Adipose tissue (AT) is the main energy store derived
from food intake in the form of triglycerides (TGs) and controls
lipid mobilization (1,2). Furthermore, AT is an active endocrine
organ, since it synthesizes and secretes several hormones,
cytokines and other bioactive factors, signaling to other metabolic
organs, such as the liver, pancreas and brain modulating systemic
metabolism, whilst also maintaining body temperature (1,2). AT
contains multiple cell types, including adipocytes, adipocytes
progenitors, endothelial cells, macrophages, fibroblasts and
leucocytes (1–3). Adipocytes, also called adipose cells
or fat cells, are the predominant cell type in AT. There are three
types of adipocytes including white, brown and beige (brite). They
differ in structure, location, abundance of mitochondria,
thermogenic gene expression and function (2,4).
White adipocytes are unilocular with a low number of mitochondria
and low oxidative rate (2). The
cells have a high capacity of energy storage in the form of TGs. In
addition, white adipocytes can prevent ectopic lipid deposition and
therefore protect organs such as skeletal muscle and the liver from
lipotoxicity (2). Brown adipocytes
are specialized cells with multilocular lipid droplets, high
numbers of mitochondria and enrichment of uncoupling protein 1
(UCP1), a high oxidative capacity and actively participate in
energy consumption via thermogenesis (2). In newborn humans, brown AT plays an
important role in thermogenesis mediated by the expression of UCP1
(5). In adult humans, it has been
found that the amount of brown AT is inversely associated to body
mass index (BMI), especially in older individuals indicating the
importance of brown AT in energy metabolism (5). Beige adipocytes are a distinct type
of adipocyte with multilocular morphology within white AT (WAT) and
extremely low UCP1 expression and are capable of thermogenesis
(2). Beige adipocytes exist mainly
in subcutaneous white fat, but a small portion in visceral fat can
be found as well. Acute cold exposure markedly triggers the
recruitment and activation of beige adipocytes (2). Based on its location in the body, WAT
can be further divided into two types of specific regional depots,
the subcutaneous depots and the visceral depots (6). Subcutaneous fat is located under the
skin in areas such as the abdomen, thighs, hips and buttocks,
however, visceral fat surrounds the intrathoraci organs, the
intraperitoneal organs, such as the greater and lesser omentum,
mesentery, mesocolon and peritoneum, and the retroperitoneal
organs, such as the pancreas, duodenum, ascending and descending
colon and kidneys (7). AT responds
to stimulation by extra nutrients via hyperplasia (proliferation)
and hypertrophy of adipocytes (8).
Excessive calories are efficiently stored in the form of neutral
TGs in AT, which results in adipose hypertrophy and subsequent
obesity (2,9). When adipocytes cannot uptake the
excess of TGs, the body synthesizes new adipocytes (hyperplasia),
which creates space for fat storage through the lipogenic pathway
(9,10). In circumstances of reduction of
food intake or an increase in energy expenditure, TGs from
adipocytes are broken down into glycerol and fatty acids through
the lipolytic pathway to provide energy. Subsequently, fatty acids
and glycerol can be transported with the blood to other organs
(11). Then, the lipids infiltrate
into multiple ectopic organs such as the skeletal muscle, heart and
liver and into the visceral adipose depots, leading to systemic
low-grade chronic inflammation (12). Moreover, with progressive adipocyte
expansion and obesity, the blood supply to adipocytes may be
reduced, leading to hypoxia, adipocyte necrosis and macrophage
infiltration into AT (13,14). During this process, AT produces and
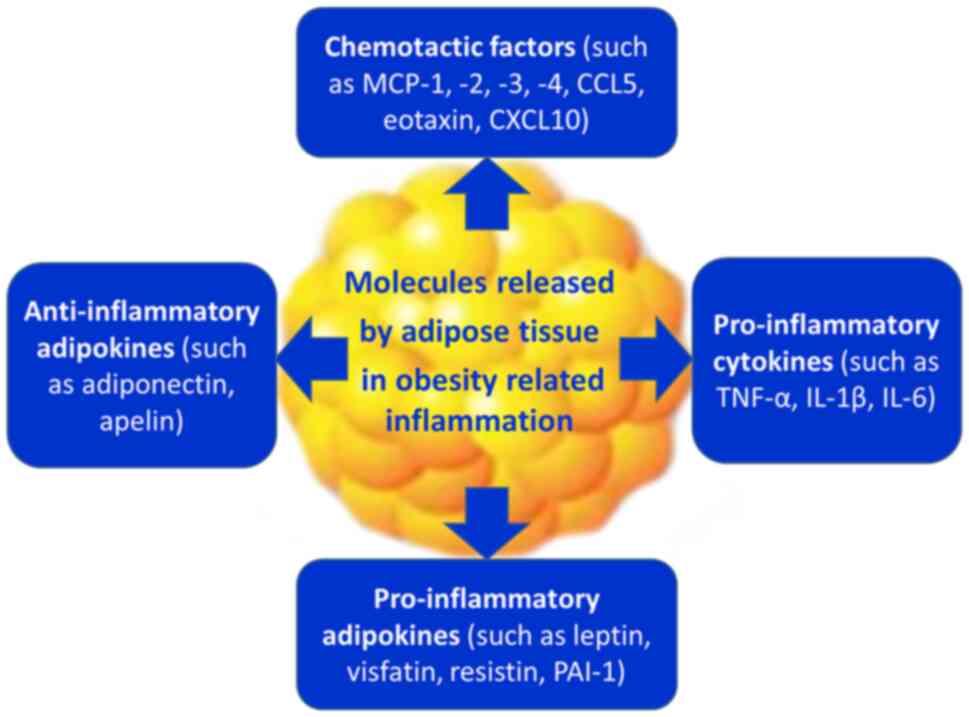
releases a variety of pro-inflammatory and anti-inflammatory
factors as well, including adipokines such as leptin, adiponectin
and resistin, as well as cytokines and chemokines, such as TNF-α,
IL-6 and monocyte chemoattractant protein (MCP) −1 (11,14–16).
The biological action of adipokine is mainly mediated by binding to
their cell surface receptors on the cell membrane of target cells
activating appropriate intracellular signaling pathways (2).
Obesity is defined by the National Institute of
Health based on the BMI, calculated as the weight of a patient in
kilograms divided by the square of height in meters, with BMI
values >30 causing concern (17). Subcutaneous AT depots seem to be
negatively associated with cardiovascular risk factors, while
higher levels of visceral AT have been highly associated with
cardio-metabolic disease (18,19).
Unhealthy expansion of adipocytes is associated with abdominal
obesity, promotion of the obesity-associated metabolic
complications, recruitment of macrophages and other immune cells,
promotion of systemic inflammation and accumulation of visceral fat
(20,21). Indeed, fat accumulation
intra-abdominally in men is associated with higher risk for
cardiometabolic diseases, independent of BMI (22–24).
In addition, abdominal visceral fat is also a strong predictor of
mortality in obese women (25). A
number of human studies have shown that omental adipocyte size
positively associates with insulin resistance (IR) (26,27).
Notably, individuals of certain ethnic backgrounds, regardless of
the present country of residence and citizenship, show
predisposition to central obesity and significant obesity-related
medical complications (28–30).
Indeed, several studies demonstrated that South Asian, Japanese and
Chinese obese populations have a greater risk for IR, type 2
diabetes mellitus (T2DM) and cardiovascular diseases (CVDs) than
Caucasians (31,32). Depending on the degree and duration
of weight gain, obesity can progressively cause and/or exacerbate a
wide spectrum of co-morbidities, including T2DM, hypertension,
dyslipidemia, CVDs, non-alcoholic fatty liver disease (NAFLD),
kidney diseases, respiratory disorders, sleep apnea,
musculoskeletal disorders, osteoarthritis, sub-fertility,
psychosocial problems and certain types of cancers (33,34)
(Table I).
It has become evident that the presence of excessive
AT enhances lipogenesis and activates the innate immune system
(17,34,36–50).
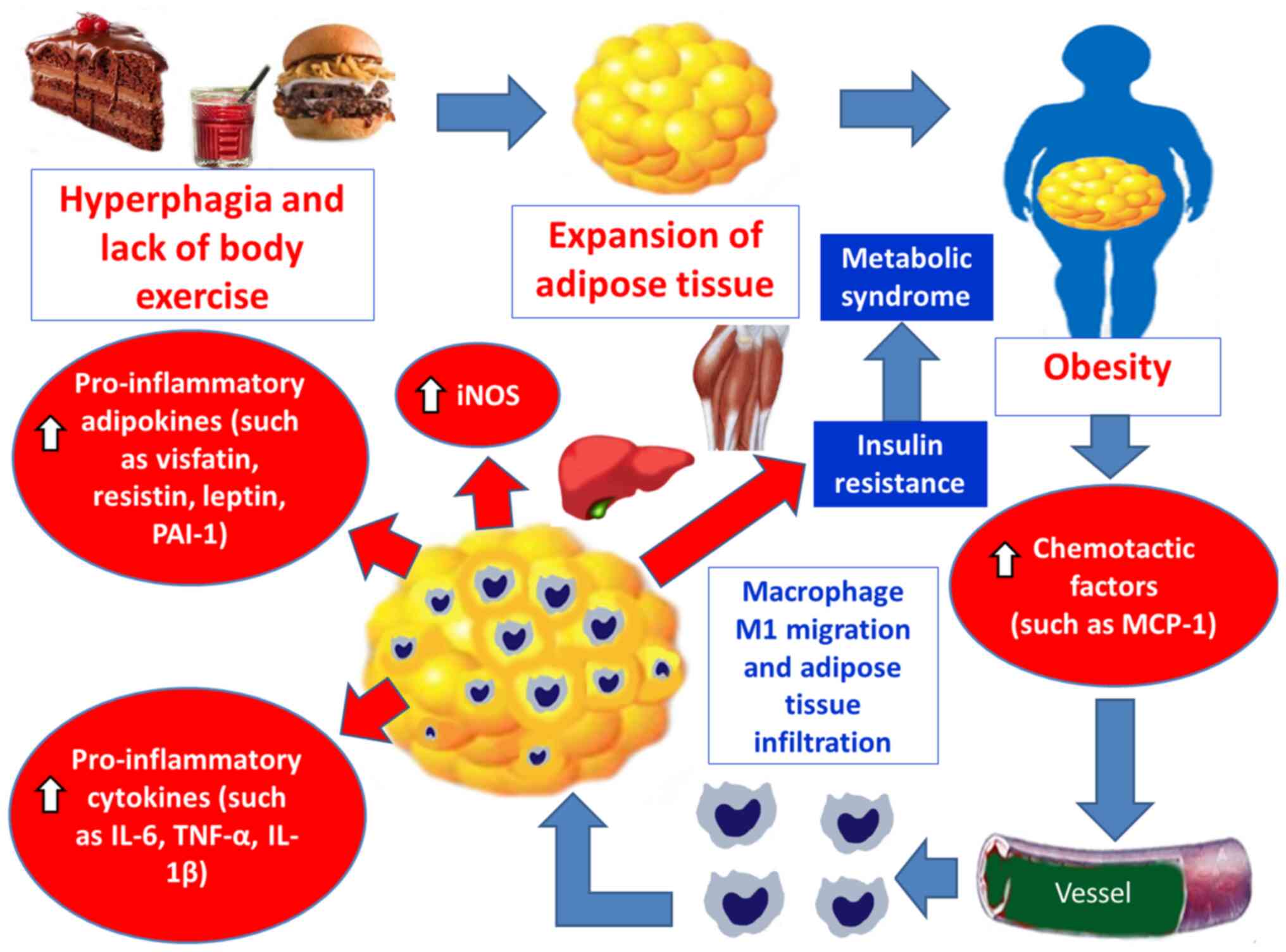
Compiling evidence suggests that AT, during the course of excessive
fat accumulation, in obese patients, and the expansion of the fat
mass, produces several chemotactic factors, such as MCP-1, −2, −3
and −4, RANTES [or chemokine CC motif ligand 5 (CCL5)], eotaxin
[chemokine CC motif ligand 11 (CCL11)] and interferon γ-induced
protein 10 [chemokine CXC motif ligand (CXCL10)] (17,34,36–50).
In response to such stimuli, monocytes are recruited from the
blood, transmigrate and infiltrate into AT depots, through adhesion
processes to endothelial cells, increasing the number of activated
pro-inflammatory M1 macrophages. In turn, the growing population of
pro-inflammatory M1 macrophages enhances the inflammatory changes
and secretes pro-inflammatory cytokines (such as TNF-α, IL-1β,
IL-6, IL-8, IL-12 and IL-23), pro-inflammatory adipokines [such as
leptin, plasminogen activator inhibitor type 1 (PAI-1), visfatin
and resistin] and inducible nitric oxide synthase (iNOS) (17,34,36–50).
The initiation of a low-grade inflammation in AT of obese
individuals contributes to an increase of leptin, visfatin,
resistin and PAI-1 and to a decrease of adiponectin (17,34,36–50).
This status leads to IR in adipocytes, which generates free fatty
acids (FFAs) in serum, impairs glucose metabolism and favors
hepatic, muscular and AT accumulation of fats and glucose (17,34,36–50).
These events promote higher mitochondrial and peroxisomal
oxidation, which results in the production of free radicals (FRs),
OS, mitochondrial DNA injury, depletion of adenosine triphosphate
(ATP) and finally, lipotoxicity (17,34,36–50).
Cellular damage leads to high production of pro-inflammatory
cytokines, such as TNF-α, which generates further reactive oxygen
species (ROS) in tissues and increases the lipid peroxidation rate.
An imbalance between the antioxidant capacity and the production of
FR induces OS and promotes a systemic low-grade inflammation
(17,34,36–50)
(Fig. 3).
MCP-1 or CCL2 is a 13-kDa pro-inflammatory
chemokine. MCP-1 is a member of the MCP family consisting of at
least four members (MCP-1, −2, −3, −4) and it exerts its action by
binding to its chemokine receptor, C-C motif chemokine receptor 2
(CCR2), which is a CC motif receptor (51). The CCR2A isoform is expressed by
mononuclear cells and vascular smooth muscle cells (VSMCs), while
CCR2B is expressed by monocytes and natural killer cells (52). MCP-1 plays a role in the
recruitment, migration and infiltration of monocytes, microglia and
memory T lymphocytes to sites of infection and injury (53–56).
MCP-1 is secreted predominately by macrophages and endothelial
cells (52). Also, MCP-1 is
produced from adipocytes and its expression is higher in visceral
AT (VAT) than in subcutaneous AT (STAT). The release of MCP-1 is
inhibited by adiponectin (57).
There is a close relationship between MCP-1 and the number of
resident macrophages in adipocytes (57–59).
Plasma levels of MCP-1 are markedly elevated in obesity and T2DM
(52,58,60–63).
In obesity, the production of MCP-1 by adipocytes results in
recruitment of monocytes and activation of macrophages, which
causes AT inflammation (56,64).
It has been suggested that obesity-associated inflammation in WAT
is the causal factor of systemic IR (65). In addition, serum MCP-1 levels are
higher in patients with atherosclerosis and the expression of mRNA
MPC-1 is increased in atherosclerotic lesions as well (66–68).
Also, inhibition of MPC-1 expression or its receptor (CCR2) reduces
the extent of atheroma formation in hypercholesterolemic mice
(55,66,69–71).
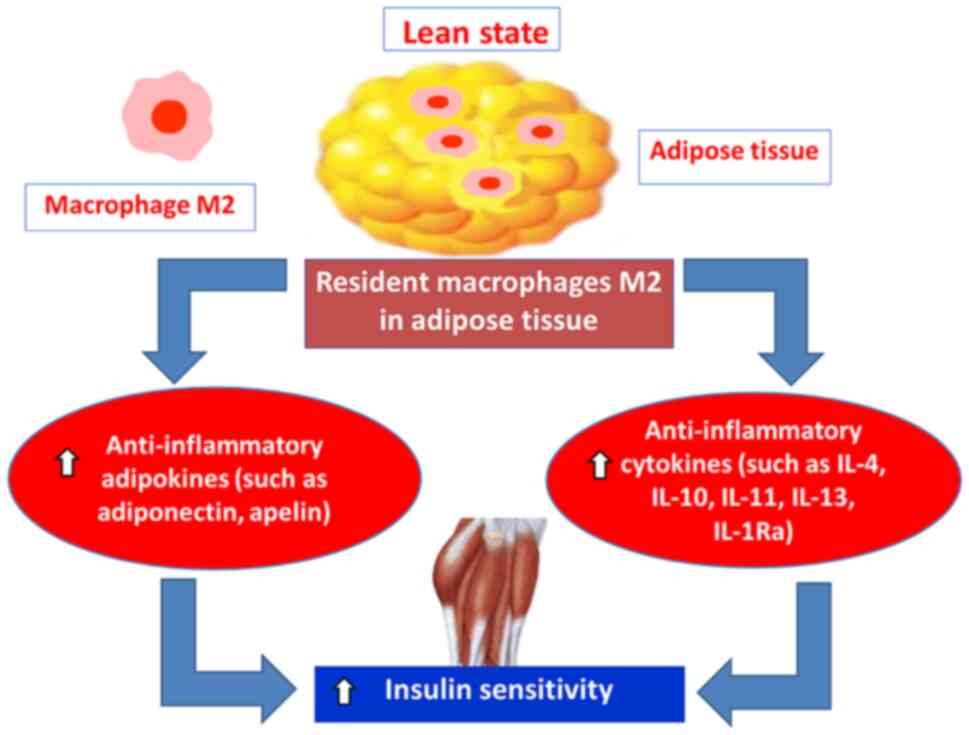
Adiponectin belongs to adipokines and is a 30-kDa
peptide secreted only by AT and in particular in large amounts by
adipocytes of white AT (2,72–74).
It acts via two receptors (ADIPOR1 and ADIPOR2) that elicit AMPK
signaling and may be modulated by T-cadherin (75). Adiponectin has been described as a
main anti-inflammatory adipokine (76). Its anti-inflammatory actions are
partly due to its ability to reduce TNF-α activity, via suppression
of adiponectin-induced NFκB (77).
Also, adiponectin was shown to directly decrease production of
pro-inflammatory cytokines TNF-α and IL-6 by macrophages (78). Additionally, adiponectin inhibits
generation of ROS induced by high glucose and oxidized low-density
lipoprotein (LDL) via a cAMP/PKA-dependent pathway (48,79,80).
High ROS levels in adipocytes suppress adiponectin expression and
secretion (81,82). Accordingly, there is an inverse
association between human serum adiponectin levels and systemic OS
(83). Adiponectin increases
insulin sensitivity in multiple tissues via up-regulation in
insulin signaling. This insulin-sensitizing effect of adiponectin
seems to be mediated by increased fatty acid oxidation through
AMP-activated protein kinase and peroxisome proliferator-activating
receptor-α (PPARα) activation (84,85).
In addition, adiponectin reduces glucose content in tissues, by
trasfering cytoplasmic glucose trasporter type 4 (GLUT4) toward the
surface of the cytoplasmic membrane (73). Also, adiponectin inhibits
gluconeogenesis within the liver (86). Adiponectin exhibits cardiovascular
protection by suppressing inflammatory processes occurring in the
early phases of atherosclerosis and microangiopathy through
inhibition of the adhesion of monocytes to blood vessel endothelial
cells, as a result of down-regulated expression of adhesion
proteins, decreasing of the transformation of macrophages into foam
cells and down-regulating intimal smooth muscle cell proliferation
(2,48,77,87–91).
It also enhances nitrogen oxide (NO) synthesis in endothelial cells
and stimulates angiogenesis (73,80,92,93).
Adiponectin synthesis is regulated by insulin and
insulin-like growth factor-1, which leads to increased
concentration of this adipokine (73,94).
However, its synthesis is inhibited by pro-inflammatory cytokines,
such as TNF-α and IL-6. This suggests that obesity and IR are
important factors contributing to low levels of serum adiponectin
(95). Also, low serum levels of
adiponectin are found in obese, insulin-resistant individuals with
related pathologies including T2DM, dyslipidemia and CVDs (89,93,96–99).
On the other hand, weight loss increases adiponectin concentrations
(73). Hence, elevated levels of
adiponectin may lead to the decrease in the risk of T2DM (100).
Apelin is a short peptide hormone, which belongs to
adipokines and is produced by adipocytes in proportion to the
present amount of fat; it plays an important role in energy
metabolism and is considered to be linked with obesity and diabetes
(39,101,102). Apelin exerts its effects by
binding with angiotensin II protein J (APJ) receptor (101,103). Apelin promotes brown adipocyte
development through the phosphatidylinositol 3-kinase (PI3K)/Akt
and AMPK signaling pathways (102). Also, apelin is able to increase
the browning in white adipocytes (102,104). In addition, it has been found
that apelin relieves the TNF-α suppression on brown adipogenesis
(102). Apelin stimulates glucose
uptake, increases insulin sensitivity and regulates lipolysis and
fatty acid oxidation (101). It
has been found that serum apelin levels are increased in obesity.
This may be due either to potential resistance to apelin or to its
attempt to delay or reduce tissue IR (105). Nevertheless, current literature
suggests that apelin administration protects diabetic and/or obese
mice (101) by lowering glucose
levels and hence, it may have a therapeutic role against obesity
and related metabolic diseases (104). Apelin with its activity on
enthothelial APJ receptors may additionally improve nitric oxide
(NO) release and endothelium-dependent vasodilation (39,106). Apelin has antioxidant effects
because it suppresses ROS production and release in AT and improves
the antioxidant state in OS-related conditions (107). Apelin promotes the synthesis of
antioxidant enzymes via Ras/Raf/mitogen-activated protein kinase
(MAPK)/ERK and AMP-activated protein kinase (AMPK) pathways
(104), suppresses the expression
of pro-oxidant enzymes via the same pathway, and increases
mitochondrial oxidative capacity (104,108–111).
Leptin belongs to adipokines and is a 16-KDa
peptide, encoded by the obese (ob) gene. It is mainly secreted by
WAT and its secretion increases according to the volume of AT and
to the TGs stored in adipocytes (2,48,112–116). When body energy stores are
adequate, leptin suppresses food intake, regulating appetite,
energy balance and causing satiety (2,48,112–114). The sensation of satiety of leptin
is achieved by crossing the blood-brain barrier and targeting the
hypothalamus (2,112,117–119). Hypothalamic leptin signaling is
mediated by leptin receptor and downstream processes, including
JAK2/STAT3 pathway (120).
However, obesity is associated with increased leptin protein and
mRNA levels compared with lean controls. The failure of the
elevated leptin levels to correct the metabolic complications seen
in obesity, is mainly related to leptin resistance, in tissues with
decreased sensitivity to leptin (3,35,39,121–123). The influence of leptin on IR is
not yet fully understood (35).
However, it has been found that IR is associated with elevated
serum leptin levels (35). A
possible explanation is that leptin resistance causes reduction of
lipid oxidation, which leads to lipid accumulation and IR (124,125). Also, leptin in obese humans
causes an increase in blood pressure, through sympathetic
activation at vascular and/or renal levels (126). Leptin has pro-inflammatory
actions, which are related to structural and functional
similarities with the cytokine IL-6 (115). Also, leptin promotes OS and
endothelial cell dysfunction and activation, increases phagocytic
activity by macrophages and induces the production of
pro-inflammatory cytokines such as TNF-α, IL-6, IL-2 and IL-1
(2,127,128). In addition, it has been found
that leptin administration increases c-reactive protein (CRP)
levels, which confirms further its inflammatory effects (123).
Visfatin also known as nicotinamide
phosphoribosyltranferase and pre-B-cell colony enhancing factor
(129,130) is a 52 kDa adipokine, which is
predominantly expressed in human VAT (131), an area of fat tissue, whose
accumulation is strongly associated with an enhanced cardiovascular
(CV) risk (48,129). Visfatin is also secreted by
macrophages, bone marrow, skeletal muscles and various organs
including the liver, lungs, brain, heart and pancreas (49,129). However, a specific receptor for
visfatin has not been identified yet (93). Several authors have suggested that
visfatin levels increase with obesity, T2DM, MetS or CVDs (132–134). However, other studies have shown
conflicting results regarding the relationship of visfatin with
MetS (135,136). It has been found that weight loss
decreases visfatin levels in obese patients (134). Moreover, leucocytes from obese
patients produce higher amounts of visfatin compared with lean
patients (137). Visfatin levels
beyond a threshold appear to be associated with IR and
obesity-related vascular disorders (130,138). Specifically, visfatin appears to
contribute to the release of pro-inflammatory cytokines IL-1β,
IL-6, IL-8 and TNF-α, through a regulation of the JAK2/STAT3 and
IKN/NF-kB signaling pathways, promoting inflammation (129,131,139–144). Moreover, in experimental studies,
it has been found that visfatin induces endothelial dysfunction,
via the NF-κB pathway, in the vascular endothelium and promotes the
proliferation of human VSMCs (129,138). Additionally, visfatin induces
NF-κB pathway dependent OS, and blockade of this pathway, via
selective IκΒ Kinase (IKK-2) inhibition, leads to a partial
reduction in OS, which it is independent of the MAPK/ERK signaling
pathway (138,145).
Resistin is a 12.5 kDa adipokine, which in human AT
is secreted predominantly in macrophages (93). Resistin is also known as
adipocyte-secreted factor or Found in Inflammatory Zone 3 (93). The resistin receptor remains
unknown, but resistin binding to the Toll-like receptor 4, adenylyl
cyclase-associated protein 1 receptor and G protein-coupled
receptors was proposed to mediate resistin inflammatory responses
in human cells (93,146,147). Resistin has also been associated
with the inflammatory response, by promoting activation of the
pro-inflammatory cytokines IL-6, IL-1β and TNF-α (16,131,148–150). Moroeover, resistin upregulates
several adhesion molecules, through NF-κB, in vascular endothelial
cells (148,151). In animal models, resistin
promotes IR, but in humans there are conflicting reports about the
potency of resistin in metabolic diseases (93,152,153). Several studies indicated that
increased serum resistin levels are associated with increased
obesity, visceral fat, IR and T2DM (154–157), while other studies failed to
reach to such conclusions (158,159). Also, resistin generates OS, which
activates MAPK signaling and inhibits endothelial nitric oxide
synthase (eNOS) gene expression (160). Moreover, resistin reduces NO
production, by inducing the proliferation of VSMCs, and causes
endothelial dysfunction (160).
In turn, reduction of NO availability results in impaired
vasodilation, increased vascular permeability, endothelial cell
adhesion and damage leading to CVDs (160,161).
PAI-1, also known as SERPINE1 is a physiological
inhibitor of tissue plasminogen activators (tPAs) (162). Increased PAI-1 activity is
associated with reduced fibrinolytic activity and thus, increased
risk for thrombus formation and CVDs (163). PAI-1 is synthesized in AT,
especially in visceral fat, as well as in preadipocytes,
fibroblasts, vascular endothelial cells and in immune cells
(2,163–165). Increased PAI-1 plasma levels have
been found in obese patients and reduced levels were achieved with
weight loss (129,166). In experimental studies, using
obese mouse models with MetS, it has been found that the deletion
of PAI-1 inhibited carotid artery atherosclerosis (167), while pharmacological PAI-1
inhibition attenuated atherosclerosis by inhibiting macrophage
accumulation and eliminating senescent cells in the atherosclerotic
plaques (168). In human studies,
PAI-1 was found to be associated with IR, MetS and atherosclerosis
in obesity (163,169). Also, in animal studies using mice
fed with a high-fat diet, a deficiency of PAI-1 led to a decrease
of body weight gain and improvement of IR (170). In addition, adipocyte hypertrophy
in obesity may create local hypoxic areas, which activates
hypoxia-inducible factor-1α (HIF-1α). The increase in HIF-1α causes
an increased expression of several pro-inflammatory cytokines, such
as TNF-α, IL-6, IL-1β and ROS, leading to higher PAI-1 expression
in adipocytes (171).
TNF-α is a 26-kDa pro-inflammatory cytokine produced
by macrophages, adipocytes and vascular endothelial cells, in
response to chronic inflammatory activity (37,85,172,173). TNF-α exists in two forms,
membrane-bound (mTNF-α) and free soluble (fTNF-α). TNF-α is
synthesized as a transmembrane monomer, which afterwards can be
cleaved by TNF-α converting enzyme, to yield the 17-kDa soluble
form (52). Both forms exist as
trimers that have biological activity. TNF-α has two distinct TNF-α
receptors, TNF-R1 and TNF-R2, that are similar in their
extracellular ligand-binding domains but differ markedly in their
intracellular signaling domains (52,85).
The majority of signaling in AT is downstream of the TNFR1
(52). TNF-α is a pro-inflammatory
cytokine characterized by various biological effects including
metabolic, inflammatory, proliferative and necrotic (173). TNF-α directly impairs peripheral
glucose uptake, by increasing serine phosphorylation of insulin
receptor substrate 1 (IRS-1), inhibits GLUT 4 translocation to the
plasma membrane and results in peripheral IR (173,174). TNF-α also potentially increases
lipolysis in human adipocytes by regulating hormone-sensitive
lipase in adipocytes, resulting in increased circulating FFA levels
and peripheral IR in obesity (173). Expression of TNF-α is increased
in obesity and IR in humans (37),
whilst treatment with TNF-α induces IR in AT (48). Serum TNF-α levels are decreased
during weight loss (175). TNF-α
is a part of a complex inflammatory network and is capable of
initiating cytokine cascade activation that involves both
synergistic and inhibitory reactions, which control the synthesis
and expression of other cytokines, hormones and their receptors
(176). ROS production can also
be induced by TNF-α binding to its TNF-R1 receptor promoting NF-κB,
ERK, p38MAPK and FADD/pro-caspase-8 signaling pathways (177–179). TNF-α causes systemic acute-phase
response via the release of other pro-inflammatory cytokines, such
as IL-6 and the reduction of anti-inflammatory adiponectins
(48). TNF-α also increases the
induction of OS and the production of superoxide anions (180).
IL-6 is produced by numerous different cell types,
including adipocytes, endothelial cells, pancreatic β-cells,
macrophages and monocytes (14,200–202). There are two signaling pathways
for IL-6 including its classical signaling mechanism involving the
binding of IL-6 to its receptor complex (IL6-Ra) that subsequently
interacts with an IL-6ST signaling protein (also known as
glycoprotein 130, gp130) at the plasma membrane, and the
non-classical signaling mechanism which is related to the
interaction of the IL-6ST protein with a soluble form of the
IL-6-binding receptor (203).
Both IL-6 signaling pathways lead to activation of the JAK1-STAT3
pathway (203). Its actions can
be beneficial or harmful to the organism, depending on the site of
action, the magnitude of production and the duration of the
response (204,205). In summary, its beneficial actions
are related to its production by skeletal muscles, in response to
physical exercise, contributing to lipid metabolism and enhancement
of insulin sensitivity in muscles (204,205), as well as appetite suppression
(206). Other actions of IL-6
include immune response and hematopoiesis (14,200–202). It participates in the regulation
of neural differentiation, maturation and function and in energy
homeostasis (14,200–202). In obesity, serum IL-6 levels are
elevated and 10–35% of IL-6 produced is attributed to WAT (207). In obesity, high caloric intake in
combination with reduced energy expenditure is directly related to
changes in the physiology and morphology of WAT (207). In particular, WAT expansion is
observed through an increase in the number or size of adipocytes
(208,209). Therefore, in the WAT of obese
patients there is an infiltration of immune cells, including T
cells and macrophages (19). Both
adipocytes, as well as immune cells in WAT, are the main sources of
increased circulating IL-6 levels. The VAT secretes a number of
substances that further promote IL-6 expression and releases ~2-3
times more IL-6 concentrations than STAT (19). Chronic exposure to high serum IL-6
levels has been associated with an elevated likelihood of impaired
glucose tolerance, T2DM, high blood pressure and obesity (48), while is also positively associated
with IR in obesity (210,211). Furthermore, exogenous IL-6
administration causes IR in humans, while weight loss results in
IL-6 decrease and bariatric surgery improves IR (212–215). Thus, if the elevated plasma IL-6
levels in obesity are considered, it could be suggested that
chronic low-grade inflammation in obesity links IL-6 as causal
factor for IR through the progressive tissue-infiltration by immune
cells (216).
Insulin is an anabolic, peptide hormone, secreted
by the pancreatic β-cells of the islets of Langerhans, in response
to high blood glucose levels and controls the metabolism of
carbohydrates, proteins and fats by stimulating the absorption of
glucose from the blood into lipid cells, skeletal muscle cells and
the liver, for ATP production or storage as glycogen and TGs
(217). More specifically, in fed
states, the exogenous glucose uptake increases the circulating
glucose levels and stimulates insulin secretion (123,217,218–220). Also, other nutrients from food
such as FAs and amino acids increase insulin secretion, which
stimulates lipogenesis and protein synthesis (218,221). Under fasting conditions,
lipolysis is induced from stored TGs in AT and supplies i) glycerol
for hepatic glucose production (gluconeogenesis) and ii) FAs for
β-oxidation (2,220). In the liver, insulin suppresses
hepatic gluconeogenesis (220).
Also, insulin reduces the rate of breakdown of glycogen in muscles
and liver (glycogenolysis), retaining normal glucose levels
(222). Regarding the effect of
insulin on lipid metabolism, insulin inhibits lipolysis
(antilipolytic action), increases hepatic lipid synthesis for
subsequent TGs storage in AT (223) and stimulates glucose uptake into
the skeletal muscles, heart and AT (220). In order to exert its effects,
insulin binds to its receptor (IF), a tyrosine kinase receptor. A
reduction in insulin signaling triggers IR that could affect the
metabolic actions of insulin (224). If a decrease of the blood glucose
levels is not achieved by insulin, then pancreatic β-cells increase
insulin release resulting in hyperinsulinemia, which is the key for
IR (225). Therefore,
hyperinsulinemia often precedes the development of marked IR and
fat mass gain (226).
Insulin signaling is initiated by the
phosphorylation/activation of the cytoplasmatic insulin tyrosine
kinase receptor that is associated with the activation of two main
signaling pathways: i) PI3K/AKT [also known as protein kinase B
(PKB)] pathway; and ii) the MAPK pathway (2,227).
In healthy subjects, by simultaneously stimulating these distinct
pathways (PI3K and MAPK), insulin couples metabolic and hemodynamic
homeostasis.
In the PI3K-AKT/PKB pathway the binding of insulin
to its cell surface receptor activates the lipid kinase, PI3K,
binding to its Src homology 2 domain, which activates several
phosphatidylinositol-(3,4,5)-triphosphate-dependent serine/threonine
kinases, including AKT/PKB (2).
Ultimately, these signaling events result in the translocation of
the insulin-dependent GLUT4 from its cytoplasmic storage vesicle to
the plasma membrane, leading to an increase in glucose uptake
(2). The PI3K-AKT signaling
pathway regulates the metabolic insulin actions by promoting
glucose utilization, protein synthesis and lipogenesis (228).
MAPK activation triggers a cascade that regulates
the effects of insulin on mitogenesis, growth and differentiation
and is not implicated in the metabolic actions of insulin (2). Also, MAPK-dependent insulin signaling
pathway controls secretion of the vasoconstrictor, endothelin-1,
from endothelium (63).
When a decrease of blood glucose levels is not
achieved by insulin, then pancreatic β-cells increase the release
of insulin, resulting in hyperinsulinemia, which is the key for IR
(224,225). Therefore, hyperinsulinemia often
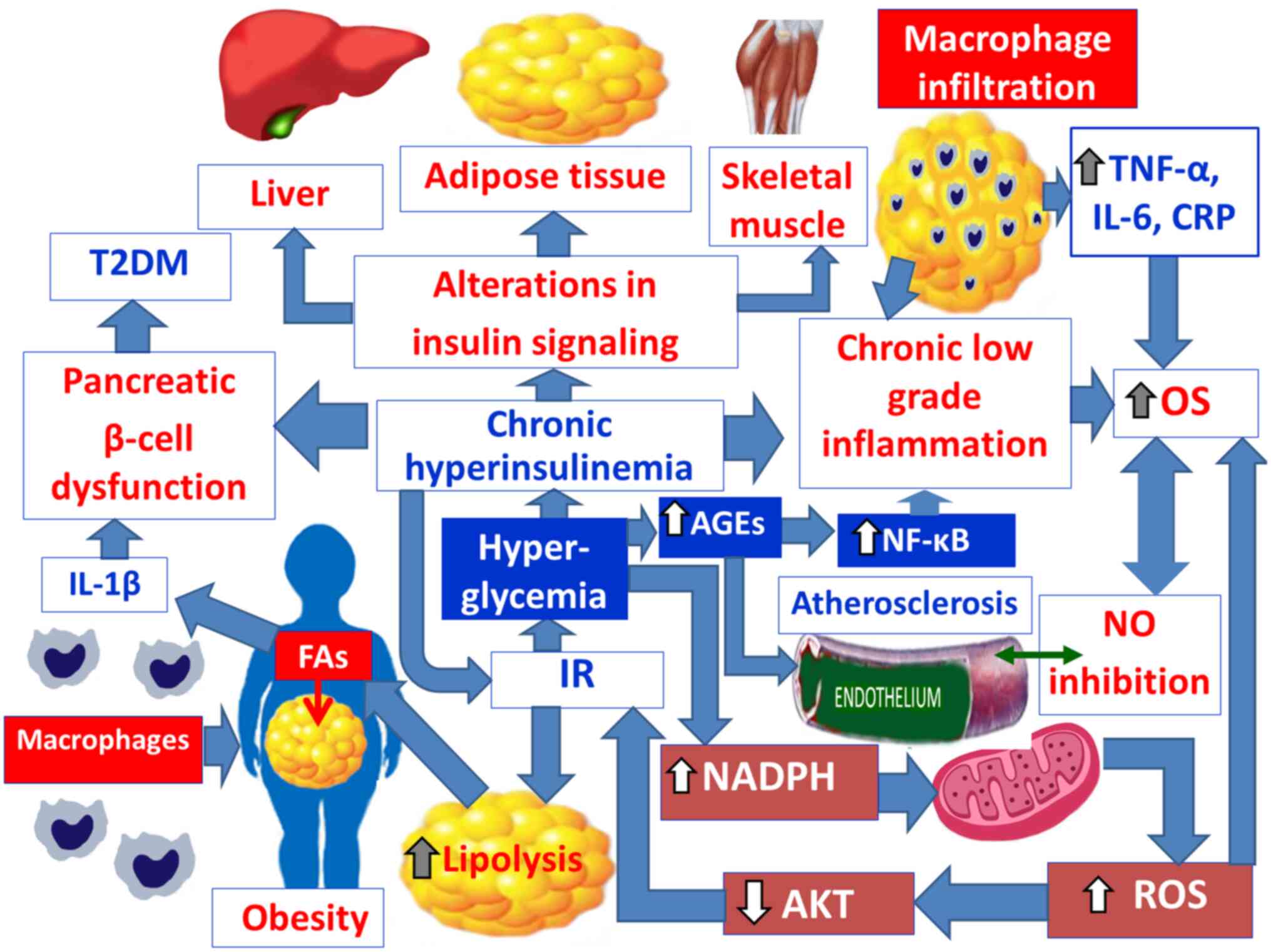
precedes the development of marked IR and fat mass gain (226,229) (Fig.
4). The IR in AT, skeletal muscles and liver is commonly linked
with obesity, which is a pathophysiological factor of T2DM
(230–233). When IR develops in fat tissues,
insulin-mediated inhibition of lipolysis is impaired leading to
increased lipolysis (123,234)
(Fig. 4). The resulting increase
in circulating FAs in turn worsen IR, causing alterations in the
insulin signaling cascade in different organs, thus creating a
vicious cycle (235,236) (Fig.
4). Moreover, in IR, there is a reduced insulin ability to
suppress glycogenolysis in hepatocytes and myocytes (234). In IR, FFAs, in muscles, affect
IRS-1-associated PI3K activity, leading to decreased GLUT4
translocation to the surface and reduced glucose uptake (235). In parallel, in IR the FFAs act on
the liver to promote gluconeogenesis. Therefore, insulin-resistant
individuals fail to inhibit hepatic glucose production and
hyperglycemia. This results in a hyperinsulinemic state to maintain
normal glucose levels (123).
However, this compensation eventually fails, leading to decreased
insulin levels, which is further exacerbated by the lipotoxic
effect of FFAs on pancreatic β-cells (123,236,237). Additionally, in IR, the FFAs act
on the liver and promote lipogenesis (Fig. 4). It is essential to note that
visceral lipolysis increases the supply of FFAs, directly to the
liver, via the splanchnic circulation, thus making visceral fat
deposits more important contributors to IR than subcutaneous fat
(123). Together with this, there
is increased de novo hepatic TG synthesis and a disruption
of β-oxidation in hepatic mitochondria (238–241). This net leads to increased
hepatic very low-density lipoproteins (VLDLs) secretion and
hypertriglyceridemia (238–241). The hepatic accumulation of TGs
and the toxic levels of FFAs results in hepatic lipotoxicity
(238–241). This mechanism contributes to the
production of ROS and the development of NAFLD, which is associated
with the development of MetS, which may progress to the more
serious non-alcoholic steatohepatitis (NASH) with subsequent
hepatic fibrosis, cirrhosis and cancer (225,242). In fact, there is a by-directional
relationship between obesity-related IR and NAFLD, since
obesity-related IR causes fatty liver and excessive hepatic fat
accumulation, promotes IR and weight gain (243). In addition, high concentrations
of FFAs increase cholesterol esters and triglyceride (TG) synthesis
and subsequently the production of VLDLs rich in TGs (244,245). These in turn, activate
cholesterol ester transfer protein, promote TG transfer from VLDL
to high-density lipoprotein (HDL), increase HDL clearance and
decrease its concentrations (244,245). Moreover, triglyceride-rich LDL,
formed after exchange with LDL cholesterol ester, becomes
hydrolyzed by lipoprotein lipase or hepatic lipase, leading to
cholesterol-depleted small dense LDL particles (244,245). All these alterations in
lipoprotein concentrations constitute the hallmark of atherogenic
dyslipidemia, caused by IR, in MetS (244,245). Another contribution of IR to MetS
is the development of hypertension caused partly by the loss of the
vasodilatory effect of insulin and by FFA-induced vasoconstriction
due to ROS production and the subsequent scavenging of NO (246). Other mechanisms involve the
increased sympathetic stimulation and the renin-induced sodium
reabsorption in the kidneys (247). Finally, the contribution of IR,
to the promotion of atherogenic processes and the increase of CVD
risk, is the development of a higher serum viscosity and a
pro-thrombotic state, caused by the increased levels of fibrinogen
and PAI-1 (34,165,169,248–253). IR and IL-6 produced during the
acute phase reaction contribute to elevated fibrinogen
concentrations (34,254). Fibrinogen is synthesized by
hepatocytes and holds a pivotal role in the coagulation cascade,
being a major determinant of plasma viscosity and platelet
aggregation, whilst potentially plays a pro-inflammatory role in
vascular wall disease (34,254,255).
In parallel, PAI-1 is elevated in IR, obesity and MetS (34,256). PAI-1 regulates the endogenous
fibrinolytic system and constitutes the main inhibitor of
fibrinolysis by binding and inactivating the tPA (34,248,257–260). Therefore, elevated PAI-1 levels
lead to decreased clearance of clots (34,248,257–260). Enhanced AT expression of PAI-1
has been reported in obesity, particularly in VAT (34,261), whilst there is an inverse
relationship between PAI-1 activity and adiponectin in overweight
and obese women (34,259,260).
MetS is a complex disorder defined by a cluster of
clinical and metabolic conditions that occur together and increase
the risk for IR, T2DM, dyslipidemia, CVDs, prothrombotic state and
stroke (262–264). According to the International
Diabetes Federation (IDF), metabolic syndrome (MetS) is
characterized as the presence of three or more of the following
features: i) obesity; ii) hyperglycemia; iii) hypertension; iv) low
HDL cholesterol levels; and/or v) hypertriglyceridemia (263). Obesity is the most frequently
observed component of MetS (265). It has been established that
patients with MetS are five times more likely to develop T2DM and
have a 2–3 times higher risk of CVDs (stroke and myocardial
infarction), compared with healthy subjects (264,266,267). In addition, MetS has been
associated with other clinical conditions, such as hepatic
steatosis and NAFLD, hypogonadism, polycystic ovarian syndrome,
obstructive sleep apnea, vascular dementia, Alzheimer's disease and
carcinomas, especially breast, pancreatic and colorectal cancers
(218,268,269). Obesity is the most frequently
observed component of MetS (265). The IDF estimates that MetS
affects almost a quarter of the adult general population in Western
societies (269–271). The prevalence of MetS in men does
not differ before and after 50 years of age, but women >50
years, show a sharp increase in prevalence (272). It is associated with higher
mortality risk in younger adults than in elders (273). Accumulating evidence indicates
that dysregulation of the production a wide range of adipocytokines
and cytokines, due to excessive accumulation of body fat,
participates in the pathogenesis of obesity associated MetS
(128,163,169). The link between PAI-1 and MetS
with obesity is well established. Increased PAI-1 serum levels are
associated with the development of IR, MetS, atherosclerosis and
thrombosis in obese patients (128,163,169). Treatment with TNF-α contributes
to the development of IR in AT (175). In patients with MetS, chronic
exposure to increased IL-6 levels is related to the development of
IR by depletion of GLUT4 and disruption of insulin signaling
(210,211). Leptin is also involved in the
pathophysiology of MetS (274).
High plasma levels of leptin are directly associated with IR, MetS
and lipid accumulation due to leptin resistance (35,39,124,125). Moreover, visfatin plays a central
role in MetS. Elevated visfatin serum levels are associated to IR,
T2DM and decreased function of pancreatic β-cells (129,275). Conversely, a protective role of
adiponectin against MetS has been reported, since it directly
attenuates production of IL-6 and TNF-α, by macrophages, through
its ability to suppress NF-κB activation (78,276). In addition, apelin reduces MetS
risk. It has been demonstrated that apelin stimulates glucose
uptake, increases insulin sensitivity and regulates lipolysis in
patients with MetS (101). OS is
also critically involved in the pathogenesis of MetS and in the
progression of its complications (277–280). In patients with MetS there are
higher levels of oxidative markers, as well as reduced antioxidant
defenses (277). In obesity, the
chronic low-grade inflammation, produced by adipocytes exacerbates
OS (35) (Fig. 4). Visceral fat accumulation induces
an increase in mitochondrial and peroxisomal oxidation of FAs, and
the production of ROS (35,281,282).
Furthermore, visceral fat accumulation causes over-consumption of
oxygen, which generates FRs in the mitochondrial respiratory chain
(35,281,282). In addition, a lipid-rich diet can
alter oxygen metabolism and generate ROS (35,281,282). Moreover, high ROS production and
a decrease in antioxidant capacity leads to a reduction in the
bioavailability of vasodilators, particularly NO, and an increase
in endothelium-derived contractile factors, favoring
atherosclerotic disease (35,281,282) (Fig.
4). With regards to hypertension, elevated OS, in vascular wall
leads to vasoconstriction, vascular remodeling, inflammation and
fibrosis which results in hypertension and atherosclerosis
(268,277,283). Regarding NAFLD, it has been
documented that elevated OS appears to be a key mechanism in
promoting liver injury and liver inflammation in NAFLD (284). Finally, it has been found that
dyslipidemia is associated with higher ROS release and lower eNOS
synthesis (277).
T2DM is a heterogeneous, chronic metabolic
disorder, characterized by elevated blood glucose levels with a
high prevalence, up to 90%, of all diagnosed diabetic cases in
adults (285,286). IR leads to hyperglycemia and over
time to T2DM (286). It has been
found that the relative risk of T2DM, in adult men and women,
increases for a BMI, >24 kg/m2 in men, and 22
kg/m2 in women (34).
Women with T2DM are 3–4 times more prone to CVDs compared with 2–3
times in men with T2DM (287).
Obesity is an important independent risk factor for IR and T2DM
(288–291). IR is responsible for the
development of hyperglycemia and over time may evolve to T2DM. IR
alone is not capable of causing an increase in blood sugar
(292), since the pancreas has
mechanisms to adapt, by increasing the mass of β-cells and the
ability to produce insulin (292,293). Thus, despite reduced peripheral
insulin sensitivity, blood sugar levels could retain stable
(292). The cellular mechanisms
involved in the pathogenesis of obesity-associated T2DM include: i)
alterations in the insulin signaling; ii) pancreatic β-cell
dysfunction and failure; and iii) chronic low-grade inflammation
and increased OS (294). The
mechanisms interrelating obesity to the pathogenesis of T2DM are
depicted in Fig. 4. Obesity causes
generalized IR in AT, liver and skeletal muscles and is associated
with increased insulin secretion and chronic hyperinsulinemia,
which promotes further weight gain (292,295,296). Therefore, there is a
bi-directional relationship between obesity and hyperinsulinemia.
Insulin resistant conditions in T2DM could be caused by signaling
defects at a number of levels of the insulin-signaling cascade in
metabolic tissues, such as liver, AT and skeletal muscles (292,243,297). In addition, in IR and T2DM, the
liver fails to suppress glycogenolysis and gluconeogenesis, despite
compensatory hyperinsulinemia, and is associated with accelerated
glucose synthesis and fasting hyperglycemia (292,243,297). In T2DM, the IR in skeletal
muscles is associated with postprandial hyperglycemia, since in
these patients, skeletal muscles exhibit decreased insulin
sensitivity, which results in impaired insulin-stimulated glucose
uptake (298). As aforementioned,
IR does not necessarily imply T2DM. A harmful mechanism for the
functionality of β-cells is ‘glucotoxicity’. Glucotoxicity is
dependent on the duration and degree of hyperglycemia, in which the
elevated glucose levels, characteristic of T2DM, contribute to
desensitization of pancreatic β-cells in insulin secretion
(292,294). Another mechanism that contributes
to further loss of β-cells and pancreatic dysfunction is
‘lipotoxicity’. It is directly related to fat occuring obesity and
is accompanied by an underlying predisposition to T2DM and
increased serum FFA levels (292,299). FFAs are elevated in the plasma of
patients with T2DM, due to uncontrolled lipolysis, by
insulin-sensitive lipase, in adipocytes (300). The high levels of FFAs impair the
function of pancreatic β-cells and the glucose-induced insulin
secretion (300) (Fig. 4). Another mechanism, related to the
disruption of pancreatic β-cells, is their low antioxidant defense,
since they do not express, in high ratio, antioxidant enzymes,
which make them susceptible to oxidative damage (301). Finally, due to the accumulated
fat during obesity, increased levels of cytokines from macrophages
are observed, such as IL-1β (302), which is also responsible for the
deficiency of insulin secretion from β-cells (34) (Fig.
4). In addition to BMI, there is a strong association between
abdominal obesity (central obesity) and the incidence of T2DM
(24,303–308). Abdominal obesity is associated
with the following conditions that may lead to systemic
inflammation and IR: i) increased levels of glucose and
non-esterified fatty acids; ii) hormonal imbalance with increased
leptin levels and decreased adiponectin levels; and iii) increased
secretion of cytokines and pro-inflammatory substances from fat
cells (309,310). In more detail, in terms of the
secretion of cytokines and pro-inflammatory substances from
adipocytes, the driving force is the excess visceral fat that
triggers the cascade of inflammation (309,310). In response to the secretion of
these substances, mononuclear cells migrate from blood circulation
to the AT, and they differentiate into macrophages (309,310). Macrophages secrete cytokines,
thus in obesity an increased secretion of TNF-α and IL-6 is
observed (309,310) (Fig.
4). Secretion of TNF-α is responsible for tissue inflammation,
due to its role in the generation of ROS, and activation of
transcription-induced pathways, and for IR, in peripheral tissues
and adipocytes, which is important for the onset of T2DM (309,310). The increase in IL-6 levels
contributes to the prevention of the binding of insulin to its
receptor, through the induction of proteins associated with it
(310–312), as well as the induction of CRP,
which like TNF-α, is associated with IR (309,310) (Fig.
4). Additionally, hyperglycemia, characteristic in T2DM, is
associated with the generation of advanced glycation end products
(AGEs), binding to their receptors. AGEs are responsible for the
up-regulation of the transcription factor NF-κB, which is a
mediator of inflammation and immunity (313), while AGEs also block the activity
of NO in the vascular endothelium and promote the production of ROS
(313) (Fig. 4). Finally, a key role for chronic
inflammation and OS in obesity related T2DM is mitochondrial
dysfunction (314–316). Specifically, due to the increased
concentration of sugars, glycolysis and the Krebs cycle are induced
and cause an increased flow of NADH and FADH2 (flavin adenine
dinucleotide; reduced form) (314–316). They act as electron donors to the
mitochondrial chain, resulting in the accumulation of electron
donors in coenzyme Q (314–316). This results in the production of
mitochondrial superoxide radical (FR), an important source of ROS
from adipocytes (314–316). Chronic exposure to high
intracellular ROS levels in adipocytes ultimately causes
mitochondrial dysfunction and perpetuates AT inflammation, together
with impairment of the AKT signaling pathway that induces IR
(314–316) (Fig.
4).
In patients with T2DM, the high blood glucose
levels induce cytokine IL-1β production and secretion, in the
β-cells of the pancreatic islets of Langerhans, leading to
pro-inflammatory immune responses, β-cell dysfunction, decreased
β-cell proliferation and increased β-cell apoptosis (198,317,318). Therefore, short-term IL-1
receptor (IL-1R) blockage could lead to improvements in both
metabolic and inflammatory parameters in patients with MetS and
T2DM and may represent a potential targeted therapeutic approach
for these patients. For instance, Larsen et al tested the
effects of anakinra therapy in two studies (319,320). In their first study, Larsen et
al (319) examined the
effects of anakinra treatment, in a double-blind, parallel-group
trial with 70 patients with T2DM, that were randomly assigned to
receive a placebo, or 100 mg of anakinra, subcutaneously, once
daily. Anakinra is a human recombinant IL-1Ra, which prevents
signal dunsduction of IL-1α and IL-1β (321,322). Anakinra is approved by the Food
and Drug Administration (FDA) for the treatment of rheumatoid
arthritis in adults and neonatal onset multisystem inflammatory
disease (323). During a 13-week
treatment period, anakinra administration improved glycemia and
pancreatic β-cell secretory function, compared with the placebo
group. This occurred by reducing the glycated hemoglobin levels
(HbA1c), the ratio of proinsulin to insulin (marker of pancreatic
β-cell dysfunction and reduced insulin secretory capacity), the
serum levels of systemic inflammatory markers (IL-6 and CRP), and
enhanced C-peptide and insulin secretion (319) (Table II). Furthermore, Larsen et
al (320) in a 39-week
follow-up study examined the durability of anakinra administration
on the management of T2DM and found maintenance of increased
insulin secretion and reduction of insulin requirements (320). These findings suggest that IL-1R
blockade with anakinra may improve glucose control and β-cell
secretory function for a long period (319,321). Van Asseldonk et al
(324) in a randomized,
double-blind, crossover study examined the effects of anakinra in
nondiabetic, obese individuals, with MetS, at a dose of 150 mg,
subcutaneously, once daily for a 4-week treatment period. The
authors found that anakinra administration compared with the
placebo group, led to a significantly lower degree of inflammation
by reducing the circulating CRP levels and the number of leukocytes
accompanied by a significant increase in the disposition index and
improvement in pancreatic β-cell function (324) (Table II). Nevertheless, anakinra did not
significantly improve insulin sensitivity (324) (Table II). Also, van Poppel et al
(325) assessed the effects of
anakinra therapy in another double-blind, randomized,
placebo-controlled crossover study, involving 16 subjects, with
impaired glucose tolerance, assigned to receive 150 mg anakinra
daily, for 8 weeks. A significant improvement in the first-phase
insulin secretion and pancreatic β-cell function was found
(325) (Table II). In line with these findings,
Cucak et al (326)
evaluated in female non-obese diabetic (NOD) mice, the effects of
SER140, which is a 10-amino-acid peptide antagonist of IL-1β
receptors (IL-1Ra), on the progression of diabetes and pancreatic
β-cell changes. The study consisted of an 8-week treatment period.
The results of this study showed a reduction in the incidence of
diabetes, by >50%, compared with the control group, a decrease
in non-fasting plasma glucose concentrations and an increase in
plasma insulin levels. Additionally, SER140 administration changed
the immune-endocrine dynamics in the NOD mouse pancreas. The
authors suggested that the SER140 treatment can postpone the onset
of diabetes in female NOD mice by competing with IL-1β for IL-1β
receptors (IL-1R) (326).
Other promising IL-1β blocking therapies have
demonstrated antidiabetic potential as well. Cavelti-Weder et
al (327) evaluated the
safety and biological activity of gevokizumab in patients with
T2DM. Gevokizumab is a recombinant human monoclonal anti-IL-1β
antibody. In this study a total of 98 patients with T2DM
participated, 17 patients to the control group and 81 patients to
the gevokizumab treatment group, at increasing doses. It was found
that gevokizumab treatment was safe and led to significant
reduction in HbA1c values (−0.85%) after 3 months, accompanied by
augmented C-peptide secretion, increased insulin sensitivity and
decreased CRP levels (327)
(Table II). Rissanen et al
(328) evaluated the effects of a
single dose of canakinumab, a recombinant human monoclonal antibody
targeting circulating IL-1β in patients with impaired glucose
tolerance or T2DM treated with insulin and metformin. The authors
found a trend towards increased insulin secretion (Table II). In another study conducted
with 551 metformin-treated patients, with T2DM, Hensen et al
(329) assessed the safety,
tolerability and effects of different monthly doses of canakinumab
(5, 15, 50 or 150 mg). The authors found that canakinumab treatment
was safe and well tolerated. In addition, canakinumab (50 mg) led
to a reduction in HBA1c values compared with the placebo group
(329) (Table II). These findings suggest that
monthly adjuvant treatment, with 50 mg canakinumab, on
metformin-treated patients, with T2DM, could potentially improve
pancreatic β-cell function (329). However, Ridker et al
(330) did not find alterations
in HbA1c, glucose and insulin levels after canakinumab treatment in
patients with T2DM with high cardiovascular risk (Table II). By contrast, cankinumab
significantly reduced inflammation markers such as CRP, IL-6 and
fibrinogen (330) (Table II). Choudhury et al
(331) examined the effects of
cankinumab in patients with atherosclerotic cardiovascular disease
and T2DM or impaired glucose tolerance for 12 months and found
reduction in inflammation markers (hsCRP, IL-6) compared with the
control group (Table II).
Furthermore, Noe et al (332) found similar results (Table II). In addition, Sloan-Lancaster
et al (333) examined the
effects of LY2189102 in the treatment of patients with T2DM.
LY2189102 is a recombinant human monoclonal antibody (IgG4) that
binds to IL-1β with high affinity and neutralizes its activity by
forming a complex with circulating IL-1β. The authors demonstrated
that the weekly subcutaneous administration of LY2189102 for 12
weeks can reduce postprandial glycemic levels and improve
anti-inflammatory effects in patients with T2DM (Table II). In a canakinumab
anti-inflammatory thrombosis outcomes study, IL-1β inhibition by
canakinumab did not show long-term (over a median period of 3.7
years) benefits in the reduction of HbA1c values in patients prior
to myocardial infarction with or without pre-diabetes or T2DM
(334) (Table II). In addition, canakinumab
administration was ineffective in reducing the occurrence of new
onset T2DM (334) (Table II). Also, the development of
vaccines against IL-1β represents a treatment option for
IL-1β-dependent diseases such as T2DM (327).
Since obese humans have increased circulating
levels of TNF-α and TNF-α levels, in human AT, is positively
associated with BMI and hyperinsulinemia (317,335), their levels have been proposed to
play a role in the development and pathogenesis of IR and T2DM
(317,336). Indeed, van den Oever et al
(337) in their study found that
adalimumab administration in patients with RA or OA and IR improves
IR and pancreatic β-cell function (337) (Table II). Adalimumab binds with
specificity to TNF-α and inhibits its interaction with the p55 and
p75 cell surface TNF receptors (337). Also, infliximab is a chimeric
monoclonal antibody that binds TNF-α with high affinity and
neutralizes TNF-α. Kiortsis et al (338) found that the administration of
infliximab in patients with RA or ankylosing spondylitis and IR
improved insulin sensitivity (Table
II). Similar results were also found by Gonzalez-Gay et
al (339) in insulin
resistant patients, with RA (Table
II). In addition, Haida et al (340) demonstrated that infliximab
treatment prevents hyperglycemia and liver gluconeogenesis, in high
fat diet-fed mice. Moreover, infliximab seems to ameliorate
TNF-α-induced IR, in 3T3-L1 adipocytes, in vitro, by
improving the insulin signaling pathway, via inhibition of protein
tyrosine phosphatase 1B (341).
Additionally, Abdelhamid et al (342) showed that infliximab
administration in rats reduces TGs, increases HDL-c levels and
reverses fructose-induced adiponectin resistance. Therefore, the
authors suggested that infliximab may affect the manifestation of
MetS. However, infliximab failed to affect MetS-mediated
hyperglycemia, hypertension and the elevated peroxidation levels,
as the levels of malondialdehyde dictate (342). Bernstein et al (343) investigated the effects of the
inhibition of TNF-α with entanercept (a TNF-α blocker), in patients
with MetS, for a 4-week treatment period. The authors concluded
that etanercept reduced CRP levels (Table II). Also, Lo et al
(344) randomized obese patients
with MetS on etanercept and demonstrated increased circulating
levels of total adiponectin but the ratio of high molecular weight
adiponectin (HMWA) to total adiponectin was reduced (Table II); HMWA is the most biologically
active form of the adipokine and is thought to mediate insulin
sensitivity (345). Conversely,
Stanley et al (346) found
that the administration of etanercept, on obese individuals, with
MetS, increased the ratio of HMWA to total adiponectin (Table II). Paquot et al (347) in their clinical trial failed to
show an improvement in insulin sensitivity after TNF-α
neutralization, following a single intravenous administration of a
TNF receptor antagonist (Ro45-2081; a soluble TNF-receptor-IgG
fusion protein) in obese insulin resistant patients (Table II). Moreover, Dominguez et
al (348) failed to reverse
vascular and metabolic IR, after short-term etanercept treatment,
in obese patients with T2DM (Table
II). This evidence may support the hypothesis that AT TNF-α,
which is not secreted in the systemic circulation may act in an
autocrine or paracrine manner. Therefore, the anti-TNF-α agents may
not reach the AT microcirculation, which is markedly impaired in
T2DM (347,349,350). Thus, anti-TNF-α therapy may fail
to improve insulin sensitivity in such cases. In summary, treatment
with anti-TNF-α agents in patients with T2DM did not yield
consistent results for glucose and HbA1c reduction. Ruscitti et
al (351) investigated the
effects of anti-IL-1 treatment with anakinra compared with TNF-α
inhibitors, such as etanercept, adalimumad, infliximad,
certolizumab pegol or golimumab, in patients with RA and T2DM in an
open label, prospective, controlled, parallel-group trial. The
authors found that anakinra reduced HbA1c values compared with
TNF-α inhibitors after a 6 month treatment period and also reduced
antidiabetic drugs defined as the reduction of administered
dosages, change from combination therapy to monotherapy or
discontinuation of anti-diabetic drugs (351). However, after the mean follow-up
of 18 months anakrinra had no effects in HbA1c values compared with
TNF-α inhibitors but continued to reduce the use of antidiabetic
drugs. On the contrary, an increase of anti-diabetic therapies was
needed in participants treated with TNF-α inhibitors to reduce
HbA1c levels (351) (Table II).
Diacerein is both an IL-1βR blocker and a TNF-α
antagonist by its active metabolite, rhein (352). Ramos-Zavala et al
(352) found that diacerein
administration in patients with T2DM increased insulin secretion
and decreased fasting glucose levels (Table II). In addition, Cardoso et
al (353) found that
diacerein administration reduced HbA1c values in patients with T2DM
(Table II). These findings are in
agreement with the studies reported from Tres et al
(354) (Table II) and Jangsiripornpakorn et
al (355) (Table II). In patients with T2DM and
chronic kidney disease, intervention with diacerein improves the
metabolic control of T2DM and reduces nighttime blood pressure but
has no effects in glomerular filtration rate and urinary
albumin/creatinine ratio (356)
(Table II).
Drugs targeting immune cell infiltration have been
tested for anti-inflammatory and anti-obesity therapy as well. Di
Prospero et al (357)
evaluated the safety, tolerability, pharmacokinetics and
pharmacodynamics of JNJ-41443532, a CCR2 antagonist, in a small
sample size, double-blind, placebo-controlled, randomized,
multicenter study for 4-weeks, in patients with T2DM. The authors
found that JNJ-41443532 treatment was well tolerated in patients
with T2DM and showed modestly improved glycemic parameters compared
with the placebo group (357)
(Table II). Also, Mulder et
al (358) examined in male
mice whether propagermanium, an inhibitor of CCR2, could attenuate
tissue inflammation and NASH development. The results of this study
showed that early propagermanium intervention was more effective
than late intervention in attenuating IR, WAT inflammation and NASH
development (358). In addition,
Huh et al (359)
investigated the effects of PF4178903, an antagonist for dual CCRs,
CCR2 and CCR5, on obesity and IR, in high fat diet fed mice. The
authors demonstrated that the dual CCR2 and CCR5 antagonist,
PF4178903, attenuated metabolic dysfunction, induced by a high-fat
diet. There was a decrease in body weight gain, blood glucose
levels, lipid levels, adipocyte size and systemic inflammation, and
an improvement in glucose tolerance and insulin sensitivity
(359). Particularly, PF4178903
significantly shifted the M1 macrophage phenotype towards the M2
phenotype, in high fat diet-induced obesity, suggesting that the
dual CCR2 and CCR5 blockade regulates macrophage polarization in AT
macrophages (359).
Sarilumab is a human anti-IL-6 receptor (IL-6R)
monoclonal IgG1 antibody that targets both the membrane-bound and
soluble IL-6 receptor forms (364,365). Therefore, sarilumab blocks both
the cis- and trans-inflammatory signaling cascades of IL-6 and
reduces the activity of pro-inflammatory cytokines and inflammation
(364,365). In particular, sarilumab has been
shown to reduce HbA1c values after a 24 week treatment period
compared with adalimumab in patients with rheumatoid arthritis with
or without T2DM (366) (Table II).
Pharmacological elevation of plasma levels of
adiponectin could become a promising therapeutic strategy in
countering-balacing obesity-associated T2DM (370). The thiazolidinediones (TZDs) are
agonists of peroxisome proliferation activating receptor-γ (PPARγ),
with TZDs such as troglitazone, rosiglitazone, glitazone and
pioglitazone having been shown to increase the activation of PPARγ,
elevate serum adiponectin concentrations, restore lipogenic
function and decrease inflammation (371–373). TZDs also block the ability of
TNF-α to inhibit insulin signaling through increased serine
phosphorylation of IRS-1 (374).
Wolf et al (375)
demonstrated in vitro that adiponectin displays potent
immunosuppressive effects inducing the production of
anti-inflammatory cytokines IL-10 and IL-1Ra in myeloid cell types.
In addition, IL-10 can inhibit the production of pro-inflammatory
mediators by macrophages, including IL-1, IL-2, IL-6, IL-12,
interferon gamma (INFγ) and TNF-α (375,376). Furthermore, adiponectin rapidly
up-regulates IL-10 and subsequently increases the levels of tissue
inhibitor metalloproteinase-1 (an inhibitor of matrix
metalloproteinases) in human macrophages preventing the degradation
of the ECM (78). Although, TZDs
are effective in controlling glycemia and IR, and are not
associated with hypoglycemia, when used as monotherapy (377,378), there are some serious safety
concerns that must be considered when selecting TZDs for the
treatment of metabolic disorders (370). For example, troglitazone was
removed from the market after the FDA received reports of 94 cases
of troglitazone-induced liver failure (379). Also, pioglitazone usage increases
the risk of bladder cancer (380)
and edema in T2DM (381,382). Another important safety issue of
TZDs, is their risk for heart failure due to fluid retention
(383). Moreover, glitazone has
been associated with macular edema of the retina that leads to
vision loss (384,385). Additionally, TZDs decrease bone
density and therefore increase the bone fracture risk (386). Apart from the aforementioned side
effects of TZDs, treatment with TZDs is associated with a rise in
body weight due to increased fat mass and fluid retention in
patients with T2DM (387,388).
Obesity has evolved to an epidemic condition that
causes health impairment by increasing the risk of developing other
relevant conditions, such as MetS, IR, T2DM, hypertension,
atherosclerosis, dyslipidemia, CVDs, respiratory disorders and
several types of cancer. The molecular and pathophysiological
mechanisms linking visceral obesity and MetS are mediated by
chronic low-grade inflammation and OS, but they are not fully
understood. Particularly, obesity results in a pro-inflammatory
state in the adipocytes characterized by increased recruitment,
accumulation and AT infiltration of M1 macrophages with a
consequent release of highly pro-inflammatory cytokines, such as
IL-1β, IL-6 and TNF-α, and pro-inflammatory adipokines, such as
PAI-1, visfatin, resistin and leptin. The polarization of
macrophages toward the M1 pro-inflammatory state results in reduced
adiponectin levels. Accumulating evidence indicates that obesity
related factors, such as a high-calorie diet, sedentary lifestyle,
AT micro-environment and gut microbiota deregulation, exacerbate
chronic tissue inflammation. To date, clinical studies, which
tested the safety, tolerability and efficacy of molecular therapies
targeting obesity-associated inflammation, have shown hopeful
results by enhancing insulin sensitivity and improving metabolic
function and IR, but they still remain unsatisfactory with poor
treatment outcomes and in numerous cases are accompanied with
serious side effects. Therefore, new efficacious and safe molecular
targeted agents need to be discovered.
Not applicable.
Funding: No funding was received.
Not applicable.
FNV and PTN conceptualized the study; FNV created
all the figures; FNV, MNV, VKV and PTN wrote and edited the
manuscript. All authors read and approved the final version of the
manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Sethi JK and Vidal-Puig AJ: Thematic
review series: Adipocyte biology. Adipose tissue function and
plasticity orchestrate nutritional adaptation. J Lipid Res.
48:1253–1262. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Luo L and Liu M: Adipose tissue in control
of metabolism. J Endocrinol. 231:R77–R99. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jung UJ and Choi MS: Obesity and its
metabolic complications: The role of adipokines and the
relationship between obesity, inflammation, insulin resistance,
dyslipidemia and nonalcoholic fatty liver disease. Int J Mol Sci.
15:6184–6223. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Curat CA, Miranville A, Sengenè C, Diehl
M, Tonus C, Busse R and Bouloumié A: From blood monocytes to
adipose tissue-resident macrophages: Induction of diapedesis by
human mature adipocytes. Diabetes. 53:1285–1292. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cypess AM, Lehman S, Williams G, Tal I,
Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, et al:
Identification and importance of brown adipose tissue in adult
humans. N Engl J Med. 360:1509–1517. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nauli AM and Matin S: Why do men
accumulate abdominal visceral fat? Front Physiol. 10:14862019.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kahn CR, Wang G and Lee KY: Altered
adipose tissue and adipocyte function in the pathogenesis of
metabolic syndrome. J Clin Invest. 129:3990–4000. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Blüher M: Adipose tissue dysfunction in
obesity. Exp Clin Endocrinol Diabetes. 117:241–250. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tan CY and Vidal-Puig A: Adipose tissue
expandability: The metabolic problems of obesity may arise from the
inability to become more obese. Biochem Soc Trans. 36:935–940.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sebo ZL and Rodeheffer MS: Assembling the
adipose organ: Adipocyte lineage segragation and adipogenesis in
vivo. Development. 146:dev1720982019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lafontan M and Langin D: Lipolysis and
lipid modilization in human adipose tissue. Prog Lipid Res.
48:275–297. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Frayn KN: Adipose tissue as a buffer for
daily lipid flux. Diabetologia. 45:1201–1210. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cinti S, Mitchell G, Barbatelli G, Murano
I, Ceresi E, Faloia E, Wang S, Fortier M, Greenberg AS and Obin MS:
Adipocyte death defines macrophage location and function in adipose
tissue of obese mice and humans. J Lipid Res. 46:2347–2355. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ellulu MS, Patimah I, Khazaai H, Rahmat A
and Abed Y: Obesity and inflammation: The linking mechanism and the
complications. Arch Med Sci. 13:851–863. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hotamisligil GS: Inflammation and
metabolic disorders. Nature. 444:860–867. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fantuzzi G: Adipose tissue, adipokines,
and inflammation. J Allergy Clin Immunol. 115:911–919. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weir CB and Jan A: BMI classification
percentile and cut off points. StatPearls Treasure Island, FL:
StatPearls Publishing; 2020
|
|
18
|
Marcadenti A and de Abreu-Silva EO:
Different adipose tissue depots: Metabolic implications and effects
of surgical removal. Endocrinol Nutr. 62:458–464. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fuster JJ, Ouchi N, Gokce N and Walsh K:
Obesity-induced changes in adipose tissue microenvironment and
their impact on cardiovascular disease. Circ Res. 118:1786–1807.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Osborn O and Olefsky JM: The cellular and
signaling networks linking the immune system and metabolism in
disease. Nat Med. 18:363–374. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gustafson B, Hedjazifar S, Gogg S,
Hammarstedt A and Smith U: Insulin resistance and impaired
adipogenesis. Trends Endocrinol Metab. 26:193–200. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kuk JL, Katzmarzyk PT, Nichaman MZ, Church
TS, Blair SN and Ross R: Visceral fat in an independent predictor
of all-cause mortality in men. Obesity (Silver Spring). 14:336–341.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Klein J, Permana PA, Owecki M, Chaldakov
GN, Böhm M, Hausman G, Lapière CM, Atanassova P, Sowiński J,
Fasshauer M, et al: What are subcutaneous adipocytes really good
for? Exp Dermatol. 16:45–70. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cameron AJ, Magliano DJ and Soderberg S: A
systemic review of the impact of including both waist and hip
circumference in risk models for cardiovascular diseases, diabetes
and mortality. Obes Rev. 14:86–94. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Koster A, Murphy RA, Eiriksdottir G,
Aspelund T, Sigurdsson S, Lang TF, Gudnason V, Launer LJ and Harris
TB: Fat distribution and mortality: The AGES-Reykjavik study.
Obesity (Silver Spring). 23:893–897. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Arner P, Andersson DP, Thörne A, Wirén M,
Hoffstedt J, Näskybd E, Thorell A and Rydén M: Variations in the
size of the major omentum are primarily determined by fat cell
number. J Clin Endocrinol Metab. 98:E897–E901. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chait A and den Hartigh LJ: Adipose tissue
distribution, inflammation and its metabolic consequences,
including diabetes and cardiovascular disease. Front Cardiovasc
Med. 25:222020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
James WP: Assessing obesity: Are ethnic
differences in body mass index and waist classification criteria
justified? Obes Rev. 6:179–181. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
James WP, Rigby N and Leach R: Obesity and
the metabolic syndrome: The stress on society. Ann N Y Acad Sci.
1083:1–10. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
El-Sayed AM, Scarborough P and Galea S:
Ethnic inequalities in obesity among children and adults in the UK:
A systematic review of the literature. Obes Rev. 12:e516–e534.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Barnett AH, Dixon AN, Bellary S, Hanif MW,
O'Hare JP, Raymond NT and Kumar S: Type 2 diabetes and
cardiovascular risk in the UK south Asian community. Diabetologia.
49:2234–2246. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Misra A and Khurana L: Obesity-related
non-communicable diseases: South Asians vs White Caucasians. Int J
Obes (Lond). 35:167–187. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pi-Sunyer X: The medical risks of obesity.
Postgrad Med. 121:21–33. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kyrou I, Randeva HS, Tsigos C, Kaltsas G,
Weickert MO, Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos
G, et al: Clinical problems caused by obesity. Endotext [Internet]
South Dartmouth (MA): MDText.com, Inc; 2018
|
|
35
|
Khanna D, Khanna S, Khanna P, Kahar P and
Patel BM: Obesity: A chronic low-grade inflammation and its
markers. Cureus. 14:e227112022.PubMed/NCBI
|
|
36
|
Bobbert T, Rochlitz H, Wegewitz U, Akpulat
S, Mai K, Weickert MO, Möhlig M, Pfeiffer AFH and Spranger J:
Changes of adiponectin oligomer composition by moderate weight
reduction. Diabetes. 54:2712–2719. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hotamisligil GS, Shargill NS and
Spiegelman BM: Adipose expression of tumor necrosis factor-α:
Direct role in obesity-linked insulin resistance. Science.
259:87–91. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mohlig M, Weickert MO, Ghadamgadai E,
Machlitt A, Pfüller B, Arafat AM, Pfeiffer AFH and Schöfl C:
Adipocyte fatty acid-binding protein is associated with marker of
obesity, but is an unlikely link between obesity, insulin
resistance and hyperandrogenism in polycystic ovary syndrome women.
Eur J Endocrinol. 157:195–200. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fonseca-Alaniz MH, Takada J, Alonso-Vale
MI and Lima FB: Adipose tissue as an endocrine organ: From theory
to practice. J Pediatr. 83:192–203. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Maury E and Brichard SM: Adipokine
dysregulation, adipose tissue inflammation and metabolic syndrome.
Mol Cell Endocrinol. 314:1–16. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gregor MF and Hotamilsigil GS:
Inflammatory mechanisms in obesity. Annu Rev Immunol. 29:415–445.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Weickert MO, Hodges P, Tan BK and Randeva
HS: Neuroendocrine and endocrine dysfunction in the
hyperinsulinemic PCOS patient: The role of metformin. Minerva
Endocrinol. 37:25–40. 2012.PubMed/NCBI
|
|
43
|
Randeva HS, Tan BK, Weickert MO, Lois K,
Nestler JE, Sattar N and Lehnert H: Cardiometabolic aspects of the
polycystic ovary syndrome. Endocr Rev. 33:812–841. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Makki K, Froguel P and Wolowczuk I:
Adipose tissue in obesity-related inflammation and insulin
resistance. Cells, cytokines and chemokines. ISRN Inflamm.
2013:1392392013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sivakumar K, Bari MF, Adaikalakoteswari A,
Guller S, Weickert MO, Randeva HS, Grammatopoulos DK, Bastie CC and
Vatish M: Elevated fetal adispin/acylation-stimulating protein
(ASP) in obese pregnancy: Novel placental secretion via Hofbauer
cells. J Clin Endocrinol Metab. 98:4113–4122. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
von Loeffelholz C, Mohlig M, Arafat AM,
Isken F, Spranger J, Mai K, Randeva HS, Pfeiffer AFH and Weickert
MO: Circulation vaspin is unrelated to insulin sensitivity in a
cohort of nondiabetic humans. Eur J Endocrinol. 162:507–513. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Elmasry SA, Al-Azzawi MA, Ghoneim AH, Nasr
MY and AboZaid MMN: Role of oxidant-antioxidant imbalance in the
pathogenesis of chronic obstructive pulmonary disease. Egypt J
Chest Dis Tuberc. 64:813–820. 2015. View Article : Google Scholar
|
|
48
|
Marseglia L, Manti S, D'Angelo G, Nicotera
A, Parisi E, Di Rosa G, Gitto E and Arrigo T: Oxidative stress in
obesity: A critical component in human diseases. Int J Mol Sci.
16:378–400. 2015. View Article : Google Scholar
|
|
49
|
Reilly SM and Saltiel AR: Adapting to
obesity with adipose tissue inflammation. Nat Rev Endocrinol.
13:633–643. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Rheinheimer J, de Souza BM, Cardoso NS,
Bauer AC and Crispim D: Current role of the NLRP3 inflammasome on
obesity and insulin resistance: A systematic review. Metabolism.
74:1–9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Deshmane SL, Kremley S, Amini S and Sawaya
BE: Monocyte chemoattractant protein-1 (MCP-1): An overview. J
Interferon Cytokine Res. 29:313–326. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Taylor EB: The complex role of adipokines
in obesity, inflammation and autoimmunity. Clin Sci (Lond).
135:731–752. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Matsushima K, Larsen CG, DuBois GC and
Oppenheim JJ: Purification and characterization of a novel monocyte
chemotactic and activating factor produced by a human
myolomonocytic cell line. J Exp Med. 169:1485–1490. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Rollins BJ: Chemokines. Blood. 90:909–928.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kanda H, Tateya S, Tamori Y, Kotani K,
Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K and
Kasuga M: MCP-1 contributes to macrophage infiltration into adipose
tissue, insulin resistance, and hepatic steatosis in obesity. J
Clin Invest. 116:1494–1505. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Singh S, Anshita D and Ravichandiran V:
MCP-1: Function, regulation, and involvement in disease. Int
Immunopharmacol. 101((PtB)): 1075982021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Dietze-Schroeder D, Sell H, Uhlig M,
Koener M and Eckel J: Autocrine action of adiponectin on human fat
cells prevents the release of insulin resistance-inducing factors.
Diabetes. 54:2003–2011. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Christiansen T, Richelsen B and Bruun JM:
Monocyte chemoattractant protein-1 is produced in isolated
adipocytes, associated with adiposity and reduced after weight loss
in morbid obese subjects. Int J Obes (Lond). 29:146–150. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Cancello R, Henegar C, Viguerie N, Taleb
S, Poitou C, Rouault C, Coupaye M, Pelloux V, Hugol D, Bouillot JL,
et al: Reduction of macrophage infiltration and chemoattractant
gene expression changes in white adipose tissue of morbidly obese
subjects after surgery-induced weight loss. Diabetes. 54:2277–2286.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Piemonti L, Calori G, Mercalli A, Lattuada
G, Monti P, Garancini MP, Constantio F, Ruotolo G, Luzi L and
Perseglin G: Fasting plasma leptin, tumor necrosis factor-alpha
receptor 2, and monocyte chemoattracting protein 1 concentration in
a population of glucose-tolerant and glucose-intolerant women:
Impact on cardiovascular mortality. Diabetes Care. 26:2883–2889.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Simeoni E, Hoffman MM, Winkelman BR, Ruiz
J, Fleury S, Boehm BO, März W and Vassalli G: Association between
the A-2518G polymorphism in the monocyte chemoattractant protein-1
gene and insulin resistance and type 2 diabetes mellitus.
Diabetologia. 47:1574–1580. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Herder C, Baumert J, Thorand B, Koenig W,
de Jager W, Meisinger C, Illig T, Martin S and Kolb H: Chemokines
as risk factors for type 2 diabetes: Results from the MONICA/KORA
Augsburg study, 1984–2002. Diabetologia. 49:921–929. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kim CS, Park HS, Kawada T, Kim JH, Lim D,
Hubbard NE, Kwon BS, Rrickson KL and Yu R: Circulating levels of
MCP-1 and IL-8 are elevated in human obese subjects and associated
with obesity-related parameters. Int J Obes (Lond). 30:1347–1355.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Shimobayashi M, Albert V, Woelnerhanssen
B, Frei IC, Weissenberger D, Meyer-Gerpach AC, Clement N, Moes S,
Colombi M, Meier JA, et al: Insulin resistance causes inflammation
in adipose tissue. J Clin Invest. 128:1538–1550. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhu B, Guo X, Xu H, Jiang B, Li H, Wang Y,
Yin O, Zhou T, Cai JJ, Glaser S, et al: Adipose tissue inflammation
and systemic insulin resistance in mice with diet-induced obesity
is possibly associated with disruption of PFKFB3 in hematopoietic
cells. Lab Invest. 101:328–340. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ylä-Herttuala S, Lipton A, Rosenfeld ME,
Särkioja T, Yoshimura T, Leonard EJ, Witztum JL and Steinberg D:
Expression of monocyte chemoattractant protein 1 in macrophage-rich
areas of human and rabbit atherosclerotic lesions. Proc Natl Acad
Sci USA. 88:5252–5256. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Arakelyan A, Petrkova J, Hermanova Z,
Boyajyan A, Lukl J and Petrek M: Serum levels of the MCP-1
chemokine in patients with ischemic stroke and myocardiac
infarction. Mediators Inflamm. 14:175–179. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sukumar D, Partridge C, Wang X and Shapses
SA: The high serum monocyte chemoattractant protein-1 in obesity is
influenced by high parathyroid hormone and not adiposity. J Clin
Endocrinol Metab. 296:1852–1858. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Takeya M, Yoshimura T, Leonard EJ and
Takahashi K: Detection of monocyte chemoattractant protein-1 in
human atherosclerotic lesions by an anti-monocyte chemoattractant
protein-1 monoclonal antibody. Hum Pathol. 24:534–539. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Gu L, Οkada Y, Clinton SK, Gerard C,
Sukhova GK, Libby P and Rollins BJ: Absence of monocyte
chemoattractant protein-1 reduces atherosclerosis in low density
lipoprotein receptor-defined mice. Mol Cell. 2:275–281. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Boring M, Gosling J, Cleary M and Charo
IF: Decreased lesion in CCR2-/- mice reveals a role for chemokines
in the initation of atherosclerosis. Nature. 394:894–897. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Deng Y and Scherer PE: Adipokines as novel
biomarkers and regulators of the metabolic syndrome. Ann N Y Acad
Sci. 1212:E1–E19. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Dutheil F, Gordon BA, Naughton G, Crendal
E, Courteix D, Chaplais E, Thivel D, Lac G and Benson AC:
Cardiovascular risk of adipokines: A review. J Inter Med Res.
46:2082–2095. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Szumilas K, Szumilas P,
Słuczanowska-Głąbowsk S, Zgutka K and Pawlik A: Role of adiponectin
in the pathogenesis of Rheumatoid arthritis. Int J Mol Sci.
21:82652020. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Adolph TE, Grander C, Grabherr F and Tilg
H: Adipokines and non-alcoholic fatty liver disease: Multiple
interactions. Int J Mol Sci. 18:16492017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Neumann UH, Chen S, Tam YY, Baker RK,
Covey SD, Dullis PP and Kieffer TJ: IGFBP2 is neither sufficient
nor necessary for the physiological actions of leptin on glucose
homeostasis in male ob/ob mice. Endocrinology. 155:716–725. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zorena K, Jachimowicz-Duda O, Ślęzak D,
Robakowska M and Mrugacz M: Adipokines in obesity. Potential lind
to metabolic disorders and chronic complications. Int J Mol Sci.
21:35702020. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Kumada M, Kihara S, Ouchi N, Kobayashi H,
Okamoto Y, Ohashi K, Maeda K, Nagaretani H, Kishida K, Maeda N, et
al: Adiponectin specifically increased tissue inhibitor of
metalloproteinase-1 through interleukin-10 expression in human
macrophages. Circulation. 109:2046–2049. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Quedraogo R, Wu X, Xu SQ, Fuchsel L,
Motoshima H, Mahadev K, Hough K, Scalia R and Goldstein BJ:
Adiponectin suppression of high-glucose-induced reactive oxygen
species in vascular endothelial cells: Evidence for involvement of
a cAMP signaling pathway. Diabetes. 55:1840–1846. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yuan F, Li YN, Liu YH, Yi B, Tian JW and
Liu FY: Adiponectin inhibits the generation of reactive oxygen
species induced by high glucose and promotes endothelial NO
synthase formation in human mesangial cells. Mol Med Rep.
6:449–453. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Furukawa S, Fujita T, Shimabukuro M, Iwaki
M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M and
Shimomura I: Increased oxidative stress in obesity and its impact
on metabolic syndrome. J Clin Invest. 114:1752–1761. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Castro JR, Grune T and Speckmann B: The
two faces of reactive oxygen species (ROS) in adipocyte function
and dysfunction. Biol Chem. 397:709–724. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Fujita K, Nishizawa H, Funahashi T,
Shimomura I and Shimabukuro M: Systemic oxidative stress is
associated with visceral fat accumulation and the metabolic
syndrome. Circulation. 70:1437–1442. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Kanazawa I, Yamaguchi T, Yano S, Yamauchi
M, Yamamoto M and Sugimoto T: Adiponectin and AMP kinase activator
stimulate proliferation, differentiation, and mineralization of
osteoblastic MC3T3-E1 cells. BMC Cell Biol. 8:512007. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Xie C and Chen Q: Adipokines: New
therapeutic target for osteoarthritis? Curr Reumatol Rep.
21:712020.
|
|
86
|
Gamberi Τ, Μagherini F, Modesti A and
Fiaschi T: Adiponectin signaling pathways in liver diseases.
Biomedicines. 6:522018. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Ouchi N, Kihara S, Arita Y, Nishida M,
Matsuyama A, Okamoto Y, Ishigami M, Kuriyama H, Kishida K,
Nishizawa H, et al: Adipocyte-derived plasma protein, adiponectin,
suppresses lipid accumulation and class A scavenger receptor
expression in human monocyte-derived macrophages. Circulation.
103:1057–1063. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Matsuda M, Shimomura I, Sata M, Arita Y,
Nishida M, Maeda N, Kumada M, Okamoto Y, Nagaretani H, Nishizawa H,
et al: Role of adiponectin in preventing vascular stenosis. The
missing link of adipo-vascular axis. J Biol Chem. 277:37487–37491.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Kadowaki T and Yamauchi T: Adiponectin and
adiponectin receptors. Endocr Rev. 26:439–451. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Zha N, Wu X and Gao P: Adiponectin and its
receptors in diabetic kidney disease: Molecular mechanisms and
clinical potential. Endocrinol. 158:2022–2034. 2017. View Article : Google Scholar
|
|
91
|
Alnaggar ARLR, Sayed M, El-Deena KE, Gomma
M and Hamed Y: Evaluation of serum adiponectin levels in diabetic
nephropathy. Diabetes Metab Syndr. 13:128–131. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Ouedraogo R, Gong Y, Berzins B, Wu X,
Mahadev K, Hough K, Chan L, Goldstein BJ and Scalia R: Adiponectin
deficiency increases leukocyte-endothelium interactions via
up-regulation of endothelial cell adhesion molecules in vivo. J
Clin Invest. 117:1718–1761. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Abella V, Scotece M, Conde J, López V,
Lazzaro V, Pino J, Gómez-Rein O and Gualillo O: Adipokines,
metabolic syndrome and rheumatic diseases. J Immunol Res.
2014:3437462014. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Stefan N and Stumvoll M: Adiponectin-its
role in metabolism and beyond. Horm Metab Res. 34:469–474. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Weyer C, Funahashi T, Tanaka S, Hotta K,
Matsuzawa Y, Pratley RE and Tataranni PA: Hypoadiponectinemia in
obesity and type 2 diabetes: Close association with insulin
resistance and hyperinsulinemia. J Clin Endocrinol Metab.
86:1930–1935. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Arita Y, Kihara S, Ouchi N, Takahashi M,
Maed K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, et
al: Paradoxical decrease of an adipose-specific protein,
adiponectin, in obesity. Biochem Biophys Res Commun. 257:79–83.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Scherer PE: Adipose tissue. From lipid
storage compartment to endocrine organ. Diabetes. 55:1537–1545.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Trujillo ME and Scherer PE: Adipose
tissue-derived factors: Impact on health and disease. Endocr Rev.
27:762–778. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Oh DK, Ciaraldi T and Henry RR:
Adiponectin in health and disease. Diabetes Obes Metab. 9:282–289.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Li S, Shin HJ, Ding EL and van Dam RM:
Adiponectin levels and risk of type 2 diabetes: A systematic review
and meta-analysis. JAMA. 302:179–188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Li C, Cheng H, Adhikari BK, Wang S, Yang
N, Liu W, Sun J and Wang Y: The role of apelin-APJ system in
diabetes and obesity. Front Endocrinol (Lausanne). 2022:132022.
|
|
102
|
Al-Mansoori L, Al-Jaber H, Price MS and
Elrayess MA: Role of inflammatory cytokines, growth factors and
adipokines in adipogenesis and insulin resistance. Inflammation.
45:31–44. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Vykoukal D and Davies MG: Vascular biology
of metabolic syndrome. J Vasc Surg. 54:819–831. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Than A, He HL, Chua SH, Xu D, Sun L, Leow
MKS and Chen P: Apelin enhances brown adipogenesis and browning of
white adipocytes. J Biol Chem. 290:1469–14691. 2015. View Article : Google Scholar
|
|
105
|
Yamamoto T, Habata Y, Matsumoto Y,
Yasuhara Y, Hashimoto T, Hamajyo H, Anayama H, Fujii R, Fuse H,
Shintani Y and Mori M: Apelin-transgenic mice exhibit a resistance
against diet-induced obesity by increasing vascular mass and
mitochondrial biogenesis in skeletal muscle. Biochim Biophys Acta.
1810:853–862. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Mughal A and O'Rourke ST: Vascular effects
on apelin: Mechanisms and therapeutic potential. Pharmacol Ther.
190:139–147. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Yamazaki S, Sekiguchi A, Uchiyama A,
Fujiwara C, Inoue Y, Yokoyama Y, Ogino S, Torii R, Hosoi M, Akai R,
et al: Apelin/APJ signaling suppresses the pressure ulcer formation
in cutaneous ischemia-perfusion injury mouse model. Sci Rep.
10:13492020. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Attané C, Foussal C, Gonidec SL, Benani A,
Daviaud D, Wanecq E, Guzmán-Ruiz R, Dray C, Bezaire V, Rancoule C,
et al: Apelin treatment increases complete fatty acid oxidation,
mitochondrial oxidative capacity and biogenesis in muscle of
insulin-resistant mice. Diabetes. 61:310–320. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Lay SL, Simard G, Martinez MC and
Andriantsitohaina R: Oxidative stress and metabolic pathologies:
From an adipocentric point of view. Oxid Med Cell Longev.
2014:9085392014.PubMed/NCBI
|
|
110
|
Kim S, Kim S, Hwang AR, Choi HC, Lee JY
and Woo CH: Apelin-13 inhibits methylglyoxal-induced unfolded
protein responses and endothelial dysfuction via regulating AMPK
pathway. Int J Mol Sci. 21:40692020. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Fibbi B, Marroncini G, Naldi L and Peri A:
The Yin and Yang effects of the apelinergic system in oxidative
stress. Int J Mol Sci. 24:47452023. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Zhang Y, Proenca R, Maffei M, Barone M,
Leopold L and Friedman JM: Positional cloning of the mouse obes
gene and its human homologue. Nature. 372:425–432. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Halaas JL, Gajiwala KS, Maffei M, Cohen
SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK and Friedman JM:
Weight-reducing effects of the plasma protein encoded by the obese
gene. Science. 269:543–546. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Caro JF, Sinha MK, Kolaczynski JW, Zhang
PL and Considine RV: Leptin: The tale of an obesity gene. Diabetes.
45:1455–1462. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Fantuzzi G and Faggioni R: Leptin in the
regulation of immunity, inflammation and haematopoiesis. J Leukoc
Biol. 68:437–446. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Münzberg H and Morrison CD: Structure,
production and signaling of leptin. Metabolism. 64:13–23. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Trayhurn P, Duncan JS, Hoggard N and
Rayner DV: Regulation of leptin production: A dominant role for the
sympathetic nervous system? Proc Nutr Soc. 57:413–419. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Dieguez C, Vazquez MJ, Romero A, Lopez M
and Nogueiras R: Hypothalamic control of lipid metabolism: Focus on
leptin, ghrelin and melanocortins. Neuroendocrinology. 94:1–11.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Morton GJ and Schwartz MW: Leptin and the
central nervous system control of glucose metabolism. Physiol Rev.
91:389–411. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Sahu A: Leptin signaling in the
hypothalamus: Emphasis on energy homeostasis and leptin resistance.
Front Neuroendocrinol. 24:225–253. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Blaszczak AM, Jalilvand A and Hsueh WA:
Adipocytes, innate immunity and obesity: A mini-review. Front
Immunol. 12:6507682021. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Obradovic M, Sudar-Milovanovic E, Soskic
S, Essack M, Arya S, Stewart AJ, Gojobori T and Isenovic ER: Leptin
and obesity: Role and clinical implication. Front Endocrinol
(Lausanne). 12:5858872021. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Fahed G, Aoun L, Zerdan MB, Allam S,
Zerdan MB, Bouferraa Y and Assi HI: Metabolic syndrome: Updates on
pathophysiology and management in 2021. Int J Mol Sci. 23:7862022.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
van den Hoek AM, Teusink B, Voshol PJ,
Havekes LM, Romijn JA and Piji H: Leptin deficiency per se dictates
body composition and insulin action in ob/ob mice. J
Neuroendocrinol. 20:120–127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Zhang H, Xie H, Zhao Q, Xie GQ, Wu XP,
Liao EY and Luo XH: Relationships between serum adiponectin,
apelin, leptin, resistin, visfatin levels and bone mineral density,
and bone biochemical markers in post-menopausal Chinese women. J
Endocrinol Invest. 33:707–711. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Aizawa-Abe M, Ogawa Y, Masuzaki H, Ebihara
K, Satoh N, Iwai H, Matsuoka N, Hayashi T, Hosoda K, Inoue G, et
al: Pathophysiological role of leptin in obesity-related
hypertension. J Clin Investig. 105:1243–1252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Ferri C, Desideri G, Valenti M, Bellini C,
Pasin M, Santucci A and De Mattia G: Early up-regulation of
endothelial adhesion molecules in obese hypertensive men.
Hypertension. 34:568–573. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Hukshorn CJ, Lindeman JH, Toet KH, Saris
WH, Eilers PH, Westerterp-Plantenga MS and Kooistra T: Leptin and
the proinflammatory state associated with human obesity. J Clin
Endocrinol Metab. 89:1773–1778. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Kim JE, Kim JS, Jo MJ, Cho E, Ahn SY, Kwon
YJ and Ko GJ: The roles and associated mechanisms of adipokines in
development of metabolic syndrome. Molecules. 27:3342022.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Romacho T, Valencia I, Ramos-González MR,
Vallejo S, López-Esteban M, Lorenzo O, Cannata P, Romero A,
Hipólito-Luengo AS, Gómez-Cerezo JF, et al: Visfatin/eNampt induces
endothelial dysfunction in vivo: A role for toll-like receptor 4
and NLRP3 inflammasome. Sci Rep. 10:53862020. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Toussirot E: Mini review: The contribution
of adipokines to joint inflammation in inflammatory rheumatic
diseases. Front Endocrinol (Lausanne). 11:6065602020. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Catalán V, Gómez-Ambrosi J, Rodríguez A,
Ramírez B, Silva C, Rotellar F, Cienfuegos JA, Salvador J and
Frühbeck G: Association of increased visfatin/PBEF/NAMPT
circulating concentrations and gene expression levels in peripheral
blood cells with lipid metabolism and fatty liver in human morbid
obesity. Nutr Metab Cardiovasc Dis. 21:245–253. 2011.PubMed/NCBI
|
|
133
|
Chang YH, Chang DM, Lin KC, Shin SJ and
Lee YJ: Visfatin in overweight/obesity, type 2 diabetes mellitus,
insulin resistance, metabolic syndrome and cardiovascular diseases:
A meta-analysis and systemic review. Diabetes Metab Res Rev.
27:515–527. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Martos-Moreno GA, Kratzch J, Korner A,
Barrios V, Hawkins F, Kiess W and Argente J: Serum visfatin and
vispin levels in prepubertal childres: Effect of obesity and weitht
loss after beharior modifications on their secretion and
relationship with glucose metabolism. Int J Obes (Lond).
35:1355–1362. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Olszanecka-Glinianowicz M, Kocełak P,
Nylec M, Chudek J and Zahorska-Markiewicz B: Circulating visfatin
level and visfatin/insulin ration in obese women with metabolic
syndrome. Arch Med Sci. 8:214–218. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
de Luis DA, Aller R, Sagrado MG, Conde R,
Izaola O and de la Fuente B: Serum visfatin levels and metabolic
syndrome criteria in obese female subjects. Diabetes Metab Res Rev.
29:576–581. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Friebe D, Neef M, Kratzch J, Erbs S,
Dittrich K, Garten A, Petzold-Qunque S, Blüher S, Reinehr T,
Stumvoll M, et al: Leucocytes are a major source of circulating
nicotinamide phorsphoribosyltransferase (NAMPT)/pre-B cell colony
(PBEF)/visfatin linking obesity and inflammation in humans.
Diabetologia. 54:1200–1211. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Kim SR, Bae YH, Bae SK, Choi KS, Yoon KH,
Koo TH, Jang HO, Yun Il, Kim KW, Kwon YG, et al: Visfatin enhances
ICAM-1 and VCAM-1 expression through ROS-dependent NF-κΒ activation
in endothelial cells. Biochim Biophys Acta. 1783:886–895. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Patel SD, Rajala MW, Rossetti L, Scherer
PE and Shapiro L: Disulfide-dependent multimeric assembly of
resistin family hormones. Science. 304:1154–1158. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Oki K, Yamane K, Kamei N, Nojima H and
Kohno N: Circulatin visfatin level is correlated with inflammation,
but not with insulin resistance. Clin Endocrinol (Oxf). 67:796–800.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Moschen AR, Kaser A, Enrich B, Mosheimer
B, Theurl M, Niederegger H and Tigl H: Visfatin an adipocytokine
with proinflammatory and immunomodulating properties. J Immunol.
178:1748–1758. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Krysiak R, Handzlik-Orlik G and Okopien B:
The role of adipokines in connective tissue diseases. Eur J Nutr.
51:513–528. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Heo YJ, Choi SE, Jeon JY, Han SJ, Kim DJ,
Kang Y, Lee KW and Kim HJ: Visfatin induces inflammation and
insulin resistance via the NF-κΒ and STAT3 signaling pathways in
hepatocytes. J Diabet Res. 2019:40216232019. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Francisco V, Sanz MJ, Real JT, Marques P,
Capuozzo M, Eldjoudi DA and Gualillo O: Adipokines in non-alcoholic
fatty liver disease: Are we on the road toward new biomarkers and
therapeutic targets? Biology (Basel). 11:12372022.PubMed/NCBI
|
|
145
|
Oita RC, Ferdinando D, Wilson S, Bunce C
and Mazzatti DJ: Visfatin induces oxidative stress in
differentiated C2C12 myotubes in an Akt- and MAPK-independent,
NFκΒ-dependent manner. Pflugers Arch. 459:619–630. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Lee S, Lee HC, Kwon YW, Lee SE, Cho Y, Kim
J, Lee S, Kim JY, Lee J, Yang HM, et al: Adenylyl
cyclase-associated protein 1 (CAP1) is a receptor for human
resistin and mediated inflammatory actions of human monocytes. Cell
Metab. 19:484–497. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Li Y, Yang Q, Cai D, Guo H, Fang J, Cui H,
Gou L, Deng J, Wang Z and Zuo Z: Resistin, a novel host defence
peptide of innate immunity. Front Immunol. 12:6998072021.
View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Kawanami D, Maemura K, Takeda N, Harada T,
Nojiri T, Imai Y, Manabe I, Utsunomiya K and Nagai R: Direct
reciprocal effects of resistin and adiponectin on vascular
endothelial cells: A new insight into adipocytokine-endothelial
cell interactions. Biochem Biophys Res Commun. 314:415–419. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Bokarewa M, Nagaev I, Dahlberg L, Smith U
and Tarkowski A: Resistin, an adipokine with potent proimflammatory
properties. J Immunol. 174:5789–5795. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Agaev I, Bokarewa M, Tarkowski A and Smith
U: Human resistin is a systemic immune-derived proinflammatory
cytokine targeting both leukocytes and adipocytes. PLoS One.
1:e312006. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Ebihara T, Matsumoto H, Matsubara T,
Matsuura H, Hirose T, Shimizu K, Ogura H, Kang S, Tanaka T and
Shimazu T: Adipocytokine profile reveals resistin forming a
prognostic-related cytokine network in the acute phase of sepsis.
Shock. 56:718–726. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Heilbronn LK, Rood J, Janderova L, Albu
JB, Kelley DE, Ravussin E and Smith SR: Relationship between serum
resistin concentrations and insulin resistance in nonobese, obese,
and obese diabetic subjects. J Clin Endocrinol Metabol.
89:1844–1848. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Steppan CM, Bailey ST, Bhat S, Brown EJ,
Banerjee RR, Wright CM, Patel HR, Ahima RS and Lazar MA: The
hormone resistin links obesity to diabetes. Nature. 409:307–312.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Vidal-Puig A and O'Rahilly S: Resistin: A
new link between obesity and insulin resistance? Clin Endocrinol
(Oxf). 55:437–438. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
McTernan CL, McTernan PG, Harte AL, Levick
PL, Barnet AH and Kumar S: Resistin, central obesity, and type 2
diabetes. Lancet. 359:46–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Wang H, Chu WS, Hemphill C and Elbein SC:
Hunan resistin gene: Molecular scanning and evaluation of
association with insulin sensitivity and type 2 diabetes in
Caucasians. J Clin Endocrinol Metabol. 87:2520–2524. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Osawa H, Yamada K, Onuma H, Murakami A,
Ochi M, Kawata H, Nishimiya T, Niiya T, Shimizu I, Nishida W, et
al: The G/G genotype of resistin single-nucleotide polymorphism at
−420 increases type 2 diabetes mellitus susceptibility by inducing
promoter activity through specific binding of Sp1/3. Am J Hum
Genet. 75:678–686. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
158
|
Kielstein JT, Becker B, Graf S, Brabant G,
Haller H and Fliser D: Increased resistin blood levels are not
associated with insulin resistance in patients with renal disease.
Am J Kidney Dis. 42:62–66. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Patel L, Buckels AC, Kinghorn IJ, Mourdock
PR, Holbrook JD, Plumpton C, Macphee CH and Smith SA: Resistin is
expressed in human macrophages and directly regulated by PPAR gamma
activators. Biochem Biophys Res Commun. 300:472–476. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Chen C, Jiang J, Lü JM, Chai H, Wang X,
Lin PH and Yao Q: Resistin descreases expression of endothelial
nitric oxide synthase through oxidative stress in human coronary
artery endothelial cells. Am J Physiol Heart Circ Physiol.
299:H193–H201. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Acquarone E, Monacelli F, Borghi R,
Nencioni A and Odetti P: Resistin: A reappraisal. Mech Ageing Dev.
178:46–63. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Iwaki T, Urano T and Umemura K: PAI-1,
progress in understanding the clinical problem and its aetiology.
Br J Heamatol. 157:291–298. 2012. View Article : Google Scholar
|
|
163
|
Para I, Albu A and Porojan MD: Adipokines
and arterial stiffness in obesity. Medicina (Kaunas). 57:6532021.
View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Mertens I and Van Gaal LF: Obesity,
haemostasis and the fibrinolytic system. Obes Rev. 3:85–101. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Juhan-Vague I, Alessi MC, Mavri A and
Morange PE: Plasminogen activator inhibitor-1, inflammation,
obesity, insulin resistance and vascular risk. J Thromb Haemost.
1:1575–1579. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Tschoner A, Sturm W, Engl J, Kaser S,
Laimer M, Laimer E, Klaus A, Patsch JR and Ebenbichler CF:
Plasminogen activator inhibitor 1 and visceral obesity during
pronounced weight loss after bariatric surgery. Nutr Metab
Cardiovasc Dis. 22:340–346. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Khoukaz HB, Ji Y, Braet DJ, Vadali M,
Abdelhamid AA, Emal CD, Lawrence DA and Fay WP: Drug targeting of
plasminogen activator inhibitor-1 inhibits metabolic dysfunction
and atherosclerosis in murine model of metabolic syndrome.
Arterioscler Thromb Vasc Biol. 40:1479–1490. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Eitzman DT, Westrick RJ, Xu Z, Tyson J and
Grinsburg D: Plasminogen activator inhibitor-1 deficiency protects
against atherosclerosis progression in the mouse carotid artery.
Blood. 96:4212–4215. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Alessi MC and Juhan-Vague I: PAI-1 and the
metabolic syndrome: Links, causes and consequences. Arterioscler
Thromb Vasc Biol. 16:2200–2207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Ma LJ, Mao SL, Taylor KL, Kanjanabuch T,
Guan Y, Zhang Y, Brown NJ, Swift LL, McGuinness OP, Wasserman DH,
et al: Prevention of obesity and insulin resistance in mice lacking
plasminogen activator inhibitor 1. Diabetes. 53:336–346. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Gleeson LE, Sheedy FJ, Palsson-McDermott
EM, Triglia D, O'Leary SM, O'Sullivan MP, O'Neill LA and Keane J:
Cytting edge: Mycobacterium tuberculosis induces aerobic glycolysis
in human alveolar macrophages that is required for control of
intracellular bacillary replication. J Immunol. 196:2444–2449.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Shoelson SE, Herrero L and Naaz A:
Obesity, inflammation and insulin resistance. Gastroenterology.
132:2169–2180. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Stojsavljević S, Palčić MG, Jukić LV,
Duvnjak LS and Duvnjak M: Adipokines and proinflammatory cytokines,
the key mediators in the pathogenesis of nonalcoholic fatty liver
disease. World J Gastroenterol. 20:18070–18091. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Plomgaard P, Bouzakri K, Krogh-Madsen R,
Mittendorfer B, Zierath JR and Pedersen BK: Tumor necrosis
factor-alpha induces skeletal muscle insulin resistance in healthy
human subjects via inhibition of Akt substrate 160 phosphorylation.
Diabetes. 54:2936–2945. 2005. View Article : Google Scholar
|
|
175
|
Ruan H and Lodish HF: Insulin resistance
in adipose tissue: Direct and indirect effects of tumor necrosis
factor-α. Cytokine Growth Factor Rev. 14:447–455. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Illei GG and Lipsky PE: Novel,
antigen-specific therapeutic approaches to autoimmuneinflammatory
diseses. Curr Opin Immunol. 12:712–718. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Chandel NS, Schumacker PT and Arch RH:
Reactive oxygen species are downstream products of TRAF-mediated
signal trasduction. J Biol Chem. 276:42728–42736. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Micheau O and Tschopp J: Induction of TNF
receptor I-mediated apoptosis via two sequential signaling
complexes. Cell. 114:181–190. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Campbell J, Ciesielski CJ, Hunt AE,
Horwood NJ, Beech JT, Hayes LA, Denys A, Feldmann M, Brennan FM and
Foxwell BMJ: A novel mechanism for TNF-alpha regulation by p38
MAPK: Involvement of NF-kappa B with implications for therapy in
rheumatoid arthritis. J Immunol. 173:6928–6937. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Wang B and Trayhurn P: Acute and prolonged
effects of TNF-alpha on the expression and secretion of
inflammation-related adipokines by human adipocytes differentiated
in culture. Pflugers Arch. 452:418–427. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Rider P, Carmi Y, Guttman O, Braiman A,
Cohen I, Voronov E, White MR, Dinarello CA and Apte RN: IL-1α and
IL-1β recruit different myeloid cells and promote different stages
of sterile inflammation. J Immunol. 187:4835–4843. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Thornberry NA, Bull HG, Calaycay JR,
Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner
JR and Aunins J: A novel heterodimeric cysteine protease is
required for interleukin-1 beta processing in monocytes. Nature.
356:768–774. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Akdis M, Arab A, Altunbulakli C, Azkur K,
Costa RA, Crameri R, Duan S, Eiwegger T, Eljaszewicz A, Ferstl R,
et al: Interleukins (from IL-1 to IL-38), interferons, transforming
growth factor β, and TNF-α: Receptors, functions, and roles in
diseases. J Allergy Clin Immunol. 138:984–1010. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Ghabari M, Maragheh SM, Aghazadeh A,
Mehrjuyan SR, Hussen BM, Shadbad MA, Dastmalchi N and Safaralizadeh
R: Interleukin-1 in obesity-related low-grade inflammation: From
molecular mechanisms to therapeutic strategies. Int
Immunopharmacol. 96:1077652021. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Speaker KJ and Fleshner M: Interleukin-1
beta: A potential link between stress and the development of
visceral obesity. BMC Physiol. 12:1–15. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Bruun JM, Pedersen SB, Kristensen K and
Richelsen B: Effects of pro-inflammatory cytokines and chemokines
on leptin production in human adipose tissue in vitro. Mol Cell
Endocrinol. 190:91–99. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Gonzalez RR and Leavis P: Leptin
upregulates β3-integrin expression and interleukin-1β upregulates
leptin and leptin receptor expression in human endometrial
epithelial cell cultures. Endocrine. 16:21–28. 2021. View Article : Google Scholar
|
|
188
|
Müller G, Ertl J, Gerl M and Preidisch G:
Leptin impairs metabolic actions of insulin in isolated rat
adipocytes. J Biol Chem. 272:10585–10593. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Moschen AR, Molnar C, Enrich B, Geiger S,
Ebenbichler CF and Tilg H: Adipose and liver expression of
interleukin (IL)-1 family members in morbid obesity and effects of
weight loss. Mol Med. 17:840–845. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Shaul ME, Bennett G, Strissel KJ,
Greenberg AS and Obin MS: Dynamic, M2-like remodeling phenotypes of
CD11c+ adipose tissue macrophages during high-fat diet-induced
obesity in mice. Diabetes. 59:1171–1181. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Schoettl T, Fischer IP and Ussar S:
Heterogeneity of adipose tissue in development and metabolic
function. J Exp Biol. 221 (Pt Suppl 1):jeb1629582018. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Buechler C, Krautbauer S and Eisinger K:
Adipose tissue fibrosis. World J Diabetes. 6:548–553. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Shikama Y, Aki N, Hata A, Nishimura M,
Oyadomari S and Funaki M: Palmitate-stimulated monocytes induce
adhesion molecule expression in endothelial cells via IL-1
signaling pathway. J Cell Physiol. 230:732–742. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Miura K, Kodama Y, Inokuchi S, Scnabl B,
Aoyama T, Ohnishi H, Olefsky JM, Brenner DA and Seki E: Toll-like
receptor 9 promotes steatohepatitis by induction of
interleukin-1beta in mice. Gastroenterology. 139:323–334. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Gao D, Madi M, Ding C, Fok M, Steele T,
Ford C, Hunter L and Bing C: Interleukin-1β mediates
macrophage-induced impairemnet of insulin signalin in human primary
adipocytes. Am J Physiol Endocrinol Metab. 307:E289–E304. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Zhou R, Tardivel A, Thorens B, Choi I and
Tschopp J: Thioredoxin-interacting protein links oxidative stress
to inflammasome activation. Nat Immunol. 11:136–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Calabrese L, Fiocco Z, Satoh TK, Peris K
and French LE: Therapeutic potential of targeting interleukin-1
family cytokines in chronic inflammatory skin diseases. Br J
Dermatol. 186:925–941. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Maedler K, Sergeev P, Ris F, Oberholzer J,
Holler-Jemelka H, Spinas GA, Kaiser N, Halban PA and Donath MY:
Glucose-induced beta cell production of IL-1beta contributes to
glucotoxicity in human pancreatic islets. J Clin Invest.
110:851–860. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Wang Q, Zhang H, Zhao B and Fei H:
IL-1beta caused pancreatic beta-cells apoptosis is mediated in part
by endoplasmic reticulum stress via the induction of endoplasmic
reticulum Ca2+ release through the c-Jun N-terminal kinase pathway.
Mol Cell Biochem. 324:183–190. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Brichory FM, Misek DE, Yim AM, Krause MC,
Giordano TJ, Beer DG and Hanash SM: An immune response manifested
by the common occurrence of annexins I and II aytoantibodies and
high circulating levels of IL-6 in lung cancer. Proc Natl Acad Sci
USA. 98:9824–9829. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Scheller J, Chalaris A, Schmidt-Arras D
and Rose-John S: The pro- and anti-inflammatory properties of the
cytokine interleukin-6. Biochim Biophys Acta. 1813:878–888. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Tanaka T, Narazaki M and Kishimoto T: IL-6
in inflammation, immunity and disease. Cold Spring Harb Perspect
Biol. 6:a0162952014. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
IL6R Genetics Consortium Emerging Risk
Factors and Collaboration, . Sarwar N, Butterworth AS, Freitag DF,
Gregson J, Willeit P, Gorman DN, Gao P, Saleheen D, Rendon A, et
al: Interleukin-6 receptor pathways in coronary heart disease: A
collaborative meta-analysis of 82 studies. Lancet. 379:1205–1213.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Elhage R, Clamens S, Besnard S, Mallat Z,
Tedgui A, Arnal J, Maret A and Bayard F: Involvement of
interleukin-6 in atherosclerosis but not in the prevention of fatty
streak formation by 17beta-estradiol in apolipoprotein E-deficient
mice. Atherosclerosis. 156:315–320. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Schieffer B, Selle T, Hilfiker A,
Hilfiker-Kleiner D, Grote K, Tietge UJF, Trautwein C, Luchtefeld M,
Schmittkamp C, Heeneman S, et al: Impact of interleukin-6 on plaque
development and morphology in experimental atherosclerosis.
Circulation. 110:3493–3500. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Mishra D, Richard JE, Maric I, Porteiro B,
Häring M, Kooijman S, Musovic S, Eerola K, López-Ferreras L, Peris
E, et al: Parabrachial interleukin-6 reduces body weight and food
intake and increases thermogenesis to regulate energy metabolism.
Cell Rep. 26:3011–3026. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
Rosen ED and Spiegelman BM: What we talk
about when we talk about fat. Cell. 156:20–44. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Mohamed-Ali V, Goodrick S, Rawesh A, Katz
DR, Miles JM, Yudkin JS, Klein S and Coppack SW: Subcutaneous
adipose tissue related interleukin-6, but not tumor necrosis
factor-alpha, in vivo. J Clin Endocrinol Metab. 82:4196–4200. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Sopasakis VR, Sandqvist M, Gustafson B,
Hammerstedt A, Schmelz M, Yang X, Jansson PA and Smith U: High
local concentrations and effects on differentiation implicate
interleukin-6 as a paracrine regulator. Obes Res. 12:454–460. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Fernandez-Real JM and Ricart W: Insulin
resistance and chronic cardiovascular inflammatory sundrome. Endocr
Rev. 24:278–301. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Charles BA, Doumatey A, Huang H, Zhou J,
Chen G, Shriner D, Adeyemo A and Rotimi CN: The roles of IL-6,
IL-10 and IL-1RA in obesity and insulin resistance in
African-Americans. J Clin Endocrinol Metab. 96:E2018–E2022. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Tsigos C, Papanicolaou DA, Kyrou I,
Defensor R, Mitsiadis CS and Chrousos GP: Dose-dependent effects of
recombinant human interleukin-6 on glucose regulation. J Clin
Endocrinol Metab. 82:4167–4170. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Pradhan AD, Manson JE, Rifai N, Buring JE
and Ridker PM: C-reactive protein, interleukin 6, and risk of
developing type 2 diabetes mellitus. JAMA. 286:327–334. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Bastard JP, Maachi M, van Nhieu JT, Jardel
C, Bruckert E, Grimaldi A, Robert JJ, Capeau J and Hainque B:
Adipose tissue IL-6 content correlates with resistance to insulin
activation of glucose uptake both in vivo and in vitro. J Clin
Endocrinol Metab. 87:2084–2089. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Kopp HP, Kopp CW, Festa A, Krzyzanowaska
K, Kriwanek S, Minar E, Roka R and Schernthaner G: Impact of weight
loss on inflammatory proteins and their association with the
insulin resistance syndrome in morbidly obese patients.
Arterioscler Throm Vasc Biol. 23:1042–1047. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Xu E, Pereira MMA, Karakasilioti I,
Theurich S, Al-Maarri M, Rappl G, Waisman A, Wenderlich FT and
Brüning JC: Temporal and tissue-specific requirements for
T-lympocyte IL-6 signaling in obesity-associated inflammation and
insulin resistance. Nat Commun. 8:148032017. View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Wondmkum YT: Obesity, insulin resistance,
and type 2 diabetes: Associations and therapeutic implications.
Diabetes Metab Syndr Obes. 13:3611–3616. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Fu Z, Gilbert ER and Liu D: Regulation of
insulin synthesis and secretion and pancreatic Beta-cell
dysfunction in diabetes. Curr Diabetes Rev. 9:25–53. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Röder PV, Wu B, Liu Y and Han W:
Pancreatic regulation of glucose homeostasis. Exp Mol Med.
48:e2192016. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Fazakerley DJ, Krycer JR, Kearney AL,
Hocking SL and James DE: Muscle and adipose tissue insulin
resistance: Malady without mechanism? J Lipid Res. 60:1720–1732.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Newsholme P and Krause M: Nutritional
regulation of insulin secretion: Implications for diabetes. Clin
Biochem Rev. 33:35–47. 2012.PubMed/NCBI
|
|
222
|
Dashty M: A quick look at biochemistry:
Carbohydrate metabolism. Clin Biochem. 46:1339–1352. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Samuel VT and Shulman GI: The pathogenesis
of insulin resistance: Intergrating signaling pathways and
substrate flux. J Clin Invest. 126:12–22. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Taniguchi CM, Emanuelli B and Kahn CR:
Critical nodes in signaling pathways: Insights into insulin action.
Nat Rev Mol Cell Biol. 7:85–96. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Merry TL, Hedges CP, Masson SW, Laube B,
Pöhlmann D, Wueest S, Walsh ME, Arnold M, Langhans W, Konrad D, et
al: Partial impairment of insulin receptor expression mimics
fasting to prevent diet-induced fatty liver diseasee. Nat Commun.
11:20802020. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Shanik MH, Xu Y, Skrha J, Dankner R, Zick
Y and Roth J: Insulin resistance and hyperinsulinemia: Is
hyperinsulinemia the cart or the horse? Diabetes Car. 31 (Suppl
2):S262–S268. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Braccini L, Ciraolo E, Campa CC, Perino A,
Longo DL, Tibolla G, Pregnolato M, Cao Y, Tassone B, Damilano F, et
al: PI3K-C2γ is a Rab5 effector selectively controlling endosomal
Akt2 activation downstream of insulin signalling. Nat Commun.
6:74002015. View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Huang X, Liu G, Guo J and Su Z: The
PI3K/AKT pathway in obesity and type 2 diabetes. Int J Biol Sci.
14:1483–1496. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Gray SL, Donald C, Jetha A, Covey SD and
Kieffer TJ: Hyperinsulinemia precedes insulin resistance in mice
lacking pancreatic beta-cell leptin signaling. Endocrinology.
151:4178–4186. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Wild S, Roglic G, Green A, Sicree R and
King H: Global prevalence of diabetes: Estimates for the year 2000
and projections for 2030. Diabetes Care. 27:1047–1053. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Smith BW and Adams LA: Nonalcoholic faty
liver disease and diabetes mellitus: Pathogenesis and treatment.
Nat Rev Endocrinol. 7:456–465. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
232
|
Willians AJK and Long AE: Following the
fate of the failing β-cell: New insights from first-phase insulin
responses. Diabetes. 62:3990–3992. 2013. View Article : Google Scholar
|
|
233
|
Gariani K, Philippe J and Jornayvaz FR:
Non-alcoholic fatty liver disease and insulin resistance: From
bench to bedside. Diabetes Metab. 39:16–26. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Zhao J, Wu Y, Rong X, Zheng C and Guo J:
Anti-lipolysis induced by insulin in diverse pathophysiologic
conditions of adipose tissue. Diabetes Metab Syndr Obes.
13:1575–1585. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
235
|
Griffin ME, Marcucci MJ, Cline GW, Bell K,
Barucci N, Lee D, Goodyear LJ, Kraegen EW, White MF and Shulman GI:
Free fatty acid-induced insulin resistance is associated with
activation of protein kinase C theta and alterations in the insulin
signaling cascade. Diabetes. 48:1270–1274. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
236
|
Boden G and Shulman GI: Free fatty acids
in obesity and type 2 diabetes: Defining their role in the
development of insulin resistance and beta-cell dysfunction. Eur J
Clin Invest. 32:14–23. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
237
|
Unger RH and Zhou YT: Lipotoxicity of
beta-cells in obesity and in other causes of fatty acid spillover.
Diabetes. 50 (Suppl 1):S118–S121. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
238
|
Bugianesi E, Gastaldelli A, Vanni E,
Gambino R, Cassader M, Baldi S, Ponti V, Pagano G, Ferranini E and
Rizzetto M: Insulin resistance in non-diabetic patients with
non-alcoholic fatty liver disease: Sites and mechanisms.
Diabeltologia. 48:634–642. 2005. View Article : Google Scholar
|
|
239
|
Perry RJ, Samuel VT, Petersen KF and
Shulman GI: The role of hepatic insulin resistance and type 2
diabetes. Nature. 510:84–91. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
240
|
Brown MS and Goldstein JL: Selective
versus total insulin resistance: A pathogenic paradox. Cell Metab.
7:95–96. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
241
|
Kim GT, Kim SJ, Park SH, Lee D and Park
TS: Hepatic expression of the serine palmitoyltansferase subunit
Sptlc2 reduces lipid droplets in the liver by activating VLDL
secretion. J Lipid Atheroscler. 9:291–303. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
242
|
Hall JE, da Silva AA, do Carmo JM,
Dubinion J, Hamza S, Munusamy S, Smith G and Stec DE:
Obesity-induced hypertension: Role of sympathetic nervous system,
leptin, and melanocortins. J Biol Chem. 285:17271–17276. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
243
|
Weickert MO and Pfeiffer AFH: Signalling
mechanisms linking hepatic glucose and lipid metabolism.
Diabetologia. 4:1732–1741. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
244
|
Murakami T, Michelagnoli S, Longhi R,
Gianfranceschi G, Pazzucconi F, Calabresi L, Sirtori CR and
Franceschini G: Triglycerides are major determinants of cholesterol
esterification/transfer and HDL remodeling in human plasma.
Arterioscler Thromb Vasc Biol. 15:1819–1828. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
245
|
Eisenberg S, Gavish D, Oschry Y, Fainaru M
and Deckelbaum RJ: Abnormalities in very low, low and high density
lipoproteins in hypertriglyceridemia. Reversal toward normal with
bezafibrate treatment. J Clin Invest. 74:470–482. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
246
|
Tripathy D, Mohanty P, Dhindsa S, Syed T,
Ghanim H, Aljada A and Dandona P: Elevation of free fatty acids
induces inflammation and impairs vascular reactivity in healthy
subjects. Diabetes. 52:2882–2887. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
247
|
Esler M, Rumantir M, Wiesner G, Kaye D,
Hastings J and Lambert G: Sympathetic nervous system and insulin
resistance from obesity to diabetes. Am J Hypertens. 14((11 Pt 2)):
304S–309S. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
248
|
Samad F and Ruf W: Inflammation, obesity
and thrombosis. Blood. 122:3415–3422. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
249
|
Ernst E and Resch KL: Fibrinogen as a
cardiovascular risk factor: A meta-analysis and review of the
literature. Ann Intern Med. 118:956–963. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
250
|
Kannel WB, Wolf PA, Castelli WP and
D'Agostino RB: Fibrionogen and risk of cardiovascular disease. The
Framingham study. JAMA. 258:1183–1186. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
251
|
Nieuwdorp M, Stroes ES, Meijers JC and
Buller H: Hypercoagulability in the metabolic syndrome. Curr Opin
Pharmacol. 5:155–159. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
252
|
Raynaud E, Perez-Martin A, Brun JF,
Aïssa-Benhaddad A, Fédou C and Mercier J: Atherosclerosis.
150:365–370. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
253
|
Tabrez S, Jabir NR, Shakil S and Alama MN:
Association of plasma fibrinogen level with insulin resistance in
angiographically confirmed coronary artery disease patients. Crit
Rev Eukaryot Gene Expr. 29:277–285. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
254
|
Bryk-Wiązania AH and Undas A:
Hypofribrinolysis in type 2 diabetes and its clinical implications:
From mechanisms to pharmacological modulation. Cardiovasc Diabetol.
20:1912021. View Article : Google Scholar : PubMed/NCBI
|
|
255
|
Davalos D and Akassoglou K: Fibrinogen as
a key regulator of inflammation in disease. Semin Immunopathol.
34:43–62. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
256
|
Nawaz SS and Siddiqui K: Plasminogen
activator inhibitor-1 mediated downregulation of adiponectin in
type 2 diabetic patients with metabolic syndrome. Cytokine X.
4:1000642002. View Article : Google Scholar : PubMed/NCBI
|
|
257
|
Chen R, Yan J, Liu P, Wang Z and Wang C:
Plasminogen activator inhibitor links obesity and thrombotic
cerebrovascular diseases: The roles of PAI-1 and obesity on stroke.
Metab Brain Dis. 32:667–673. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
258
|
Matsuzawa Y: The metabolic syndrome and
adipocytokines. FEBS Lett. 580:2917–2921. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
259
|
Mertens I, Ballaux D, Funahashi T,
Matsuzawa Y, Van der Planken M, Verrijken A, Ruge JB and Gaal LFV:
Inverse relationship between plasminogen activator inhibitor-I
activity and adiponectin in overweight and obese women.
Interrelationship with visceral adipose tissue, insulin resistance,
HDL-chol and inflammation. Thromb Haemost. 94:1190–1195. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
260
|
Shimomura I, Funahashi T, Takahashi M,
Maeda K, Kotani K, Nakamura T, Yamashita S, Miura N, Fukuda Y,
Takemura K, et al: Enhanced expression of PAI-1 in visceral fat:
Possible contributor to vascular disease in obesity. Nat Med.
2:800–803. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
261
|
Kaji H: Adipose tissue-derived plasminogen
activator inhibitor-1 function and regulation. Compr Physiol.
6:1873–1896. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
262
|
Reaven GM: Banting lecture 1988. Role of
insulin resistance in human disease. Diabetes. 37:1595–1607. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
263
|
Alberti KG, Zimmet P and Shaw J; IDF
Edipemiology Task Force Consensus Group, : The metabolic syndrome-a
new world-wide definition. Lancet. 366:1059–1062. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
264
|
Zafar U, Khaliq S, Ahmad HU, Manzoor S and
Lone KP: Metabolic syndrome: An update on diagnostic criteria,
pathogenesis, and genetic links. Hormones (Athens). 17:299–313.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
265
|
Spiegelman BM and Flier JS: Obesity and
the regulation of energy balance. Cell. 104:531–543. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
266
|
Mottilo S, Filion KB, Genest J, Joseph L,
Pilote L, Poirier P, Rinfret S, Schiffrin EL and Eisenberg MJ: The
metabolic syndrome and cardiovascular risk a systematic review and
meta-analysis. J Am Coll Cardiol. 56:1113–1132. 2010. View Article : Google Scholar
|
|
267
|
Weiss R, Bremer AA and Lusting RH: What is
metabolic syndrome, and why are children getting it? Ann N Y Acad
Sci. 1281:123–140. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
268
|
Grundy SM: Metabolic syndrome pandemic.
Arteroscler Thromb Vasc Biol. 28:629–636. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
269
|
Ford ES, Li C and Zhao G: Prevalence and
correlates of metabolic syndrome based on a harmonious definition
among adults in the US. J Diabetes. 2:180–193. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
270
|
Kahn R, Buse J, Ferrannini E and Stern M:
The metabolic syndrome: Time for a critical appraisal. Joint
statement from the Americal diabetes association and the European
association for the study of diabetes. Diabetologia. 48:1684–1699.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
271
|
Pi-Sunyer X: The metabolic syndrome: How
to approach differing definitions. Med Clin North Am. 91:1025–1040.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
272
|
Chung G, Jung HS and Kim HJ:
Sociodemographic and health characteristics associated with
metabolic syndrome in men and women aged ≥50 Years. Metab Sundr
Relat Disord. 19:159–166. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
273
|
Hydrie MZ, Shera AS, Fawwad A, Basit A and
Hussain A: Prevalence of metabolic syndrome in urban Pakistan
(Karachi): Comparison of newly proposed international diabetes
federation and modified adult treatment panel III criteria. Metab
Syndr Relat Disord. 7:119–124. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
274
|
Rosenbaum M, Sy M, Pavlovich K, Leibel RL
and Hirsch J: Leptin reverses weight loss-induced changes in
regional neural activity responses to visual food stimuli. J Clin
Invest. 118:2583–2591. 2008.PubMed/NCBI
|
|
275
|
Imai SI: Nicotinamide
phosphoribosyltrasferase (Nampt): A link between NED biology,
metabolism, and disease. Curr Pharm Des. 15:20–28. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
276
|
Lago F, Dieguez C, Gomez-Reino G and
Gulillo O: Adipokines as emerging mediators of immune response and
inflammation. Nat Clin Pract Rheumatol. 3:716–724. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
277
|
Hopps E, Noto D, Caimi G and Averna MR: A
novel comoponent of the metabolic syndrome: The oxidative stress.
Nutr Metab Cardiovasc Dis. 20:72–77. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
278
|
Vona R, Gambardella L, Cittadini C,
Straface E and Pietraforte D: Biomarkers of oxidative stress in
metabolic syndrome and associated dieseases. Oxid Med Cell Longev.
2019:82672342019. View Article : Google Scholar : PubMed/NCBI
|
|
279
|
Schieber M and Chandel NS: ROS function in
redox signaling and oxidative stress. Curr Biol. 24:R453–R462.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
280
|
Juan CA, de la Lastra JM, Plou FJ and
Pérez-Lebeña EP: The chemistry of reactive oxygen species (ROS)
revisited: Outlining their role in biological macromolecules (DNA,
lipids and proteins) and induced pathologies. Int J Mol Sci.
22:46422021. View Article : Google Scholar : PubMed/NCBI
|
|
281
|
Grattagliano I, Palmieri VO, Portincasa P,
Moschetta A and Palasciano G: Oxidative stress-induced risk factors
associated with the metabolic syndrome: A unifying hypothesis. J
Nutr Biochem. 19:491–504. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
282
|
Fernández-Sánchez A, Madrigal-Santillán E,
Bautista M, Esquivel-Soto J, Morales-González A, Esquivel-Chirino
C, Durante-Montiel I, Sánchez-Rivera G, Valadez-Vega C and
Morales-González JA: Inflammation, oxidative stress, and obesity.
Int J Mol Sci. 12:3117–3132. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
283
|
Sena CM, Leadro A, Azul L, Seiça R and
Perry G: Vascular oxidative stress: Impact and therapeutic
approaches. Front Physiol. 9:16682018. View Article : Google Scholar : PubMed/NCBI
|
|
284
|
Smirne C, Croce E, Di Benedetoo D,
Cantaluppi V, Comi C, Sainaghi PP, Minisini R, Grossini E and
Pirisi M: Oxidative stress in non-alchoholic fatty liver disease.
Livers. 2:30–76. 2022. View Article : Google Scholar
|
|
285
|
Field AE, Coakley EH, Must A, Spadano JL,
Laird N, Dietz WH, Rimm E and Golditz GA: Imact of overweight on
the risk of developing common chronic diseases during a 10-year
period. Arch Intern Med. 161:1581–1586. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
286
|
Rother KI: Diabetes treatment-Bridging the
devide. N Engl J Med. 356:1499–1501. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
287
|
Norhammar A and Schenck-Gustafsson K: Type
2 diabetes and cardiovascular disease in women. Diabetologia.
56:1–9. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
288
|
Chan JM, Rimm EB, Colditz GA, Stampfer MJ
and Willett WC: Obesity, fat distribution, and weight gain as risk
factors for clinical diabetes in men. Diabetes Care. 17:961–969.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
289
|
Colditz GA, Willett WC, Rotnitzky A and
Manson JE: Weight gain as a risk factor for clinical diabetes
mellitus in women. Ann Intern Med. 122:481–486. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
290
|
Wannamethee SG and Shaper AG: Weight
change and duration of overweight and obesity in the incidence of
type 2 diabetes. Diabetes Care. 22:1266–1272. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
291
|
Schienkiewitz A, Schulz MB, Hoffmann K,
Kroke A and Boeing H: Body mass index history and risk of type 2
diabetes: Results from the European Prospective Investigation into
cancer nutrition (EPIC)-Potsdam study. Am J Clin Nutr. 84:427–433.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
292
|
DeFronzo RA: Pathogenesis of type 2
diabetes mellitus. Med Clin North Am. 88:787–835. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
293
|
Ferrannini E, Gastaldelli A, Miyazaki Y,
Matsuda M, Mari A and DeFronzo RA: Beta-cell function in subjects
spanning the range from normal glucose tolerance to overt diabetes:
A new analysis. J Clin Endocrinol Metab. 90:493–500. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
294
|
Panenin F, Castantino S and Cosentino F:
Insulin resistance, diabetes, and cardiovascular risk. Curr
Atheroscler Rep. 16:4192014. View Article : Google Scholar : PubMed/NCBI
|
|
295
|
Abdul-Ghami MA and DeFronzo RA:
Phathophysiology of prediabetes. Curr Diab Rep. 9:193–199. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
296
|
Reaven GM: Insulin resistance: The link
between obesity and cardiovascular disease. Med Clin North Am.
95:875–892. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
297
|
DeFronzo RA, Ferrannini E and Simonson DC:
Fasting hyperglycemia in non-insulin-dependent diabetes mellitus:
contributions of excessive hepatic glucose production and impaired
tissue glucose uptake. Metabolism. 38:387–395. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
298
|
Shulman GI, Rothman DL, Jue T, Stein P,
DeFronzo RA and Shulman RG: Quatitation of muscle glycogen
synthesis in normal subjects and subjects with
non-insulin-dependent diabetes by 13C nuclear magnetic resonance
spectroscopy. N Engl J Med. 322:223–228. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
299
|
McGarry JD: Banting lecture 2001:
Dysregulation of fatty acid metabolism in the etiology of type 2
diabetes. Diabetes. 51:7–18. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
300
|
Kashyap S, Belfort R, Gastaldelli A,
Pratipanawatr T, Berria R, Pratipanawatr W, Bajaj M, Madarino L,
DeFronzo R and Cusi K: A substained increase in plasma free fatty
acids impairs insulin secretion in nondiabetic sujects genetically
predisposed to develop type 2 diabetes. Diabetes. 52:2461–2474.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
301
|
Lei XG and Vatamaniuk MZ: Two tales of
antioxidant enzymes on β cells and diabetes. Antioxid Redox Signal.
14:489–503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
302
|
Krebs M, Krssaak M, Bernroider E,
Anderwald C, Brehm A, Meyerspeer M, Nowotny P, Roth E, Waldhäusl W
and Roden M: Mechanism of amino acid-induced skeletal muscle
insulin resistance in humans. Diabetes. 51:599–605. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
303
|
Pi-Sunyer FX: The epidemiology of central
fat distribution in relation to disease. Nutr Rev. 62((7 Pt2)):
S120–S126. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
304
|
Despres JP: Intra-abdominal obesity: An
untreated risk factor for type 2 diabetes and cardiovascular
disease. J Endocrinol Invest. 29 (3 Suppl):S77–S82. 2006.
|
|
305
|
Klein S, Allison DB, Heymsfield SB, Kelley
DE, Leibel RL, Nomas C and Kahn R; Association for Weight
Management and Obesity Prevention; NASSO, the Obesity Society;
American Society for Nutrition, : American Diabetes Association:
Waist circumference and cardiometabolic risk: A consensus statement
from shaping America's health: Association for weight management
and obesity pevention; NAASO, the obesity society; the American
society for nutrition; and the American diabetes association.
Diabetes Care. 30:1647–1652. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
306
|
Ashwell M, Gumn P and Gibson S:
Waist-to-height is a better screening tool than waist cincumference
and BMI for adult cardiometabolic risk factors: Systemic review and
meta-analysis. Obes Rev. 13:275–286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
307
|
Kouli GM, Panagiotakos DB, Kyrou I,
Georgousopoulou EN, Chryoshoou C, Tsigos C, Tousoulis D and
Pitsavos C: Visceral adiposity index and 10-year cardiovascular
disease incidence. The ATTICA study. Nutr Metab Cardiovasc Dis.
27:881–889. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
308
|
Weiss R: Fat distribution and storage: How
much, where, and how? Eur J Endocrinol. 157 (Suppl 1):S39–S45.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
309
|
Montague CT and O'Rahilly S: The perils of
portliness: Causes and consequences of viscelar adiposity.
Diabetes. 49:883–888. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
310
|
Yang X and Smith U: Adipose tissue
distribution and risk of metabolic disease: Does
thiazolidinedione-induced adipose tissue redistribution provide a
clue to the anwer? Diagetologia. 50:1127–1139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
311
|
Peraldi P and Spiegelman B: TNF-α and
insulin resistance: Summary and future prospects. Mol Cell Biochem.
182:169–175. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
312
|
Pickup JC: Inflammation and activated
innate immunity in the pathogenesis of type 2 diabetes. Diabetes
Care. 27:813–823. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
313
|
Han CY: Roles of reactive oxygen species
on insulin resistance in adipose tissue. Diabetes Metab J.
40:272–279. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
314
|
Yan LJ: Redox imbalance stress in diabetes
mellitus: Role of the polyol pathway. Animal Model Exp Med. 1:7–13.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
315
|
Dutta BJ, Singh S, Seksaria S, Gupta GD
and Singh A: Inside the diabetic brain: Insulin resistance and
molecular mechanism associated with cognitive impairment and its
possible therapeutic strategies. Pharmacol Res. 182:1063582022.
View Article : Google Scholar : PubMed/NCBI
|
|
316
|
Li H, Ren J, Li Y, Wu Q and Wei J:
Oxidative stress: The nexus of obesity and cognitive dysfunction in
diabetes. Front Endocrinol (Lausanne). 14:11340252023. View Article : Google Scholar : PubMed/NCBI
|
|
317
|
Emanuela F, Grazia M, Marco DR, Paola LM,
Giorgio F and Marco B: Inflammation as a link between obesity and
metabolic syndrome. J Nutr Metab. 2012:4763802012. View Article : Google Scholar : PubMed/NCBI
|
|
318
|
Böni-Schnetzler M and Meier DT: Islet
inflammation in type 2 diabetes. Semin Immunopathol. 41:501–513.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
319
|
Larsen CN, Faulenbach A, Vaag Α, Vølund A,
Ehses JA, Seifert B, Mandrup-Poulsen T and Donath MY:
Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N
Engl J Med. 356:1517–1526. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
320
|
Larsen CM, Faulenbach M, Vaag A, Ehses JA,
Donath MY and Mandrup-Poulsen T: Sustained effects of interleukin-1
receptor antagonist treatment in type 2 diabetes. Diabetes Care.
32:1663–1668. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
321
|
Infante M, Padilla N, Alejandro R, Caprio
M, Della-Morte D, Fabbri A and Ricordi C: Diabetes-modifying
antirheumatic drugs: The roles of DMARDs as glucose-lowering
agents. Medicina (Kaunas). 58:5712022. View Article : Google Scholar : PubMed/NCBI
|
|
322
|
Powers NE, Swartzwelter B, Marchetti C, de
Graaf DM, Lerchner A, Schlapschy M, Datar R, Binder U, Edwards CK
III, Skerra A and Dinarell CA: PASylation of IL-1 receptor
antagonist (IL-1Ra) retains IL-1 blockade and extends its duration
in mouse urate crystal-induced peritonitis. J Biol Chem.
295:868–882. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
323
|
Tegtmeyer K, Atassi G, Zhao J, Maloney NJ
and Lio PA: Off-Label studies on anakinra in dermatology: A review.
J Dermatolog Treat. 33:73–86. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
324
|
van Asseldonk EJ, Stienstra R, Koenen TB,
Joosten LA, Netea MG and Tack CJ: Treatment with Anakinra improves
disposition index but not insulin sensitivity in nondiabetic
subjects with the metabolic syndrome: A randomized, double-blind,
placebo-controlled study. J Clin Endocrinol Metab. 96:2119–2126.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
325
|
van Poppel PCM, van Asseldonk EJP, Holst
JJ, Vilsbøll T, Netea MG and Tack CJ: The interleukin-1 receptor
antagonist anakinra improves first-phase insulin secretion and
insulinogenic index in subjects with impaired glucose tolerance.
Diabetes Obes Metab. 16:1269–1273. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
326
|
Cucak H, Hansen G, Vrang N, Skarsfeldt T,
Steiness E and Jelsing J: The IL-1β receptor antagonist SER140
postpones the onset of diabetes in female nonobese diabetic mice. J
Diabetes Res. 2016:74846012016. View Article : Google Scholar : PubMed/NCBI
|
|
327
|
Cavelti-Weder C, Babians-Brunner A, Keller
C, Stahel MA, Kurz-Levin M, Zayed H, Solinger AM, Mandrup-Poulsen
T, Dinarello CA and Donath MY: Effects of gevokizumab on glycemia
and inflammatory markers in type 2 diabetes. Diabetes Care.
35:1654–1662. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
328
|
Rissanen A, Howard CP, Botha J and Thuren
T; Global Investigators, : Effects of anti-IL-1β antibody
(canakinumab) on insulin secretion rates in impaired glucose
tolerance or type 2 diabetes: Results of a randomized
placebo-controlled trial. Diabetes Obes Metab. 14:1088–1096. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
329
|
Hensen J, Howard CP, Walter V and Thuren
T: Impact of interleukin-1β antibody (canakinumab) on glycemic
indicators in patients with type 2 diabetes mellitus: Results of
secondary endpoints from a randomized, placebo-controlled trial.
Diabetes Metab. 39:524–531. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
330
|
Ridker PM, Howard CP, Walter V, Everett B,
Libby P, Hensen J and Thuren T; on the behalf of CANTOS Pilot
Investigative Group, : Effects of Interleukin-1β Inhibition with
Canakinumab on Hemoglobin A1c, Lipids, C-Reactive Protein,
Interleukin-6, and Fibrinogen: A Phase IIb Randomized,
Placebo-Controlled Trial. Circulation. 126:2739–2748. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
331
|
Choudhury RP, Birks JS, Manii V, Biasiolli
L, Robson MD, L'Allier PL, Gingras MA, Alie N, McLaughlin MA,
Basson CT, et al: Artherial effects of canakinumab in patients with
atherosclerosis and type 2 diabetes or glucose intolerance. J Am
Coll Cardiol. 68:1769–1780. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
332
|
Noe A, Howard C, Thuren T, Taylor A and
Skerjanec A: Pharmacokinetic and pharmacodynamics characteristics
of single-dose canakinumab in patients with type 2 diabetes
mellitus. Clin Ther. 36:1625–1637. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
333
|
Sloan-Lancaster J, Abu-Raddad E, Polzer J,
Miller JW, Schere JC, De Gaetano A, Berg JK and Landschulz WH:
Double blind, randomized study evaluating the glycemic and
anti-inflammatory effects of subcutaneous LY2189102, a neutralizing
IL-1 antibody, in patients with type 2 diabetes. Diabetes Care.
36:2239–2246. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
334
|
Everett BM, Donath MY, Pradhan AD, Thuren
T, Pais P, Nicolau JC, Glynn RJ, Libby P and Ridker PM:
Anti-inflimmatory therapy with canakinumad for the prevention and
management of diabetes. J Am Coll Cardiol. 71:2392–2401. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
335
|
Olson NC, Callas PW, Hanley AJG, Festa A,
Haffner SM, Wagenknecht LE and Tracy RP: Circulating levels of
TNF-α are associated with impaired glucose tolerance, increased
insulin resistance, and ethnicity: The insulin resistance
atherosclerosis study. J Clin Endocrinol Metab. 97:1032–1040. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
336
|
Wascher TC, Lindeman JHN, Sourij H,
Kooistra T, Pacini G and Roden M: Chronic TNF-α neutralization does
not improve insulin resistance or endothelial function in ‘healthy’
men with metabolic syndrome. Mol Med. 17:189–193. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
337
|
van den Oever IAM, Baniaamam M, Simsek S,
Raterman HG, van Denderen JC, van Eijk IC, Peters MJL, van der
Horst-Bruinsma IE, Smulders YM and Nurmohamed MT: The effect of
anti-TNF treatment on body composition and insulin resistance in
patients with rheumatoid arthritis. Rheumatol Int. 41:319–328.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
338
|
Kiortsis DN, Mavridis AK, Vasakos S, Nikas
SN and Drosos AA: Effects of infliximab treatment on insulin
resistance in patients with rheumatoid arthritis and ankylosing
spondylitis. Ann Rheum Dis. 64:765–766. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
339
|
Gonzalez-Gay MA, De Matias JM,
Gonzalez-Juanatey C, Garcia-Porrua G, Sanchez-Andrade A, Martin J
and Llorca J: Anti-tumor necrosis factor-alpha blockade improves
insulin resistance in patients with rheumatoid arthritis. Clin Exp
Rheumatol. 24:83–86. 2006.PubMed/NCBI
|
|
340
|
Haida KS, Bertachini G, Tavoni T,
Guilhermetti M, Loures MR and Bazotte RB: Infliximab treatment
prevents hyperglycemia and the intensification of hepatic
gluconeogenesis in an animal model of high fat diet-induced liver
glucose overproduction. Braz Arch Biol Technol. 55:389–393. 2012.
View Article : Google Scholar
|
|
341
|
Méndez-García LA, Trejo-Millán F,
Martínez-Reyes CP, Majarrez-Reyna AN, Esquivel-Velázquez M,
Melendez-Mier G, Islas-Andrade S, Rojas-Bernbé A, Kzhyshkowska J
and Escobedo G: Infliximab ameriorates tumor necrosis
factor-alpha-induced insulin resistance by attenuating PTP1B
activation in 3T3L1 adipocytes in vitro. Scan J Immunol.
88:e127162018. View Article : Google Scholar : PubMed/NCBI
|
|
342
|
Abdelhamid YA, Elyamany MF, Al-Shorbagy MY
and Badary OA: Effects of TNF-α antagonist infliximad on
fructose-induced metabolic syndrome in rats. Hum Exp Toxicol.
40:801–811. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
343
|
Bernstein LE, Berry J, Kim S, Canavan B
and Grinspoon SK: Effects of etanercept in patients with the
metabolic syndrome. Arch Intern Med. 166:902–908. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
344
|
Lo J, Bernstein LE, Canavan B, Torriani M,
Jackson MB, Ahima RS and Grinspoon SK: Effects of TNF-alpha
neutralization on adipocytokines and skeletal muscle adiposity in
the metabolic syndrome. Am J Physiol Endocrinol Metabol.
293:E102–E109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
345
|
Bravo C, Cataldo LR, Galgani J, Parada J
and Santos JL: Leptin/Adiponectin ratios using either total or high
molecular weight adiponectin as biomarkers of systemic insulin
sensitivity in normoglycemic women. Diabetes Res.
2017:90310792017.PubMed/NCBI
|
|
346
|
Stanley TL, Zanni MV, Johnsen S, Rasheed
S, Makimura H, Lee H, Khor VK, Ahima RS and Grinspoon SK: TNF-α
antagonism with etanercept decreases glucose and increases the
proportion of high molecular weight adiponectin in obese subjects
with features of the metabolic syndrome. J Clin Endocrinol Metab.
96:E146–E150. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
347
|
Paquot N, Castillo MJ, Lefèbvre PJ and
Scheen AJ: No increased insulin sensitivity after a single
intravenous administration of a recombinant human tumor necrosis
factor receptor: Fc fusion protein in obese insulin-resistant
patients. J Clin Endocrinol Metab. 85:1316–1319. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
348
|
Dominguez H, Storgaard H, Rask-Madsen C,
Hermann TS, Ihlemann N, Nielsen DB, Spohr C, Kober L, Vaag A and
Torp-Pedersen C: Metabolic and vascular effects of tumor necrosis
factor-α blockade with etanercept in obese patients with type 2
diabetes. J Vasc Res. 42:517–525. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
349
|
Ronti T, Lupattelli G and Mannarino E: The
endocrine function of adipose tissue: An update. Clin Endocrinol
(Oxf). 64:355–365. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
350
|
Hu D, Russell RD, Remash D, Greenaway T,
Rattigan S, Squibb KA, Jones G, Ross RM, Roberts CK, Premilovac D,
et al: Are the metabolic benefits of resistance in type 2 diabetes
linked to improvement in adipose tissue microvascular blood flow?
Am J Physiol Endocrinol Metab. 315:E1242–E1250. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
351
|
Ruscitti P, Berardicurti O, Cipriani P and
Giacomelli R; TRACK Study Group, : Benefits of anakinra versus TNF
inhibitors in rheumatoid arthritis and type 2 diabetes: Long-term
findings from participants furtherly followed-up in the TRACK
study, a multicentre, open-label, randomized, controlled trial.
Clin Exp Rheumatol. 39:403–406. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
352
|
Ramos-Zavala MG, Gonzalez-Ortiz M,
Martinez-Abundis E, Robles-Cervantes JA, Gonzalez-Lopez R and
Santiago-Hernandez NJ: Effect of diacerein on insulin secretion and
metabolic control in drug-naïve patients with type 2 diabetes.
Diabetes Care. 34:1591–1594. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
353
|
Cardoso CRL, Leite NC, Carlos FO, Loureiro
AA, Viegas BB and Salles GF: Efficacy and safety of diacerein in
patients with inadequately controlled type 2 diabetes: A randomized
controlled trial. Diabetes Care. 40:1356–1363. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
354
|
Tres GS, Fuchs SC, Piovesan F,
Koehler-Santos P, Pereira FD, Camey S, Lisboa HK and Moreira LB:
Effect of diacerein on metabolic control and inflammatory markers
in patients with type 2 diabetes using antidiabetic agents: A
randomized controlled trial. J Diabetes Res. 2018:42465212018.
View Article : Google Scholar : PubMed/NCBI
|
|
355
|
Jangsiripornpakorn J, Srisuk S, Chailurkit
L, Nimitphong H, Saetung S and Ongphiphadhanakul B: The
glucose-lowering effect of low-dose diacerein and its
responsiveness metabolic markers in uncontrolled diabetes. BMC Res
Notes. 15:912022. View Article : Google Scholar : PubMed/NCBI
|
|
356
|
Piovesan F, Tres GS, Moreira LB, Andrades
ME, Lisboa HK and Fucks SC: Effects of diacerein on renal function
and inflammatory cytokines in participants with type 2 diabetes
mellitus and chronic kidney disease: A randomized controlled trial.
PLoS One. 12:e01865542017. View Article : Google Scholar : PubMed/NCBI
|
|
357
|
Di Prospero NA, Artis E, Andrade-Gordon P,
Johnson DL, Vaccaro N, Xi L and Rothenberg P: CCR2 antagonism in
patients with type 2 diabetes mellitus: A randomized,
placebo-controlled study. Diabetes Obes Metab. 16:1055–1064. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
358
|
Mulder P, van den Hoek AM and Kleemann R:
The CCR2 inhibitor propagermanium attenuates diet-induced insulin
resistance, adipose tissue inflammation and non-alcoholic
steatohepatitis. PLoS One. 12:e01697402017. View Article : Google Scholar : PubMed/NCBI
|
|
359
|
Huh JH, Kim HM, Lee ES, Kwon MH, Lee BR,
Ko HJ and Chung CH: Dual CCR2/5 antagonist attenuates
obesity-induced insulin resistance by regulating macrophage
recruitment and M1/M2 status. Obesity (Silver Spring). 26:378–386.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
360
|
Tuttle KR, Brosius FC III, Adler SG,
Kretzler M, Mehta RL, Tumlin JA, Tanaka Y, Haneda M, Liu J, Silk
ME, et al: JAK1/JAK2 inhibition by baricitinib in diabetic kidney
disease: Results from a phase 2 randomized controlled clinical
trial. Nephrol Dial Transplant. 33:1950–1959. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
361
|
Faghihimani E, Amnorroaya A, Rezvanian H,
Adibi P, Ismail-Beigi F and Amini M: Salsalate improves glycemic
control in patients with newly diagnosed type 2 diabetes. Acta
Diabetol. 50:537–543. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
362
|
Goldfine AB, Fonseca V, Jablonski KA, Chen
YD, Tipton L, Staten MA and Steven E; Targeting Inflammation Using
Salsalate in Type 2 Diabetes Study Team, : Salicylate (Salsalate)
in patients with type 2 diabetes: A randomized trial. Ann Intern
Med. 159:1–12. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
363
|
Li D, Zhong J, Zhang Q and Zhang J:
Effects of anti-inflammatory therapies on glycemic control in type
2 diabetes mellitus. Front Immunol. 14:11251162023. View Article : Google Scholar : PubMed/NCBI
|
|
364
|
Raimondo MG, Biggioggero M, Crotti C,
Becciolini A and Favalli EG: Profile of sarilumab and its potential
in the treatment of rheumatoid arthritis. Drug Design Dev Ther.
11:1593–1603. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
365
|
Klinder A, Waletzko-Hellwig J, Sellin ML,
Seyfarth-Sehlke A, Wolfien M, Prehn F, Bader R and Jonitz-Heincke
A: Effects of the interleukin-6 receptor blocker sarilumab on
metabolic activity and differentiation capacity of primary human
osteoblasts. Pharmaceutics. 14:13902022. View Article : Google Scholar : PubMed/NCBI
|
|
366
|
Genovese MC, Burmester GR, Hagino O,
Thangavelu K, Iglesias-Rodriguez M, John GT, González-Gay MA,
Mandrup-Poulsen T and Fleischmann R: Interleukin-6 receptor
blockade or TNFα inhibition for reducing glycaemia in patients with
RA and diabetes: Post hoc analyses of three randomised, controlled
trials. Arthritis Res Ther. 22:2062020. View Article : Google Scholar : PubMed/NCBI
|
|
367
|
Drutskaya MS, Efimou GA, Kruglou AA and
Nedospasou SA: Can we design a better anti-cytokine therapy? Semin
Arthritis and Rhematism. 49:S39–S42. 2019.PubMed/NCBI
|
|
368
|
Nosenko MA, Atretkhany KSN, Mokhonov VV,
Vasilenko EA, Kruglov AA, Tillib SV, Drutskaya MS and Nedospasov
SA: Moduatation of bioavailability of proinflammatory cytokines
produced by myeloid cells. Semin Arthritis Rheum. 49:S39–S42. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
369
|
Velikova TV, Kabakchieva PP, Assyov YS and
Georgiev TA: Targeting inflammatory cytokines to improve type 2
diabetes control. Biomed Res Int. 2021:72974192021. View Article : Google Scholar : PubMed/NCBI
|
|
370
|
Achari A and Jain SK: Adiponectin, a
therapeutic target for obesity, diabetes, and endothelial
dysfunction. Int J Mol Sci. 18:13212017. View Article : Google Scholar : PubMed/NCBI
|
|
371
|
Yamauchi T, Kamon J, Waki H, Terauchi Y,
Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka T,
et al: The fat-derived hormone adiponectin reverses insulin
resistance associated with both lipoatrophy and obesity. Nat Med.
7:941–946. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
372
|
Combs TP, Wagner JA, Berger J, Doebber T,
Wang WJ, Zhang BB, Tanen M, Berg AH, O'Rahilly S, Savage DB, et al:
Induction of adipocyte complement-related protein of 30 kilodaltons
by PPRgamma agonists: A potential mechanism of insulin
sensitization. Endocrinology. 143:998–1007. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
373
|
Kolak M, Yki-Järvinen H, Kannisto K,
Tiikkainen M, Hamsten A, Eriksson P and Fisher RM: Effects of
chronic rosiglitazone therapy on gene expression in human adipose
tissue in vivo in patients with type 2 diabetes. J Clin Endocrinol
Metab. 92:720–724. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
374
|
Peraldi P, Xu M and Spiegelman BM:
Thiazolidinediones block tumor necrosis factor-alpha-incuded
inhibition of insulin signaling. J Clin Invest. 100:1863–1869.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
375
|
Wolf AM, Wolf D, Rumpold H, Enrich B and
Tilg H: Adiponectin induces the anti-inflammatory cytokines IL-10
and IL-1RA in human leukocytes. Biochem Biophys Res Commun.
323:630–635. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
376
|
Mosser DM and Zhang X: Interleukin-10: New
perspectives on an old cytokine. Immunol Rev. 226:205–218. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
377
|
Maclsaac RJ and Jerum G: Clinical
indications for thiazolidinediones. Aust Prescr. 27:70–74. 2004.
View Article : Google Scholar
|
|
378
|
Quinn CE, Hamilton PK, Lockhart CJ and
McVeigh GE: Thiazolidinediones: Effects on insulin resistance and
the cardiovascular system. Br J Pharmacol. 153:636–645. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
379
|
Graham DJ, Green L, Senior JR and Nourjah
P: Troglitazone-induced liver failure: A case study. Am J Med.
114:299–306. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
380
|
Tuccori M, Filion KB, Yin H, Yu OH, Platt
RW and Azoulay L: Pioglitazone use and risk of bladder cancer:
Population based cohort study. BMJ. 352:i15412016. View Article : Google Scholar : PubMed/NCBI
|
|
381
|
Aronoff S, Rosenblatt S, Braithwaite S,
Egan JW, Mathisen AL and Schneider RL: Pioglitazone hydrochloride
monotherapy improves glycemic control in the treatment of patients
with type 2 diabetes: A 6-month randomized placebo-controlled
dose-response study. The pioglitazone 001 study group. Diabetes
Care. 23:1605–1611. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
382
|
Mudaliar S, Chang AR and Henry RR:
Thiazolidinediones, peripheral edema, and type 2 diabetes:
Incidence, pathophysiology, and clinical implications. Endocr
Pract. 9:406–416. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
383
|
Arnold SV, Inzucchi SE, Echouffo-Tcheugui
JB, Tang F, Lam CSP, Sperling LS and Kosiborod M: Understanding
contemporary use of thiazolidinediones. Cir Heart Fail.
12:e0058552019. View Article : Google Scholar : PubMed/NCBI
|
|
384
|
Ferris FL III and Patz A: Macular edema. A
complication of diabetic retinopathy. Surv Ophthalmol. 28:452–461.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
385
|
Ryan EH Jr, Han DP, Ramsay RC, Cantrill
HL, Bennett SR, Dev S and Williams DF: Diabetic macular edema
associated with glitazone use. Retina. 26:562–570. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
386
|
Vestergaard P: Discrepacies in bone
mineral density and fracture risk in patients with type 1 and type
2 diabetes-a meta-analysis. Osteoporos Int. 18:427–444. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
387
|
Fonseca V: Effect of thiazolidinediones on
body weight in patients with diabetes mellitus. Am J Med.
115:42–48. 2003. View Article : Google Scholar
|
|
388
|
Ko KD, Kim KK and Lee KR: Does weight gain
associated with thiazolidinedione use negatively affect
cardiometabolic health? J Obes Metab Syndr. 26:102–106. 2017.
View Article : Google Scholar : PubMed/NCBI
|