Hepatocellular carcinoma (HCC) is a common type of
cancer worldwide and the incidence is expected to exceed 1 million
cases by 2025, posing a threat to human health (1). Tumour cells, stromal cells, and
stroma constitute the tumor microenvironment (TME), which can be
depleted of certain nutrients (2,3).
Metabolic reprogramming refers to the process by which tumour cells
undergo specific metabolic changes to adapt to the hypoxic and
nutrient-deficient microenvironment, thereby enabling tumour cells
to proliferate rapidly (4).
Glucose metabolic reprogramming mainly includes changes in
glycolysis and the pentose phosphate pathway (PPP). Compared with
normal cells, which use mitochondrial oxidative phosphorylation
(OXPHOS) to produce energy, most cancer cells use glycolysis as the
main way to produce energy for their growth even in an aerobic
state. This process is known as aerobic glycolysis or the ‘Warburg
Effect’ and is the most well-studied part of glucose metabolism
(5,6). In addition to glycolysis, the PPP
also provides biomacromolecules to meet material requirements of
cancer cells for cell replication (7). Fatty acid (FA) metabolism is another
energy source that supports tumour metastasis. Lipids and
cholesterol regulate the construction of lipid rafts and
invadopodia on the cell membrane, which in turn affect tumor cell
invasion and metastasis (8).
Through deamination, amino acids can not only be oxidized and
decomposed to produce energy but also provide carbon and nitrogen
sources for the synthesis of sugars, lipids and nucleic acids
(9). Therefore, the energy
production of cancer cells can be affected by the reprogramming of
these three major metabolic pathways to inhibit the growth and
proliferation of cancer cells. The present review considers current
research progress on noncoding RNAs (ncRNAs), oncogenes, tumour
suppressors and other tumour regulatory factors affecting HCC
through metabolic reprogramming. This review focuses on glucose,
lipid and amino acid metabolism, aiming to report potential
therapeutic targets and mechanisms for HCC treatment.
In HCC cells, the glucose metabolism pathway is
reprogrammed according to the requirements of the cancer cells. In
normal cells, most glucose is converted to pyruvate by glycolysis,
which is then subjected to OXPHOS under aerobic conditions or
anaerobic oxidation to lactate under anaerobic conditions (10). However, cancer cells rely on
glycolysis to provide energy even in the presence of oxygen. This
is because although only 2 adenosine triphosphates (ATPs) are
produced per molecule of glucose through the glycolytic pathway,
the rate of glycolysis is faster than OXPHOS and thus is more
suitable for the rapid proliferation of tumour cells (11). The metabolic intermediates produced
during aerobic glycolysis are used to synthesize biological
macromolecules, while the accumulation of lactic acid can create an
acidic microenvironment which can drive tumour progression
(12).
A previous study have reported that the levels of
both glycolysis and the PPP are significantly increased in HCC
(13) and that tumor regulators,
such as ncRNAs, oncogenic factors, and tumor suppressors can
mediate this change to reprogram glycolysis (Table I) and the PPP, resulting in an
impact on HCC progression (14,15).
Glucose transporter 1 (GLUT1), the major glucose
transporter, promotes glucose uptake in numerous tissues. GLUT1 is
highly expressed in HCC tissues and the knockdown of GLUT1
expression by siRNA significantly inhibits the proliferation of HCC
cells (16). A recent study
reported that cyclin-dependent kinase 6 (CDK6) deficiency inhibited
GLUT1 expression by inhibiting the H3K27ac, H4K8ac and H3K4me1
levels on the GLUT1 enhancer, resulting in the autophagy of HCC
cells (17). Forkhead box
transcription factor M1 (FOXM1) is a key transcriptional activator
of GLUT1 (18). Basic
transcription factor 3 (BTF3) can transactivate FOXM1 to regulate
the expression of GLUT1. Furthermore, BTF3 knockdown inhibits the
proliferation of HCC cells and the glycolysis in HCC cells through
the FOXM1/GLUT1 axis (19).
Similarly, the antisense lncRNA SLC2A1-AS1 can regulate HCC cell
proliferation and metastasis by competitively binding to the
transcription factor STAT3, which inhibits the transcriptional
activation of FOXM1 (20). The
GLUT1 inhibitor BAY-876 has been previously reported to demonstrate
antitumour activity in HCC models, but its clinical application
faces challenges due to the systemic distribution of the drug at
the time of administration, which leads to insufficient drug dose
at the tumor site (21). Based on
these previous reports, CDK6, BTF3 and the lncRNA SLC2A1-AS1 are
suggested as possible novel targets for targeting GLUT1 in the
treatment of HCC. Moreover, given the effect of the lncRNA
SLC2A1-AS1 on HCC cell proliferation and metastasis, it should be
considered as a potential biomarker for predicting HCC recurrence
(20).
Hexokinase (HK), the first rate-limiting enzyme in
aerobic glycolysis, has four isoforms (HK1, HK2, HK3 and HK4). Most
normal tissues express only HK1, but HK2 is highly expressed in HCC
tissue and is directly related to patient prognosis (22). In animal models, HK2 depletion has
been shown to reduce cancer cell proliferation without obvious side
effects, indicating its potential as a therapeutic target (23). Ubiquitin Protein Ligase E3
Component N-Recognin 7 (UBR7) is an E3 ubiquitin ligase, Zhao et
al (24) reported that the
UBR7-mediated mono-ubiquitination of histone H2B promoted the
transcriptional activation of Keap1 and indirectly inhibited the
expression of HK2, thereby inhibiting aerobic glycolysis and HCC
tumorigenesis. Furthermore, numerous studies have reported that HK2
is a direct target of miR-188-5p, miR-202, circCCT3 and HuaChanSu
in HCC cells, which affect the proliferation, invasion and
migration of HCC cells by regulating the expression of HK2
(25–29). Known direct HK2 inhibitors include
2-deoxyglucose (2-DG) and 3-bromopyruvate (3-BP); however these are
not cell specific and can affect normal tissues causing
drug-related hepatotoxicity (30).
Therefore, miR-3662, miR-188-5p and miR-202 have been suggested as
potential targets for HCC treatment. It is also reported a novel
molecular mechanism by which the traditional Chinese medicine (TCM)
extract HuaChanSu delays HCC progression, supporting the use of
Huashansu in HCC treatment (28).
Pyruvate kinase muscle type (PKM) is a member of the
phosphokinase family that regulates the final rate-limiting step of
glycolysis. PKM has two isoforms PKM1 and PKM2, which are produced
by alternative splicing of PKM precursor mRNA. PKM1 has
tumor-suppressor activity (31).
Whereas PKM2 is upregulated in most tumour tissues and serves a
role in the energy metabolism of HCC by enhancing the Warburg
Effect and supporting anabolism (32). ZFP91 has been proposed as a novel
E3 ubiquitin ligase, which inhibits PKM2 isoform formation and HCC
metabolic reprogramming (33).
Moreover, transcription factor GATA6 epigenetically regulates PKM2
transcription, and the downregulation of GATA6 expression induces
glycolysis and promotes tumorigenicity, self-renewal and metastasis
in HCC cells (34). Circular
(circ)RNAs acts as a sponge for miRNAs to regulate gene expression
by affecting the transcription, the mRNA turnover and translation
(35). Under hypoxic conditions,
circMAT2B promotes glycolysis and HCC progression by acting as a
sponge for miR-338-3p upregulating PKM2 expression (36). By contrast, miR-374b antagonizes
the Warburg Effect by inhibiting PKM2 and resensitizing HCC cells
to sorafenib (37). Sorafenib is
an effective first-line therapy for patients with advanced HCC, but
sorafenib resistance is becoming increasingly common, which is
affected by the TME (38).
Targeting PKM2 can also regulate the metabolism of immune cells,
enhance the anticancer immune response and inhibit cancer growth
and metastasis (39). Therefore
ZFP91, GATA6, circMAT2B, miR-338-3p and miR-374b may serve as
useful targets for the treatment of and prevention of drug
resistance in HCC as they modulate PKM2 expression.
Lactate dehydrogenase A (LDHA) is a key enzyme in
the glycolytic conversion of pyruvate to lactate. Abnormally high
LDHA expression is closely related to the malignant progression of
numerous types of cancer (40).
Several specific inhibitors of LDHA are currently being assessed as
potential anticancer treatments (41). A recent study reported that acyl
phosphatase 1 (ACYP1) serves a tumour promoting role by activating
the c-MYC/LDHA axis to promote glycolysis. Furthermore, ACYP1
expression is associated with lenvatinib resistance; lenvatinib is
the first-line therapy for advanced HCC, however, drug resistance
limits the efficiency of lenvatinib. The targeting of ACYP1 can
exert a synergistic effect with lenvatinib to more effectively
treat HCC (42). Moreover, LDHA
has been reported to be a direct target of miR-122-5p, miR-34a and
miR-142-3p, which exhibit potential tumour suppressive effects in
HCC, providing targets for future therapeutic strategies (43–45).
The PI3K/AKT/mTOR signalling pathway is involved in
the regulation of glucose metabolism in HCC. Numerous components
have been reported to impact this pathway. Firstly, tumor
regulators can modulate the glycolytic pathway by directly
targeting this signalling pathway, thereby affecting the
development of HCC (46–48). circRHBDD1 and FA receptor CD36
directly activate the PI3K/AKT/mTOR pathway, thereby enhancing
glycolysis and ultimately promoting HCC growth and metastasis
(46,47). However, the tumour suppressor PTEN
inhibits the activation of the PI3K/AKT pathway, thus inducing
apoptosis and fighting against the development of HCC (48). Furthermore, the high expression of
circRHBDD1 in patients with HCC has been reported to limit the
efficacy of immunotherapy (46).
Secondly, the activity of the PI3K/AKT/mTOR signalling pathway can
be indirectly regulated by glycolytic metabolic enzymes, thereby
affecting the Warburg Effect. For example, LINC01554 and circRPN2
promote the ubiquitin-mediated proteasomal degradation of PKM2 and
enolase 1 (ENO1), respectively. This inhibits the Akt/mTOR
signalling pathway to decrease aerobic glycolysis and the
proliferation of HCC cells (49,50).
Lastly, the activation of the PI3K/AKT/mTOR signalling pathway can
lead to increased GLUT1 expression in HCC tumour tissue, which
accelerates glucose uptake, thereby promoting glucose metabolism
and contributing to impaired immune cell function in HCC (51).
Hypoxia-inducible factor-1α (HIF-1α) is involved in
multiple aspects of tumorigenesis and cancer progression (52). HIF-1α can activate the expression
of numerous glycolysis-related enzymes, including GLUT1, HK2, PKM2,
LDHA and ENO2, and promotes glycolysis (53), whereas miR-199a-5p can interfere
with the expression of HK2, abrogating HIF-1α-enhanced Warburg
effect in HCC (54). Moreover,
miR-3662 regulates the GLUT1, HK2, PKM2, and LDHA expression via
directly targeting HIF-1α, thereby exerts its suppressive effect on
HCC glycolysis and proliferation (55). Furthermore, PKM2 is also a
coactivator of HIF-1α, which increases the levels of HIF-1α
creating a positive feedback loop for this pathway (56). Moreover, HIF-1α can also affect the
AKT/mTOR signalling pathway. HIF-1α enhances the process that
miR-873 promotes the expression of GLUT1 and HK2 in HCC cells by
activating the AKT/mTOR pathway and promotes glycolysis,
proliferation, invasion and metastasis in HCC (57). Therefore, targeting HIF-1α may be
an effective strategy for the treatment of HCC.
The PPP is the main pathway of glucose catabolism.
Glucose outside HCC cells is transported into the cells through
GLUT1 and then phosphorylated by HK to form glucose-6-phosphate
(G6P), which can be further metabolized through the glycolytic
pathway or the PPP. The PPP is made up of an oxidative and
non-oxidative pathway. In the oxidative pathway, G6P is
dehydrogenated by the rate-limiting enzyme glucose-6-phosphate
dehydrogenase (G6PD) to produce ribose-5 phosphate and NAPDH, which
is critical to maintain redox balance of cancer cells (58). The non-oxidative pathway
metabolizes the glycolytic intermediate to supply cancer cells with
ribose 5-phosphate for nucleic acid synthesis and precursors for
amino acid synthesis (59). G6PD
has been reported to induce epithelial-mesenchymal transition by
activating the STAT3 signalling pathway, thereby promoting the
migration and invasion of HCC cells (60). miR-122 can inhibit HCC cells
proliferation by reducing G6PD activity, inhibiting the PPP
(61). The knockdown of
glucose-6-phosphate lactonase (PGLS), a cytosolic enzyme involved
in the oxidation stage of the PPP, has been shown to inhibit the
PPP and HCC progression (7).
Similarly, transketolase (TKT), a key enzyme in the non-oxidative
branch of the PPP, promotes HCC progression through significantly
increasing the level of glucose flux and NADPH, which maintaining
redox homeostasis of HCC cells (62). Oroxylin A, a small molecule
inhibitor of TKT, directly targets TKT and leads to accumulation of
glycolytic intermediates in the non-oxidative PPP, which inhibits
HCC proliferation by inducing apoptosis and cell cycle arrest,
providing a novel approach for the treatment of HCC (63). Therefore, in addition to the
rate-limiting enzymes G6PD and TKT, miR-122 and PGLS could also be
used as novel potential therapeutic targets to inhibit HCC
progression by regulating HCC cell metabolism.
In addition to glucose metabolism, alterations in
lipid metabolism are also thought to contribute to HCC progression.
Accelerated de novo FA synthesis and cholesterol
biosynthesis, as well as altered FA oxidation (FAO), contribute to
the development and progression of HCC (64). Some tumor regulators serve roles in
lipid metabolism by regulating the expression of lipid metabolism
enzymes such as lipid synthetase and FA oxidase, thereby affecting
the progression of HCC.
Sterol-regulatory element binding proteins (SREBPs)
are key transcription factors for lipid synthesis. There are three
SREBP isoforms, SREBP1a, SREBP1c and SREBP2 (65). SREBP1 transcriptionally activates
acetyl-coenzyme A carboxylase 1 (ACC1), FA synthase (FASN),
stearoyl-coenzyme A desaturase 1 (SCD1) and other lipid synthases.
mSREBP-1, the mature form of SREBP-1, is transported to the nucleus
to play a transcriptional regulatory role, where it promotes the
expression of lipid synthetase (66). The oncoprotein c-Myc can interact
with SREBP1 to further activate the expression of these lipid
synthases, thereby increasing FA synthesis to promote cancer growth
(67). Long-chain acyl-coenzyme A
synthetase 4 (ACSL4) and stomatin-like protein 2 (SLP2) increase
de novo FA synthesis and HCC progression through the
upregulation of SREBP1 and its downstream lipogenic enzymes by
c-Myc (68,69). Mitochondria continuously change
their morphology through fission and fusion processes that are
tightly regulated to meet cellular metabolic demands. Abnormal
increase of mitochondrial fission has been shown to be closely
related to the progression of cancer (70). Wu et al (71) reported that the activation of
mitochondrial fission not only increased the expression of ACC1 and
FASN in HCC cells by upregulating SREBP1 expression, but also
significantly promotes de novo FA synthesis. Furthermore,
SREBP1 can also inhibit FAO by downregulating the FA oxidase
carnitine palmitoyltransferase 1 (CPT1), thereby promoting the
proliferation and metastasis of HCC cells (71). Likewise, circPRKAA1 can increase
de novo FA synthesis by increasing the stability of mSREBP-1
(72). Caspase-3 (CASP3) mediates
SREBP2 cleavage from the endoplasmic reticulum, allowing SREBP2 to
stimulate the transcription of genes involved in cholesterol
biosynthesis (73). Conversely,
miR-612 inhibits the expression of HMG-CoA reductase (HMGCR) by
inhibiting SREBP2 transcriptional activity, thereby inhibiting
cholesterol synthesis and the formation of invadopodia, and further
inhibiting HCC migration and invasion (8).
Inhibitors targeting ACC1, FASN and SCD1 have shown
antitumour effects in xenograft models (74–76),
and a new-generation FASN inhibitor, TVB-2640, has entered clinical
trials for patients with solid tumours (77). The inhibition of SREBPs can also
inhibit tumour growth and induce cancer cell death, but the direct
inhibition of transcription factors is a challenge, as
transcription factors often make poor drug targets (78). Therefore, the aforementioned
findings indicate that ACSL4, SLP2, circPRKAA1, CASP3 and miR-612,
which can directly regulate lipid synthases and SREBPs, are
potential therapeutic targets.
HCC cells can also participate in redox homeostasis
by reprogramming amino acid metabolism to provide intermediates for
HCC biosynthesis and energy requirements. Amino acids can be
divided into two groups: Non-essential amino acids, including
glutamate, glutamine, serine, aspartic acid and proline; and
essential amino acids, including threonine, leucine and methionine.
It is shown that amino acids can act as metabolic regulators to
support cancer cell growth, with glutamine and methionine being the
most studied (9).
Glutamine is a non-essential amino acid, which is
converted to glutamate by glutaminase (GLS), before being further
converted to α-ketoglutaric acid (α-KG) and metabolized to ATP in
the citric acid cycle (TCA). GLS exist as two isozymes in mammalian
cells named GLS1 and GLS2. It was reported that GLS1 functions as a
tumor promotor in many cancer types, while GLS2 seems to act as a
tumor suppressor (79). Numerous
tumour cells use glutamine as a carbon source for energy production
and anabolism (80). The
upregulation of glutamate dehydrogenase 1 (GDH1) expression and the
silencing of oxoglutarate dehydrogenase-like (OGDHL) expression
both promote glutamine metabolism and drive α-KG synthesis, thereby
promoting TCA cycle, providing energy and biosynthetic substrates
for HCC cell growth and proliferation (81,82).
Moreover, OGDHL can activate the mTORC1 signalling pathway, which
upregulates SREBP1 expression and induces the expression of key
enzymes involved in lipogenesis, which increases lipogenesis and
HCC progression (82). c-Myc also
serves a role in glutamine metabolism in HCC cells. SMYD2 is a
protein lysine methyltransferase that stabilizes c-Myc, increases
its expression through the ubiquitin-proteasome system and further
upregulates GLS1, thereby activating glutamine metabolism and
promoting the development of HCC (83). In phase I clinical trials, the GLS
inhibitor telaglenastat has been reported to exhibits modest
single-agent activity for renal cell carcinoma, but whether it is
also effective for HCC treatment is unknown (84). Furthermore, SMYD2 targets GLS1 in
HCC, which may provide a novel direction for inhibiting glutamine
metabolism and thus inhibiting HCC progression.
The essential amino acid methionine is metabolized
to S-adenosylmethionine (SAM) through the methionine cycle by
methionine adenosyltransferase 2A (MAT2A) (85). Hung et al (86) reported that the loss of MAT2A can
lead to the inhibition of HCC growth by inducing T cell
dysfunction. Moreover, miR-203 can directly inhibit MAT2A
expression and increase SAM expression in HCC, producing a tumour
suppressor effect (87).
Tetrahydrofolate is one of the products of the methionine cycle,
and a high folate diet can promote HCC development by increasing
the expression of MAT2A to accelerate the methionine cycle
(88). Methionine restriction has
been reported to be a promising therapeutic avenue for the
treatment of numerous cancers, such as metastatic melanoma and
gastric cancer, and colorectal cancer (CRC) (89). Such restriction can be achieved in
three ways: By restricting methionine in the diet; using
methionine-depleting enzymes; and inhibiting methionine metabolism.
At present, clinical studies on methionine-restricted diet are
limited and a more promising treatment method may be the use of
methionine-depleting enzymes, such as recombinant methioninases,
which reduce serum methionine levels, resulting in insufficient
methionine supply to tumours and resulting in reduced tumour volume
(89,90). Moreover, metabolic inhibitors
including MAT2A inhibitors, can prevent tumours from using
exogenous methionine for energy, thereby inhibiting tumour growth
(91). At present, MAT2A
inhibitors are in phase I clinical trials. However, drug resistance
and off-target effects have been reported for these inhibitors
(85). Therefore, it can be
hypothesised that targeting miR-203 and individualized folic acid
diets may serve as novel strategies for HCC treatment.
HCC poses a serious threat to human health and is
the fourth leading cause of cancer-related mortality worldwide
(1). Emerging targeted therapies
have been reported to only achieve lasting clinical benefits for a
small proportion of patients; therefore, there are still major
challenges regarding the treatment of HCC, and the development of
novel effective therapeutic targets is needed (92). Metabolic reprogramming is a
characteristic of cancer cells, which ensures they can meet the
increased energy demand and maintain redox balance (93). In HCC, this reprogramming is mainly
caused by the activation of metabolic enzymes: Including glucose
metabolism-related proteins GLUT1, HK2, PKM2 and LDHA; lipid
metabolism-related enzymes ACC1 and FASN; and amino acid
metabolism-related enzymes GLS and MAT2A. The metabolism of
proteins, lipids and amino acids produces biological macromolecules
and releases energy. The expression of GLUT1, HK2, PKM2, LDHA,
ACC1, FASN, GLS and MAT2A is upregulated in HCC to promote the
metabolic process, thereby ensuring the rapid growth and
proliferation of liver cancer cells. In addition, dysregulation of
transcription factors and signalling pathways, such as the
PI3K/AKT/mTOR pathway, HIF-1α, c-Myc and SREBPs involved in lipid
metabolism, also serve a role in HCC metabolic reprogramming.
Multiple studies have reported that metabolic
inhibitors, such as the GLS inhibitor CB-839 and FASN inhibitor
TVB3664, which exert therapeutic effects in HCC by inhibiting the
metabolic reprogramming of HCC cells, are currently in clinical
studies of multiple solid tumours, including CRC and breast cancer
(94,95). In the present review, numerous
ncRNAs, oncogenes, and tumour suppressors that target glucose,
lipid, and amino acid metabolic reprogramming were summarized.
These impact the expression of key metabolism-related enzymes in
HCC cells directly or by altering the activity of transcription
factors and signalling pathways, which affect the energy and
nutrient production of HCC cells, subsequently affecting their
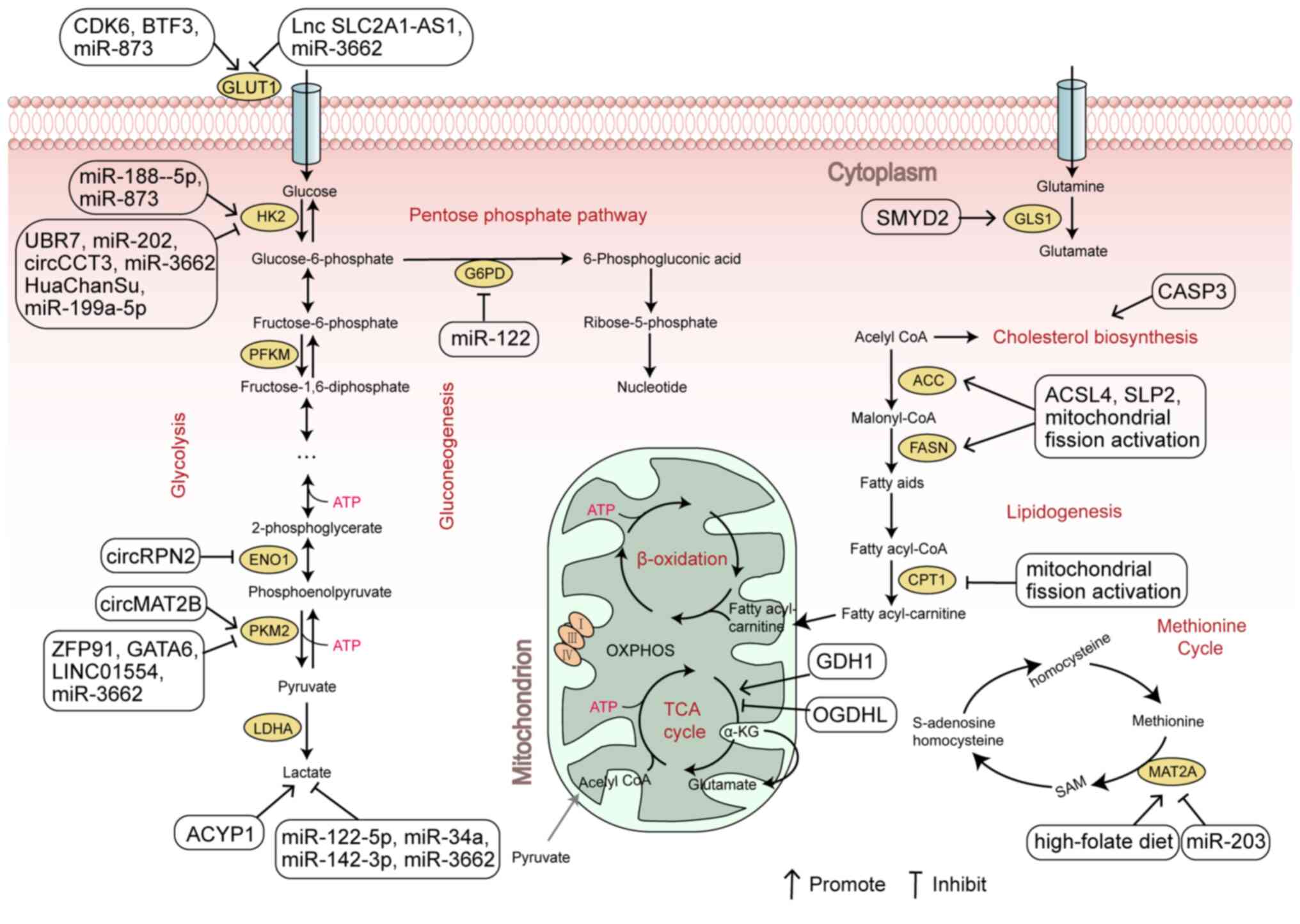
growth and proliferation (Fig. 1).
These tumour regulators can be used as promising therapeutic
targets or potential prognostic biomarkers for HCC. The oncogenes
CDK6, CD36, ACSL4 and CASP3 and the tumour suppressors GATA6, PTEN,
miR-34a, miR-122 and miR-203 are considered to have significant
effects on HCC progression. These regulators are suggested as
future targets in the treatment of HCC. Certain molecules can also
affect the sensitivity of cells to existing drugs. For example,
ACYP1 and CASP3 impact the chemoresistance of HCC cells to
lenvatinib by re-regulating the glycolytic pathway and lipid
synthesis, respectively (42,73).
Although the targeting of metabolic reprogramming in
cancer cells has therapeutic potential, its application remains
problematic. First, there are multiple isoforms of many of the
enzymes involved in metabolism, and small-molecule inhibitors may
not be able to precisely target the predominant isoform expressed
by cancer cells (96). Second,
some inhibitors are not highly specific and may target normal
tissues and cause drug-related hepatotoxicity (96). For example, a small clinical trial
reported that nearly all patients with breast, thyroid, or
non-small-cell lung cancer (NSCLC) treated with the HK2 inhibitor
2-DG had hypoglycaemic symptoms, including fatigue, sweating,
dizziness, nausea; asymptomatic QTc prolongation occurred in 72% of
patients; upper gastrointestinal bleeding occurred in 6% of
patients (97). Moreover, cancer
cells may develop resistance to metabolic pathway inhibitors
(96). To overcome these problems,
metabolic inhibitors can be combined with other targeted drugs to
improve treatment efficacy. Studies have reported that the
combination of the FASN inhibitor TVB3664 and sorafenib can improve
therapeutic efficacy in a mouse model of HCC and that the
combination of the glycolysis inhibitor 2-DG and sorafenib can
significantly decrease the viability of sorafenib-sensitive and
resistant cells (94,98). Furthermore, the combination of two
metabolic inhibitors has been shown to have a stronger anticancer
effect than the equivalent monotherapies. The combination of the
GLS inhibitor CB-839 and the ASCT2 glutamine transporter inhibitor
V-9302 can inhibit Gln metabolism and reduce extracellular
glutamine uptake, respectively, leading to a shortage of Gln supply
and GSH synthesis, thereby inhibiting HCC xenograft growth and
inducing apoptosis in vivo (95). Therefore, studies should develop
more effective metabolic inhibitors with fewer side effects. Most
of the side effects of current metabolic inhibitors are caused by
the failure to precisely target the metabolic enzyme isoform
expressed by cancer cells. Therefore, we can indirectly target the
isoform expressed by cancer cells by trying to develop inhibitors
and/or activators of the tumour regulators mentioned in this
review, especially those that have significant effects on HCC
progression, to reduce side effects. Further research is required
to assess the therapeutic potential of combination therapies which
use metabolic inhibitors with other targeted agents.
In conclusion, despite certain limitations and
difficulties, in-depth exploration of regulatory factors related to
HCC metabolic reprogramming continues to reveal options for the
development of novel and effective HCC treatment strategies,
especially the combination of metabolic inhibitors with other
targeted agents, which may help to improve the anticancer efficacy
of existing treatments.
Not applicable.
This work was supported by the National Natural Science
Foundation of China (grant no. 82274260).
Not applicable.
WG and JW performed the literature review and wrote
the manuscript. YX, HY, SY, CB and QC revised the manuscript and
provide critical review of the scientific content. YZ conceived the
work and approved the final version. All authors approved the final
version of the manuscript. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Llovet JM, Kelley RK, Villanueva A, Singal
AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, Zucman-Rossi J and
Finn RS: Hepatocellular carcinoma. Nat Rev Dis Primers. 7:62021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xing Y, Zhao S, Zhou BP and Mi J:
Metabolic reprogramming of the tumour microenvironment. FEBS J.
282:3892–3898. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Martínez-Reyes I and Chandel NS: Cancer
metabolism: Looking forward. Nat Rev Cancer. 21:669–680. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ward PS and Thompson CB: Metabolic
reprogramming: A cancer hallmark even warburg did not anticipate.
Cancer Cell. 21:297–308. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li C, Chen J, Li Y, Wu B, Ye Z, Tian X,
Wei Y, Hao Z, Pan Y, Zhou H, et al: 6-Phosphogluconolactonase
promotes hepatocellular carcinogenesis by activating pentose
phosphate pathway. Front Cell Dev Biol. 9:7531962021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu Y, Lu LL, Wen D, Liu DL, Dong LL, Gao
DM, Bian XY, Zhou J, Fan J and Wu WZ: MiR-612 regulates invadopodia
of hepatocellular carcinoma by HADHA-mediated lipid reprogramming.
J Hematol Oncol. 13:122020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vettore L, Westbrook RL and Tennant DA:
New aspects of amino acid metabolism in cancer. Br J Cancer.
122:150–156. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhuang X, Chen Y, Wu Z, Xu Q, Chen M, Shao
M, Cao X, Zhou Y, Xie M, Shi Y, et al: Mitochondrial miR-181a-5p
promotes glucose metabolism reprogramming in liver cancer by
regulating the electron transport chain. Carcinogenesis.
41:972–983. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N,
Yi P, Tang L, Pan Q, Rao S, et al: The cancer metabolic
reprogramming and immune response. Mol Cancer. 20:282021.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vaupel P, Schmidberger H and Mayer A: The
Warburg effect: essential part of metabolic reprogramming and
central contributor to cancer progression. Int J Radiat Biol.
95:912–919. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Enzo E, Santinon G, Pocaterra A, Aragona
M, Bresolin S, Forcato M, Grifoni D, Pession A, Zanconato F, Guzzo
G, et al: Aerobic glycolysis tunes YAP/TAZ transcriptional
activity. EMBO J. 34:1349–1370. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liao W, Du J, Wang Z, Feng Q, Liao M, Liu
H, Yuan K and Zeng Y: The role and mechanism of noncoding RNAs in
regulation of metabolic reprogramming in hepatocellular carcinoma.
Int J Cancer. 151:337–347. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li J, Eu JQ, Kong LR, Wang L, Lim YC, Goh
BC and Wong ALA: Targeting Metabolism in Cancer Cells and the
Tumour Microenvironment for Cancer Therapy. Molecules. 25:48312020.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Amann T, Maegdefrau U, Hartmann A, Agaimy
A, Marienhagen J, Weiss TS, Stoeltzing O, Warnecke C, Schölmerich
J, Oefner PJ, et al: GLUT1 expression is increased in
hepatocellular carcinoma and promotes tumorigenesis. Am J Pathol.
174:1544–1552. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yao J, Tang S, Shi C, Lin Y, Ge L, Chen Q,
Ou B, Liu D, Miao Y, Xie Q, et al: Isoginkgetin, a potential CDK6
inhibitor, suppresses SLC2A1/GLUT1 enhancer activity to induce
AMPK-ULK1-mediated cytotoxic autophagy in hepatocellular carcinoma.
Autophagy. 19:1221–1238. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shang R, Pu M, Li Y and Wang D: FOXM1
regulates glycolysis in hepatocellular carcinoma by transactivating
glucose transporter 1 expression. Oncol Rep. 37:2261–2269. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang P, Sun J, Sun C, Zhao H, Zhang Y and
Chen J: BTF3 promotes proliferation and glycolysis in
hepatocellular carcinoma by regulating GLUT1. Cancer Biol Ther.
24:22258842023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shang R, Wang M, Dai B, Du J, Wang J, Liu
Z, Qu S, Yang X, Liu J, Xia C, et al: Long noncoding RNA SLC2A1-AS1
regulates aerobic glycolysis and progression in hepatocellular
carcinoma via inhibiting the STAT3/FOXM1/GLUT1 pathway. Mol Oncol.
14:1381–1396. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang H, Zhang MZ, Sun HW, Chai YT, Li X,
Jiang Q and Hou J: A Novel Microcrystalline BAY-876 formulation
achieves long-acting antitumor activity against aerobic glycolysis
and proliferation of hepatocellular carcinoma. Front Oncol.
11:7831942021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
DeWaal D, Nogueira V, Terry AR, Patra KC,
Jeon SM, Guzman G, Au J, Long CP, Antoniewicz MR and Hay N:
Hexokinase-2 depletion inhibits glycolysis and induces oxidative
phosphorylation in hepatocellular carcinoma and sensitizes to
metformin. Nat Commun. 9:4462018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Garcia SN, Guedes RC and Marques MM:
Unlocking the Potential of HK2 in Cancer Metabolism and
Therapeutics. Curr Med Chem. 26:7285–7322. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao L, Kang M, Liu X, Wang Z, Wang Y,
Chen H, Liu W, Liu S, Li B, Li C, et al: UBR7 inhibits HCC
tumorigenesis by targeting Keap1/Nrf2/Bach1/HK2 and glycolysis. J
Exp Clin Cancer Res. 41:3302022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ding Z, Guo L, Deng Z and Li P: Circ-PRMT5
enhances the proliferation, migration and glycolysis of hepatoma
cells by targeting miR-188-5p/HK2 axis. Ann Hepatol. 19:269–279.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang J, Chen J, Sun F, Wang Z, Xu W, Yu Y,
Ding F and Shen H: miR-202 functions as a tumor suppressor in
hepatocellular carcinoma by targeting HK2. Oncol Lett.
19:2265–2271. 2020.PubMed/NCBI
|
|
27
|
Lv B, Zhu W and Feng C: Coptisine Blocks
Secretion of Exosomal circCCT3 from cancer-associated fibroblasts
to reprogram glucose metabolism in hepatocellular carcinoma. DNA
Cell Biol. Oct 2–2020.(Epub ahead of print). View Article : Google Scholar
|
|
28
|
Wu Q, Wang SP, Sun XX, Tao YF, Yuan XQ,
Chen QM, Dai L, Li CL, Zhang JY and Yang AL: HuaChanSu suppresses
tumor growth and interferes with glucose metabolism in
hepatocellular carcinoma cells by restraining Hexokinase-2. Int J
Biochem Cell Biol. 142:1061232022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huang J, Chen F, Zhong Z, Tan HY, Wang N,
Liu Y, Fang X, Yang T and Feng Y: Interpreting the pharmacological
mechanisms of huachansu capsules on hepatocellular carcinoma
through combining network pharmacology and experimental evaluation.
Front Pharmacol. 11:4142020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Laussel C and Léon S: Cellular toxicity of
the metabolic inhibitor 2-deoxyglucose and associated resistance
mechanisms. Biochem Pharmacol. 182:1142132020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ma WK, Voss DM, Scharner J, Costa ASH, Lin
KT, Jeon HY, Wilkinson JE, Jackson M, Rigo F, Bennett CF and
Krainer AR: ASO-Based PKM splice-switching therapy inhibits
hepatocellular carcinoma growth. Cancer Res. 82:900–915. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Israelsen WJ and Vander Heiden MG:
Pyruvate kinase: Function, regulation and role in cancer. Semin
Cell Dev Biol. 43:43–51. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen D, Wang Y, Lu R, Jiang X, Chen X,
Meng N, Chen M, Xie S and Yan GR: E3 ligase ZFP91 inhibits
Hepatocellular Carcinoma Metabolism Reprogramming by regulating PKM
splicing. Theranostics. 10:8558–8572. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tan HW, Leung CO, Chan KK, Ho DW, Leung
MS, Wong CM, Ng IO and Lo RC: Deregulated GATA6 modulates stem
cell-like properties and metabolic phenotype in hepatocellular
carcinoma. Int J Cancer. 145:1860–1873. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Panda AC: Circular RNAs Act as miRNA
Sponges. Adv Exp Med Biol. 1087:67–79. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li Q, Pan X, Zhu D, Deng Z, Jiang R and
Wang X: Circular RNA MAT2B promotes glycolysis and malignancy of
hepatocellular carcinoma through the miR-338-3p/PKM2 axis under
hypoxic stress. Hepatology. 70:1298–1316. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang M, Zhang H, Hong H and Zhang Z:
MiR-374b re-sensitizes hepatocellular carcinoma cells to sorafenib
therapy by antagonizing PKM2-mediated glycolysis pathway. Am J
Cancer Res. 9:765–778. 2019.PubMed/NCBI
|
|
38
|
Tang W, Chen Z, Zhang W, Cheng Y, Zhang B,
Wu F, Wang Q, Wang S, Rong D, Reiter FP, et al: The mechanisms of
sorafenib resistance in hepatocellular carcinoma: theoretical basis
and therapeutic aspects. Signal Transduct Target Ther. 5:872020.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen M, Liu H, Li Z, Ming AL and Chen H:
Mechanism of PKM2 affecting cancer immunity and metabolism in tumor
microenvironment. J Cancer. 12:3566–3574. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Feng Y, Xiong Y, Qiao T, Li X, Jia L and
Han Y: Lactate dehydrogenase A: A key player in carcinogenesis and
potential target in cancer therapy. Cancer Med. 7:6124–6136. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Malvi P, Rawat V, Gupta R and Wajapeyee N:
Transcriptional, chromatin, and metabolic landscapes of LDHA
inhibitor-resistant pancreatic ductal adenocarcinoma. Front Oncol.
12:9264372022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang S, Zhou L, Ji N, Sun C, Sun L, Sun J,
Du Y, Zhang N, Li Y, Liu W and Lu W: Targeting ACYP1-mediated
glycolysis reverses lenvatinib resistance and restricts
hepatocellular carcinoma progression. Drug Resist Updat.
69:1009762023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang X, Zhang P and Deng K: MYC Promotes
LDHA Expression through MicroRNA-122-5p to potentiate glycolysis in
hepatocellular carcinoma. Anal Cell Pathol (Amst).
2022:14351732022.PubMed/NCBI
|
|
44
|
Zhang HF, Wang YC and Han YD: MicroRNA-34a
inhibits liver cancer cell growth by reprogramming glucose
metabolism. Mol Med Rep. 17:4483–4489. 2018.PubMed/NCBI
|
|
45
|
Hua S, Liu C, Liu L and Wu D: miR-142-3p
inhibits aerobic glycolysis and cell proliferation in
hepatocellular carcinoma via targeting LDHA. Biochem Biophys Res
Commun. 496:947–954. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cai J, Chen Z, Zhang Y, Wang J, Zhang Z,
Wu J, Mao J and Zuo X: CircRHBDD1 augments metabolic rewiring and
restricts immunotherapy efficacy via m(6)A modification in
hepatocellular carcinoma. Mol Ther Oncolytics. 24:755–771. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Luo X, Zheng E, Wei L, Zeng H, Qin H,
Zhang X, Liao M, Chen L, Zhao L, Ruan XZ, et al: The fatty acid
receptor CD36 promotes HCC progression through activating
Src/PI3K/AKT axis-dependent aerobic glycolysis. Cell Death Dis.
12:3282021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhao C, Wang B, Liu E and Zhang Z: Loss of
PTEN expression is associated with PI3K pathway-dependent metabolic
reprogramming in hepatocellular carcinoma. Cell Commun Signal.
18:1312020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zheng YL, Li L, Jia YX, Zhang BZ, Li JC,
Zhu YH, Li MQ, He JZ, Zeng TT, Ban XJ, et al: LINC01554-Mediated
glucose metabolism reprogramming suppresses tumorigenicity in
hepatocellular carcinoma via downregulating PKM2 expression and
inhibiting Akt/mTOR signaling pathway. Theranostics. 9:796–810.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li J, Hu ZQ, Yu SY, Mao L, Zhou ZJ, Wang
PC, Gong Y, Su S, Zhou J, Fan J, et al: CircRPN2 inhibits aerobic
glycolysis and metastasis in hepatocellular carcinoma. Cancer Res.
82:1055–1069. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Li X, Zhang Y, Ma W, Fu Q, Liu J, Yin G,
Chen P, Dai D, Chen W, Qi L, et al: Enhanced glucose metabolism
mediated by CD147 contributes to immunosuppression in
hepatocellular carcinoma. Cancer Immunol Immunother. 69:535–548.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lin D and Wu J: Hypoxia inducible factor
in hepatocellular carcinoma: A therapeutic target. World J
Gastroenterol. 21:12171–12178. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Iyer NV, Kotch LE, Agani F, Leung SW,
Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY
and Semenza GL: Cellular and developmental control of O2
homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev.
12:149–162. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Guo W, Qiu Z, Wang Z, Wang Q, Tan N, Chen
T, Chen Z, Huang S, Gu J, Li J, et al: MiR-199a-5p is negatively
associated with malignancies and regulates glycolysis and lactate
production by targeting hexokinase 2 in liver cancer. Hepatology.
62:1132–1144. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chen Z, Zuo X, Zhang Y, Han G, Zhang L, Wu
J and Wang X: MiR-3662 suppresses hepatocellular carcinoma growth
through inhibition of HIF-1α-mediated Warburg effect. Cell Death
Dis. 9:5492018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Luo W, Hu H, Chang R, Zhong J, Knabel M,
O'Meally R, Cole RN, Pandey A and Semenza GL: Pyruvate kinase M2 is
a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell.
145:732–744. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhang Y, Zhang C, Zhao Q, Wei W, Dong Z,
Shao L, Li J, Wu W, Zhang H, Huang H, et al: The miR-873/NDFIP1
axis promotes hepatocellular carcinoma growth and metastasis
through the AKT/mTOR-mediated Warburg effect. Am J Cancer Res.
9:927–944. 2019.PubMed/NCBI
|
|
58
|
Kowalik MA, Columbano A and Perra A:
Emerging role of the pentose phosphate pathway in hepatocellular
carcinoma. Front Oncol. 7:872017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Stincone A, Prigione A, Cramer T, Wamelink
MM, Campbell K, Cheung E, Olin-Sandoval V, Grüning NM, Krüger A,
Tauqeer Alam M, et al: The return of metabolism: biochemistry and
physiology of the pentose phosphate pathway. Biol Rev Camb Philos
Soc. 90:927–963. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lu M, Lu L, Dong Q, Yu G, Chen J, Qin L,
Wang L, Zhu W and Jia H: Elevated G6PD expression contributes to
migration and invasion of hepatocellular carcinoma cells by
inducing epithelial-mesenchymal transition. Acta Biochim Biophys
Sin (Shanghai). 50:370–380. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Barajas JM, Reyes R, Guerrero MJ, Jacob
ST, Motiwala T and Ghoshal K: The role of miR-122 in the
dysregulation of glucose-6-phosphate dehydrogenase (G6PD)
expression in hepatocellular cancer. Sci Rep. 8:91052018.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Qin Z, Xiang C, Zhong F, Liu Y, Dong Q, Li
K, Shi W, Ding C, Qin L and He F: Transketolase (TKT) activity and
nuclear localization promote hepatocellular carcinoma in a
metabolic and a non-metabolic manner. J Exp Clin Cancer Res.
38:1542019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Jia D, Liu C, Zhu Z, Cao Y, Wen W, Hong Z,
Liu Y, Liu E, Chen L, Chen C, et al: Novel transketolase inhibitor
oroxylin A suppresses the non-oxidative pentose phosphate pathway
and hepatocellular carcinoma tumour growth in mice and
patient-derived organoids. Clin Transl Med. 12:e10952022.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Luo X, Cheng C, Tan Z, Li N, Tang M, Yang
L and Cao Y: Emerging roles of lipid metabolism in cancer
metastasis. Mol Cancer. 16:762017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Shimano H and Sato R: SREBP-regulated
lipid metabolism: Convergent physiology-divergent pathophysiology.
Nat Rev Endocrinol. 13:710–730. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Muku GE, Blazanin N, Dong F, Smith PB,
Thiboutot D, Gowda K, Amin S, Murray IA and Perdew GH: Selective Ah
Receptor Ligands Mediate Enhanced SREBP1 Proteolysis to Restrict
Lipogenesis in Sebocytes. Toxicol Sci. 171:146–158. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Li H, Chen Z, Zhang Y, Yuan P, Liu J, Ding
L and Ye Q: MiR-4310 regulates hepatocellular carcinoma growth and
metastasis through lipid synthesis. Cancer Lett. 519:161–171. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Chen J, Ding C, Chen Y, Hu W, Yu C, Peng
C, Feng X, Cheng Q, Wu W, Lu Y, et al: ACSL4 reprograms fatty acid
metabolism in hepatocellular carcinoma via c-Myc/SREBP1 pathway.
Cancer Lett. 502:154–165. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Liu Y, Sun L, Guo H, Zhou S, Wang C, Ji C,
Meng F, Liang S, Zhang B, Yuan Y, et al: Targeting SLP2-mediated
lipid metabolism reprograming restricts proliferation and
metastasis of hepatocellular carcinoma and promotes sensitivity to
Lenvatinib. Oncogene. 42:374–388. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Chan DC: Mitochondrial dynamics and its
involvement in disease. Annu Rev Pathol. 15:235–259. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Wu D, Yang Y, Hou Y, Zhao Z, Liang N, Yuan
P, Yang T, Xing J and Li J: Increased mitochondrial fission drives
the reprogramming of fatty acid metabolism in hepatocellular
carcinoma cells through suppression of Sirtuin 1. Cancer Commun
(Lond). 42:37–55. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Li Q, Yao H, Wang Y, Wu Y, Thorne RF, Zhu
Y, Wu M and Liu L: circPRKAA1 activates a Ku80/Ku70/SREBP-1 axis
driving de novo fatty acid synthesis in cancer cells. Cell Rep.
41:1117072022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Mok EHK, Leung CON, Zhou L, Lei MML, Leung
HW, Tong M, Wong TL, Lau EYT, Ng IOL, Ding J, et al:
Caspase-3-Induced Activation of SREBP2 Drives drug resistance via
promotion of cholesterol biosynthesis in hepatocellular carcinoma.
Cancer Res. 82:3102–3115. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Svensson RU, Parker SJ, Eichner LJ, Kolar
MJ, Wallace M, Brun SN, Lombardo PS, Van Nostrand JL, Hutchins A,
Vera L, et al: Inhibition of acetyl-CoA carboxylase suppresses
fatty acid synthesis and tumor growth of non-small-cell lung cancer
in preclinical models. Nat Med. 22:1108–1119. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zaytseva YY, Rychahou PG, Le AT, Scott TL,
Flight RM, Kim JT, Harris J, Liu J, Wang C, Morris AJ, et al:
Preclinical evaluation of novel fatty acid synthase inhibitors in
primary colorectal cancer cells and a patient-derived xenograft
model of colorectal cancer. Oncotarget. 9:24787–24800. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Peck B, Schug ZT, Zhang Q, Dankworth B,
Jones DT, Smethurst E, Patel R, Mason S, Jiang M, Saunders R, et
al: Inhibition of fatty acid desaturation is detrimental to cancer
cell survival in metabolically compromised environments. Cancer
Metab. 4:62016. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Stine ZE, Schug ZT, Salvino JM and Dang
CV: Targeting cancer metabolism in the era of precision oncology.
Nat Rev Drug Discov. 21:141–162. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Cheng C, Geng F, Cheng X and Guo D: Lipid
metabolism reprogramming and its potential targets in cancer.
Cancer Commun (Lond). 38:272018.PubMed/NCBI
|
|
79
|
Li B, Cao Y, Meng G, Qian L, Xu T, Yan C,
Luo O, Wang S, Wei J, Ding Y and Yu D: Targeting glutaminase 1
attenuates stemness properties in hepatocellular carcinoma by
increasing reactive oxygen species and suppressing Wnt/beta-catenin
pathway. EBioMedicine. 39:239–254. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Altman BJ, Stine ZE and Dang CV: From
Krebs to clinic: Glutamine metabolism to cancer therapy. Nat Rev
Cancer. 16:619–634. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Marsico M, Santarsiero A, Pappalardo I,
Convertini P, Chiummiento L, Sardone A, Di Noia MA, Infantino V and
Todisco S: Mitochondria-Mediated Apoptosis of HCC cells triggered
by knockdown of glutamate dehydrogenase 1: Perspective for its
inhibition through quercetin and permethylated anigopreissin A.
Biomedicines. 9:16642021. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Dai W, Xu L, Yu X, Zhang G, Guo H, Liu H,
Song G, Weng S, Dong L, Zhu J, et al: OGDHL silencing promotes
hepatocellular carcinoma by reprogramming glutamine metabolism. J
Hepatol. 72:909–923. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Xu K, Ding J, Zhou L, Li D, Luo J, Wang W,
Shang M, Lin B, Zhou L and Zheng S: SMYD2 promotes hepatocellular
carcinoma progression by reprogramming glutamine metabolism via
c-Myc/GLS1 Axis. Cells. 12:252022. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Meric-Bernstam F, Tannir NM, Iliopoulos O,
Lee RJ, Telli ML, Fan AC, DeMichele A, Haas NB, Patel MR, Harding
JJ, et al: Telaglenastat plus cabozantinib or everolimus for
advanced or metastatic renal cell carcinoma: An open-label phase I
trial. Clin Cancer Res. 28:1540–1548. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Li C, Gui G, Zhang L, Qin A, Zhou C and
Zha X: Overview of methionine adenosyltransferase 2A (MAT2A) as an
anticancer target: Structure, function, and inhibitors. J Med Chem.
65:9531–9547. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Hung MH, Lee JS, Ma C, Diggs LP, Heinrich
S, Chang CW, Ma L, Forgues M, Budhu A, Chaisaingmongkol J, et al:
Tumor methionine metabolism drives T-cell exhaustion in
hepatocellular carcinoma. Nat Commun. 12:14552021. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Simile MM, Peitta G, Tomasi ML, Brozzetti
S, Feo CF, Porcu A, Cigliano A, Calvisi DF, Feo F and Pascale RM:
MicroRNA-203 impacts on the growth, aggressiveness and prognosis of
hepatocellular carcinoma by targeting MAT2A and MAT2B genes.
Oncotarget. 10:2835–2854. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Li JT, Yang H, Lei MZ, Zhu WP, Su Y, Li
KY, Zhu WY, Wang J, Zhang L, Qu J, et al: Dietary folate drives
methionine metabolism to promote cancer development by stabilizing
MAT IIA. Signal Transduct Target Ther. 7:1922022. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Chaturvedi S, Hoffman RM and Bertino JR:
Exploiting methionine restriction for cancer treatment. Biochem
Pharmacol. 154:170–173. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kawaguchi K, Han Q, Li S, Tan Y, Igarashi
K, Miyake K, Kiyuna T, Miyake M, Chemielwski B, Nelson SD, et al:
Intra-tumor L-methionine level highly correlates with tumor size in
both pancreatic cancer and melanoma patient-derived orthotopic
xenograft (PDOX) nude-mouse models. Oncotarget. 9:11119–11125.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Wang Z, Yip LY, Lee JHJ, Wu Z, Chew HY,
Chong PKW, Teo CC, Ang HY, Peh KLE, Yuan J, et al: Methionine is a
metabolic dependency of tumor-initiating cells. Nat Med.
25:825–837. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Yang C, Zhang H, Zhang L, Zhu AX, Bernards
R, Qin W and Wang C: Evolving therapeutic landscape of advanced
hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol.
20:203–222. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Todisco S, Convertini P, Iacobazzi V and
Infantino V: TCA cycle rewiring as emerging metabolic signature of
hepatocellular carcinoma. Cancers (Basel). 12:682019. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Wang H, Zhou Y, Xu H, Wang X, Zhang Y,
Shang R, O'Farrell M, Roessler S, Sticht C, Stahl A, et al:
Therapeutic efficacy of FASN inhibition in preclinical models of
HCC. Hepatology. 76:951–966. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Jin H, Wang S, Zaal EA, Wang C, Wu H,
Bosma A, Jochems F, Isima N, Jin G, Lieftink C, et al: A powerful
drug combination strategy targeting glutamine addiction for the
treatment of human liver cancer. Elife. 9:e567492020. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Hay N: Reprogramming glucose metabolism in
cancer: can it be exploited for cancer therapy? Nat Rev Cancer.
16:635–649. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Raez LE, Papadopoulos K, Ricart AD,
Chiorean EG, Dipaola RS, Stein MN, Rocha Lima CM, Schlesselman JJ,
Tolba K, Langmuir VK, et al: A phase I dose-escalation trial of
2-deoxy-D-glucose alone or combined with docetaxel in patients with
advanced solid tumors. Cancer Chemother Pharmacol. 71:523–530.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Reyes R, Wani NA, Ghoshal K, Jacob ST and
Motiwala T: Sorafenib and 2-Deoxyglucose synergistically inhibit
proliferation of both sorafenib-sensitive and -resistant HCC cells
by inhibiting ATP production. Gene Expr. 17:129–140. 2017.
View Article : Google Scholar : PubMed/NCBI
|