Ang II (Shanghai Yuanye Biotechnology Co., Ltd.) in
PBS for 24 h in a 5% CO2 incubator at 37°C. The control
group cells were treated with an equal volume of PBS in a 5%
CO2 incubator at 37°C for 48 h (when compared with ISO)
and 24 h (when compared with Ang II).
Small interfering (si)RNA transfection
of H9c2 cells
H9c2 cells were transfected with 1 µg/ml siRICH1
using siRNA-Mate (Shanghai GenePharma Co., Ltd.) and incubated in
5% CO2 at 37°C for 6 h, according to the manufacturer's
instructions. A total of 18 h after the 6-h transfection, cells
were treated with 60 µM ISO and incubated in a 5% CO2
incubator at 37°C for 48 h. A total of 42 h after the 6-h
transfection, 3 µM Ang II was added and incubated in a 5%
CO2 incubator at 37°C for 24 h. The siRNA sequences were
as follows: siRICH1 (Shanghai GenePharma Co., Ltd.) sense,
5′-GGUGAAGAAGGAGAGCUUUTT-3′ and anti-sense,
5′-AAAGCUCUCCUUCUUCACCTT-3′; and negative control (siNC; Suzhou
GenePharma Co., Ltd.) sense, 5′-UUCUCCGAACGUGUCACGUTT-3′ and
anti-sense, 5′-ACGUGACACGUUCGGAGAATT-3′.
Plasmid transfection of H9c2
cells
H9c2 cells were transfected with 3 µg/ml
pCDNA3.1-RICH1 plasmid (TsingKe Biological Technology) with
siRNA-Mate and incubated in 5% CO2 at 37°C for 6 h to
overexpress RICH1. Cells transfected with 3 µg/ml pCDNA3.1 plasmid
(TsingKe Biological Technology) were used as a control. A total of
18 h after the 6-h transfection, cells were treated with 60 µM ISO
and incubated in a 5% CO2 incubator at 37°C for 48 h. A
total of 42 h after the 6-h transfection, 3 µM Ang II was added and
incubated in a 5% CO2 incubator at 37°C for 24 h.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from the cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), after which, 1 µg RNA was reverse transcribed into cDNA
using an ABScript III RT Master Mix (ABclonal Biotech Co., Ltd.)
according to the manufacturer's protocol. After RT, qPCR was
performed using a SYBR Green PCR kit (ABclonal Biotech Co., Ltd.)
The amplification procedure was as follows: Pre-denaturation at
95°C for 3 min, followed by 45 cycles at 95°C for 5 sec and 60°C
for 30 sec. mRNA expression levels were quantified using the
2−ΔΔCq method (30).
Data were normalized to Gapdh. Primer sequences are shown in
Table I.
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Gene | Species | Forward primer,
5′-3′ | Reverse primer,
5′-3′ |
|---|
| Nppa | Rat |
GAAGATGCCGGTAGAAGATGAG |
AGAGCCCTCAGTTTGCTTTTC |
| Nppb | Rat |
CTGGAGACTGGCTAGGACTTC |
GGTGCTGCCCCAGATGATT |
| Myh7 | Rat |
GCCCCAAATGCAGCCAT |
CGCTCAGTCATGGCGGAT |
| Gapdh | Rat |
GGTTGTCTCCTGCGACTTCA |
TGGTCCAGGGTTACTCC |
Western blotting
Total protein was extracted from H9c2 cells using
RIPA lysis buffer containing phosphatase inhibitors and protease
inhibitors (Wuhan Servicebio Technology Co., Ltd.). Protein
concentration was determined using the BCA protein assay kit (Wuhan
Servicebio Technology Co., Ltd.). Proteins (15 µg/lane) were
separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis on 10% gels and were transferred to polyvinylidene
difluoride membranes. The membranes were blocked with 5% skimmed
milk in 1X TBS-0.5% Tween 20 (Biosharp Life Sciences) for 1 h at
room temperature. The blots were then incubated with primary
antibodies against RICH1 (1:500; cat. no. sc-514438; Santa Cruz
Biotechnology, Inc.) and α-tubulin (1:2,000; cat. no. GB15201;
Wuhan Servicebio Technology Co., Ltd.) overnight at 4°C, followed
by anti-mouse horseradish peroxidase-conjugated secondary
antibodies (1:50,000; cat. no. BL001A; Biosharp Life Sciences) or
anti-rabbit horseradish peroxidase-conjugated secondary antibodies
(1:50,000; cat. no. BL003A; Biosharp Life Sciences) for 1 h at room
temperature. Protein bands were visualized using a
Chemiluminescence Kit (Biosharp Life Sciences). Bio-ID software
(Version 6.0; Bio-Rad Laboratories, Inc.) was used to semi-quantify
protein expression.
Cell viability
Cell viability was assessed using Cell Counting
Kit-8 (CCK-8; Biosharp Life Sciences). H9c2 cells were seeded in
96-well plates at 5×103 cells/well. After culturing for
24 h, cells were treated with different concentrations of ISO (0,
20, 40, 60, 80 and 100 µM) for 48 h or Ang II (0, 1, 2, 3, 4 and 5
µM) for 24 h at 37°C. The cells were then incubated with 10% CCK-8
solution in DMEM for 1 h and the absorbance was measured at 450
nm.
Cell fluorescence staining
H9c2 cells were seeded into 35-mm cell culture
dishes at 4×104 cells/dish. Cells were fixed with 4%
paraformaldehyde at 37°C for 20 min and then permeabilized with
0.1% Triton X-100 in PBS at 37°C for 15 min. Cells were incubated
with Acti-stain™ 488 phalloidin (Cytoskeleton, Inc.) in the dark
for 30 min at 37°C. ProLong™ Gold antifade reagent was mixed with
DAPI (Invitrogen; Thermo Fisher Scientific, Inc.) for 30 min at
37°C and used for nuclear staining. Images were captured using an
Olympus FV3000 confocal microscope (Olympus Corporation). Cell
surface area (CSA) was calculated using CellSens software (Version
4.2.1; Olympus Corporation).
Statistical analysis
Experiments were repeated at least three times and
statistical analyses were performed using GraphPad Prism 9
(Dotmatics). Before analysis, data were tested for normality and
homogeneity using Shapiro-Wilk and Levene's tests, respectively.
Data were analyzed by unpaired Student's t-test to compare the
differences between two groups and one-way ANOVA with Tukey's post
hoc test to compare the means among >2 groups. P<0.05 was
considered to indicate a statistically significant difference
Results
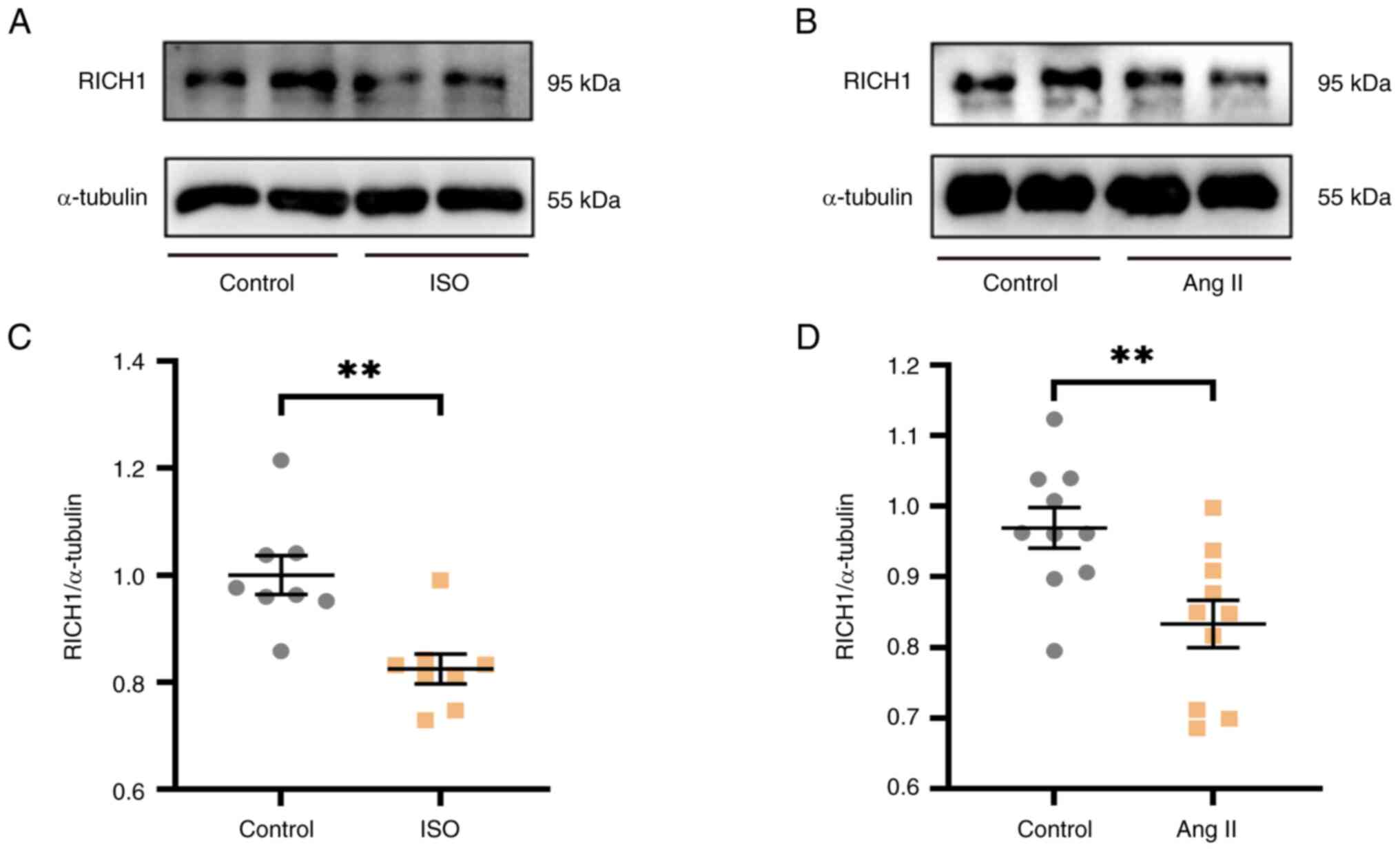
RICH1 is downregulated in ISO- or Ang
II-treated cardiomyocytes
ISO or Ang II are two common inducers of myocardial
cell hypertrophy. In this study, ISO or Ang II were used to induce
cardiomyocyte hypertrophy and to verify whether the expression
levels of RICH1 were changed under the induction of ISO or Ang II.
Notably, 0, 20, 40, 60 and 80 µM ISO, or 0, 1, 2, 3, 4 and 5 µM Ang
II were demonstrated to have no negative effect on cell viability
at the concentrations tested (Fig.
S1). According to these cell viability experiments results, the
protein expression levels of RICH1 were assessed after treatment
with ISO or Ang II at different concentrations, which have no
negative effect on cell viability. In particular, the protein
expression levels of RICH1 were significantly decreased in response
to 60 µM ISO (Fig. S2A and C) and
3 µM Ang II (Fig. S2B and D).
Therefore, these concentrations were selected for further
experimentation, and it was verified that the protein expression
levels of RICH1 were markedly decreased in 60 µM ISO-treated
(Fig. 1A and C) and 3 µM Ang
II-treated (Fig 1B and D) H9c2
cells. Therefore, it was suggested that RICH1 may serve a role in
cardiomyocyte hypertrophy.
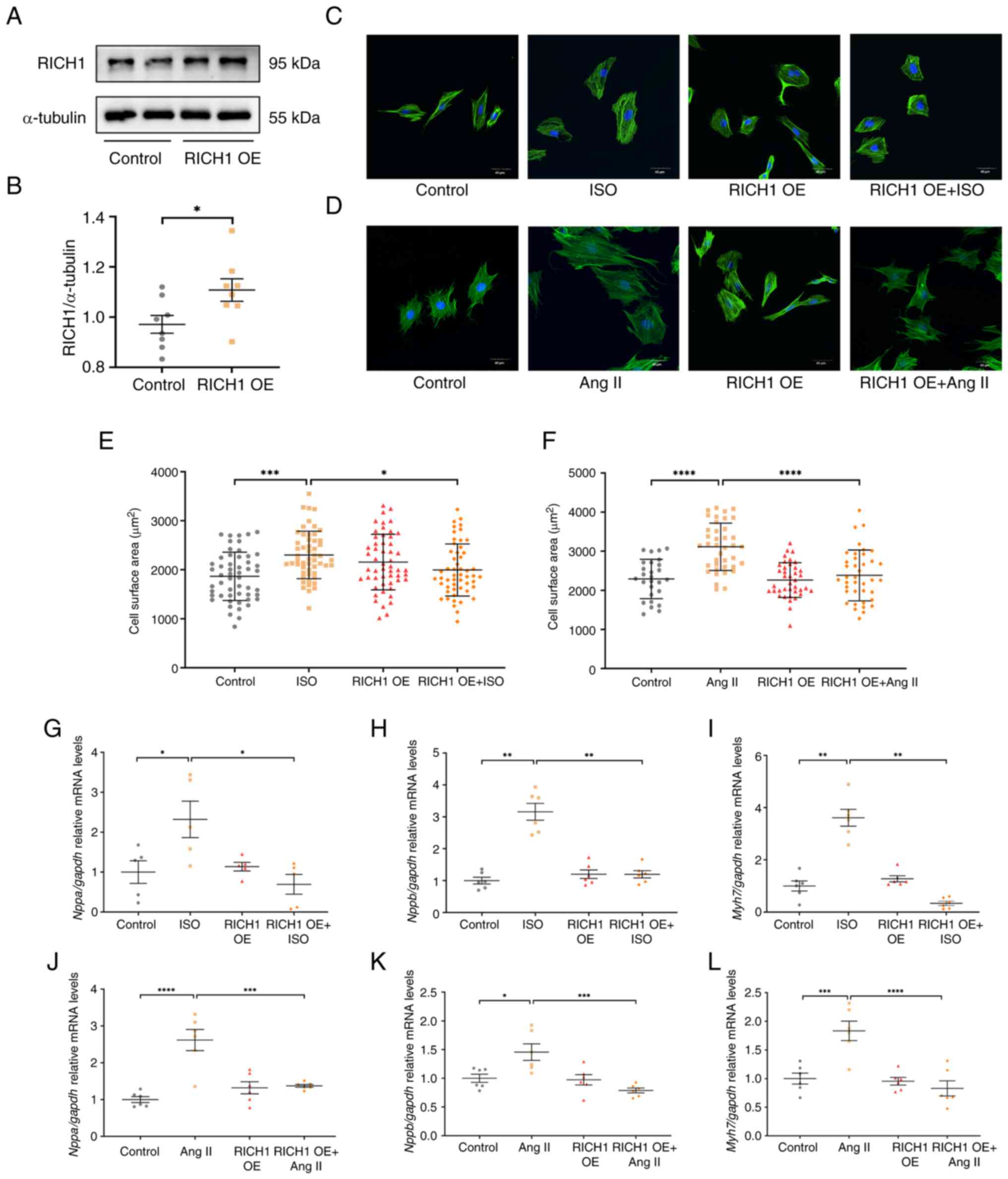
RICH1 can attenuate ISO or Ang
II-induced cardiomyocyte hypertrophy
To assess the role of RICH1 in cardiomyocyte
hypertrophy, RICH1 was overexpressed in H9c2 cells by transfecting
cells with pCDNA3.1-RICH1 plasmids. Compared with in the control
group, the protein expression levels of RICH1 were significantly
increased in the RICH1 overexpression group (Fig. 2A and B).
The increase in CSA and upregulation of
hypertrophy-related genes are two critical features of
cardiomyocyte hypertrophy. Notably, it was demonstrated that CSA
was significantly increased in ISO-treated (Fig. 2C and E) and Ang II-treated
(Fig. 2D and F) cells compared
with in the control group. There was no significant difference in
CSA between the control group and the RICH1 overexpression group
(Fig. 2C and E). Compared with in
the ISO group, overexpression of RICH1 inhibited CSA. No difference
in CSA was detected between the control group and the RICH1
overexpression group. However, the overexpression of RICH1
inhibited the CSA compared with that in the Ang II group (Fig. 2D and F).
Furthermore, the cardiac hypertrophy marker genes
Nppa, Nppb and Myh7 were assessed by RT-qPCR. It was
demonstrated that the relative mRNA expression levels of these
genes were significantly increased in ISO- or Ang II-treated cells
compared with those in the control groups, whereas they were
significantly decreased following overexpression of RICH1 in ISO-
or Ang II-treated cells compared with those in the ISO or Ang II
single treatment groups, respectively (Fig. 2G-L). These results suggested that
RICH1 can alleviate cardiomyocyte hypertrophy induced by ISO or Ang
II.
KD of RICH1 can promote ISO or Ang
II-induced cardiomyocyte hypertrophy
To further assess the role of RICH1 in ISO- and Ang
II-induced cardiomyocyte hypertrophy, siRNA was used to KD RICH1 in
H9c2 cells. First, a siRNA fragment that can induce RICH1 KD in
H9c2 cells was identified (Fig. 3A and
B). It was then demonstrated that RICH1 KD in ISO- and Ang
II-treated cells resulted in a significantly larger CSA compared
with that in ISO- and Ang II-treated cells transfected with the
siNC, respectively (Fig. 3C-F).
Similarly, the relative mRNA expression levels of the cardiac
hypertrophy-related genes Nppa, Nppb and Myh7 were
significantly higher in the siRICH1 + ISO group and siRICH1 + Ang
II groups compared with those in the siNC + ISO and siNC + Ang II
groups, respectively, as demonstrated by RT-qPCR (Fig. 3G-L). These results suggested that
siRNA-mediated RICH1 KD can exacerbate cardiomyocyte hypertrophy
induced by ISO or Ang II.
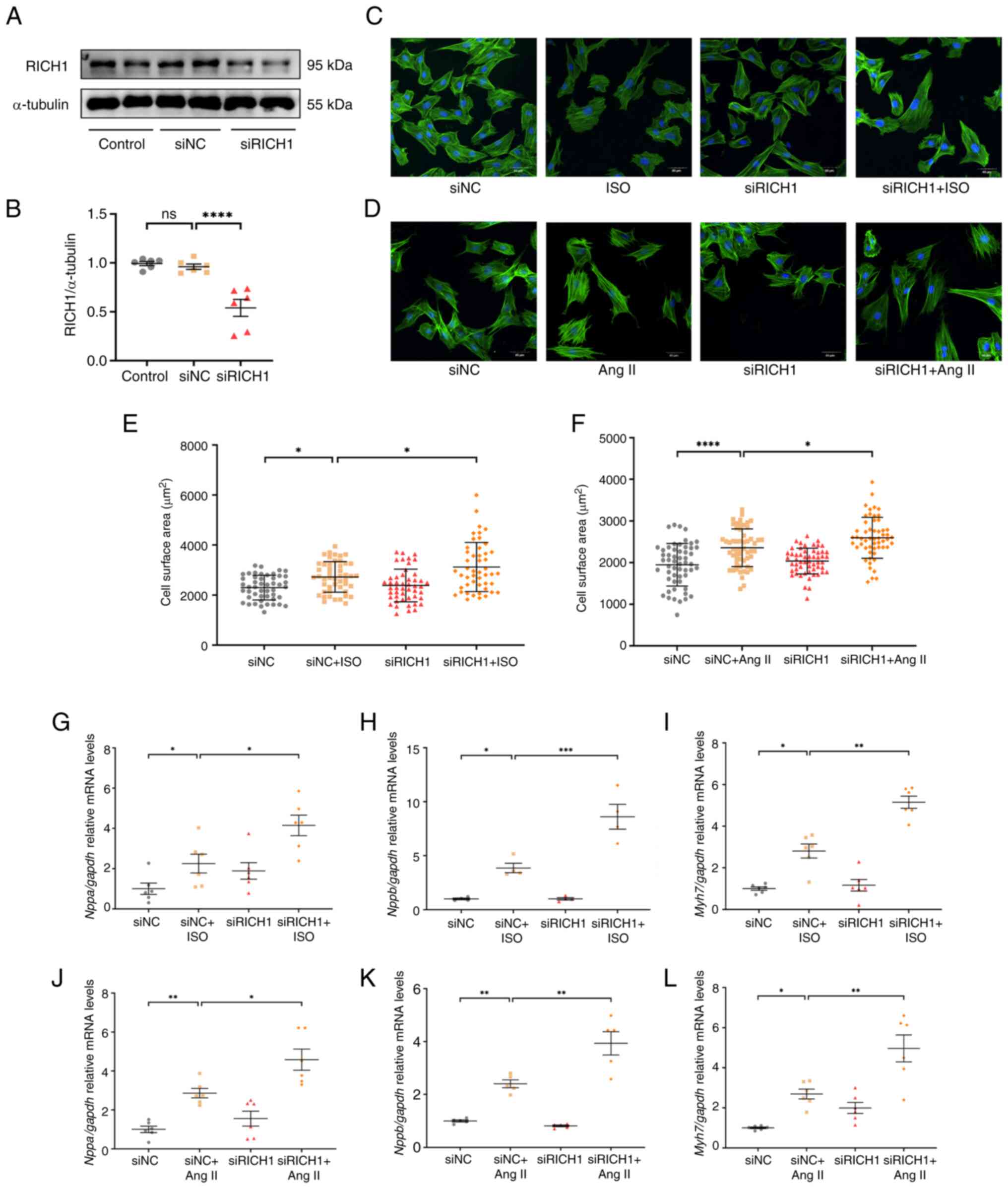 | Figure 3.siRNA-directed RICH1 knockdown
exacerbates ISO- or Ang II-induced cardiomyocyte hypertrophy. (A)
Protein expression levels of RICH1 in H9c2 cells transfected with
siRICH1 and siNC were assessed by western blotting, and (B) RICH1
expression was normalized to α-tubulin (n=6 experimental
repeats/group). Data were analyzed using one-way ANOVA with Tukey's
post hoc test and are presented as the mean ± SEM. H9c2 cells were
stained with Acti-stain™ 488 phalloidin and captured under an
Olympus FV3000 confocal microscope. Knockdown of RICH1 enhanced the
enlargement of CSA induced by (C) ISO or (D) Ang II. Scale bar, 40
µm. CSA quantification in response to (E) ISO (n=48 number of cells
assessed/group) and (F) Ang II (n=55 number of cells
assessed/group). This experiment was repeated three times. Data
were analyzed using one-way ANOVA with Tukey's post hoc test and
are presented as the mean ± SD. Expression levels of the cardiac
hypertrophy-related genes (G and J) Nppa, (H and K)
Nppb and (I and L) Myh7 treated with (G-I) ISO or
(J-L) Ang II were assessed by reverse transcription-quantitative
PCR and normalized to Gapdh (n=4-6 experimental
repeats/group). Data were analyzed using one-way ANOVA with Tukey's
post hoc test and are presented as the mean ± SEM. *P<0.05,
**P<0.01, ***P<0.001 and ****P<0.0001. CSA, cell surface
area; ISO, isoproterenol; Ang II, angiotensin II; NC, negative
control; si, small interfering; ns, not significant; RICH1, Rho
GTPase-activating protein interacting with CIP4 homologs protein
1. |
Discussion
Heart failure is the final stage of cardiovascular
diseases, which results in high morbidity and mortality, and
cardiac hypertrophy is an important stage in the occurrence of
heart failure. Inhibiting cardiac hypertrophy is of great
significance for preventing and alleviating heart failure. It has
been reported that various signaling pathways, such as the
MAPK/ERK, RhoA and PI3K/Akt/NF-κB pathways, are involved in cardiac
hypertrophy (1,4–7).
Previous studies have reported that RICH1 is a type of RhoGAP and a
key regulatory factor in numerous signaling pathways, such as ERK
and Rho signaling pathways, and diseases, including breast cancer
(26,29,31).
However, to the best of our knowledge, the relationship between
RICH1 and cardiac hypertrophy has not been reported.
In the present study, it was demonstrated that RICH1
can act as a novel suppressor of ISO- or Ang II-induced
cardiomyocyte hypertrophy. RICH1 was downregulated in ISO- or Ang
II-induced cardiomyocyte hypertrophy, and the overexpression of
RICH1 inhibited the increase in CSA and decreased the relative mRNA
expression levels of cardiac hypertrophic markers, including
Nppa, Nppb and Myh7, in ISO- or Ang II-treated H9c2
cells. By contrast, siRNA-mediated RICH1 KD exacerbated ISO- or Ang
II-induced increases in CSA, as well as the expression of genes
related to cardiomyocyte hypertrophy. The present study
demonstrated that RICH1 may be a novel target in the development of
treatments for cardiac hypertrophy. It has been reported that RICH1
can directly interact with small GTPases Cdc42 and Rac1, and serves
a key role in Cdc42/Rac1/ERK1/2 signaling (32). Notably, RICH1 can inhibit the
phosphorylation of ERK1/2 in epithelial cells through inactivating
Cdc42 and Rac1 (30). It is thus
implied that RICH1 may inhibit cardiac hypertrophy through
regulating the Cdc42/Rac1/ERK1/2 signaling pathway.
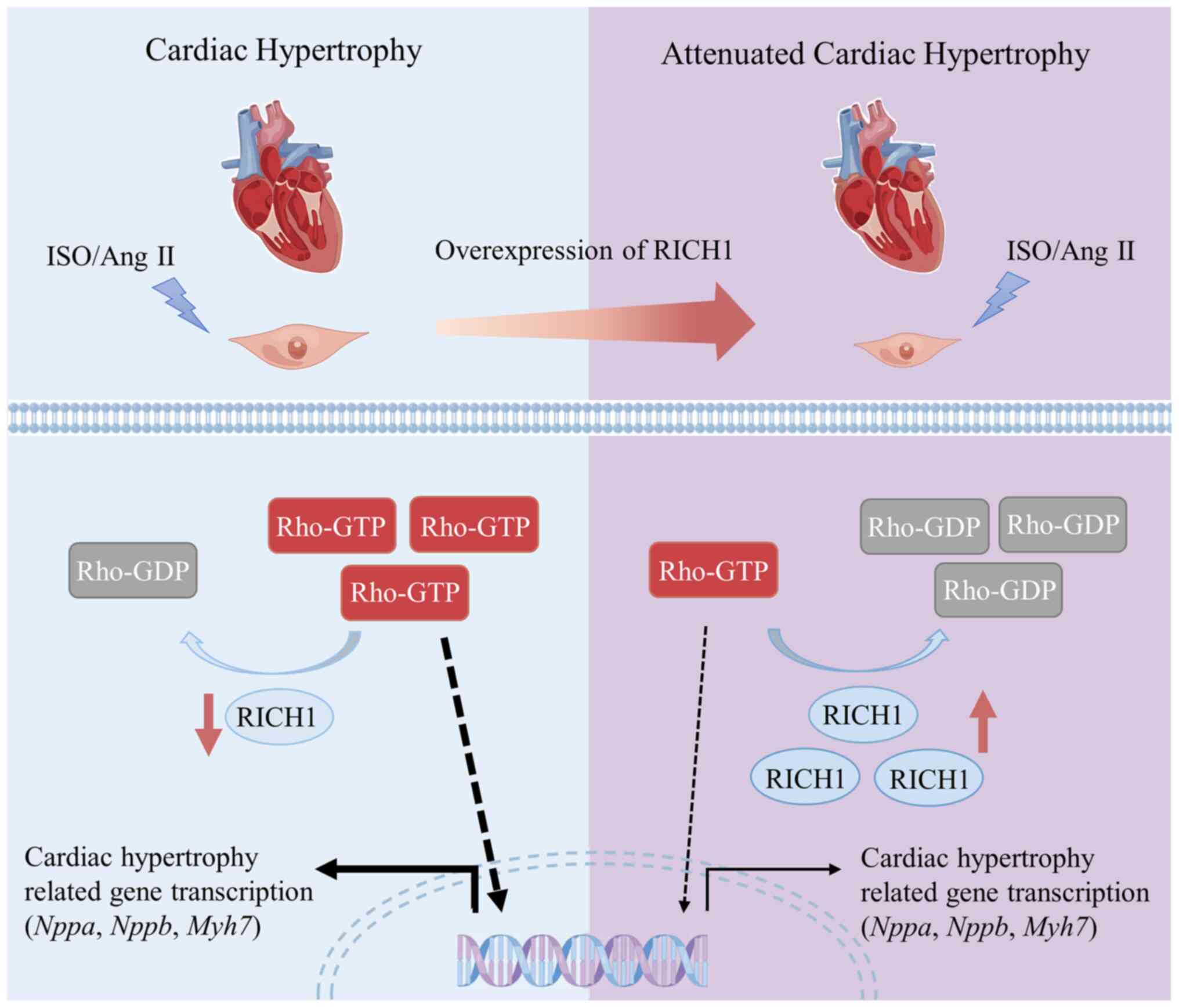
Based on previous studies and the results of the
present study, a prospective molecular mechanism by which RICH1
mediates cardiomyocyte hypertrophy has been proposed. It was
demonstrated that RICH1 is downregulated in ISO- or Ang II-induced
cardiomyocyte hypertrophy; therefore its downstream effectors, such
as Cdc42 and Rac1, may remain active, thus leading to cardiomyocyte
hypertrophy (Fig. 4). However,
RICH1 overexpression may inactivate its downstream effectors and
result in the attenuation of ISO or Ang II-induced cardiomyocyte
hypertrophy (Fig. 4). To elucidate
more detailed roles and mechanisms of RICH1 in cardiac hypertrophy,
cardiac-specific overexpression or KD of RICH1 should be assessed
in future studies.
RICH1 is highly expressed in cardiac tissue
(23), which implies that the
maintenance of high concentrations of RICH1 may be important for
myocardial structure and function; therefore, downregulation of
RICH1 may trigger myocardial remodeling. The use of RICH1 could
thus be beneficial as a prospective diagnostic indicator of
myocardial hypertrophy; however, further studies are required. For
example, it is worth testing what changes occur in downstream
proteins or signaling molecules after the dysregulation of
RICH1.
Previous studies have reported that autophagy;
actin-binding proteins, such as cofilin, Formin and CapZ; and the
MAPK/ERK signaling pathway serve roles in cardiac hypertrophy
(33–35). It has been reported that
autophagosome formation depends on small GTPases (36). In addition, small GTPases, such as
Cdc42 and Rac1, are the upstream regulators of actin-binding
proteins and the ERK signaling pathway (37,38).
Likewise, the abnormal upregulation of Rho GTPase functions can
induce cardiac hypertrophy. Fasudil, an inhibitor of ROCK, which is
a RhoA effector, can inhibit cardiac remodeling and hypertrophy
(39). In addition, vitamin D can
inhibit the expression of Rac1 and alleviate pressure
overload-induced cardiac hypertrophy (40), and statins can relieve cardiac
hypertrophy by inhibiting Rac1-mediated oxidative stress (41). Molecular switches act as a critical
and specific ‘brakes’ of Rho GTPases. RICH1 is upstream of Rho
GTPases; therefore, RICH1 may be considered a promising therapeutic
target for the treatment of cardiac hypertrophy. The present study
provides insights into the molecular mechanisms underlying cardiac
hypertrophy, which should be further assessed in future
studies.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by the Natural Science Foundation of
Hubei Province (grant no. 2022CFB843), the Xianning Science and
Technology Plan Project (grant no. 2021ZRKX024), the Hubei
University of Science and Technology School-level Fund (grant nos.
BK202121 and BK202220), the Special Project on Diabetes and
Angiopathy (grant no. 2022TNB04), the horizontal scientific
research project of Hubei University of Science and Technology
(grant no. 2022HX102) and the Scientific Research and Innovation
Team of Hubei University of Science and Technology (grant no.
2022T01).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SW, LL and CL made substantial contributions to the
conception or design of the work: acquisition, analysis and
interpretation of data; drafting the work or reviewing it
critically for important intellectual content; and finally approval
of the version to be published; they also agree to be accountable
for all aspects of the work in ensuring that questions related to
the accuracy or integrity of any part of the work are appropriately
investigated and resolved. XW completed and checked data statistics
and analysis. ZR conceived, edited and finalized the manuscript. SW
and ZR confirm the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bernardo BC, Weeks KL, Pretorius L and
McMullen JR: Molecular distinction between physiological and
pathological cardiac hypertrophy: Experimental findings and
therapeutic strategies. Pharmacol Ther. 128:191–227. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ritterhoff J and Tian R: Metabolic
mechanisms in physiological and pathological cardiac hypertrophy:
New paradigms and challenges. Nat Rev Cardiol. 20:812–829. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zeidan A, Javadov S and Karmazyn M:
Essential role of Rho/ROCK-dependent processes and actin dynamics
in mediating leptin-induced hypertrophy in rat neonatal ventricular
myocytes. Cardiovasc Res. 72:101–111. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rangrez AY, Bernt A, Poyanmehr R, Harazin
V, Boomgaarden I, Kuhn C, Rohrbeck A, Frank D and Frey N: Dysbindin
is a potent inducer of RhoA-SRF-mediated cardiomyocyte hypertrophy.
J Cell Biol. 203:643–656. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu G, Yussman MG, Barrett TJ, Hahn HS,
Osinska H, Hilliard GM, Wang X, Toyokawa T, Yatani A, Lynch RA, et
al: Increased myocardial Rab GTPase expression: A consequence and
cause of cardiomyopathy. Circ Res. 89:1130–1137. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ramos-Kuri M, Meka SH, Salamanca-Buentello
F, Hajjar RJ, Lipskaia L and Chemaly ER: Molecules linked to Ras
signaling as therapeutic targets in cardiac pathologies. Biol Res.
54:232021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tcherkezian J and Lamarche-Vane N: Current
knowledge of the large RhoGAP family of proteins. Biol Cell.
99:67–86. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Johnson DS and Chen YH: Ras family of
small GTPases in immunity and inflammation. Curr Opin Pharmacol.
12:458–463. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Reiner DJ and Lundquist EA: Small GTPases.
WormBook. 2018:1–65. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Song S, Cong W, Zhou S, Shi Y, Dai W,
Zhang H, Wang X, He B and Zhang Q: Small GTPases: Structure,
biological function and its interaction with nanoparticles. Asian J
Pharm Sci. 14:30–39. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Aikawa R, Komuro I, Nagai R and Yazaki Y:
Rho plays an important role in angiotensin II-induced hypertrophic
responses in cardiac myocytes. Mol Cell Biochem. 212:177–182. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Aikawa R, Komuro I, Yamazaki T, Zou Y,
Kudoh S, Zhu W, Kadowaki T and Yazaki Y: Rho family small G
proteins play critical roles in mechanical stress-induced
hypertrophic responses in cardiac myocytes. Circ Res. 84:458–466.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schmidt A and Hall A: Guanine nucleotide
exchange factors for Rho GTPases: Turning on the switch. Genes Dev.
16:1587–1609. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cherfils J and Zeghouf M: Regulation of
small GTPases by GEFs, GAPs, and GDIs. Physiol Rev. 93:269–309.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barbeau PA, Houad JM, Huber JS,
Paglialunga S, Snook LA, Herbst EAF, Dennis KMJH, Simpson JA and
Holloway GP: Ablating the Rab-GTPase activating protein TBC1D1
predisposes rats to high-fat diet-induced cardiomyopathy. J
Physiol. 598:683–697. 2020. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Binsch C, Barbosa DM, Hansen-Dille G,
Hubert M, Hodge SM, Kolasa M, Jeruschke K, Weiß J, Springer C,
Gorressen S, et al: Deletion of Tbc1d4/As160 abrogates cardiac
glucose uptake and increases myocardial damage after
ischemia/reperfusion. Cardiovasc Diabetol. 22:172023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bisserier M, Berthouze-Duquesnes M,
Breckler MS, Tortosa F, Fazal L, de Régibus A, Laurent AC, Varin A,
Lucas A, Branchereau M, et al: Carabin protects against cardiac
hypertrophy by blocking calcineurin, Ras, and
Ca2+/calmodulin-dependent protein kinase II signaling. Circulation.
131:390–400. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
De Windt LJ, Lim HW, Bueno OF, Liang Q,
Delling U, Braz JC, Glascock BJ, Kimball TF, del Monte F, Hajjar RJ
and Molkentin JD: Targeted inhibition of calcineurin attenuates
cardiac hypertrophy in vivo. Proc Natl Acad Sci USA. 98:3322–3327.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu J, Ismat FA, Wang T, Lu MM, Antonucci N
and Epstein JA: Cardiomyocyte-specific loss of neurofibromin
promotes cardiac hypertrophy and dysfunction. Circ Res.
105:304–311. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Richnau N, Fransson A, Farsad K and
Aspenström P: RICH-1 has a BIN/Amphiphysin/Rvsp domain responsible
for binding to membrane lipids and tubulation of liposomes. Biochem
Biophys Res Commun. 320:1034–1042. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wells CD, Fawcett JP, Traweger A, Yamanaka
Y, Goudreault M, Elder K, Kulkarni S, Gish G, Virag C, Lim C, et
al: A Rich1/Amot complex regulates the Cdc42 GTPase and
apical-polarity proteins in epithelial cells. Cell. 125:535–548.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nagy Z, Wynne K, von Kriegsheim A,
Gambaryan S and Smolenski A: Cyclic nucleotide-dependent protein
kinases target ARHGAP17 and ARHGEF6 complexes in platelets. J Biol
Chem. 290:29974–29983. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Richnau N and Aspenström P: Rich, a rho
GTPase-activating protein domain-containing protein involved in
signaling by Cdc42 and Rac1. J Biol Chem. 276:35060–35070. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pan SL, Deng YY, Fu J, Zhang YH, Zhang ZJ
and Qin XJ: ARHGAP17 enhances 5-Fluorouracil-induced apoptosis in
colon cancer cells by suppressing Rac1. Neoplasma. 69:640–647.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang L, Yang X, Wan L, Wang S, Pan J and
Liu Y: ARHGAP17 inhibits pathological cyclic strain-induced
apoptosis in human periodontal ligament fibroblasts via Rac1/Cdc42.
Clin Exp Pharmacol Physiol. 47:1591–1599. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu PR, Chiang SY, Midence R, Kao WC, Lai
CL, Cheng IC, Chou SJ, Chen CC, Huang CY and Chen RH: Wdr4 promotes
cerebellar development and locomotion through Arhgap17-mediated
Rac1 activation. Cell Death Dis. 14:522023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yi C, Troutman S, Fera D,
Stemmer-Rachamimov A, Avila JL, Christian N, Persson NL, Shimono A,
Speicher DW, Marmorstein R, et al: A tight junction-associated
Merlin-angiomotin complex mediates Merlin's regulation of mitogenic
signaling and tumor suppressive functions. Cancer Cell. 19:527–540.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ullah R, Yin Q, Snell AH and Wan L:
RAF-MEK-ERK pathway in cancer evolution and treatment. Semin Cancer
Biol. 85:123–154. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tian Q, Gao H, Zhou Y, Zhu L and Yang J,
Wang B, Liu P and Yang J: RICH1 inhibits breast cancer stem cell
traits through activating kinases cascade of Hippo signaling by
competing with Merlin for binding to Amot-p80. Cell Death Dis.
13:712022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aslan JE: Platelet Rho GTPase regulation
in physiology and disease. Platelets. 30:17–22. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang J, Wang J, Zhou YF, Ren XY, Lin MM,
Zhang QQ, Wang YH and Li X: Rich1 negatively regulates the
epithelial cell cycle, proliferation and adhesion by
CDC42/RAC1-PAK1-Erk1/2 pathway. Cell Signal. 27:1703–1712. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Luan Y, Luan Y, Feng Q, Chen X, Ren KD and
Yang Y: Emerging role of mitophagy in the heart: Therapeutic
potentials to modulate mitophagy in cardiac diseases. Oxid Med Cell
Longev. 2021:32599632021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pan C, Wang S, Liu C and Ren Z:
Actin-binding proteins in cardiac hypertrophy. Cells. 11:35662022.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yan ZP, Li JT, Zeng N and Ni GX: Role of
extracellular signal-regulated kinase 1/2 signaling underlying
cardiac hypertrophy. Cardiol J. 28:473–482. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zoppino FC, Militello RD, Slavin I,
Alvarez C and Colombo MI: Autophagosome formation depends on the
small GTPase Rab1 and functional ER exit sites. Traffic.
11:1246–1261. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sun Y, Xu C, Jiang Z and Jiang X:
DEF6(differentially exprehomolog. exacerbates pathological cardiac
hypertrophy via RAC1. Cell Death Dis. 14:4832023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li G, Wang Y, Guo XB and Zhao B: CDC42
regulates cell proliferation and apoptosis in bladder cancer via
the IQGAP3-mediated Ras/ERK pathway. Biochem Genet. 60:2383–2398.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mondaca-Ruff D, Araos P, Yañez CE, Novoa
UF, Mora IG, Ocaranza MP and Jalil JE: Hydrochlorothiazide reduces
cardiac hypertrophy, fibrosis and rho-kinase activation in
DOCA-salt induced hypertension. J Cardiovasc Pharmacol Ther.
26:724–735. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Moradi A, Maroofi A, Hemati M, Hashemzade
T, Alborzi N and Safari F: Inhibition of GTPase Rac1 expression by
vitamin D mitigates pressure overload-induced cardiac hypertrophy.
Int J Cardiol Heart Vasc. 37:1009222021.PubMed/NCBI
|
|
41
|
Liao JK: Statin therapy for cardiac
hypertrophy and heart failure. J Investig Med. 52:248–253. 2004.
View Article : Google Scholar : PubMed/NCBI
|