Introduction
Doxorubicin (Dox) is a potent anticancer drug that
carries the risk of cardiotoxicity in cancer patients (1,2). The
exact underlying mechanisms of Dox-induced cardiomyopathy (DIC)
remain elusive, making it difficult to predict or prevent the
associated adverse cardiovascular side effects. Numerous molecular
mechanisms are known to be involved in DIC, including oxidative
stress (OS) (2,3). Dox mediated an increase in reactive
oxygen species (ROS) and a decrease in endogenous antioxidants,
resulting in OS and the disruption of cell homeostasis, ultimately
leading to cell death (4). Among
the Dox-induced cellular changes, most remarkable is the dilation
of the endoplasmic reticulum (ER) (5,6).
Disruption of the ER structure is associated with changes in
protein synthesis, folding and translocation, as well as calcium
cycling and excitation-contraction coupling in the heart (7,8).
Stress and/or pathological stimuli, such as OS,
results in the accumulation of unfolded and/or misfolded proteins
in the ER, referred to as ER stress. When ER stress is mild and
transient, ER adaptive responses are activated to maintain ER
functioning and homeostasis. ER stress activates the unfolded
protein response (UPR) through three ER transmembrane sensor
proteins: i) Protein kinase-like ER kinase (PERK) (9); ii) activating transcription factor 6α
(ATF6α) (10); and iii)
inositol-requiring kinase 1α (IRE1) (11). Upon mediation of adaptive
responses, luminal ER chaperone, glucose-regulated protein 78
(GRP78) dissociates from PERK, IRE1 and ATF6α, allowing the
activation of their downstream pathways. Subsequent activation
leads to the upregulation of ER chaperones and protein foldases,
and induces the expression of various genes, including X-box
binding protein 1 (XBP1). The unconventional splicing of XBP1 mRNA
generates spliced XBP1 (XBP1s) that subsequently plays an essential
role in further upregulation of chaperones, such as
glucose-regulated protein 94 (GRP94), GRP78 and/or protein
disulfide-isomerase (PDI) (8,12).
However, when ER stress is severe, UPR fails to restore ER
homeostasis. ER stress initiates apoptotic signaling pathways via
CCAAT/enhancer homologous protein (CHOP), caspase-12 activation,
phosphorylation of c-JUN NH2-terminal kinase (JNK) and
pro-apoptotic gene expression, which play an essential role in the
progression of DIC and cardiomyocytes loss (13,14).
Empagliflozin (EMPA) is a sodium-glucose
co-transporter 2 (SGLT-2) inhibitor that is used to decrease blood
glucose levels and sodium load in diabetic patients (15). Results of the EMPA-REG OUTCOME
trial demonstrated that EMPA reduced the risk of hospitalization
for heart failure and cardiovascular death in the EMPA-REG OUTCOME
trial (16). Additional clinical
trials, including the EMPEROR-Reduced and EMPEROR-Preserved trials,
reported cardio-renal benefits of EMPA in the non-diabetic
population (17–19). As per the Canadian Cardiovascular
Society heart failure guidelines, SGLT-2 inhibitors have now been
included as first-line therapy in the setting of heart failure with
reduced ejection fraction (20).
In addition, results of pre-clinical studies demonstrated the
utility of EMPA and other SGLT-2 inhibitors in the prevention of
DIC (21,22). Dapagliflozin was reported to
attenuate DIC in a murine diabetic model (22) via inhibition of ER stress.
Moreover, Sabatino et al (21) suggested that the beneficial effects
of EMPA on DIC may be independent of its glycemic control.
Results of a previous study by the authors and other
studies demonstrated that the mitigation of ER stress reduces the
detrimental effects of Dox and cardiomyocyte loss (23–26).
However, the effects of EMPA in mitigating Dox-induced ER stress as
well as the molecular pathways involved are unknown. The present
study aimed to examine the potential role of EMPA in mitigating
Dox-mediated ER stress injury in the in vitro setting using
isolated cardiomyocytes. It was demonstrated that EMPA reduces
overall ER-stress in Dox treated cardiomyocytes. Additionally, the
present novel results confirmed that these changes were due to
increased expression of an ER transmembrane protein,
inositol-requiring kinase 1α (IRE1), which plays an essential role
in maintaining ER homeostasis and inhibiting inflammation and
ER-initiated apoptosis upon Dox insult.
Materials and methods
Cardiomyocyte isolation and
treatment
The guidelines of the Canadian Council on Animal
Care were followed for all animal procedures, and the present study
was approved [(approval no. 22-012 (AC11739)] by the Animal
Protocol Review Committee at the University of Manitoba (Winnipeg,
Canada). Male Sprague Dawley rats (n=10; ~8 weeks-old; weight,
200±10 g) were ordered from Charles River laboratories and shipped
to our facility in a climate-controlled vehicle. Rats were housed
in a temperature-regulated room (22–24°C) on a 12/12-h light-dark
cycle, and had ad libitum access to standard chow and water.
After 1 week of conditioning, rats were administered the following
anesthetic agents intraperitoneally (ketamine 90 mg/kg and xylazine
10 mg/kg) prior to euthanasia. Subsequently, the hearts were
removed for cardiomyocyte isolation using the standard Langendorff
apparatus, as previously described (23,24).
In total, ~106 cells were plated in 10-cm laminin-coated
polystyrene tissue culture plates (cat. no. CLS430167; Corning,
Inc.) with 10% fetal bovine serum (FBS; Thermo Fisher Scientific,
Inc.) and streptomycin/penicillin (100 U/ml) in M199 culture media
(Sigma-Aldrich; Merck KGaA) at 37°C in 5% CO2 with 95%
O2. After 2 h primary incubation, dead cells were
removed by washing with M199 culture media and viable cells were
incubated overnight with 0.5% FBS under the same incubation
conditions. The following morning, culture plates were randomly
divided into four treatment groups as follows: Control
(cardiomyocytes cultured in M199 +0.5% FBS); EMPA (500 nM; cat. no.
22369-1; Cayman Chemical Company); Dox (10 µM; Pfizer, Inc.); and
Dox + EMPA, for 24 h. For the combination group of Dox + EMPA,
cells were treated with EMPA for 1 h prior to the addition of
Dox.
Cell viability
Isolated cardiomyocytes were incubated for different
times (0, 6, 12 and 24 h) to determine time- and
treatment-dependent viability. An MTT
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]
assay was performed after each treatment period. Briefly,
cardiomyocytes (~104 cells/well) were plated on 96-well
plates for a specific time, and 5 mg/ml MTT was subsequently added
to each well for 2 h at 37°C. After 2 h incubation, formazan
crystals were observed. Once the crystallization was >90%, MTT
was removed, and 150 µl of dimethyl sulfoxide was added to each
well and mixed in the dark. The absorbance of each well was
subsequently recorded at 570 nm using a Cytation 5 Cell Imaging
Multi-Mode Reader (RRID:SCR_019732; BioTek Instruments, Inc.).
Protein estimation and western blot
analysis
At the end of the treatment period, cardiomyocytes
were washed with PBS and centrifuged at 12,000 × g for 10 min at
room temperature (RT). The cell pellet was resuspended in ice cold
RIPA buffer (cat. no. 89900; Thermo Fisher Scientific, Inc.) with
Pierce protease and phosphatase inhibitors, (cat. no. A32959;
Pierce; Thermo-Fisher Scientific, Inc.), 1 mM phenylmethylsulfonyl
fluoride, and DTT overnight at −20°C. After overnight incubation,
samples were sonicated using a water bath sonicator. Sonicated
samples were subsequently centrifuged at 12,000 × g for 15 min at
4°C. The supernatant was used to determine total protein
concentration as per the Bradford dye-binding method using BSA as a
standard (Bio-Rad Laboratories, Inc.) using a microwell plate
reader (Cytation 5; BioTek Instruments, Inc.). Total protein was
stored at −20°C until further analysis.
A total of 20–40 µg of protein was separated using
electrophoresis on 8–12% SDS-polyacrylamide gels at 80 mV. The
separated proteins were subsequently transferred onto a PVDF
membrane for 90 min at constant 200 mA in a cold room. Membranes
were subsequently blocked in 5% BSA and washed with Tris-buffered
saline plus 0.1% Tween-20 (TBST) blocking buffer for 1 h at RT.
After blocking, membranes were incubated with the respective
antibodies for either 1.5 h at RT or overnight at 4°C on a rocker.
Membranes were subsequently washed with TBST (3 washes 10 min each)
and incubated with horseradish peroxidase (HRP)-conjugated
anti-rabbit IgG (1:5,000; cat. no. W401B; RRID: AB_430833; Promega
Corporation) or HRP-conjugated anti-mouse IgG secondary antibodies
(1:5,000; cat. no. 170-6516; RRID: AB_11125547; Bio-Rad
Laboratories, Inc.) for 1 h at RT. Protein bands were visualized
using the Pierce ECL Plus Western Blotting Substrate (PerkinElmer,
Inc.) at different exposure times on a Chem Doc imager (BioRad
Laboratories, Inc.). Densitometric analysis was performed using
Image Lab 6.0 (Bio-Rad Laboratories, Inc.) and GAPDH was used for
normalization for each sample.
Primary antibodies were as follows: Anti-Caspase-12
(1:2,000; cat. no. ab62484; RRID: AB_955729; Abcam); anti-K-Lysine,
D-Aspartic acid, E-Glutamic acid and L-Leucine sequence (1:1,000;
cat. no. ab176333; RRID: AB_2819147; Abcam) which is used to target
GRP94, GRP78 and PDI; anti-XBP1 (1:500; cat. no. ab37152; RRID:
AB_778939; Abcam); anti-ATF6 (1:5,000; cat. no. 24169-1-AP; RRID:
AB_2876891; ProteinTech Group, Inc.); anti-PERK (1:500; cat. no.
20582-1-AP; RRID: AB_10695760; ProteinTech Group, Inc.); anti-IRE1
(1:1,500; cat. no. 20582-1-AP; RRID: AB_1069#5760; ProteinTech
Group, Inc.); anti-JNK1 + JNK2 + JNK3 (1:1,000; cat. no. ab179461;
RRID: AB_2744672; Abcam); anti-phosphorylated (p-)JNK1 + JNK2 +
JNK3 [T183 + T183 + T221 (1:500; cat. no. ab124956; RRID:
AB_10973183; Abcam); anti-sodium-glucose cotransporter-2 (SGLT2;
1:1,000; cat. no. ab37296; RRID: AB_777895; Abcam) and anti-GAPDH
(1:4,000; cat. no. 2118; RRID: AB_561053; Cell Signaling
Technology, Inc.).
Reverse transcription-quantitative
(RT-q) PCR
Aurum™ Total RNA Mini kit (cat. no. 7326820; BioRad
Laboratories, Inc.) was used to isolate total RNA in the cell
culture hood according to the manufacturer's protocols. Extracted
RNA was quantified using nanodrop (Nanodrop Lite; Thermo Fisher
Scientific, Inc.). Total RNA was reverse transcribed into cDNA (40
ng) using High-Capacity cDNA Reverse Transcription kit with RNase
Inhibitor as per manufacturer's instructions (cat. no. 4374966;
Thermo Fisher Scientific, Inc.) on the T100 Thermal Cycler
(RRID:SCR_021921; BioRad Laboratories, Inc.). RT-qPCR reactions
were prepared using 22.5 ng/µl of cDNA template and 500 nM of
forward and reverse primers to a final volume of 10 µl. RT-PCR
amplification was performed using a QuantStudio 3 Real Time PCR
System (RRID:SCR_020238; Thermo Fisher Scientific, Inc.) with Luna
Universal Master Mix (cat. no. M3003; New England Biolabs, Inc.).
The program was set up to have initial denaturation at 95°C for 60
sec, followed by 40 cycles of denaturation at 95°C for 15 sec, and
extension at 60°C for 30 sec. A continuous melt curve was
subsequently produced from 60–95°C for ~20 sec. mRNA levels were
quantified using the 2−ΔΔCq method and normalized to the
internal reference gene GAPDH (27). Primer sequences are displayed in
Table I.
 | Table I.Rat-specific primer sequences used
for reverse transcription-quantitative PCR. |
Table I.
Rat-specific primer sequences used
for reverse transcription-quantitative PCR.
| Gene name | Primer sequence
(5′à3′) |
|---|
| XBP1 | F:
ACGAGAGAAAACTCATGG |
|
| R:
ACAGGGTCCAACTTGTCC |
| IL-10 | F:
AGTGGAGCAGGTGAAGAATGA |
|
| R:
CAGTAGATGCCGGGTGGTT |
| TNFα | F:
ATGGGCTCCCTCTCATCAGT |
|
| R:
GCTTGGTGGTTTGCTACGAC |
| TGFβ | F:
GCGGACTACTACGCCAAAGA |
|
| R:
TGCTTCCCGAATGTCTGACG |
| STAT-3 | F:
TCGGAAAGTATTGTCGCCCC |
|
| R:
GACATCGGCAGGTCAATGGT |
| GAPDH | F:
CATCAACGACCCCTTCATTGACCTCA |
|
| ACTA |
|
| R:
TCCACGATGCCAAAGTTGTCATGG |
Glucose uptake assay
Glucose Uptake Assay kit (cat. no. ab136956; Abcam)
was used to record glucose uptake, according to the manufacturer's
instructions. Briefly, ~104 cells were plated in a
96-well flat bottom plate and treated as previously described. At
the end of the treatment period, cells were washed three times with
PBS and Tyrode's solution with 10 mmol/l mannitol was added for 45
min to starve cells of glucose. Subsequently, 2-deoxyglucose
(2-DG), which is structurally similar to glucose, was added to all
cells excluding the respective negative controls, and incubated for
30 min. Cells were subsequently washed with PBS to remove exogenous
2-DG and lysed using extraction buffer at −80°C. After repeating
freeze/thaw cycles, cell lysates were boiled at 80°C for 45 min.
Cell lysate was cooled on ice for 5 min and then neutralised using
neutralization buffer. Following centrifugation, the supernatant
was collected and mixed with reaction mix and 2-DG uptake assay
buffer and incubated in the dark for 40 min. Fluorescence of
accumulated 2-DG-6-phosphate (2-DG6P) was recorded with
excitation/emission at 535/587 nm using the Cytation 5 Cell Imaging
Multi-Mode Reader (RRID:SCR_019732; BioTek Instruments, Inc.).
Final 2-DG uptake was calculated using a 2-DG6P standard curve.
2′,7′-dichlorodihydrofluorescein
diacetate (H2DCFDA) assay
H2DCFDA dye (cat. no. D399; Invitrogen;
Thermo Fisher Scientific, Inc.) was used to measure overall OS in
different treatment groups. Briefly, ~104 cells were
plated in 96-well flat bottom plates and treated as previously
described. At the end of the treatment period, cells were washed
three times with PBS +0.1% FBS and 5 µM dye was added in each well.
Plates were incubated in the dark for 45 min to allow diffusion of
dye into the cell. After three washes with PBS +0.1% FBS,
fluorescence was recorded with excitation/emission at 485/535 nm
using the Cytation 5 Cell Imaging Multi-Mode Reader
(RRID:SCR_019732; BioTek Instruments, Inc.). Images were captured
at a magnification of ×20.
Statistical analysis
All experiments were performed in duplicate for each
treatment group for a total of 3–5 individual biological samples.
Data are expressed as the mean ± SEM. Comparisons between treatment
groups were carried out using one-way ANOVA, and Tukey-Kramer's
test was performed to identify differences between groups. Data
analysis was performed using GraphPad Prism (version 8.0; GraphPad
Software, Inc.; Dotmatics). P<0.05 was considered to indicate a
statistically significant difference.
Results
Expression of SGLT2 and effects of
EMPA on 2-DG uptake
As EMPA inhibits SGLT2 transporters, isolated
cardiomyocytes were treated with 10 µM Dox for 24 h with or without
EMPA, and SGLT2 protein expression was investigated using western
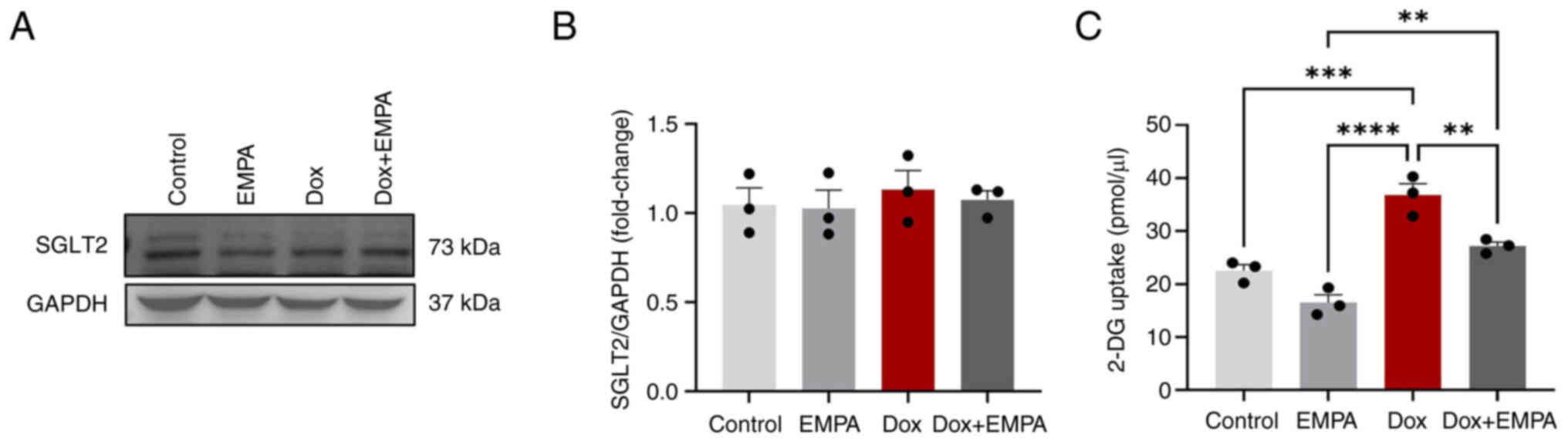
blot analysis (Fig. 1A). Although
the baseline expression of SGLT2 was detected (both the 73 kDa
SGLT-2 and its cleavage fragments) (Fig. 1A), the relative expression of SGLT2
among different treatment groups was not different from each other
(Fig. 1B). As SGLT2 was expressed
in adult rat cardiomyocytes, the primary function of EMPA was
evaluated. Subsequently, a 2-DG uptake assay was performed because
of its structural similarity to glucose. 2-DG is taken up by
glucose transporters and metabolized to 2-DG-6-phosphate (2-DG6P).
However, it cannot be further metabolized, and therefore
accumulates within cells. Using the relative fluorescence of
oxidized 2-DG6P, 2-DG6P uptake was determined amongst treatment
groups. Results of the present study demonstrated that EMPA-treated
cardiomyocytes exhibited significantly low levels of 2-DG6P uptake,
compared with Dox, and Dox + EMPA groups (Fig. 1C). Notably, the Dox group exhibited
a higher 2-DG uptake, compared with the control group (Fig. 1C). However, no significant changes
were observed between the Dox + EMPA and control groups (Fig. 1C).
Dox-induced expression of ER stress
markers and their mitigation by EMPA
ER stress leads to the overexpression of UPR
chaperones which were significantly higher following Dox treatment.
Dox treatment for 24 h increased the protein expression of GRP94,
GRP78 and PDI compared with the control and EMPA groups (Fig. 2A-D). In Dox + EMPA-treated
cardiomyocytes, increases in protein expression were significantly
inhibited following EMPA treatment, compared with the Dox group
(Fig. 2A). Notably, GRP94, RP78
and PDI expression levels were reduced following treatment with
EMPA; however, these changes were not significant.
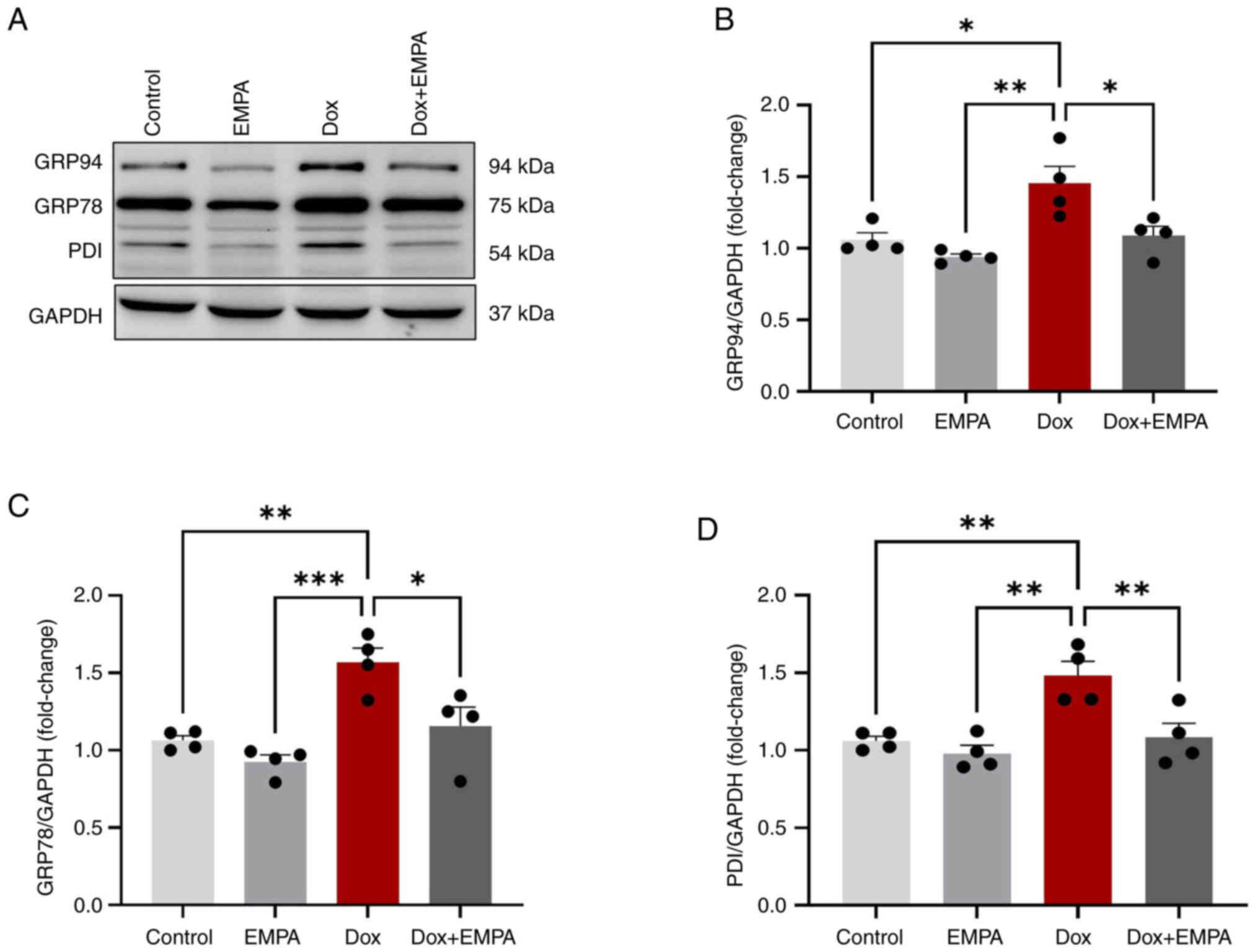 | Figure 2.Protein expression of unfolded
protein response chaperones. Cardiomyocytes were treated with
either Dox (10 µM), EMPA (500 nM) or Dox + EMPA for 24 h. (A)
K-Lysine, D-Aspartic acid, E-Glutamic acid and L-Leucine sequence
antibody was used to determine GRP94, GRP78 and PDI expression
levels. (B-D) Densitometric analysis of respective proteins using
GAPDH as the loading control. Data are presented as the mean ± SEM,
using four individual biological samples repeated in duplicate.
*P<0.05, **P<0.005 and ***P<0.0005. Dox, Doxorubicin;
EMPA, empagliflozin; PDI, protein disulfide-isomerase; GRP,
glucose-regulated protein. |
EMPA-induced changes in UPR signaling
proteins
Upon induction of ER stress, UPR signaling proteins
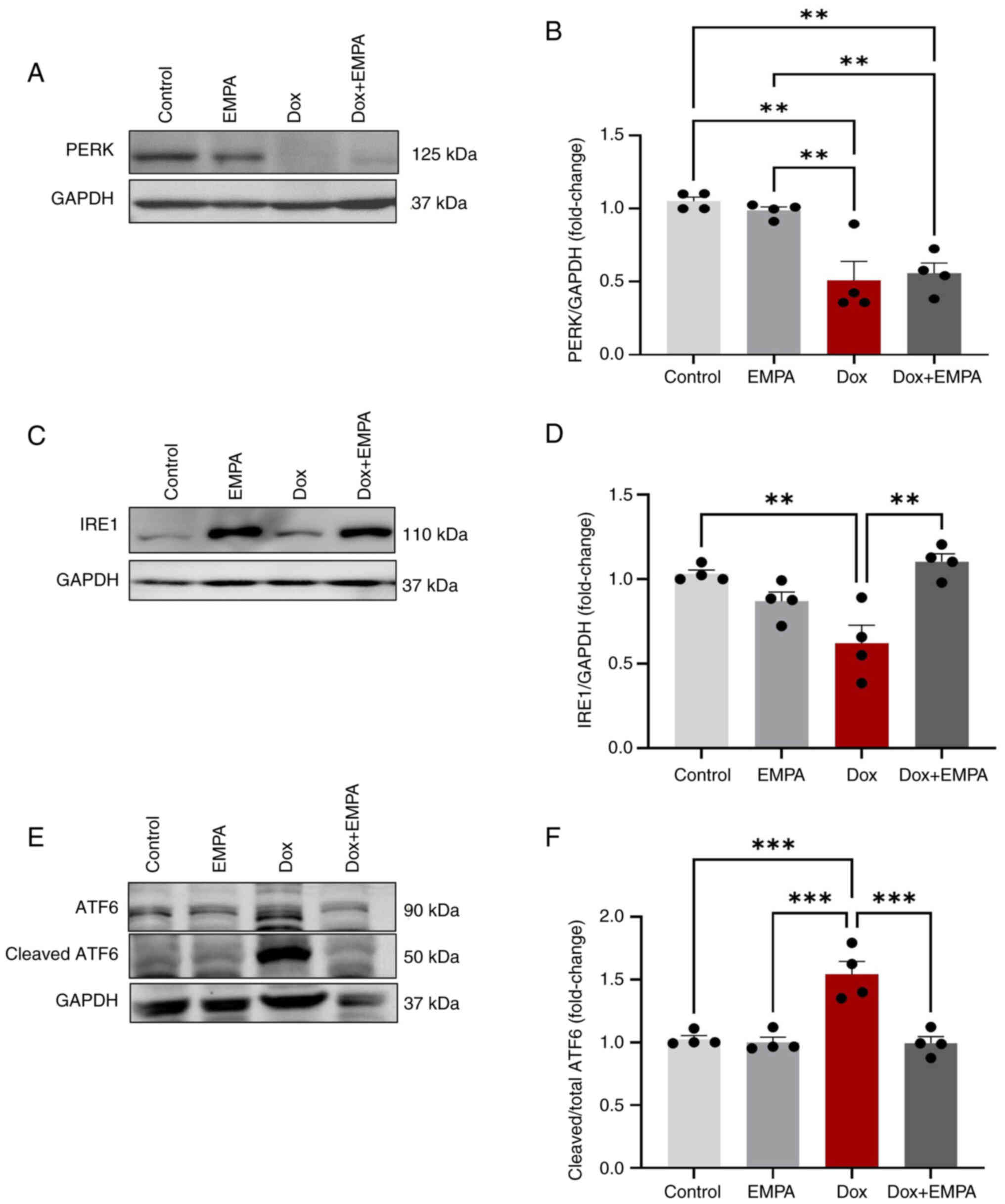
(PERK, IRE1 and ATF6α) are activated. Results of the present study
demonstrated that Dox treatment significantly reduced the
expression of PERK and IRE1, compared with the control group
(Fig. 3A-D). Notably,
pre-treatment with EMPA prevented the downregulation of IRE1 by
Dox, and the observed expression levels were not significant
compared with the control group (Fig.
3C and D). However, EMPA exerted no significant effect on PERK
expression (Fig. 3A and B) in the
Dox + EMPA group. Dox activates ATF6α cleavage, and results of the
present study revealed an increase in the activated cleaved ATF6α
fragment (Fig. 3E and F).
Increased expression of active ATF6α was prevented following
treatment with EMPA.
EMPA-induced changes in XBP1 mRNA and
protein expression levels
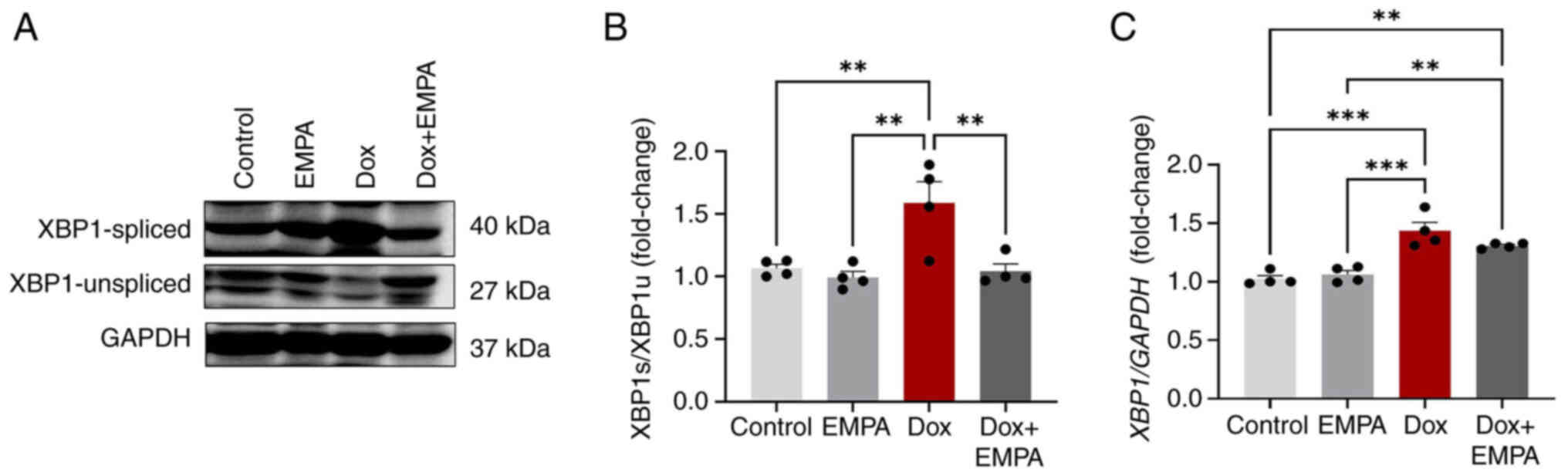
XBP1 plays an essential role in mediating UPR
function and is a transcription factor for genes required by UPR.
Dox treatment significantly increased XBP1 mRNA expression
levels, compared with the control and EMPA groups (Fig. 4C). However, pre-treatment with EMPA
exerted no effect on XBP1 mRNA, and the expression levels
remained high. Results of the present study also demonstrated that
Dox treatment caused a 1.5-fold increase in XBP1s/XBP1u protein
expression, and these levels were significantly reduced following
treatment with EMPA (Fig. 4A and
B). However, XBP1u protein expression levels remained unchanged
in all groups, excluding the Dox group. Results of the present
study demonstrated that the expression of XBP1s/XBP1u was high in
Dox-treated cardiomyocytes, and expression returned to levels
similar to that of the control following treatment with EMPA
(Fig. 4A and B).
Effects of EMPA on ER stress-induced
apoptosis
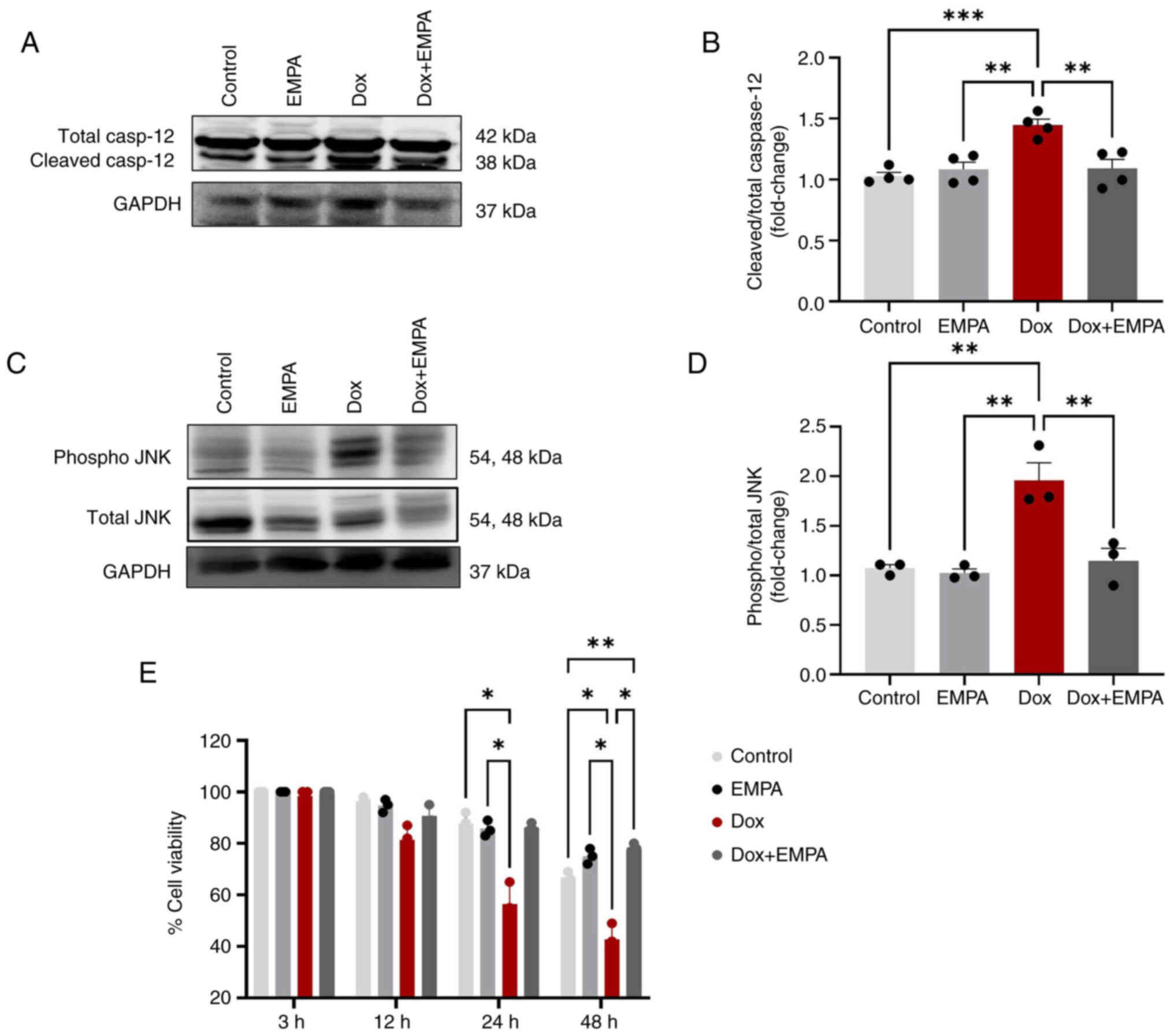
When cells fail to recover from ER stress, ER
initiates apoptosis through the JNK-pathway, leading to the
activation of caspases. Results of the present study demonstrated
that ER resident caspase-12 was activated and the levels of
cleaved/total caspase-12 were significantly higher following Dox
treatment. However, pre-treatment with EMPA reduced the activation
of caspase-12 through preventing cleavage (Fig. 5A and B). Results of the present
study also demonstrated a significant increase in the
phosphorylation of JNK (p-JNK) following treatment with Dox,
compared with the control and EMPA groups (Fig. 5C and D). However, the expression of
p-JNK was significantly reduced in the in Dox + EMPA group,
compared with the Dox group.
Moreover, the viability of cardiomyocytes was
measured at 3, 12, 24 and 48 h using an MTT assay (Fig. 5E). Results of the present study
demonstrated that Dox significantly reduced cell viability to 55%
at 24 h, and ~40% of cardiomyocytes survived for 48 h.
Pre-treatment with EMPA rescued cardiomyocytes from Dox-induced
cell death, and >80% of cardiomyocytes were viable at 24 h.
After 48 h, EMPA-treated cardiomyocytes exhibited ~75% viability,
and this was significantly higher than the control group (Fig. 5E).
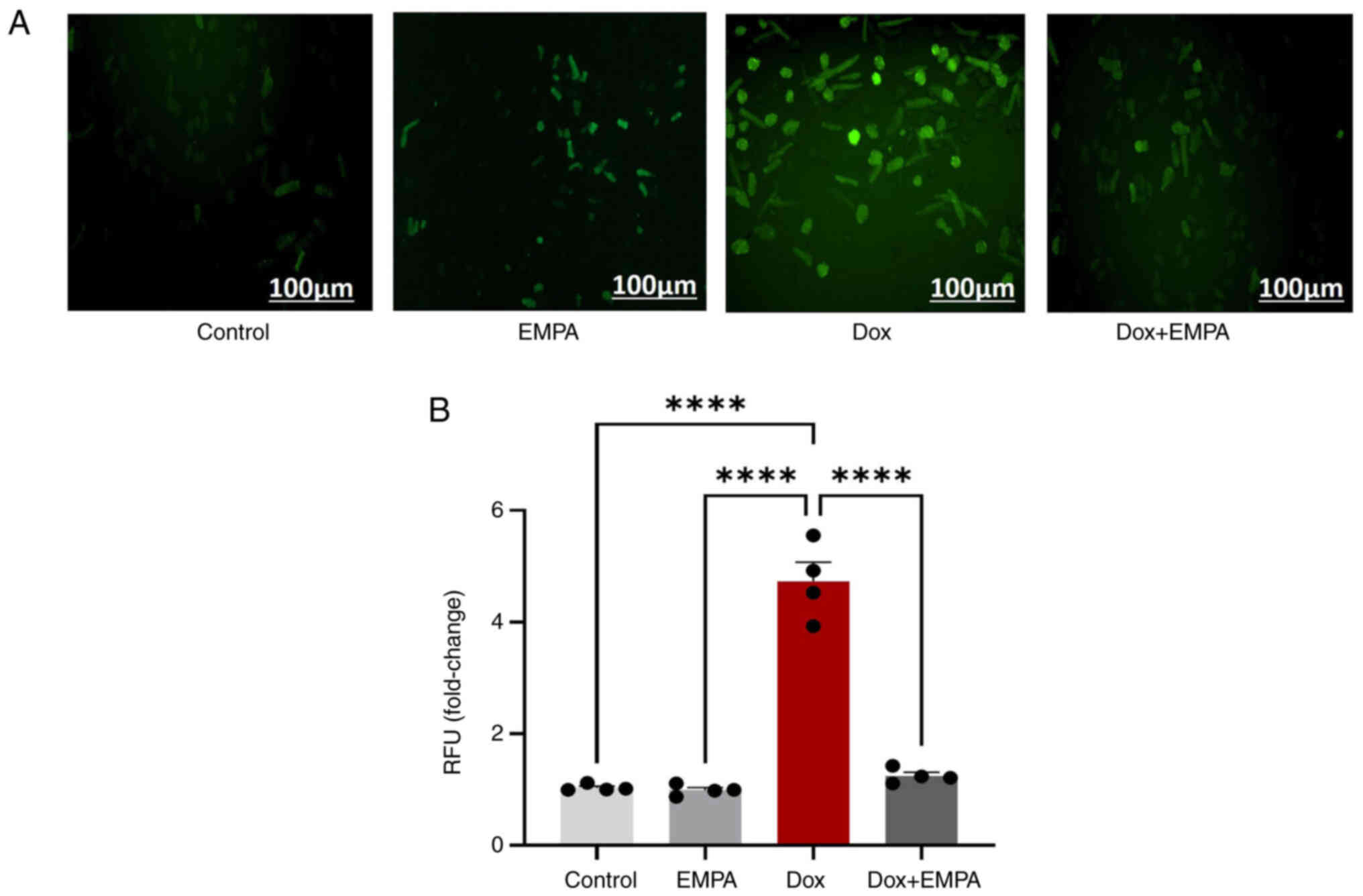
EMPA protects against Dox-induced OS
and inflammation
Dox-induced production of ROS and excessive OS lead
to cardiomyopathy. Thus, ROS production was recorded following Dox
treatment with or without EMPA. Results of the present study
demonstrated a >4-fold increase in ROS production following 24 h
Dox treatment (Fig. 6A and B), and
pre-treatment with EMPA significantly reduced OS.
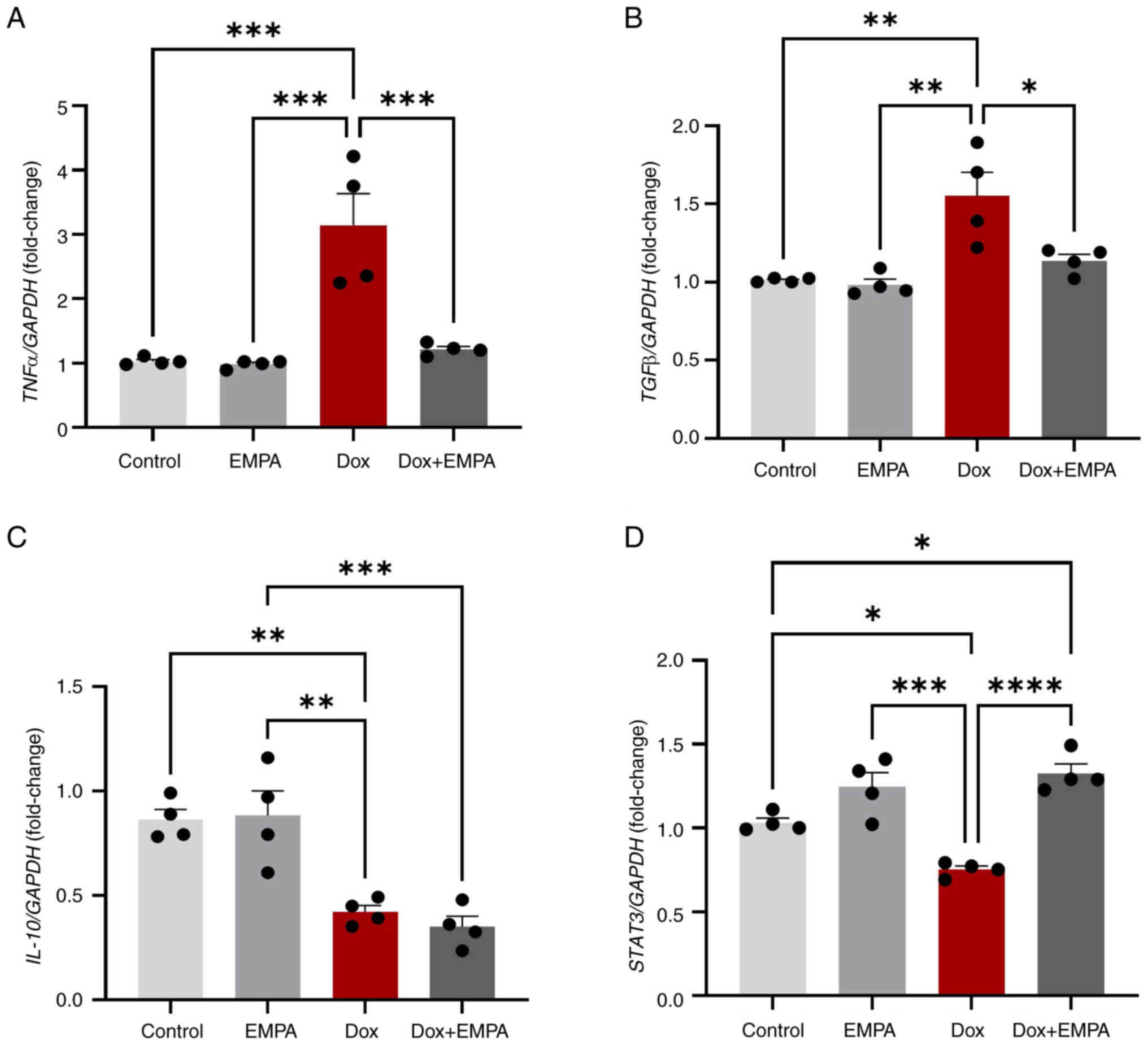
Dox-induced inflammation plays an essential role in
cardiac remodelling and the development of heart failure. Dox
treatment for 24 h significantly increased mRNA expression levels
of the pro-inflammatory cytokine, TNFα, and pro-fibrosis
cytokine, TGFβ, and reduced the expression levels of
inflammation-induced cardioprotective signal transducer and
activator of transcription-3 (STAT-3) and anti-inflammatory
cytokine, IL-10. In Dox-treated cardiomyocytes,
pre-treatment with EMPA significantly reduced the expression of
both TNFα and TGFβ, compared with the Dox group
(Fig. 7A and B). Moreover, Dox
promoted inflammation through the significant downregulation of
IL-10 and STAT-3; however, treatment with EMPA
exerted no effects on the levels of IL-10. Notably, the
levels of STAT-3 returned to baseline following treatment
with EMPA, compared with the Dox group (Fig. 7C and D).
Discussion
EMPA, an SGLT2 inhibitor, was first approved in 2014
for the treatment of diabetes. In 2022, the US Food and Drug
Administration expanded the use of EMPA for guideline-directed
medical therapy in individuals with heart failure and reduced
ejection fraction, irrespective of diabetes status (28). However, the use of EMPA in DIC and
identification of its potential cardioprotective mechanism(s) is
yet to be explored. The present study aimed to investigate the
effects of EMPA pre-treatment on Dox-treated adult primary
cardiomyocytes, with a focus on ER stress.
Numerous previous studies have demonstrated the
absence of SGLT2 expression in the heart (29,30);
however, the results of some studies have reported low expression
of SGLT2 in the heart, when compared with expression in the kidney
and liver (21,30–34).
In the present study, the protein expression of SGLT2 was measured,
and expression remained unchanged following Dox treatment in
primary cardiomyocytes. To verify the impact of EMPA on the
inhibition of glucose absorption, a glucose uptake assay was
performed. In 24 h, EMPA had reduced glucose absorption, compared
with the Dox group. However, it was not completely inhibited as
cardiomyocytes possess other glucose transporters (31,32).
Notably, Dox-treated cardiomyocytes exhibited a higher glucose
uptake than the control group, confirming the metabolic
derangements induced by Dox. Dox impairs oxidative phosphorylation
in the heart, and the persistent activation of glycolysis enables
cardiomyocytes to meet the energy demand of a failing myocardium
(35,36). The ability of EMPA to reduce
glucose uptake following Dox treatment may prevent the heart from
metabolic imbalances and the associated complications.
The intermediary role of ER stress in Dox-induced
apoptosis and myocardial dysfunction have previously been
identified (13,24,37).
The present study examined the activity of UPR transmembrane
proteins and the EMPA-mediated management of ER stress. Dox
treatment significantly increased the expression of ER stress
markers (GRP94, GRP78 and PDI), and expression of these stress
markers was reduced following treatment with EMPA. Notably, PERK
and IRE1 protein expression levels are reduced following Dox
treatment (13,24). Moreover, EMPA treatment restored
IRE1 expression levels. However, EMPA exerted no significant effect
on PERK expression. Results of a previous study demonstrated that
EMPA inhibited autophagy in myocardial I/R injury through
inhibition of the PERK pathway (38). Thus, EMPA-induced low expression of
PERK in the Dox + EMPA group, may aid in mitigating Dox-induced
autophagy. Results of the present study also demonstrated the high
expression of IRE1 (Fig. 3D). The
downstream pathway molecule of IRE1, XBP1, was explored to
further elucidate its involvement in the attenuation of overall
ER-stress. IRE1 plays an essential role in inducing the splicing of
XBP1. Splicing of XBP1 generates a highly active
XBP1s, and its negative regulator XBP1u. IRE1-XBP1 is
the most essential arm of the unfolded protein response that is
required for efficient protein folding, maturation and degradation
of the misfolded proteins (11,39,40).
When translocated to the nucleus, XBP1s aids
in the transcription of UPR-associated genes, inflammatory
responses, differentiation, and the structural and functional
expansion of ER (40,41). Results of previous studies
suggested that an increase in XBP1 levels and the associated
splicing may also occur via the ATF6α pathway (10,11,24).
Results of the present study demonstrated activation of the ATF6α
pathway through cleaved ATF6α expression, and high expression of
XBP1 levels in Dox-treated cardiomyocyte without IRE1
overexpression is indicative of this. Thus, EMPA regulated the
XBP1s/XBP1u protein ratio through downregulation of cleaved ATF6α
and upregulation of IRE1.
Results of the present study demonstrated an
induction of ER stress in Dox-treated cardiomyocytes. As prolonged
ER stress mediates cardiomyocyte apoptosis (13,24,42),
the expression of ER-initiated apoptotic markers were examined. An
overexpression of ER-resident caspase-12 and cleaved caspase-12 was
observed following Dox treatment, and these expression levels were
significantly reduced following pre-treatment with EMPA. Cleaved
caspase-12 acts as a precursor of the activation of
caspase-3-induced apoptosis (14).
EMPA exerts an inhibitory effect on caspase-induced apoptosis and
subsequent cell death in both renal and myocardial I/R injury
(38,43). Expression of the downstream
signaling molecule, JNK, and the subsequent phosphorylation is
involved in ER stress-induced apoptosis and autophagy in fibroblast
or in cancer cells via the IRE1-JNK pathway (42,44).
Although the activation of IRE1 was not observed following Dox
treatment, phosphorylation of JNK was increased, suggesting
ER-independent activation. EMPA inhibits IL-1β (45), which may play an important role in
the inhibition of inflammation, JNK-induced apoptosis and thus, the
promotion of cellular homeostasis. The effect of EMPA on apoptosis
was evident through the significant increase in the viability of
Dox-treated cardiomyocytes.
DIC is also mediated via increased OS and
inflammation that leads to cardiomyocyte apoptosis. Results of the
EMPA's effects in patients with type 2 diabetes mellitus and
coronary artery disease (EMPA-CARD) trial demonstrated that 6
months of treatment with EMPA significantly mitigated inflammation,
OS and platelet activity in patients with diabetes and coronary
artery disease (46). In the
present study, 1 h of pre-treatment with EMPA significantly reduced
OS, compared with the Dox treatment group. Results of the present
study also demonstrated a significant increase in the levels of
TNFα under Dox treatment; however, pre-treatment with EMPA
reduced TNFα levels in the Dox + EMPA group. Notably, EMPA
improved renal ischemia/reperfusion injury in non-diabetic rats
through the TNFα signaling pathway (43). Dox treatment significantly reduced
IL-10 levels, which exert anti-inflammatory and anti-oxidant
effects in the heart (47,48). Results of the present study also
revealed an increase in TGFβ mRNA levels in the Dox group,
and expression was reduced following treatment with EMPA.
Inhibition of TGFβ signalling in cardiac endothelial cells
exerts cardioprotective effects and mitigates DIC (49,50).
Results of a previous study demonstrated that inhibition of SGLT2
was beneficial in mitigating fibrosis in a model of myocardial
ischemia (51). Thus, EMPA-induced
inhibition of TGFβ expression may protect the heart from
Dox-induced fibrosis. In addition, STAT-3 plays a beneficial role
in the heart through upregulating anti-oxidants, anti-apoptotic
genes and mediating inflammation (52). Notably, STAT-3 expression
was downregulated following Dox treatment (53). However, EMPA significantly
increased STAT-3 expression levels, thus maintaining the
redox state and inflammation in Dox-treated cardiomyocytes. Another
SGLT2 inhibitor, Dapagliflozin, also reduces DIC via restoring
STAT-3 levels (54). The
beneficial effects of EMPA on the restoration of STAT-3 protein
expression may promote cardiomyocyte proliferation, maintain the
redox state, and protect the heart from cardiac inflammation.
In conclusion, ER is associated with cardiac
function in a number of cardiovascular diseases (55,56).
In vitro, EMPA mitigated ER stress in DIC via the IRE1-ATF6α
pathway, and subsequent reduced OS, inflammation and cardiomyocyte
apoptosis. Further investigations are required to evaluate the
cardioprotective role of EMPA and the associated mechanisms in both
the prevention and treatment of DIC.
Notably, the present study exhibits numerous
limitations. For example, SGLT2s are not the only glucose
transporters present in the heart, as the heart primarily relies on
GLUT1 and GLUT4 for glucose uptake. Therefore, the inhibition of
GLUT1 and GLUT4 may provide further insights into the direct role
of SGLT2 in DIC. The present study was focused on Dox-induced ER
stress and ER stress-induced apoptosis and investigated the
potential beneficial effects of EMPA in inhibiting ER stress.
However, DIC is multifaceted, and further investigations into the
cardioprotective role of EMPA in Dox-induced injury are required.
Additionally, in vivo studies are required to determine the
efficacy of EMPA in protecting heart damage, without interfering
with the anticancer properties of Dox.
Acknowledgements
Not applicable.
Funding
The present study was supported by operating grants from the
Heart and Stroke Foundation of Canada (grant no. G-19-0024241), the
Molson Women's Heart Health Program (grant no. 2003-31-94-c), and
the CANUSA grant (grant no. R510867).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AM and PKS conceived and designed the research
study. AM completed all of the experiments and acquired data. AM
and DSJ confirm the authenticity of all the raw data. PKS and DSJ
provided resources and materials. AM, AKB, PKS and DSJ contributed
to data analysis and writing and editing the manuscript. All
authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
The animal experimental procedures were performed
per the guidelines of the Canadian Council on Animal Care for all
animal procedures, including animal handling and drug
administration as approved [approval no. (22-012 (AC11739)] by the
Animal Protocol Review Committee at the University of Manitoba
(Winnipeg, Canada).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ATF
|
activating transcription factor
|
|
Dox
|
doxorubicin
|
|
DIC
|
doxorubicin-induced cardiomyopathy
|
|
EMPA
|
empagliflozin
|
|
ER
|
endoplasmic reticulum
|
|
GRP78
|
glucose-regulated protein 78
|
|
IRE
|
inositol-requiring enzyme
|
|
OS
|
oxidative stress
|
|
PDI
|
protein disulphide-isomerase
|
|
PERK
|
protein kinase-like ER kinase
|
|
ROS
|
reactive oxygen species
|
|
SGLT2
|
sodium-glucose cotransporter-2
|
|
STAT
|
signal transducer and activator of
transcription
|
|
UPR
|
unfolded protein response
|
|
XBP1
|
X-box binding protein 1
|
References
|
1
|
Lefrak EA, Pitha J, Rosenheim S and
Gottlieb JA: A clinicopathologic analysis of adriamycin
cardiotoxicity. Cancer. 32:302–314. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Singal PK and Iliskovic N:
Doxorubicin-induced cardiomyopathy. N Engl J Med. 339:900–905.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ludke AR, Sharma AK, Akolkar G, Bajpai G
and Singal PK: Downregulation of vitamin C transporter SVCT-2 in
doxorubicin-induced cardiomyocyte injury. Am J Physiol Cell
Physiol. 303:C645–C653. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Singal PK, Siveski-Iliskovic N, Hill M,
Thomas TP and Li T: Combination therapy with probucol prevents
adriamycin-induced cardiomyopathy. J Mol Cell Cardiol.
27:1055–1063. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Torti FM, Bristow MM, Lum BL, Carter SK,
Howes AE, Aston DA, Brown BW Jr, Hannigan JF Jr, Meyers FJ,
Mitchell EP, et al: Cardiotoxicity of epirubicin and doxorubicin:
Assessment by endomyocardial biopsy. Cancer Res. 46:3722–3727.
1986.PubMed/NCBI
|
|
6
|
Singal PK, Deally CM and Weinberg LE:
Subcellular effects of adriamycin in the heart: A concise review. J
Mol Cell Cardiol. 19:817–828. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Krebs J, Agellon LB and Michalak M: Ca(2+)
homeostasis and endoplasmic reticulum (ER) stress: An integrated
view of calcium signaling. Biochem Biophys Res Commun. 460:114–121.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hetz C and Papa FR: The unfolded protein
response and cell fate control. Mol Cell. 69:169–181. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Harding HP, Zhang Y, Bertolotti A, Zeng H
and Ron D: Perk is essential for translational regulation and cell
survival during the unfolded protein response. Mol Cell. 5:897–904.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Glembotski CC: Roles for ATF6 and the
sarco/endoplasmic reticulum protein quality control system in the
heart. J Mol Cell Cardiol. 71:11–15. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yoshida H, Matsui T, Yamamoto A, Okada T
and Mori K: XBP1 mRNA is induced by ATF6 and spliced by IRE1 in
response to ER stress to produce a highly active transcription
factor. Cell. 107:881–891. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bertolotti A, Zhang Y, Hendershot LM,
Harding HP and Ron D: Dynamic interaction of BiP and ER stress
transducers in the unfolded-protein response. Nat Cell Biol.
2:326–332. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bagchi AK, Malik A, Akolkar G, Zimmer A,
Belló-Klein A, De Angelis K, Jassal DS, Fini MA, Stenmark KR and
Singal PK: Study of ER stress and apoptotic proteins in the heart
and tumor exposed to doxorubicin. Biochim Biophys Acta Mol Cell
Res. 1868:1190392021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hitomi J, Katayama T, Taniguchi M, Honda
A, Imaizumi K and Tohyama M: Apoptosis induced by endoplasmic
reticulum stress depends on activation of caspase-3 via caspase-12.
Neurosci Lett. 357:127–130. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hsia DS, Grove O and Cefalu WT: An update
on sodium-glucose co-transporter-2 inhibitors for the treatment of
diabetes mellitus. Curr Opin Endocrinol Diabetes Obes. 24:73–79.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zinman B, Wanner C and Lachin JM; EMPA-REG
OUTCOME Investigators, : Empagliflozin, cardiovascular outcomes,
and mortality in type 2 diabetes. N Engl J Med. 373:2117–2128.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Butler J, Anker SD, Filippatos G, Khan MS,
Ferreira JP, Pocock SJ, Giannetti N, Januzzi JL, Piña IL, Lam CSP,
et al: Empagliflozin and health-related quality of life outcomes in
patients with heart failure with reduced ejection fraction: The
EMPEROR-reduced trial. Eur Heart J. 42:1203–1212. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Packer M, Anker SD, Butler J, Filippatos
G, Pocock SJ, Carson P, Januzzi J, Verma S, Tsutsui H, Brueckmann
M, et al: Cardiovascular and Renal outcomes with empagliflozin in
heart failure. N Engl J Med. 383:1413–1424. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Anker SD, Butler J, Filippatos G, Ferreira
JP, Bocchi E, Böhm M, Brunner-La Rocca HP, Choi DJ, Chopra V,
Chuquiure-Valenzuela E, et al: Empagliflozin in heart failure with
a preserved ejection fraction. N Engl J Med. 385:1451–1461. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
McDonald M, Virani S, Chan M, Ducharme A,
Ezekowitz JA, Giannetti N, Heckman GA, Howlett JG, Koshman SL,
Lepage S, et al: CCS/CHFS heart failure guidelines update: Defining
a new pharmacologic standard of care for heart failure with reduced
ejection fraction. Can J Cardiol. 37:531–546. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sabatino J, De Rosa S, Tammè L, Iaconetti
C, Sorrentino S, Polimeni A, Mignogna C, Amorosi A, Spaccarotella
C, Yasuda M and Indolfi C: Empagliflozin prevents
doxorubicin-induced myocardial dysfunction. Cardiovasc Diabetol.
19:662020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chang WT, Lin YW, Ho CH, Chen ZC, Liu PY
and Shih JY: Dapagliflozin suppresses ER stress and protects
doxorubicin-induced cardiotoxicity in breast cancer patients. Arch
Toxicol. 95:659–671. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bagchi AK, Malik A, Akolkar G, Jassal DS
and Singal PK: Endoplasmic reticulum stress promotes iNOS/NO and
influences inflammation in the development of doxorubicin-induced
cardiomyopathy. Antioxidants (Basel). 10:18972021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Malik A, Bagchi AK, Jassal DS and Singal
PK: Interleukin-10 mitigates doxorubicin-induced endoplasmic
reticulum stress as well as cardiomyopathy. Biomedicines.
10:8902022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhao L and Zhang B: Doxorubicin induces
cardiotoxicity through upregulation of death receptors mediated
apoptosis in cardiomyocytes. Sci Rep. 7:447352017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yarmohammadi F, Rezaee R, Haye AW and
Karimi G: Endoplasmic reticulum stress in doxorubicin-induced
cardiotoxicity may be therapeutically targeted by natural and
chemical compounds: A review. Pharmacol Res. 164:1053832021.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Heidenreich PA, Bozkurt B, Aguilar D,
Allen LA, Byun JJ, Colvin MM, Deswal A, Drazner MH, Dunlay SM,
Evers LR, et al: 2022 AHA/ACC/HFSA Guideline for the Management of
Heart Failure: A Report of the American College of
Cardiology/American Heart Association Joint Committee on Clinical
Practice Guidelines. Circulation. 145:e89–e1032. 2022. View Article : Google Scholar
|
|
29
|
Di Franco A, Cantini G, Tani A, Coppini R,
Zecchi-Orlandini S, Raimondi L, Luconi M and Mannucci E:
Sodium-dependent glucose transporters (SGLT) in human ischemic
heart: A new potential pharmacological target. Int J Cardiol.
243:86–90. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen J, Williams S, Ho S, Loraine H, Hagan
D, Whaley JM and Feder JN: Quantitative PCR tissue expression
profiling of the human SGLT2 gene and related family members.
Diabetes Ther. 1:57–92. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhou L, Cryan EV, D'Andrea MR, Belkowski
S, Conway BR and Demarest KT: Human cardiomyocytes express high
level of Na+/glucose cotransporter 1 (SGLT1). J Cell Biochem.
90:339–346. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kashiwagi Y, Nagoshi T, Yoshino T, Tanaka
TD, Ito K, Harada T, Takahashi H, Ikegami M, Anzawa R and Yoshimura
M: Expression of SGLT1 in human hearts and impairment of cardiac
glucose uptake by phlorizin during ischemia-reperfusion injury in
mice. PLoS One. 10:e01306052015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Quagliariello V, De Laurentiis M, Rea D,
Barbieri A, Monti MG, Carbone A, Paccone A, Altucci L, Conte M,
Canale ML, et al: The SGLT-2 inhibitor empagliflozin improves
myocardial strain, reduces cardiac fibrosis and pro-inflammatory
cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc
Diabetol. 20:1502021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nakano D, Akiba J, Tsutsumi T, Kawaguchi
M, Yoshida T, Koga H and Kawaguchi T: Hepatic expression of
sodium-glucose cotransporter 2 (SGLT2) in patients with chronic
liver disease. Med Mol Morphol. 55:304–315. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bertero E and Maack C: Metabolic
remodelling in heart failure. Nat Rev Cardiol. 15:457–470. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Asnani A, Shi X, Farrell L, Lall R, Sebag
IA, Plana JC, Gerszten RE and Scherrer-Crosbie M: Changes in citric
acid cycle and nucleoside metabolism are associated with
anthracycline cardiotoxicity in patients with breast cancer. J
Cardiovasc Transl Res. 13:349–356. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fu HY, Sanada S, Matsuzaki T, Liao Y,
Okuda K, Yamato M, Tsuchida S, Araki R, Asano Y, Asanuma H, et al:
Chemical endoplasmic reticulum chaperone alleviates
doxorubicin-induced cardiac dysfunction. Circ Res. 118:798–809.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang CC, Li Y, Qian XQ, Zhao H, Wang D,
Zuo GX and Wang K: Empagliflozin alleviates myocardial I/R injury
and cardiomyocyte apoptosis via inhibiting ER stress-induced
autophagy and the PERK/ATF4/Beclin1 pathway. J Drug Target.
30:858–872. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hetz C: The unfolded protein response:
Controlling cell fate decisions under ER stress and beyond. Nat Rev
Mol Cell Biol. 13:89–102. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee AH, Iwakoshi NN and Glimcher LH: XBP-1
regulates a subset of endoplasmic reticulum resident chaperone
genes in the unfolded protein response. Mol Cell Biol.
23:7448–7459. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yoshida H, Oku M, Suzuki M and Mori K:
pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded
protein response activator pXBP1(S) in mammalian ER stress
response. J Cell Biol. 172:565–575. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kato H, Nakajima S, Saito Y, Takahashi S,
Katoh R and Kitamura M: mTORC1 serves ER stress-triggered apoptosis
via selective activation of the IRE1-JNK pathway. Cell Death
Differ. 19:310–320. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ala M, Khoshdel MRF and Dehpour AR:
Empagliflozin enhances autophagy, mitochondrial biogenesis, and
antioxidant defense and ameliorates renal ischemia/reperfusion in
nondiabetic rats. Oxid Med Cell Longev. 2022:11970612022.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Haberzettl P and Hill BG: Oxidized lipids
activate autophagy in a JNK-dependent manner by stimulating the
endoplasmic reticulum stress response. Redox Biol. 1:56–64. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pirklbauer M, Sallaberger S, Staudinger P,
Corazza U, Leierer J, Mayer G and Schramek H: Empagliflozin
inhibits IL-1beta-mediated inflammatory response in human proximal
tubular cells. Int J Mol Sci. 22:50892021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gohari S, Reshadmanesh T, Khodabandehloo
H, Karbalaee-Hasani A, Ahangar H, Arsang-Jang S, Ismail-Beigi F,
Dadashi M, Ghanbari S, Taheri H, et al: The effect of EMPAgliflozin
on markers of inflammation in patients with concomitant type 2
diabetes mellitus and coronary ARtery disease: The EMPA-CARD
randomized controlled trial. Diabetol Metab Syndr. 14:1702022.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bagchi AK, Surendran A, Malik A, Jassal
DS, Ravandi A and Singal PK: IL-10 attenuates OxPCs-mediated lipid
metabolic responses in ischemia reperfusion injury. Sci Rep.
10:121202020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dhingra S, Sharma AK, Arora RC, Slezak J
and Singal PK: IL-10 attenuates TNF-alpha-induced NF kappaB pathway
activation and cardiomyocyte apoptosis. Cardiovasc Res. 82:59–66.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sun Z, Schriewer J, Tang M, Marlin J,
Taylor F, Shohet RV and Konorev EA: The TGF-β pathway mediates
doxorubicin effects on cardiac endothelial cells. J Mol Cell
Cardiol. 90:129–138. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li J, Deane JA, Campanale NV, Bertram JF
and Ricardo SD: Blockade of p38 mitogen-activated protein kinase
and TGF-beta1/Smad signaling pathways rescues bone marrow-derived
peritubular capillary endothelial cells in adriamycin-induced
nephrosis. J Am Soc Nephrol. 17:2799–2811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Li G, Zhao C and Fang S: SGLT2 promotes
cardiac fibrosis following myocardial infarction and is regulated
by miR-141. Exp Ther Med. 22:7152021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lim CP and Fu XY: Multiple roles of STAT3
in cardiovascular inflammatory responses. Progress in Molecular
Biology and Translational Science. Vol 106. Shenolikar S: Academic
Press; pp. 63–73. 2012, View Article : Google Scholar : PubMed/NCBI
|
|
53
|
de Oliveira Santos TC, Pereira G, Coutinho
AGG, Dos Santos Silva HP, Lima MMS, Dias FAL, de Almeida DC,
Resende E, Silva DT, Perez RF and Pereira RL: STAT-3 signaling role
in an experimental model of nephropathy induced by doxorubicin. Mol
Cell Biochem. 478:981–989. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chang WT, Shih JY, Lin YW, Chen ZC, Kan
WC, Lin TH and Hong CS: Dapagliflozin protects against
doxorubicin-induced cardiotoxicity by restoring STAT3. Arch
Toxicol. 96:2021–2032. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Dufey E, Sepúlveda D, Rojas-Rivera D and
Hetz C: Cellular mechanisms of endoplasmic reticulum stress
signaling in health and disease. 1. An overview. Am J Physiol Cell
Physiol. 307:C582–C594. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gaut JR and Hendershot LM: The
modification and assembly of proteins in the endoplasmic reticulum.
Curr Opin Cell Biol. 5:589–595. 1993. View Article : Google Scholar : PubMed/NCBI
|