Introduction
Colorectal cancer (CRC) is one of the most prevalent
malignancies worldwide, with nearly 2 million new cases reported
annually (1). Although treatment
strategies incorporating surgery and chemotherapy are used for CRC,
up to 23% of patients still experience distant recurrence, known as
metachronous metastasis (2). Such
patients often have a poor prognosis, with 5-year survival rate
<30% (3,4). Moreover, the effectiveness of current
first-line chemotherapy in preventing metachronous metastasis of
CRC is limited (5,6). This calls for in-depth
characterization of the molecular profile of distant recurrence of
CRC following surgery to guide the development of more effective
interventions for high-risk patients.
KRAS is an oncogene involved in the development and
progression of CRC, with ~40% of CRC cases harboring KRAS mutation
(7). Evidence from previous
investigations has indicated that KRAS mutation drives
epithelial-mesenchymal transition (EMT), promoting CRC migration
and invasion (8,9). KRAS mutation might serve as a risk
factor for metachronous metastasis in CRC (10–12).
Therefore, a novel risk assessment model and targeted therapy
tailored for patients with KRAS mutation are needed to offer new
perspectives for prevention of metachronous metastasis in CRC.
KRAS has long been considered an undruggable target.
However, studies have led to the development of KRAS inhibitors
(13,14). Adagrasib, a small molecule which
traps KRASG12C in the inactive state (GDP-bound), has
been demonstrated to be efficacious in a CRC clinical trial
(15). A recent study developed a
pan-KRAS inhibitor, BI2865, which binds to a residue in the switch
II binding pocket (His95) present only in KRAS, sparing other RAS
family proteins, which showed satisfactory results in a
pre-clinical trial (16). However,
whether such KRAS inhibitors prevent CRC metastasis remains to be
clarified. Existing literature suggests that KRAS mutations drive
EMT via TGF-β signaling in CRC (8,17).
Thus, it was hypothesized that KRAS inhibitors suppress CRC
metastasis by restraining EMT.
The present study aimed to verify the association
between KRAS mutation and metachronous metastasis of CRC and
develop a tool for identifying patients with high-risk CRC who are
likely to experience metachronous metastasis, as well as assess the
potential of KRAS inhibitors on preventing CRC metachronous
metastasis and explore the underlying mechanism.
Materials and methods
Meta-analysis
A systemic literature review was performed on the
PubMed database (pubmed.ncbi.nlm.nih.gov), Embase (embase.com) and
Cochrane library (cochrane.org). The search was conducted using the
following terms: [(KRAS) or (K-RAS)] and [(colorectal cancer) or
(colon cancer) or (rectal cancer)] and [(metachronous metastasis)
or (distant recurrence) or (distant relapse)]. The study was
performed in line with the guidelines of PRISMA (18). Inclusion criteria were as follows:
i) Study included patients who were initially diagnosed with stage
I–III CRC and underwent curative surgery; ii) KRAS mutational
status was provided and iii) follow-up information including
distant recurrence after surgery was reported. Duplicates,
non-English literature, reviews, editorials, case reports,
abstracts and studies unrelated to CRC were excluded (Fig. S1A). A systematic full-text review
was conducted on eligible literature, to collect information,
including the first author name, publication date, sample size,
KRAS mutation rate and metachronous survival rate. The odds ratio
(OR) and 95% confidence interval (CI) were utilized as the outcome
measures. The OR >1 indicated that the KRAS mutation was a risk
factor for metachronous metastasis. Heterogeneity was examined
using I2 and Cochran's Q statistics. An I2
value >50% indicated substantial heterogeneity. Statistical
significance was determined based on P<0.05 obtained by the Wald
test. The results were presented using a forest plot. Publication
bias in the studies was determined using a funnel plot. Figures
were plotted with R (Version:4.0.3; R-project.org).
Data acquisition
Clinical information, KRAS mutation status and
transcriptome sequencing data of 1,001 patients with CRC were
downloaded from the Omics Study of CRC (COCC) in China
(icgc-argo.org/page/114/cgcc) cohort of International Cancer Genome
Consortium Accelerating Research in Genomic Oncology (ICGC-ARGO)
project. Moreover, clinical data and KRAS mutational status were
derived from the MSKCC (Memorial Sloan-Kettering Cancer Center)
cohort reported by Chatila et al (19) and Sidra-LUMC (Leiden University
Medical Center) AC-ICAM (Atlas and Compass of
Immune-Cancer-Microbiome interactions) cohort data reported by
Roelands et al (20) were
retrieved from cbioportal (cbioportal.org). Transcriptome
sequencing data were downloaded from the Gene Expression Omnibus
(GEO; accession no. GSE209746;
ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE209746) and Genotypes and
Phenotypes (accession no. phs002978.v1.p1;
ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs002978.v1.p1).
Transcriptome data of 42 KRAS-MUT CRC cell lines were acquired from
the Cancer Cell Line Encyclopedia (CCLE) database
(sites.broadinstitute.org/ccle/datasets).
Model construction
To construct the metachronous metastatic risk
prediction model for patients with KRAS-mutant CRC, 286 cases with
KRAS mutation from the ICGC-ARGO cohort were selected as the
training cohort. In addition, 77 KRAS-mutant cases from the
Sidra-LUMC AC-ICAM cohort and 32 KRAS-mutant cases from the MSKCC
cohort with eligible gene expression data were enrolled as
independent validation cohorts. Inclusion criteria were as follows:
i) Patients with stage I–III CRC who underwent curative surgery and
ii) CRC with KRAS-mutation. Cases with no sequencing data and
incomplete clinical information were excluded. In the training
cohort, Lasso-Cox regression was performed to identify
metastasis-associated genes and establish KRAS
metachronous-metastasis (KMM) score (KMM score=∑i
coefficient (genei) × expression (genei)).
The patients were stratified into high- and low-risk groups based
on the unsupervised KMM score cutoff (ARGO, −0.2184; Sidra-LUMC
AC-ICAM, −0.0362; MSKCC, −31.1379) determined by the Youden index.
Kaplan-Meier, differential gene expression and gene set enrichment
analysis (GSEA) were performed using R (Version:4.0.3;
r-project.org) to compare differences in metastasis, gene
expression and pathway activation pattern between high- and
low-risk groups.
Cell culture
The cell lines were purchased from Meisen Chinese
Tissue Culture Collections (Zhejiang Meisen Cell Technology, Ltd.)
and authenticated using short tandem repeat analysis. The cell
lines were confirmed to be KRAS-mutant CRC (SW480: KRAS-G12V;
HCT116: KRAS-G13D; SW837: KRAS-G12C; DLD1: KRAS-G13D; HCT15:
KRAS-G13D) making them suitable models for testing KRAS inhibitors
(21). The cells were cultured in
DMEM (SW480, HCT116 and SW837) or RPMI-1640 (both Gibco; Thermo
Fisher Scientific, Inc.) (DLD1 and HCT15) supplemented with 10%
fetal bovine serum (HyClone, Cytiva) and 1% penicillin-streptomycin
(Gibco; Thermo Fisher Scientific, Inc.) in 5% CO2
environment at 37°C. Monthly mycoplasma testing was performed on
the cells. BI-2865 (1 µM), adagrasib (60 nM), oxaliplatin (1 µM)
and 5-fluorouracil (5 µM) were purchased from TargetMol. After drug
addition, all the cells were incubated at 37°C for 48 h. The human
recombinant TGF-β1 protein was purchased from MedChemExpress.
Colony formation assay
SW837 cells were dissociated by digestion with
trypsin, resuspended in complete culture DMEM as single cells and
seeded into six-well plates (20,000 cells/well) at 37°C. The medium
was changed every 3 days and the cells were cultured for 12 days at
37°C. The cell colonies were fixed with 20% methanol for 15 min at
room temperature, stained with crystal violet (0.5%) for 15 min at
room temperature and then imaged. The colony is defined to consist
of at least 50 cells. Colonies were quantified using ImageJ
(Version:1.53q; imagej.net/ij).
Wound healing assay
The cells (HCT116, SW480, DLD1, HCT15 and SW837)
were seeded in a six-well plate at a density of 5×105
cells/well and incubated overnight at 37°C to form a 100% confluent
monolayer. Complete medium was replaced with serum-free medium
(RPMI-1640 for DLD1 and HCT15; DMEM for HCT116, SW480 and SW837) as
previously described (22). A
scratch was created on the cell surface using a 200-µl pipette tip.
After incubating at 37°C for 48 h, wounds were photographed under
Olympus IX73 phase-contrast inverted light microscopy on 40×
magnification and images were analyzed using ImageJ (Version:1.53q;
imagej.net/ij).
Transwell assay
A total of 1×105 cells (HCT116, SW480,
DLD1, HCT15 and SW837) was resuspended in 200 µl serum-free medium
(RPMI-1640 for DLD1 and HCT15; DMEM for HCT116, SW480 and SW837)
and seeded into the upper chamber of a Transwell plate (8 µm pore
size; Corning, Inc.). The upper chamber was pre-coated with
Matrigel (1:20, cat. no. 356234; Corning, Inc.) at 37°C for 30 min.
Next, 500 µl medium (RPMI-1640 for DLD1 and HCT15; DMEM for HCT116,
SW480 and SW837) containing 10% fetal bovine serum (HyClone,
Cytiva) was added to the lower chamber. After incubation at 37°C
for 48 h, cells were fixed with 4% paraformaldehyde for 15 min at
room temperature and then stained with 1% crystal violet solution
for 15 min at room temperature. A total of five randomly selected
fields of view/plate was visualized under Olympus IX73 inverted
light microscope on 200× magnification. Finally, the images were
processed using ImageJ (Version:1.53q; imagej.net/ij).
Organoid culture
The source and culture of organoids was performed
using a previously published method (23).
Cell viability assay
The cells (HCT116, SW480, DLD1, HCT15 and SW837)
were cultured in 96-well plates (2,000 cells per well, with three
replicates for each group. After incubating at 37°C for 24 h, cells
were treated with varying concentrations of drugs and incubated at
37°C for 96 h. To test the IC50 of HCT116, SW480, DLD1 and HCT15,
the gradient concentrations of oxaliplatin and 5-fluorouracil are 1
nM, 10 nM, 100 nM, 500 nM, 1 µM, 5 µM, 10 µM, 50 µM, 100 µM and 1
mM. To test the IC50 of SW837, concentrations of adagrasib are 100
pM, 1 nM, 5 nM, 10 nM, 50 nM, 100 nM, 500 nM, 1 µM, 5 µM and 10 µM.
The concentration of adagrasib for growth curve assay (SW837) is
500 nM. The number of viable cells was determined using the
CellTiter-Glo Luminescent Cell Viability Assay (cat. no. #G7573,
Promega Corporation), following the manufacturer's protocol.
Establishment of cell lines with
acquired resistance to adagrasib
1×10^6 SW837 cells were cultured with 60 nM
adagrasib at 37°C for 72 h. Next, 50% confluent cells were treated
with adagrasib at IC50 (60 nM) for 72 h, followed by a 96 h
incubation at 37°C. This cycle was repeated once and the remaining
cells were cultured with adagrasib (60 nM) for 3 passages. The
resistant cells were continuously cultured in the presence of
adagrasib (60 nM) at 37°C for 72 h and the passaged parental cells
were utilized for cellular assays.
Western blotting
Total protein of cells was extracted using RIPA cell
lysis buffer, containing 150 mM NaCl, 50 mM Tris-HCl, 0.5% sodium
deoxycholate, 200 mM NaF, 200 mM PMSF, 1.0% NP-40 and 1 mM EDTA,
supplemented with a protease inhibitor cocktail (Roche
Diagnostics). Protein lysate was determined using BCA method. Equal
amounts of protein (15 µg/lane), boiled with a pre-stained protein
marker (cat. no. M221, Beijing Kangrun Chengye Biotechnology Co.,
Ltd.) was subjected to 10% SDS-PAGE and then transferred to PVDF
membranes for immunoblotting analysis. After blocking with TBST
(0.1% Tween-20) containing 5% non-fat dry milk powder for 2 h at
room temperature, the membranes were incubated overnight at 4°C
with primary antibodies. The membranes were incubated with
secondary HRP-conjugated antibodies at room temperature for 2 h and
then protein bands were visualized with ECL) western-blotting kit
(Tanon, Cat. No.: 180-5001) using ChemiDoc (Bio-Rad Laboratories,
Inc.) system. The density of the bands was determined using ImageJ
(Version:1.53q; imagej.net/ij). The antibodies are presented in
Table SI.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted from the cells (SW480,
HCT116 and SW837) using the RNeasy Mini kit (Qiagen GmbH) according
to the manufacturer's instructions. A total of 1 µg extracted RNA
was reverse-transcribed to cDNA using the RT kit (cat. no.
#AT341-02, Transgen Biotech Co., Ltd.) before qPCR using the PCR
kit (cat. no. #N30920, Transgen Biotech Co., Ltd.) on a Biorad CFX
Real-time PCR system. Each qPCR experiment was performed in
triplicate and the average values were calculated for each gene.
18S rRNA served as the internal control for normalization. The
relative expression level of mRNA was calculated using
2−ΔΔCq. The primer sequences are provided in Table SII.
RNA sequencing (seq)
Total RNA of SW837 cells was isolated using the
RNAeasy Mini kit (Qiagen GmbH, cat. no. 74104) following the
manufacturer's instructions. Integrity of RNA was verified using
4200 Bioanalyzer (Agilent). Subsequently, mRNA libraries were
prepared using the TruSeq Stranded Total RNA Sample Preparation kit
on the Ribo Zero Gold (Illumina, Inc.). Concentration of mRNA
libraries were measured using Qubit 3.0 fluorometer dsDNA HS Assay
(Thermo Fisher Scientific). 0.5 nM prepared libraries were loaded
on the NovaSeq 6000 platform (Illumina, Inc.) and sequenced under
150 bp paired end mode using NovaSeq 6000 S4 Reagent Kit v1.5 (300
cycles, cat. No. 20028312). For RNA-seq analysis, raw sequencing
reads were first processed with fastp (version 0.12.5;
github.com/OpenGene/fastp) to remove low-quality sequences and
adapters. The cleaned reads were aligned to the human reference
genome (GRCh38, hg38) using STAR (version 2.7.0f;
github.com/alexdobin/STAR) under default parameters (24,25).
The gene expression levels were quantified using RNA-seq by
Expectation-Maximization (26).
Differentially expressed genes were identified by normalizing raw
counts and applying the DESeq2 package (version 1.44.0;
bioconductor.org/packages/release/bioc/html/DESeq2.html), based on
the threshold of |log2 fold-change|>1 and an adjusted P-value
<0.05. Pathway enrichment analysis was conducted using GSEA
desktop software (version 4.3.3;
gsea-msigdb.org/gsea/downloads.jsp), focusing on Kyoto Encyclopedia
of Genes and Genomes and Hallmark gene sets (27–29).
Animal experiments
A total of 12 male BALB/c nude mice (age, 6 weeks;
weight, 18–22 g were purchased from Beijing Vital River Laboratory
Animal Technology Co., Ltd. and housed in the Biological Resource
Centre of the sixth affiliated hospital, Sun Yat-sen University,
Guangzhou, China. The mice were given ad libitum access to food and
water under a 12/12-h dark/light cycle with 23–25°C temperature,
55~60% controlled humidity. 5×106 SW480 cells were
treated with either DMSO or BI2865 (1 µM) at 37°C for 96 h. Cells
were washed with PBS and cultured in a complete DMEM (Gibco; Thermo
Fisher Scientific, Inc.) at 37°C for 48 h and 2×106
cells in 50 µl sterile saline were injected into the spleen of nude
mice (6 mice/group). Mice were palpated every 2 days to monitor
tumor progression. Animal health and behavior (signs of distress
including hunched posture, ruffled coat, lack of appetite and
dehydration) were monitored daily. Mice demonstrating ≥15% weight
loss within 12 weeks were euthanized. No animal reached the humane
endpoint during the experiment. Body weight of the animals was
measured every 2 days. After 12 weeks, mice were euthanized by
CO2 inhalation using a flow rate of 3 l/min for 3 min.
After respiratory arrest, mice were continually exposed to
CO2 for ≥15 min to ensure death. Following animal death
confirmation (loss of heartbeat), livers were collected,
photographed and preserved in 4% paraformaldehyde at room
temperature for 24 h for further hematoxylin-eosin staining. Fixed
liver samples were embedded in paraffin and then sliced into 5 µm
sections. The sections were stained using hematoxylin for 8 min and
eosin for 5 min at room temperature. After staining, the sections
were scanned using PANNORAMIC DESK II DW (3DHISTECH, Hungary)
system. Scanned images were processed using 3DHISTECH CaseViewer
(version: 2.4.0, 3dhistech.com). The animal experiment was
conducted in compliance with animal protocols approved by the
Animal Research Committee of the Sixth Affiliated Hospital of Sun
Yat-sen University, Guangzhou, China (approval no.
SYSU-IACUC-2022-080601).
Statistical analysis
In survival analysis, clinical data obtained from
the ICGC-ARGO, MSKCC and Sidra-LUMC AC-ICAM databases were
screened. The exclusion criteria were as follows: i) Cases without
KRAS mutation status; ii) patients initially diagnosed with stage
IV CRC; iii) distant recurrence information missing and iv) local
recurrence. The remaining patients were included in subsequent
analysis of metachronous-metastasis free survival (MFS; Fig. S2A-C). Metachronous metastasis was
defined as distant metastasis following initial diagnosis or
primary surgery (30). To
investigate the impact of chemotherapy on metachronous metastasis
in CRC, propensity score matching was performed to eliminate bias
toward clinical decision-making of chemotherapy caused by
postoperative pathological staging. Matched cases were included in
the subsequent survival analysis. The Kaplan-Meier method was
employed to estimate cumulative MFS rates; P-values, hazard ratio
(HR) and 95% CI were assessed using log-rank test. R
(Version:4.0.3; r-project.org) was utilized to construct
Kaplan-Meier curves. Propensity score matching was performed using
SPSS 26.0 (IBM Corp.).
All experiments were replicated three times and data
are presented as the mean ± SD. Statistical differences between two
groups were determined using two-tailed unpaired Student's t test.
Multiple groups were compared with two-way ANOVA followed by
Dunnett's multiple comparisons or one-way ANOVA followed by
Bonferroni's post hoc test. All statistical analyses were performed
using GraphPad Prism software (version: 9.5.0; Dotmatics).
P<0.05 was considered to indicate a statistically significant
difference.
Results
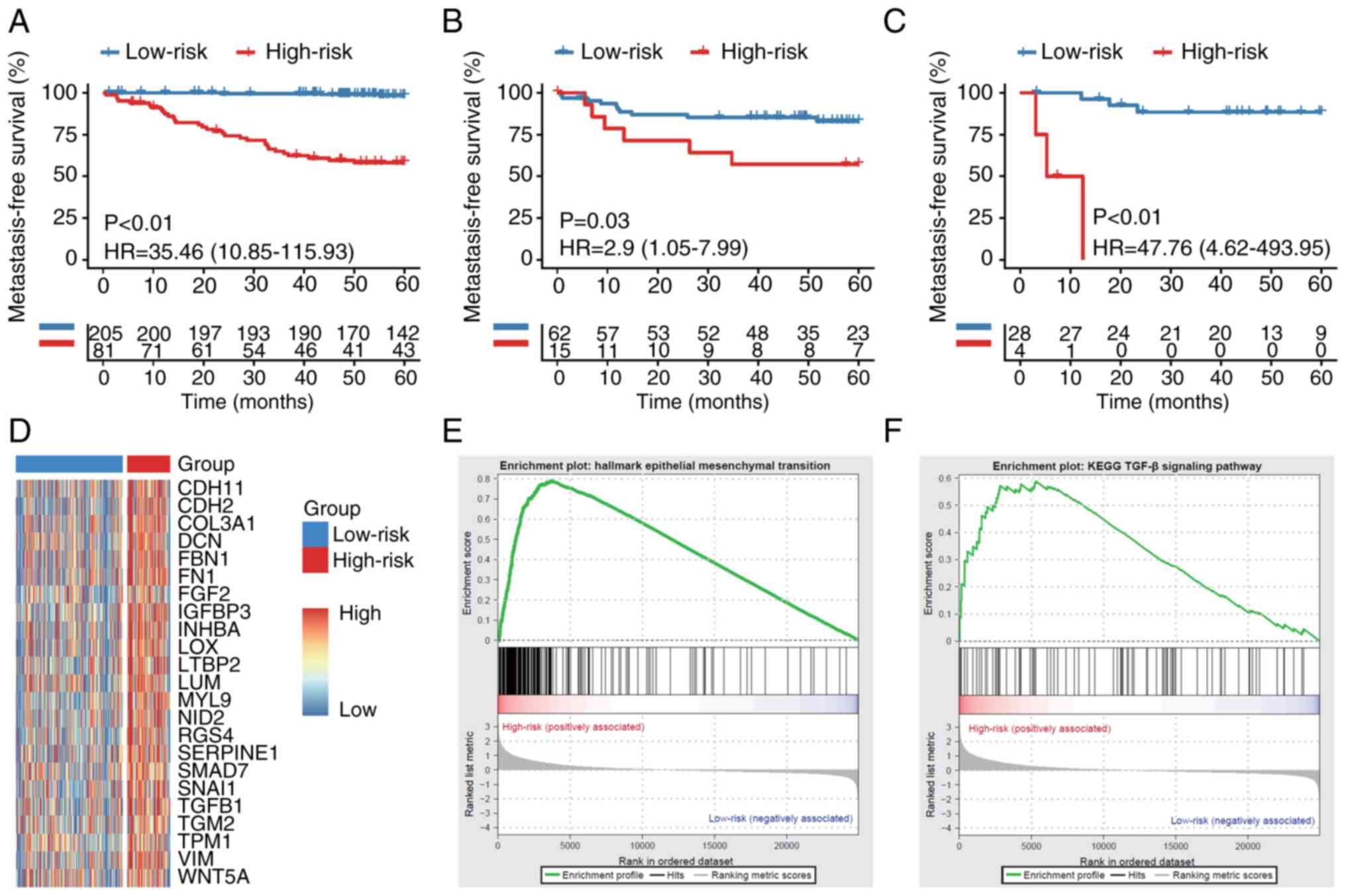
KRAS mutation is a risk factor for CRC
metachronous metastasis
KRAS mutation is a predictive factor for
metachronous metastasis of CRC (31–33).
The present study included meta-analysis of the available
literature. The initial search identified 210 records (Fig. S1A), which were screened, leading
to the exclusion of 117 articles. A review of reference lists of
the remaining articles resulted in 11 relevant articles which were
pooled for the meta-analysis (Fig.
1A). These articles comprised 1,296 patients, of whom 387 had
mutant KRAS (10,12,32,34–41).
The results from these studies were generally consistent, with KRAS
positively associated with CRC metachronous metastasis (Fig. 1A). Analysis of the funnel plot did
not find significant publication bias (Fig. S1B). Further investigations using
ICGC-ARGO database indicated that the KRAS mutation was negatively
associated with postoperative MFS of patients with CRC (Figs. 1B and S2A; Table
SIII). Similar trends were observed in two other online
databases, however differences were not significant (Figs. 1C and D and S2B and C; Table SIII) (19,20).
Comparison of MFS between patients with or without chemotherapy
following propensity score matching (Table SIV) showed that adjuvant
chemotherapy significantly prevented distant recurrence in KRAS-WT
CRC, while the efficacy was markedly limited in patients with
KRAS-MUT (Fig. 1E-J). These
results demonstrate that the KRAS mutation may be a risk factor for
CRC metachronous metastasis and first-line chemotherapy does not
significantly improve metastasis-free survival in patients with
KRAS-MUT CRC after curative surgery.
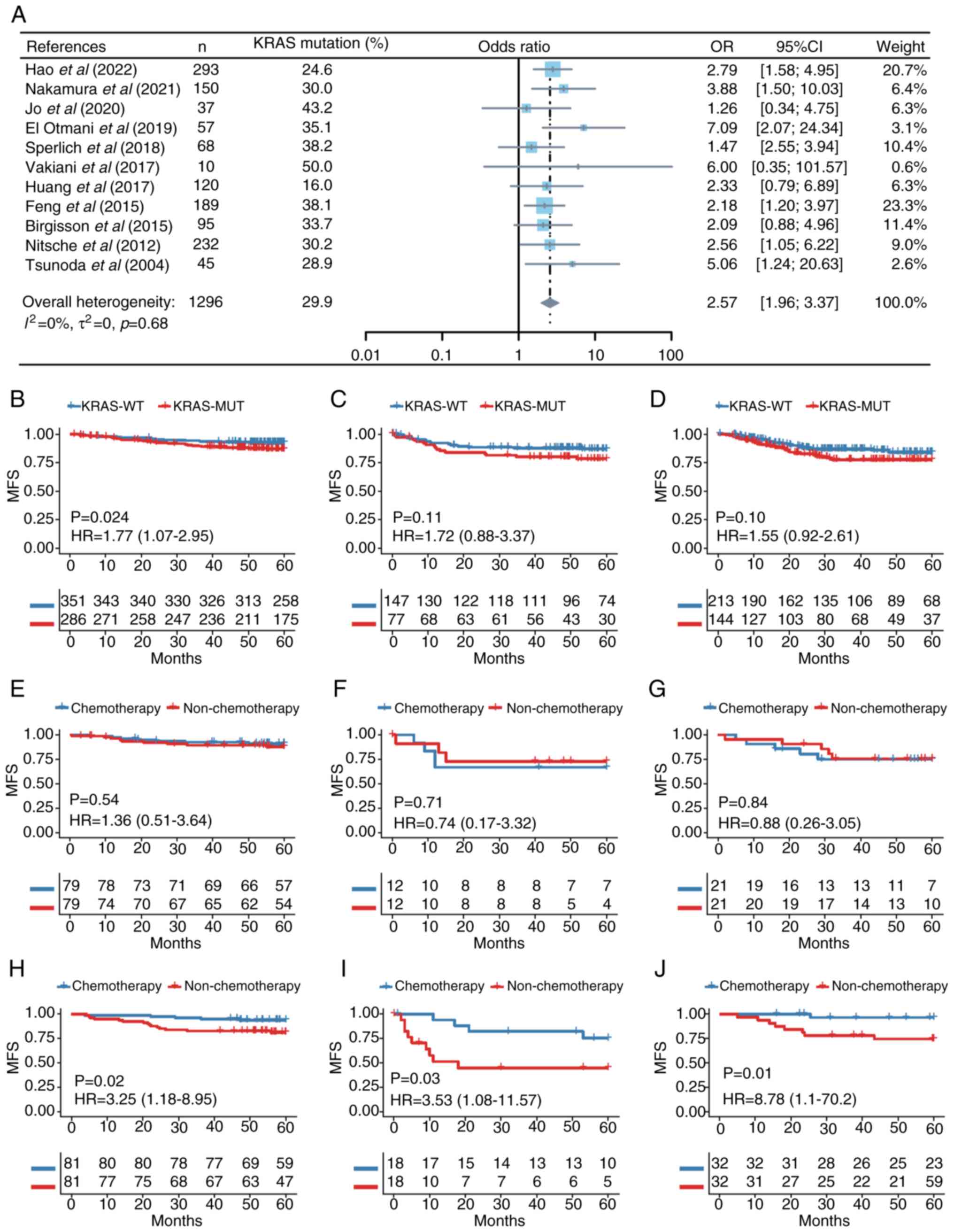 | Figure 1.KRAS mutation is associated with CRC
metachronous metastasis. (A) Association between KRAS mutation
status and CRC metachronous metastasis in 11 studies. A
random-effects model with inverse-variance method was used in the
meta-analysis. MFS between patients with KRAS-MUT and KRAS-WT CRC
in (B) ARGO, (C) Sidra-LUMC AC-ICAM and (D) MSKCC cohort.
Comparison of MFS between patients with KRAS-MUT with or without
chemotherapy in (E) ARGO, (F) Sidra-LUMC AC-ICAM and (G) MSKCC
cohort. Comparison of MFS between patients with KRAS-WT with or
without chemotherapy in (H) ARGO, (I) Sidra-LUMC AC-ICAM and (J)
MSKCC cohort. CRC, colorectal cancer; MFS, metachronous-metastasis
free survival; MUT, mutant; WT, wild-type; ARGO, Accelerating
Research in Genomic Oncology; LUMC AC-ICAM, Leiden University
Medical Center Atlas and Compass of Immune-Cancer-Microbiome
interactions; MSKCC, Memorial Sloan-Kettering Cancer Center; HR,
hazard ratio. |
Machine-learning method stratifies
KRAS-MUT CRC based on the risk of metachronous metastasis
A metastatic risk prediction model consisting of 36
genes (Fig. S3A-C) was
constructed and validated using complete RNA sequencing data and
follow-up information from 395 KRAS-MUT CRC cases across three
cohorts. Using KMM score, patients in the training cohort (COCC,
n=286) and two validation cohorts (Sidra-LUMC AC-ICAM, n=77; MSKCC,
n=32) were categorized as high- or low-risk. Kaplan-Meier curve
analysis showed that patients in the high-risk group had
significantly worse MFS (Fig.
2A-C). Furthermore, differential gene expression analysis
demonstrated significantly higher expression of genes associated
with the epithelial-mesenchymal transition (EMT) and TGF-β pathway
in the high-compared with the low-risk group (Fig. 2D) in the ICGC-ARGO cohort. GSEA
revealed that EMT and TGF-β pathways were also highly activated in
the high-risk group (Fig. 2E).
However, significant differences were not observed in the heatmaps
of MSKCC and AC-ICAM due to the relatively small sample sizes in
these cohorts (Fig. S3D). These
findings suggested that the highly activated EMT and TGF-β pathways
may contribute to tumor metastasis in patients with KRAS-MUT CRC,
which can be identified by KMM signature.
KRAS inhibitors suppress migration and
invasion ability of KRAS-MUT CRC cells
Using the aforementioned KMM scoring model, the
KRAS-MUT CRC cell lines were classified based on the gene
expression profile in the CCLE database (Fig. 3A). Based on the scoring results,
two KRAS-MUT CRC cell lines with high (HCT116 and SW480) and low
KMM score (DLD1 and HCT15) were selected to evaluate their
sensitivity to chemotherapy and KRAS inhibitors. KMM score
stratification was not associated with chemo-sensitivity (Fig. S4). CRC cell lines were treated
with 5-fluorouracil and oxaliplatin to assess the effect of
first-line chemotherapy on migration and invasion ability. Wound
healing and Transwell experiments showed that chemotherapy
significantly decreased the migration and invasion ability of KMM
low-risk CRC cell lines (Fig.
S5A-D) but had no significant effect on KMM high-risk CRC cell
lines (Fig. S5E-H). SW620 was a
KMM low-risk cell line, although it shares the same origin as SW480
(42). GSEA using GSE228010
dataset indicated that the EMT pathway was more enriched in SW480
compared with SW620 (Fig. S6A).
Furthermore, unlike the EMT morphology of SW480, SW620 exhibited a
typical epithelial morphology during cell culture (Fig. S6C).
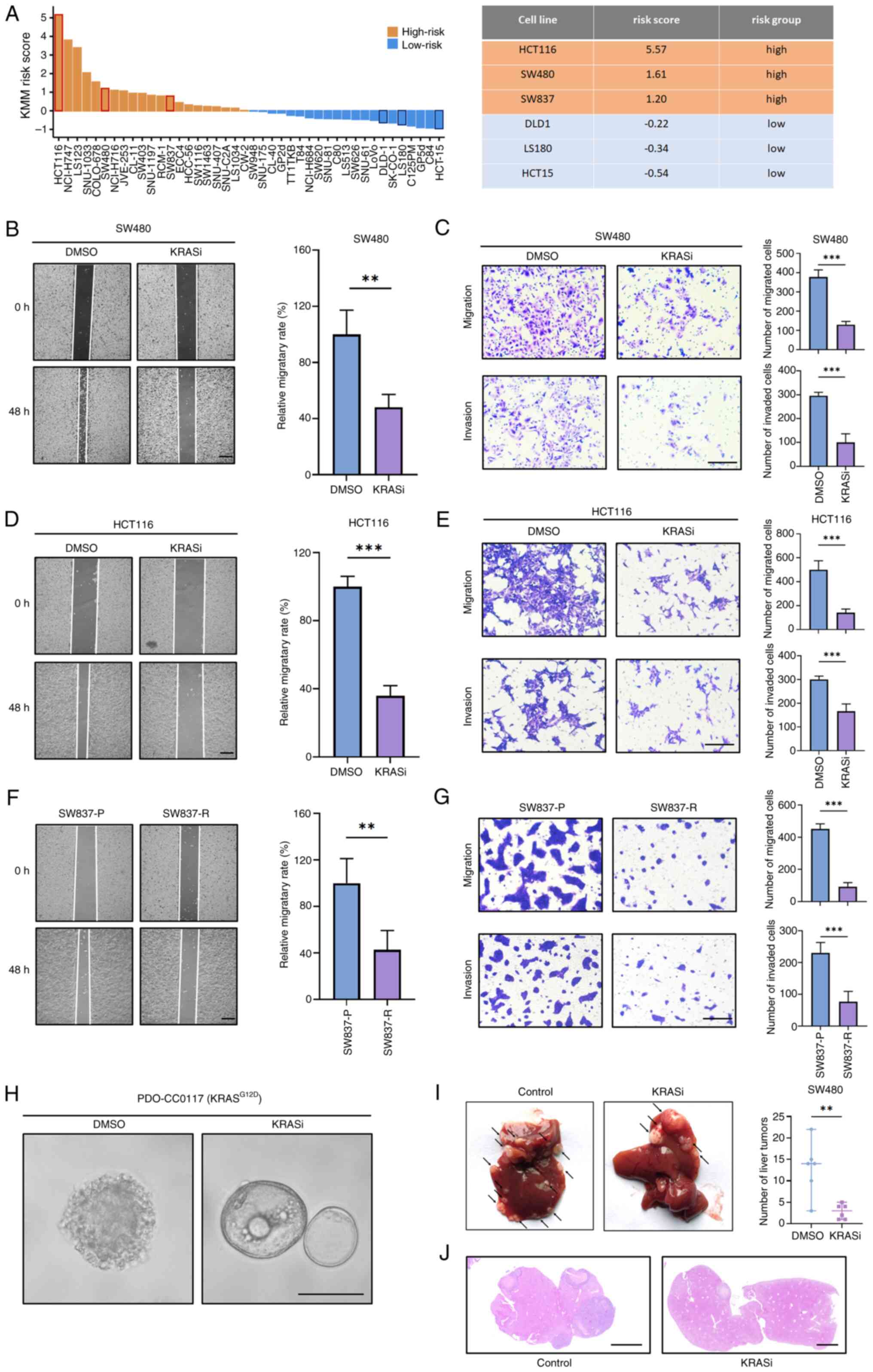 | Figure 3.KRAS inhibitors inhibit migration and
invasion of CRC. (A) KMM score of Cancer Cell Line Encyclopedia CRC
cell lines. Effect of BI-2865 on the migration and invasion
phenotype of SW480 cells was examined using (B) wound healing and
(C) Transwell assays. Scale bar, 200 µm. Effect of BI-2865 on the
migration and invasion phenotype of HCT116 cells was examined using
(D) wound healing and (E) Transwell assays. Scale bar, 200 µm. (F)
Migration and (G) invasion of SW837-P and SW837-R. Scale bar, 200
µm. (H) Effects of BI-2865 treatment of KRASG12D CRC
organoid PDO-CC0117. The cystic structure was maintained and
spike-like features were decreased. Scale bar, 200 µm. (I)
Representative liver metastasis of SW480 cells treated with or
without BI-2865 before trans-splenic injection into nude mice.
Arrows indicate metastatic nodules. (J) Hematoxylin-eosin staining
of liver tissue samples. Scale bar, 2 mm. **P<0.01,
***P<0.001. KRASi, KRAS inhibitor; CRC, colorectal cancer; KMM,
KRAS metachronous metastasis; P, parental; R, resistant. |
BI-2865 is a recently designed pan-KRAS inhibitor
that inhibits mutant-KRAS (KRAS-G12V in SW480 and KRAS-G13D in
HCT116) (16). Wound healing and
Transwell assay demonstrated that BI-2865 effectively inhibited
migration and invasion capacity of SW480 cells (Fig. 3B and C) and HCT116 cells (Fig. 3D and E). BI2865 treatment of SW480
and HCT116 cells transformed the mesenchymal morphology of the
cells to an epithelial-like morphology, suggesting KRAS inhibitor
may promote mesenchymal-epithelial transition (Fig. S6). Since BI-2865 has not been
tested in clinical settings, results were compared with US. Food
and Drug Administration-approved KRAS inhibitor adagrasib (43). Long-term in vitro treatment
of SW837, a KMM high-risk CRC cell line harboring KRAS-G12C
mutation (Fig. 3A), was performed
to simulate the extended application of adagrasib in clinical
scenarios (Fig. S7A). Acquired
resistance to KRAS inhibitors is a challenge during treatment of
CRC (44). Prolonged treatment
induced resistance, characterized by increased IC50 and maintenance
of p-ERK levels under the relatively high dosage of adagrasib
compared with SW837-parental (P; Fig.
S7B-E) cells. Although it exhibited acquired resistance to
adagrasib, SW837-resistant (R) cell line showed significantly lower
migration and invasion ability compared with SW837-P cells
(Fig. 3F-G), indicating that
adagrasib stabilized tumor cells and prevented metastasis. BI-2865
was used to treat previously established organoid, PDO-CC0117,
harboring KRASG12D mutation (21). Although treatment-naive PDO-CC0117
organoids exhibited a solid structure with spike-formation, which
indicated migration activity, those treated with BI-2865 had
relatively normal cystic morphology, suggesting that the KRAS
inhibitor exerted stabilizing effects (Fig. 3H). Effect of the KRAS inhibitor on
the metastatic ability of CRC was assessed in vivo. Body
weight of the animals was measured every 2 days after intrasplenic
tumor injection (Fig. S8). No
metastasis outside the liver was noted in any mice. BI2865-treated
group had significantly reduced liver metastases compared with the
control group (Fig. 3I and J).
Collectively, these results indicate that KRAS inhibitors suppress
the metastatic ability of high-risk CRC by inhibiting metachronous
metastasis of KRAS-MUT CRC.
KRAS inhibitors suppress
TGF-β-mediated pathway in CRC cell lines
KRAS-MUT enhances migration and invasion
capabilities of CRC via EMT and TGF-β signaling pathway (8,9,45,46).
GSEA using the GSE228010 dataset demonstrated that the BI-2865
treatment decreases the EMT and TGF-β pathways in SW480 and HCT116
cells (16), as verified by
RT-qPCR (Fig. 4A and B, D and E).
Enrichment analysis using GSE116823 dataset showed that KRAS
knockdown inhibited EMT and TGF-β in HCT116 (Fig. S9A and B). Consistent with RT-qPCR,
western blot revealed that BI-2865 can decrease levels of EMT
markers such as N-cadherin, vimentin, fibronectin and MMP1 while
upregulating the levels of E-cadherin (Fig. 4C). Given that KRAS regulates EMT
via the TGF-β pathway, cells were treated with TGF-β1, which
partially rescued the inhibitory effect of BI-2865 on migration and
invasion capacity of SW480 and HCT116 cells (Fig. 4F-I).
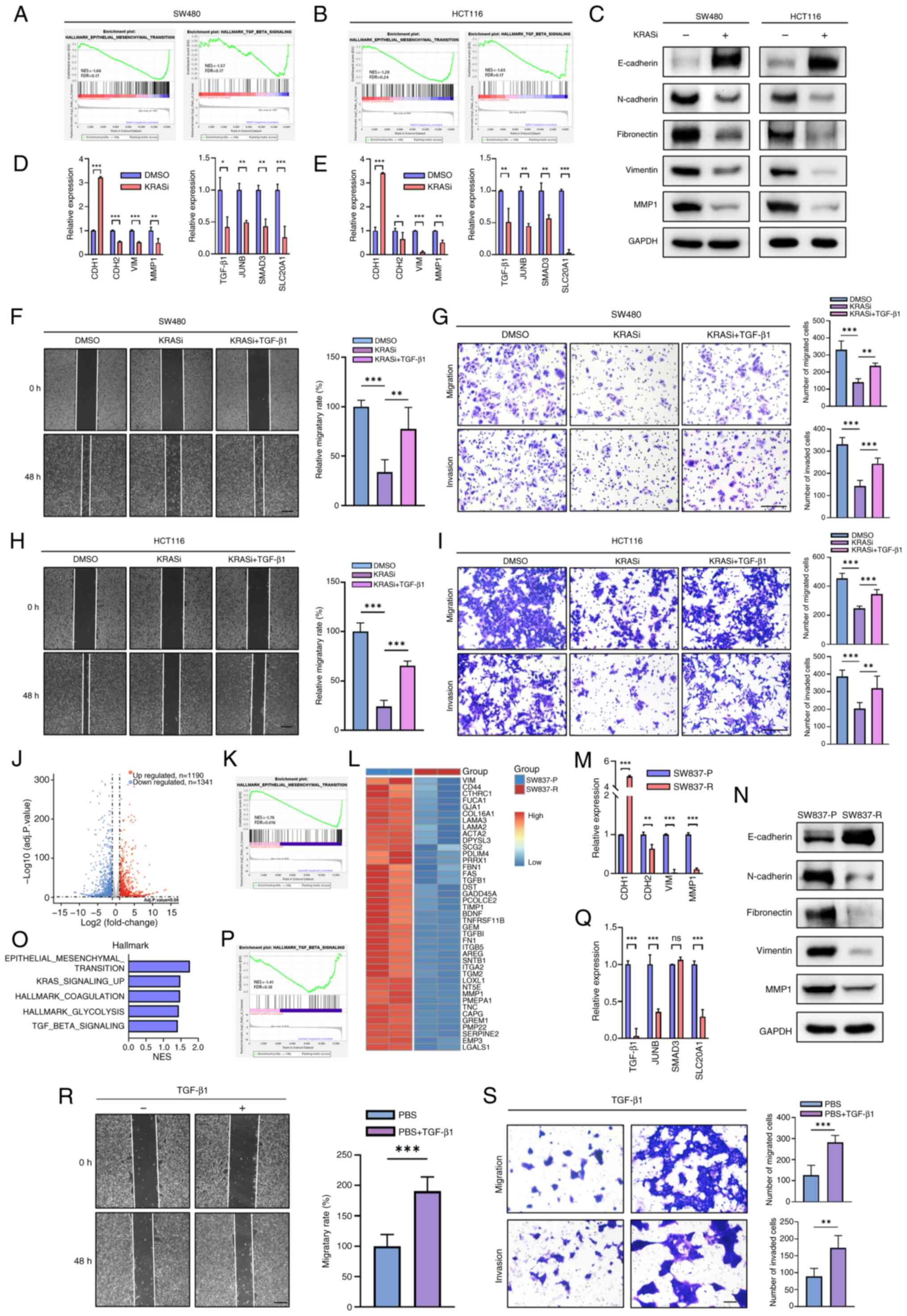 | Figure 4.KRAS inhibitor abrogates TGF-β and
EMT pathways in colorectal cancer cell lines. Gene Set Enrichment
Analysis of Hallmark in GSE228010 showing enrichment of the EMT and
TGF-β signaling pathways following BI-2865 treatment in (A) SW480
and (B) HCT116 cells. (C) Protein expression of EMT markers in
SW480 and HCT116 following treatment with DMSO or BI-2865.
Validation of EMT- and TGF-β-associated genes by reverse
transcription-quantitative PCR in (D) SW480 and (E) HCT116 cell
lines. Migration and invasion of SW480 cells was investigated using
(F) wound healing assay and (G) Transwell assay. Scale bar, 200 µm.
Migration and invasion of HCT116 cells was investigated using (H)
scratch wound healing assay and (I) Transwell assay. Scale bar, 200
µm. (J) Differentially expressed genes in SW837-P and SW837-R
cells. (K) Hallmark pathway analysis indicated that downregulated
genes in SW837-R were enriched in EMT. (L) Significantly
downregulated EMT genes. (M) Relative mRNA levels of EMT genes in
SW837-P/R cells. (N) Western blot analysis of EMT markers in
SW837-P/R cells. (O) Top five down-regulated enrichment pathways in
SW837-R and SW837-P cells. (P) Hallmark pathway analysis indicated
that downregulated genes in SW837-R were enriched in TGF-β
pathways. (Q) Relative mRNA levels of TGF-β-related genes in
SW837-P/R cells. Migration and invasion capacity of SW837-R cells
assessed by (R) wound healing and (S) Transwell assay. *P<0.05,
**P<0.01, ***P<0.001. EMT, epithelial-mesenchymal transition;
KRASi, KRAS inhibitor; P, parental; R, resistant; NES, normalized
enrichment score; FDR, false discovery rate; VIM, vimentin; ns, not
significant; CDH, cadherin; SLC20A1, solute carrier family 20
member 1. |
Transcriptomes of SW837-P and SW837-R were compared
using RNA-seq, which identified 2,531 differentially expressed
genes (Fig. 4J). Pathway
enrichment analysis indicated significant downregulation of the EMT
and TGF-β pathways in SW837-R compared with SW837-P cells (Fig. 4K and L, O and P). These findings
were validated by RT-qPCR (Fig. 4M and
Q). Western blot analysis confirmed downregulation of
N-cadherin, fibronectin, vimentin and MMP1, along with the
upregulation of E-cadherin in SW837-R (Fig. 4N). The results indicated that,
although acquired resistance occurred, prolonged treatment with
adagrasib downregulated EMT and TGF-β pathways in SW837 and
decreased migration and invasion abilities, and these effects were
rescued by exogenous TGF-β1 administration (Fig. 4R and S). Collectively, these
results indicated that KRAS inhibitors may inhibit CRC metastatic
potential by suppressing TGF-β-mediated EMT.
Discussion
CRC is one of the most prevalent malignancies
accounting for 10% of new cancer cases worldwide (1). Although several treatments, including
surgery, neoadjuvant chemotherapy and radiotherapy have been
developed for the treatment of non-stage IV CRC, up to 23% of
patients develop metachronous metastasis (2). The present study found that the KRAS
mutation is a risk factor for CRC metachronous metastasis and
constructed a model for assessing metachronous metastasis risk in
patients with KRAS-MUT CRC. In vitro and in vivo
experiments indicated that KRAS inhibitors effectively suppressed
the metastatic potential of high-risk CRC, providing a novel
approach for CRC adjuvant therapy (Fig. 5).
KRAS is an oncogene with the highest mutation
frequency in CRC, with a cumulative mutation rate of 49% reported
in a recent study (47). Ample
evidence suggests an association between KRAS mutation and CRC
metastasis, with KRAS-MUT associated with higher incidence of stage
IV CRC compared with wild-type (48,49).
However, whether KRAS-MUT increases the risk of CRC metachronous
metastasis is unclear. Studies have indicated that KRAS mutations
are associated with metachronous lung metastasis but not liver
metastasis in CRC (50,51). The present meta-analysis revealed
that KRAS-MUT was a risk factor for CRC metachronous metastasis,
highlighting the role of KRAS in metastasis. As a single factor,
KRAS mutation is not a robust predictive factor, and thus, the
association between KRAS and metastasis is highly variable
(52). Therefore, the present
analyzed public omics databases to develop a tool for predicting
metachronous metastasis risk in KRAS-MUT CRC, which showed good
results. However, to improve the performance of the KMM tool, which
was primarily based on gene expression omics, future studies need
to incorporate clinical data, radiomics, genomics and proteomics
information to establish a multiomics model.
The treatment of KRAS-MUT CRC is a clinical
challenge due to the lack of a specific therapeutic approach. KRAS
mutations may decrease the efficacy of chemotherapy (53) and cause EGFR-targeted therapy
resistance (54). Moreover, the
KRAS-MUT can reduce tumor-infiltrated T lymphocytes, resulting in
the establishment of a suppressive anti-tumor immune
microenvironment in CRC, suggesting that immunotherapy may not be a
preferred option for KRAS-MUT CRC (55). Thus, alternative adjuvant treatment
options should be explored for management of KMM high-risk CRC. In
recent years, several KRAS inhibitors, including adagrasib and
BI-2865, have been developed, some of which effectively treat
KRAS-MUT CRC (56). However,
current KRAS inhibitors are only approved for the treatment of
metastatic CRC, which is not responsive to first-line treatment
(43). To the best of our
knowledge, no study has reported their ability to prevent tumor
metastasis (57). The present
study demonstrated that adagrasib, an FDA-approved KRASG12C
inhibitor, and pan-KRAS inhibitor BI-2865, which targets multiple
mutational subtypes, inhibited migration and invasion capabilities
of KRAS-MUT CRC. To the best of our knowledge, the present study is
the first to provide preclinical evidence for the benefits of KRAS
inhibitors and offers a prophylactic strategy for management of
metachronous metastasis in KRAS-MUT CRC. Since the potency of
adagrasib and BI-2865 against KRAS-MUT but not KRAS-wild-type
cancer has been well documented (16,58),
the present study did not further validate the efficacy of KRAS
inhibitors in KRAS wild-type CRC. Microsatellite status is a
commonly used clinical signature in CRC (59,60).
HCT116 is an microsatellite instability-high CRC, while SW480 and
SW837 are microsatellite stable) CRC (61). All three cell lines are KMM
high-risk. KRAS inhibitors suppressed migration and invasion
capability in both MSI (HCT116) and MSS (SW480 and SW837) CRC cell
lines, indicating microsatellite instability status did not affect
phenotype.
EMT affects metastatic behavior of colorectal cancer
(62). EMT level in primary tumors
may be a risk factor for distant relapse (63), which is consistent with the present
finding that EMT was enriched in the KMM high-risk group. The
oncogenic KRAS drives EMT in CRC (64). However, a recent study suggested
that long-term KRAS inhibition induces EMT reactivation in lung
cancer, implying that the relationship between KRAS and EMT may
exhibit different patterns depending on cancer heterogeneity
(65). KRAS signaling exerts
crosstalk with TGF-β in CRC (66).
Meanwhile, as a canonical upstream regulator of EMT, inhibition of
TGF-β pathway blocks EMT in CRC (8). Therefore, it was hypothesized that
the KRAS inhibitor can suppress EMT via KRAS-mediated TGF-β
signaling; the results confirmed this hypothesis, indicating that
KRAS inhibitors prevent the metastatic potential of CRC. SW480 has
a SMAD4 mutation while HCT116 has a TGFBR2 mutation, both of which
are key factors in the TGF-β signaling pathway (67,68).
Nevertheless, studies have shown EMT phenotype of SW480 and HCT116
can be activated by either exogenous SMAD4 or TGF-β protein,
indicating that the TGF-β signaling pathway is functional in these
cell models (67,69). Thus, it is reasonable to use KRAS
inhibitors to suppress this pathway to inhibit EMT and
invasion/metastasis.
In conclusion, KRAS mutation is a risk factor for
metachronous metastasis. The present study constructed a scoring
model for predicting the risk of distant relapse in KRAS-MUT CRC.
KRAS inhibitors suppressed the EMT process that drives the
migration and invasion properties of CRC by blocking the TGF-β
signaling pathway, offering a potential prophylactic treatment for
metachronous metastasis.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by National Natural Science
Foundation of China (grant no. 82203072) and Tianshan Talents
Leading Medical Talents in Guangdong Province Cooperative Expert
Studio (grant no. KSYJ2022001).
Availability of data and materials
The data generated in the present study may be found
in the Gene Expression Omnibus database at the National Center for
Biotechnology Information under accession number PRJNA1085403 or at
the following URL: ncbi.nlm.nih.gov/bioproject/1085403.
Authors' contributions
YG, XW and FG conceived and designed the study. YG
and CH wrote the manuscript and analyzed data. KC, GL and XH
performed experiments. DC, ZY, ZC, PH and YC analyzed data and
reviewed the manuscript. PH and ZY confirm the authenticity of all
the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was conducted in accordance with
the Declaration of Helsinki and approved by the Ethics Committee of
the Sixth Affiliated Hospital, Sun Yat-sen University (approval no.
2024ZSLYEC-192). The animal experiment was conducted in compliance
with animal protocols approved by the Institutional Animal Care and
Use Committee at Sun Yat-sen University (approval no.
SYSU-IACUC-2022-080601).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD, Wagle NS and Jemal
A: Cancer statistics, 2023. CA Cancer J Clin. 73:17–48. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cohen R and Platell CF: Metachronous
colorectal cancer metastasis: Who, what, when and what to do about
it. J Surg Oncol. 129:71–77. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hansdotter P, Scherman P, Nikberg M,
Petersen SH, Holmberg E, Rizell M, Naredi P and Syk I; COLOFOL
study group, : Treatment and survival of patients with metachronous
colorectal lung metastases. J Surg Oncol. 127:806–814. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Reboux N, Jooste V, Goungounga J,
Robaszkiewicz M, Nousbaum JB and Bouvier AM: Incidence and survival
in synchronous and metachronous liver metastases from colorectal
cancer. JAMA Netw Open. 5:e22366662022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Martin J, Petrillo A, Smyth EC, Shaida N,
Khwaja S, Cheow HK, Duckworth A, Heister P, Praseedom R, Jah A, et
al: Colorectal liver metastases: Current management and future
perspectives. World J Clin Oncol. 11:761–808. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pfannschmidt J, Dienemann H and Hoffmann
H: Surgical resection of pulmonary metastases from colorectal
cancer: A systematic review of published series. Ann Thorac Surg.
84:324–338. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Biller LH and Schrag D: Diagnosis and
treatment of metastatic colorectal cancer: A review. JAMA.
325:669–685. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Boutin AT, Liao WT, Wang M, Hwang SS,
Karpinets TV, Cheung H, Chu GC, Jiang S, Hu J, Chang K, et al:
Oncogenic Kras drives invasion and maintains metastases in
colorectal cancer. Genes Dev. 31:370–382. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lemieux E, Cagnol S, Beaudry K, Carrier J
and Rivard N: Oncogenic KRAS signalling promotes the Wnt/β-catenin
pathway through LRP6 in colorectal cancer. Oncogene. 34:4914–4927.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Feng Q, Liang L, Ren L, Chen J, Wei Y,
Chang W, Zhu D, Lin Q, Zheng P and Xu J: A specific KRAS codon 13
mutation is an independent predictor for colorectal cancer
metachronous distant metastases. Am J Cancer Res. 5:674–688.
2015.PubMed/NCBI
|
|
11
|
Ilm K, Kemmner W, Osterland M, Burock S,
Koch G, Herrmann P, Schlag PM and Stein U: High MACC1 expression in
combination with mutated KRAS G13 indicates poor survival of
colorectal cancer patients. Mol Cancer. 14:382015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hao M, Li H, Wang K, Liu Y, Liang X and
Ding L: Predicting metachronous liver metastasis in patients with
colorectal cancer: Development and assessment of a new nomogram.
World J Surg Oncol. 20:802022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Herzberg BO and Manji GA: KRAS: druggable
at last. Oncologist. 28:283–286. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Abubaker J, Bavi P, Al-Haqawi W, Sultana
M, Al-Harbi S, Al-Sanea N, Abduljabbar A, Ashari LH, Alhomoud S,
Al-Dayel F, et al: Prognostic significance of alterations in KRAS
isoforms KRAS-4A/4B and KRAS mutations in colorectal carcinoma. J
Pathol. 219:435–445. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nusrat M and Yaeger R: KRAS inhibition in
metastatic colorectal cancer: An update. Curr Opin Pharmacol.
68:1023432023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim D, Herdeis L, Rudolph D, Zhao Y,
Böttcher J, Vides A, Ayala-Santos CI, Pourfarjam Y, Cuevas-Navarro
A, Xue JY, et al: Pan-KRAS inhibitor disables oncogenic signalling
and tumour growth. Nature. 619:160–166. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ji Q, Liu X, Han Z, Zhou L, Sui H, Yan L,
Jiang H, Ren J, Cai J and Li Q: Resveratrol suppresses
epithelial-to-mesenchymal transition in colorectal cancer through
TGF-β1/Smads signaling pathway mediated Snail/E-cadherin
expression. BMC Cancer. 15:972015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Moher D, Liberati A, Tetzlaff J and Altman
DG; Group P, : Preferred reporting items for systematic reviews and
meta-analyses: The PRISMA statement. PLoS Med. 6:e10000972009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chatila WK, Kim JK, Walch H, Marco MR,
Chen CT, Wu F, Omer DM, Khalil DN, Ganesh K, Qu X, et al: Genomic
and transcriptomic determinants of response to neoadjuvant therapy
in rectal cancer. Nat Med. 28:1646–1655. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Roelands J, Kuppen PJK, Ahmed EI, Mall R,
Masoodi T, Singh P, Monaco G, Raynaud C, de Miranda NFCC, Ferraro
L, et al: An integrated tumor, immune and microbiome atlas of colon
cancer. Nat Med. 29:1273–1286. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Medico E, Russo M, Picco G, Cancelliere C,
Valtorta E, Corti G, Buscarino M, Isella C, Lamba S, Martinoglio B,
et al: The molecular landscape of colorectal cancer cell lines
unveils clinically actionable kinase targets. Nat Commun.
6:70022015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim WK, Kwon Y, Jang M, Park M, Kim J, Cho
S, Jang DG, Lee WB, Jung SH, Choi HJ, et al: β-catenin activation
down-regulates cell-cell junction-related genes and induces
epithelial-to-mesenchymal transition in colorectal cancers. Sci
Rep. 9:184402019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu Z, Deng P, Chen Y, Liu S, Chen J, Yang
Z, Chen J, Fan X, Wang P, Cai Z, et al: Inhibition of the
PLK1-coupled cell cycle machinery overcomes resistance to
oxaliplatin in colorectal cancer. Adv Sci (Weinh). 8:e21007592021.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen S, Zhou Y, Chen Y and Gu J: fastp: An
ultra-fast all-in-one FASTQ preprocessor. Bioinformatics.
34:i884–i890. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dobin A, Davis CA, Schlesinger F, Drenkow
J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR: STAR:
Ultrafast universal RNA-seq aligner. Bioinformatics. 29:15–21.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li B and Dewey CN: RSEM: Accurate
transcript quantification from RNA-Seq data with or without a
reference genome. BMC Bioinformatics. 12:3232011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kanehisa M, Furumichi M, Sato Y, Matsuura
Y and Ishiguro-Watanabe M: KEGG: Biological systems database as a
model of the real world. Nucleic Acids Res. gkae9092024.(Epub ahead
of print). View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liberzon A, Birger C, Thorvaldsdóttir H,
Ghandi M, Mesirov JP and Tamayo P: The molecular signatures
database (MSigDB) hallmark gene set collection. Cell Syst.
1:417–425. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Engstrand J, Stromberg C, Nilsson H,
Freedman J and Jonas E: Synchronous and metachronous liver
metastases in patients with colorectal cancer-towards a clinically
relevant definition. World J Surg Oncol. 17:2282019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kuan TC, Chang SC, Lin JK, Lin TC, Yang
SH, Jiang JK, Chen WS, Wang HS, Lan YT, Lin CC, et al:
Prognosticators of long-term outcomes of TNM stage II colorectal
cancer: Molecular patterns or clinicopathological features. World J
Surg. 43:3207–3215. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jo P, Bernhardt M, Nietert M, König A,
Azizian A, Schirmer MA, Grade M, Kitz J, Reuter-Jessen K, Ghadimi
M, et al: KRAS mutation status concordance between the primary
tumor and the corresponding metastasis in patients with rectal
cancer. PLoS One. 15:e02398062020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin CC, Lin JK, Lin TC, Chen WS, Yang SH,
Wang HS, Lan YT, Jiang JK, Yang MH and Chang SC: The prognostic
role of microsatellite instability, codon-specific KRAS, and BRAF
mutations in colon cancer. J Surg Oncol. 110:451–457. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tsunoda A, Iijima T, Tsunoda Y, Nakao K,
Miyaki M and Kusano M: Association of K-ras mutations with liver
metastases from colorectal carcinoma. Anticancer Res. 24:2471–2476.
2004.PubMed/NCBI
|
|
35
|
Vakiani E, Shah RH, Berger MF,
Makohon-Moore AP, Reiter JG, Ostrovnaya I, Attiyeh MA, Cercek A,
Shia J, Iacobuzio-Donahue CA, et al: Local recurrences at the
anastomotic area are clonally related to the primary tumor in
sporadic colorectal carcinoma. Oncotarget. 8:42487–42494. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sperlich A, Balmert A, Doll D, Bauer S,
Franke F, Keller G, Wilhelm D, Mur A, Respondek M, Friess H, et al:
Genetic and immunological biomarkers predict metastatic disease
recurrence in stage III colon cancer. BMC Cancer. 18:9982018.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nitsche U, Rosenberg R, Balmert A,
Schuster T, Slotta-Huspenina J, Herrmann P, Bader FG, Friess H,
Schlag PM, Stein U and Janssen KP: Integrative marker analysis
allows risk assessment for metastasis in stage II colon cancer. Ann
Surg. 256:763–771. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nakamura Y, Yokoyama S, Matsuda K, Tamura
K, Mitani Y, Iwamoto H, Mizumoto Y, Murakami D, Kitahata Y and
Yamaue H: Preoperative detection of KRAS mutated circulating tumor
DNA is an independent risk factor for recurrence in colorectal
cancer. Sci Rep. 11:4412021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Huang SC, Huang SF, Chen YT, Chang Y, Chiu
YT, Chang IC, Wu HI and Chen JS: Overexpression of MutL homolog 1
and MutS homolog 2 proteins have reversed prognostic implications
for stage I–II colon cancer patients. Biomed J. 40:39–48. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
El Otmani I, El Agy F, El Baradai S,
Bouguenouch L, Lahmidani N, El Abkari M, Benajah DA, Toughrai I, El
Bouhaddouti H, Mouaqit O, et al: Analysis of molecular pretreated
tumor profiles as predictive biomarkers of therapeutic response and
survival outcomes after neoadjuvant therapy for rectal cancer in
moroccan population. Dis Markers. 2020:84593032020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Birgisson H, Edlund K, Wallin U, Påhlman
L, Kultima HG, Mayrhofer M, Micke P, Isaksson A, Botling J,
Glimelius B and Sundström M: Microsatellite instability and
mutations in BRAF and KRAS are significant predictors of
disseminated disease in colon cancer. BMC Cancer. 15:1252015.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Verhagen MP, Xu T, Stabile R, Joosten R,
Tucci FA, van Royen M, Trerotola M, Alberti S, Sacchetti A and
Fodde R: The SW480 cell line as a model of resident and migrating
colon cancer stem cells. iScience. 27:1106582024. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dhillon S: Adagrasib: First approval.
Drugs. 83:275–285. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Awad MM, Liu S, Rybkin II, Arbour KC,
Dilly J, Zhu VW, Johnson ML, Heist RS, Patil T, Riely GJ, et al:
Acquired resistance to KRASG12C inhibition in cancer. N
Engl J Med. 384:2382–2393. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Flum M, Dicks S, Teng YH, Schrempp M,
Nyström A, Boerries M and Hecht A: Canonical TGFβ signaling induces
collective invasion in colorectal carcinogenesis through a Snail1-
and Zeb1-independent partial EMT. Oncogen. 41:1492–1506. 2022.
View Article : Google Scholar
|
|
46
|
Sánchez-Tilló E, Pedrosa L, Vila I, Chen
Y, Győrffy B, Sánchez-Moral L, Siles L, Lozano JJ, Esteve-Codina A,
Darling DS, et al: The EMT factor ZEB1 paradoxically inhibits EMT
in BRAF-mutant carcinomas. JCI Insight. 8:e1646292023. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lee JK, Sivakumar S, Schrock AB, Madison
R, Fabrizio D, Gjoerup O, Ross JS, Frampton GM, Napalkov P,
Montesion M, et al: Comprehensive pan-cancer genomic landscape of
KRAS altered cancers and real-world outcomes in solid tumors. NPJ
Precis Oncol. 6:912022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Huang D, Sun W, Zhou Y, Li P, Chen F, Chen
H, Xia D, Xu E, Lai M, Wu Y and Zhang H: Mutations of key driver
genes in colorectal cancer progression and metastasis. Cancer
Metastasis Rev. 37:173–187. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Meng M, Zhong K, Jiang T, Liu Z, Kwan HY
and Su T: The current understanding on the impact of KRAS on
colorectal cancer. Biomed Pharmacother. 140:1117172021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Pereira AAL, Rego JFM, Morris V, Overman
MJ, Eng C, Garrett CR, Boutin AT, Ferrarotto R, Lee M, Jiang ZQ, et
al: Association between KRAS mutation and lung meta-stasis in
advanced colorectal cancer. Br J Cancer. 112:424–428. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tie J, Lipton L, Desai J, Gibbs P,
Jorissen RN, Christie M, Drummond KJ, Thomson BN, Usatoff V, Evans
PM, et al: KRAS mutation is associated with lung metastasis in
patients with curatively resected colorectal cancer. Clin Cancer
Res. 17:1122–1130. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Guo L, Wang Y, Yang W, Wang C, Guo T, Yang
J, Shao Z, Cai G, Cai S, Zhang L, et al: Molecular profiling
provides clinical insights into targeted and immunotherapies as
well as colorectal cancer prognosis. Gastroenterology.
165:414–428.e7. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Iseas S, Sendoya JM, Robbio J, Coraglio M,
Kujaruk M, Mikolaitis V, Rizzolo M, Cabanne A, Ruiz G, Salanova R,
et al: Prognostic impact of an integrative landscape of clinical,
immune, and molecular features in non-metastatic rectal cancer.
Front Oncol. 11:8018802022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Misale S, Di Nicolantonio F,
Sartore-Bianchi A, Siena S and Bardelli A: Resistance to anti-EGFR
therapy in colorectal cancer: From heterogeneity to convergent
evolution. Cancer Discov. 4:1269–1280. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lal N, White BS, Goussous G, Pickles O,
Mason MJ, Beggs AD, Taniere P, Willcox BE, Guinney J and Middleton
GW: KRAS mutation and consensus molecular subtypes 2 and 3 are
independently associated with reduced immune infiltration and
reactivity in colorectal cancer. Clin Cancer Res. 24:224–233. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Caughey BA and Strickler JH: Targeting
KRAS-mutated gastrointestinal malignancies with small-molecule
inhibitors: A new generation of breakthrough therapies. Drugs.
84:27–44. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Erlanson DA and Webster KR: Targeting
mutant KRAS. Curr Opin Chem Biol. 62:101–108. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hallin J, Engstrom LD, Hargis L, Calinisan
A, Aranda R, Briere DM, Sudhakar N, Bowcut V, Baer BR, Ballard JA,
et al: The KRASG12C Inhibitor MRTX849 provides insight
toward therapeutic susceptibility of KRAS-mutant cancers in mouse
models and patients. Cancer Discov. 10:54–71. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Popat S, Hubner R and Houlston RS:
Systematic review of microsatellite instability and colorectal
cancer prognosis. J Clin Oncol. 23:609–618. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Storojeva I, Boulay JL, Heinimann K,
Ballabeni P, Terracciano L, Laffer U, Mild G, Herrmann R and
Rochlitz C: Prognostic and predictive relevance of microsatellite
instability in colorectal cancer. Oncol Rep. 14:241–249.
2005.PubMed/NCBI
|
|
61
|
Berg KCG, Eide PW, Eilertsen IA,
Johannessen B, Bruun J, Danielsen SA, Bjørnslett M, Meza-Zepeda LA,
Eknæs M, Lind GE, et al: Multi-omics of 34 colorectal cancer cell
lines-a resource for biomedical studies. Mol Cancer. 16:1162017.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Vu T and Datta P: Regulation of EMT in
colorectal cancer: A culprit in metastasis. Cancers (Basel).
9:1712017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wang K, Song K, Ma Z, Yao Y, Liu C, Yang
J, Xiao H, Zhang J, Zhang Y and Zhao W: Identification of
EMT-related high-risk stage II colorectal cancer and
characterisation of metastasis-related genes. Br J Cancer.
123:410–417. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Chu PC, Lin PC, Wu HY, Lin KT, Wu C,
Bekaii-Saab T, Lin YJ, Lee CT, Lee JC and Chen CS: Mutant KRAS
promotes liver metastasis of colorectal cancer, in part, by
upregulating the MEK-Sp1-DNMT1-miR-137-YB-1-IGF-IR signaling
pathway. Oncogene. 37:3440–3455. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Adachi Y, Kimura R, Hirade K, Yanase S,
Nishioka Y, Kasuga N, Yamaguchi R and Ebi H: Scribble
mis-localization induces adaptive resistance to KRAS G12C
inhibitors through feedback activation of MAPK signaling mediated
by YAP-induced MRAS. Nat Cancer. 4:829–843. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Vivekanandhan S and Mukhopadhyay D:
Genetic status of KRAS influences transforming growth factor-beta
(TGF-β) signaling: An insight into neuropilin-1 (NRP1) mediated
tumorigenesis. Semin Cancer Biol. 54:72–79. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Pohl M, Radacz Y, Pawlik N, Schoeneck A,
Baldus SE, Munding J, Schmiegel W, Schwarte-Waldhoff I and
Reinacher-Schick A: SMAD4 mediates mesenchymal-epithelial reversion
in SW480 colon carcinoma cells. Anticancer Res. 30:2603–2613.
2010.PubMed/NCBI
|
|
68
|
Lee J, Ballikaya S, Schönig K, Ball CR,
Glimm H, Kopitz J and Gebert J: Transforming growth factor beta
receptor 2 (TGFBR2) changes sialylation in the microsatellite
unstable (MSI) Colorectal cancer cell line HCT116. PLoS One.
8:e570742013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hawinkels LJAC, Paauwe M, Verspaget HW,
Wiercinska E, van der Zon JM, van der Ploeg K, Koelink PJ, Lindeman
JH, Mesker W, ten Dijke P and Sier CF: Interaction with colon
cancer cells hyperactivates TGF-β signaling in cancer-associated
fibroblasts. Oncogene. 33:97–107. 2014. View Article : Google Scholar : PubMed/NCBI
|