Introduction
Neuroblastoma (NBL) is the most common extracranial
solid tumor in children and a major cause of neoplastic death in
infancy. It originates from undifferentiated cells of the
sympathetic nervous system. Based on its cellular and biological
heterogeneity, NBL behavior can range from low-risk cancers with a
tendency toward spontaneous regression or maturation, to high-risk
cancers with extensive growth, early metastasis and a poor
prognosis (1). Treatment of
high-risk neuroblastomas (HR NBL) usually fails despite intensive
therapy, which includes megatherapy followed by hematopoietic
progenitor cell transplantation, biotherapy and immunotherapy.
Treatment failure is due to drug resistance that arises in the
majority of patients who initially responded well to chemotherapy.
The necessity to develop new treatment modalities is
indisputable.
An increasing body of information indicates that
epigenetic modifications are associated with cancer onset and
progression. This awareness has led to prolific research into drugs
that interfere with the epigenome (2,3).
Histone deacetylase inhibitors (HDACi) represent such a group of
compounds since histones are the main protein components of
chromatin and have an indispensable role in gene regulation. Cancer
cell histones are frequently hypo-acetylated, due to overexpression
of histone deacetylases (HDACs), and are often connected with
impaired gene transcription in tumors (4), including dysregulation of genes
responsible for growth control and apoptosis. Consequently
inhibition of HDACs can reactivate gene transcription and restore
the balance between pro- and anti-apoptotic genes and eventually
lead to apoptosis (5). HDAC
inhibition also decompacts chromatin structure making the DNA
structure more available to other cytotoxic agents that target DNA.
Despite advances in understanding, the mode of anti-tumor action of
HDACi is complex and still not completely understood (6,7).
Valproic acid (VPA) has been studied as an
anti-cancer drug excessively over the past years because it can be
taken orally, is well tolerated by patients and there is cumulative
experience coming from its use as an anti-epileptic drug. Although
earlier reports showed the cytotoxic potential of VPA on NBL cells
in vitro and in vivo (8,9), the
studies were carried out solely under normoxic conditions and
little was known about its anti-tumor activity under hypoxic
conditions.
Hypoxic areas are common in solid tumors. Hypoxia
arises as a consequence of pathological microcirculation within the
tumor. Rapid tumor growth can outstrip its own blood supply and
therefore cancer cells are exposed to oxygen deprivation (chronic
hypoxia) (10). Another factor that
contributes to tumor hypoxia is the poor quality of the newly
developing tumor vessels, which often display severe structural
abnormalities. Whereas normal vasculature shows a hierarchical
branching pattern, tumor blood vessels are often tortuous in
appearance with uneven diameters, branch irregularity and form
arterio-venous shunts. These vessels are more susceptible to
thrombosis and on occasion collapse, which ultimately leads to
acute hypoxia within the tumor mass (11).
Hypoxia also induces adaptational changes in cells
that are otherwise physiological, in the sense that they are normal
and noncancerous; however, due to regional hypoxia these cells
contribute to chemo- and radio-resistance in hypoxic cancer cells
(12–14). Notably, hypoxia-induced resistance
is not limited to only conventional chemotherapy but it can also
decrease the efficiency of targeted therapy, as documented with
imatinib in cases of chronic myeloid leukemia (15). Additionally, hypoxia induces genomic
instability that leads to progressive transformation of cancer
cells into more malignant phenotypes (16). The presence of hypoxic regions
within the tumor mass correlates with more aggressive phenotypes,
lower response rates and a decline in overall disease survival
(17–19).
In our study, we addressed the issue of whether
hypoxia promotes resistance to VPA and if apoptosis pathways differ
between normoxic and hypoxic conditions, with respect to VPA
treatment.
Materials and methods
Cell lines and chemicals
The UKF-NB-3 cell line was established from bone
marrow metastases of HR NBL with MYCN amplification. The
line was kindly provided by Professor J. Cinatl Jr. (Institute for
Medical Virology, Hospital of the Johann Wolfgang Goethe
University, Frankfurt, Germany). Cells were grown in Iscove’s
modified Dulbecco’s medium (IMDM) with 10% fetal calf serum (PAA
Laboratories, Pasching, Austria). The SK-N-AS cell line was derived
from bone marrow metastasis of a female patient with HR NBL.
SK-N-AS, with normal diploid MYCN status, was purchased from
the European Collection of Cell Cultures (ECACC, Salisbury, UK) and
was cultivated according to the manufacturer’s instructions. The
CDDP-resistant sub-line, designated UKF-NB-3CDDP was
also kindly provided by Professor J. Cinatl Jr.
SK-N-ASCDDP was prepared in our laboratory by incubation
of parental cells with increasing concentrations of CDDP. Solutions
of CDDP (EBEWE Pharma Ges.m.b.H. Nfg. KG, Unterach, Austria) were
prepared according to the manufacturer’s instructions.
CDDP-resistant cell lines were cultivated in a medium containing 1
μg/ml of CDDP. Valproic acid (dissolved in distilled water) and
trichostatin A (dissolved in DMSO) were purchased from Sigma
Chemical Co. (St. Louis, MO, USA). The specific caspase-8
inhibitor, Z-IETD-FMK (specific caspase-8 inhibitor), was obtained
from R&D Systems, Inc. (Minneapolis, MN, USA). It was dissolved
in DMSO and was used at a final concentration of 2 μM, as
recommended by producer. All other chemicals used in experiments
were of analytical purity or better.
Hypoxic environment
A hypoxia chamber purchased from Billups-Rothenberg
(Del Mar, CA, USA) was prepared with an atmosphere containing 1%
O2, 5% CO2, and 94% N2. Controls
were grown at 5% CO2 and all samples were grown at
37°C.
Annexin V/propidium iodide labeling
Annexin V, a phospholipid-binding protein with a
high affinity for phosphatidyl serine, was used to measure
apoptosis and viability. Apoptosis was determined using an Annexin
V-FITC Apoptosis Detection kit according to manufacturer
instructions (Biovision, Mountain View, CA, USA). Cells were washed
in PBS and resuspended in a ‘binding buffer’ after incubation with
different compounds, under normoxic and/or hypoxic conditions, as
described below. Cells were incubated with Annexin V and propidium
iodide for 10 min at room temperature and then analyzed using flow
cytometry (FACSCalibur, BD, San Jose, CA, USA). Data obtained from
flow cytometry were evaluated using the same technique described in
a study by Bossy-Wetzel (20).
TUNEL assay
Apoptotic cells were determined using an ApoDirect
DNA Fragmentation Assay kit per manufacturer’s instructions
(Biovision). Cells were fixed with 1% paraformaldehyde and then
incubated with terminal deoxynucleotidyl transferase and FITC-dUTP
for 60 min at 37°C and counter-stained with propidium iodide. Cells
were then analyzed using flow cytometry.
Western blot was used to determine the
expression of BID protein
Cells were homogenized in RIPA buffer. Protein
concentrations were assessed using the DC protein assay (Bio-Rad,
Hercules, CA, USA) with serum albumin as a standard. 10–45 μg of
extracted proteins were subjected to SDS-PAGE electrophoresis on a
10% gel. After migration, proteins were transferred to a
nitrocellulose membrane and incubated with 5% non-fat milk to block
non-specific binding. The membranes were then exposed to specific
anti-BID (1:1000, AbCam, Cambridge, UK ) rabbit monoclonal
antibodies overnight at 4°C. Membranes were washed and exposed to
peroxidase-conjugated anti-IgG secondary antibody (1:3000,
Bio-Rad), and the antigen-antibody complex was visualized using an
enhanced chemiluminescence detection system according to the
manufacturer’s instructions (Immun-Star HRP Substrate, Bio-Rad).
The resulting films (MEDIX XBU, Foma, Hradec Králové, Czech
Republic) were scanned with a computerized image-analyzing system
(ElfoMan 2.0, Ing. Semecký, Prague, Czech Republic).
Caspase activity
Caspase-8 activity was measured using a caspases-8
assay kit according to manufacturer’s instructions (Biovision).
Briefly, cells were lysed in cell lysis buffer after incubation
with VPA. Total protein (200 μg) were added to the reaction buffer,
which contained IETD-pNA colorimetric substrate, and incubated for
2 h at 37°C. Hydrolyzed pNA was detected using a VersaMax plate
reader (Molecular Device Inc., Sunnyvale, CA, USA) at 405 nm.
Real-time PCR analysis
Total RNA was extracted from cells lines using
TRIzol reagent (Invitrogen, Carlsbad, CA, USA). The quality of the
isolated RNA was verified using horizontal agarose gel
electrophoresis and RNA quantity was measured using a BioMate 3
UV-Vis Spectrophotometer (Thermo Scientific, Waltham, MA, USA).
Complementary DNA was synthesized from 500 ng of RNA using random
hexamers and MultiScribe reverse transcriptase (Applied Biosystems,
Foster City, CA, USA). RT-PCR was performed using assays for
vascular endothelial growth factor (VEGF), carbonic anhydrase-9
(CA9) and β-2-microglobulin (B2M) purchased from Generi Biotech
(Hradec Kralove, Czech Republic). B2M was used as a reference gene.
Relative expression and statistical significance were determined
using REST-MCS software (Dr Michael Pfaffl, Germany) using the
technique described by Pfaffl (21).
Results
VPA induces apoptosis under both normoxic
and hypoxic conditions
We set up dose and time course experiments in order
to prove efficacy of VPA under hypoxic and normoxic conditions.
Concentrations of VPA ranged from 0.5 to 10 mM. Cells were grown
under normoxic conditions for 24 h after plating and then VPA was
added. Plates were then put into the hypoxia chamber, while control
cells stayed under normoxic conditions. Apoptosis was determined
using Annexin V (An) and propidium iodide (PI) staining at 24, 48
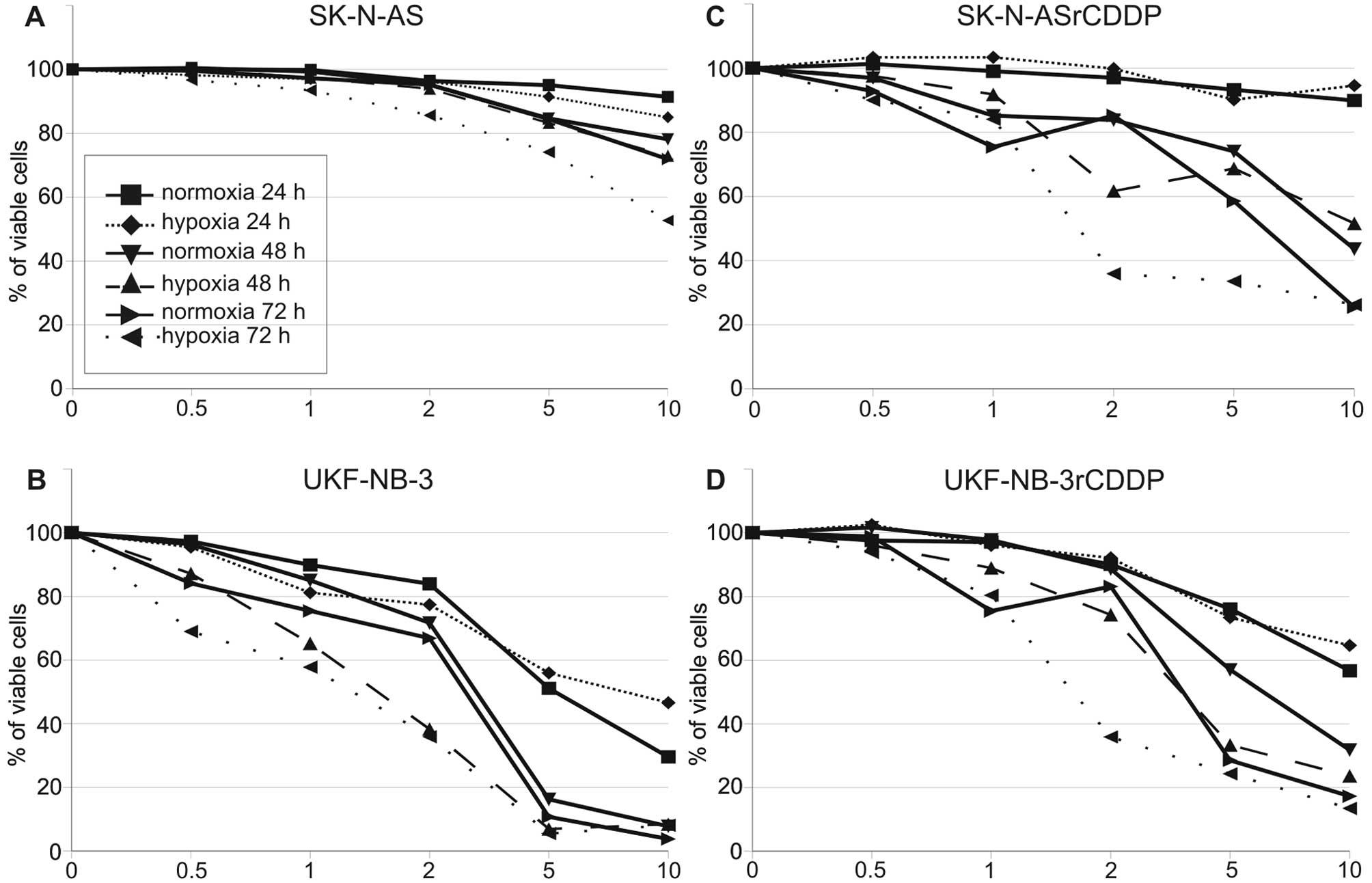
and 72 h after addition of VPA. We observed time- and
dose-dependent apoptosis. UKF-NB-3 showed higher sensitivity to VPA
compared to SK-N-AS (Fig. 1A and
B). We did not observe any hypoxia induced resistance to VPA.
Moreover, slightly more Annexin positive/propidium iodide negative
cells (early apoptotic) and Annexin positive/propidium iodide
positive cells (late apoptotic or necrotic) were seen under hypoxic
conditions in both cell lines (Table
I). For instance, 13.4% Annexin V single positive
(An+/PI−) cells were observed after treatment
with 5 mM VPA under normoxic conditions whereas 19.0%
An+/PI− cells were observed in the hypoxia
SK-N-AS cell line. Although the higher number of apoptotic cells,
under hypoxic conditions, was not statistically significant, this
trend was clearly obvious in all cell lines tested. This result
indicates that VPA promotes apoptosis irrespective of oxygen
tension and therefore should be equally efficient throughout the
entire tumor volume. We performed the same experiments with cell
lines resistant to cisplatin, which had been derived from SK-N-AS
and UKF-NB-3, and obtained similar results (Fig. 1C and D).
 | Table IPercentage of apoptotic cells
measured as An+/PI− cells. |
Table I
Percentage of apoptotic cells
measured as An+/PI− cells.
| Control | 24 h | 48 h | 72 h |
|---|
|
|
|
|
|
|---|
| N (%) | H (%) | N (%) | H (%) | N (%) | H (%) | N (%) | H (%) |
|---|
| SK-N-AS (5 mM) | 1.6 | 1.1 | 5.59 | 6.63 | 13.86 | 14.91 | 13.37 | 19.02 |
| SK-N-ASrCDDP (5
mM) | 2.59 | 3.99 | 7.51 | 11.51 | 13.61 | 16.82 | 40.63 | 53.47 |
| UKF-NB-3 (2
mM) | 6.3 | 6.66 | 10.13 | 9.29 | 21.49 | 20.70 | 19.39 | 16.57 |
| UKF-NB-3rCDDP (2
mM) | 2.89 | 2.73 | 7.81 | 7.43 | 9.12 | 16.60 | 7.22 | 25.49 |
We also evaluated apoptosis using TUNEL assay in
order to validate the data using an independent method. Both
SK-N-AS and UKF-NB-3 cell lines revealed higher number of apoptotic
cells (TUNEL positive) under hypoxic conditions than under normoxic
conditions. The TUNEL results therefore supported the data obtained
using An/PI staining (data not shown).
VPA has a synergistic effect with
cisplatin
As mentioned in a previous section, VPA is capable
of overcoming hypoxia resistance; however, its overall toxicity to
NBL cells is quite poor considering that clinically achievable
concentrations are <1 mM. Thus, we addressed the issue of
whether small concentrations of VPA, which are clinically well
tolerated, could be useful in overcoming hypoxia induced resistance
to chemotherapeutic agents, such as cisplatin (CDDP), which are
commonly used in HR NBL therapy.
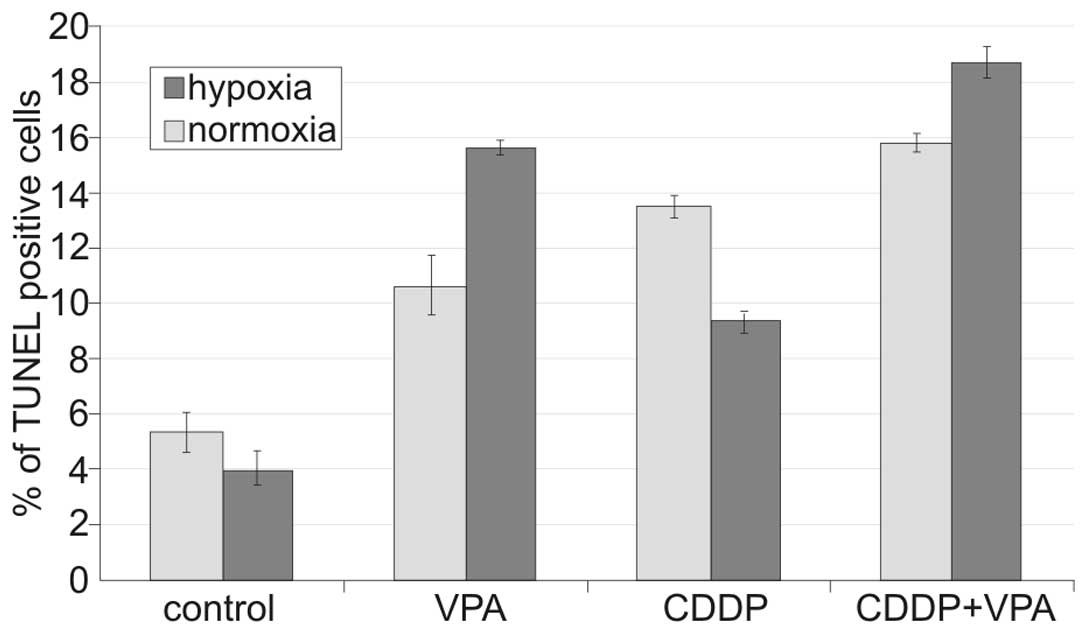
Cells were treated with lower concentrations of VPA
(1 mM) or CDDP (1 μM) alone and in combination. Apoptosis was
assessed 24 h after administration of the drugs using a TUNEL
assay. The degree of apoptosis induced by CDDP alone was diminished
by hypoxic conditions, while VPA alone was more efficient under
hypoxic conditions than under normoxic conditions. Cells
administered as combination of VPA and CDDP showed a higher degree
of apoptosis under hypoxic conditions (Fig. 2), suggesting not merely a
synergistic effect for VPA and CDDP, but the added ability of VPA
to overcome hypoxia-induced resistance to CDDP.
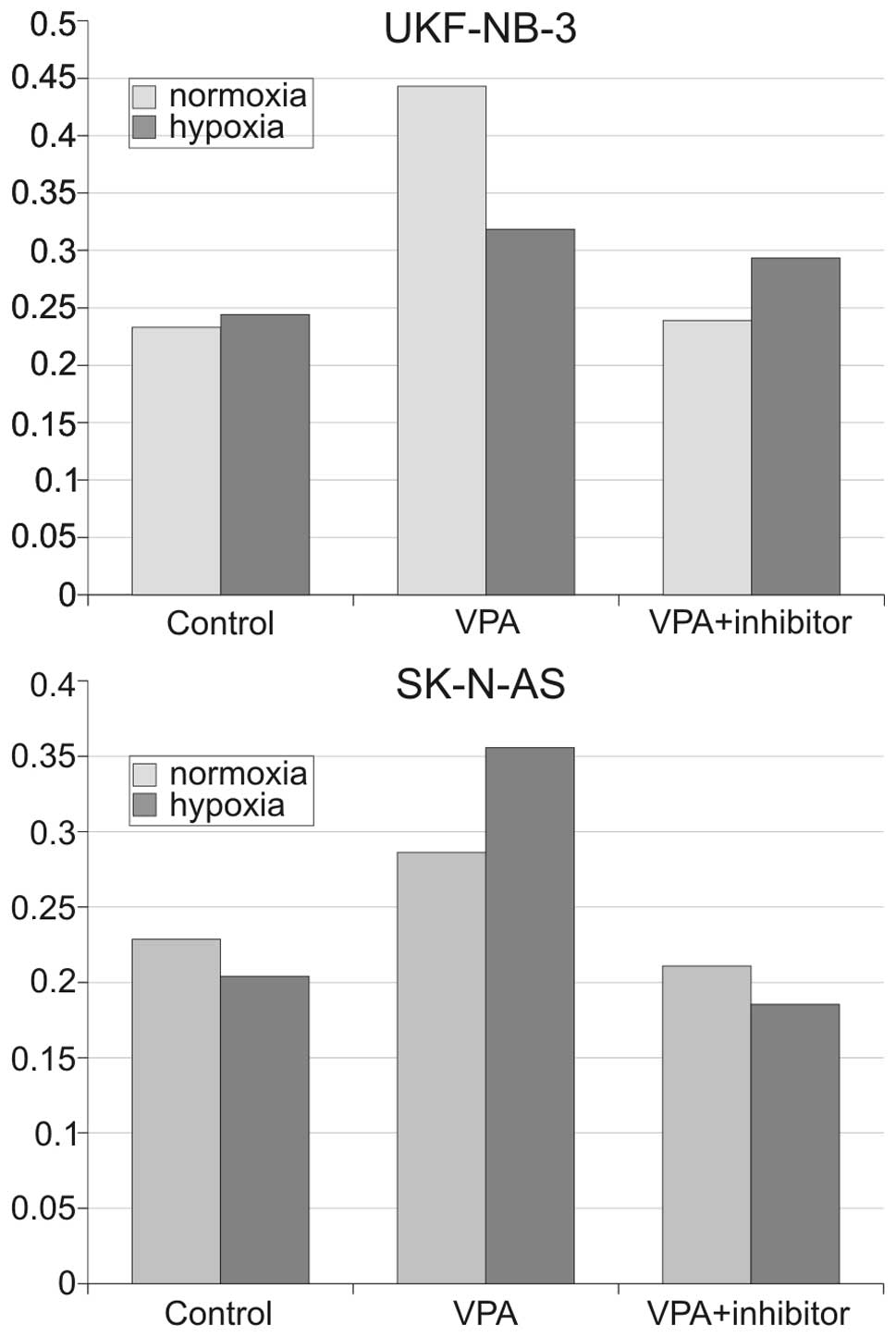
VPA activates caspase-8
To clarify whether VPA activates the
receptor-mediated apoptotic pathway, we determined the activity of
caspase-8. Cells were grown for 24 h and then 2 mM VPA was added to
UKF-NB-3 cells and 5 mM was added to SK-N-AS cells. Caspase-8
activity was determined after 48 h of treatment. VPA increased the
activity of caspase-8 in both cells lines (Fig. 3). Of note, caspase-8 activity was
higher under hypoxic conditions in the SK-N-AS line, albeit only
slightly. This discovery supports the above mentioned observations
that showed VPA to be more effective under hypoxic conditions. This
result also suggests that caspase-8 is the first caspase activated
in the apoptotic cascade during VPA treatment, which is why we
focused on the cleavage of the pro-apoptotic BID protein. Since BID
is the substrate for caspase-8, its cleavage would clearly
demonstrate the presence of activated caspase-8.
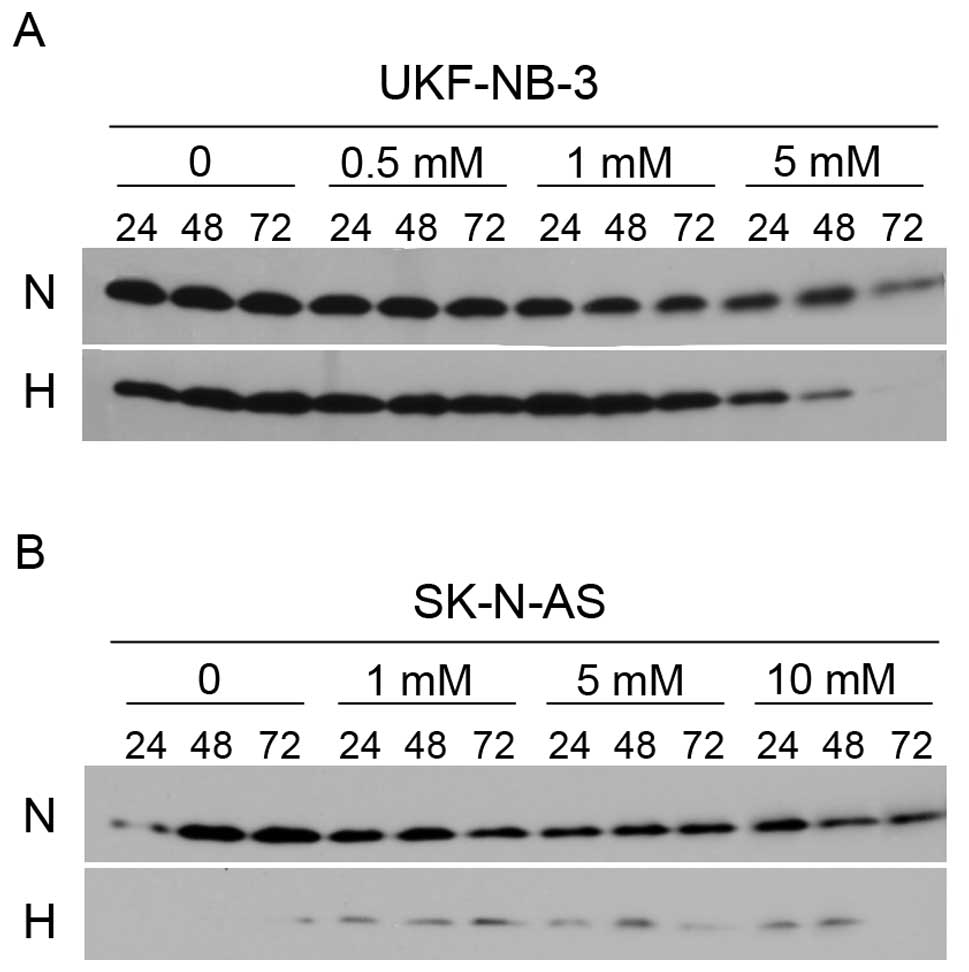
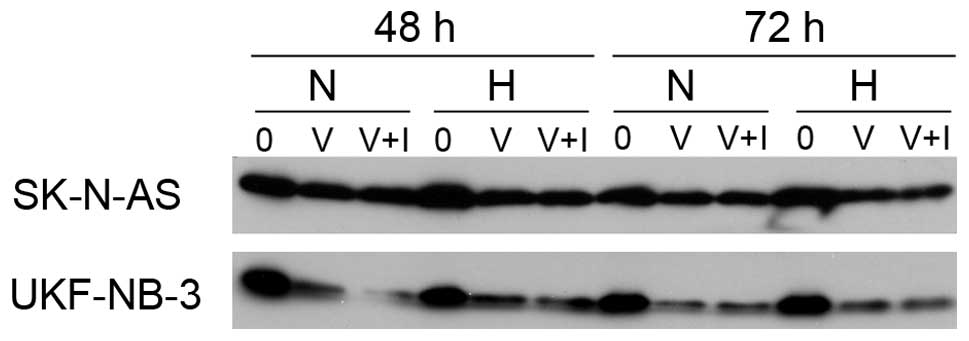
VPA initiates cleavage of BID
We addressed the question whether BID is cleaved to
its active form, which could consecutively activate the
mitochondrial apoptotic pathway. Cells were treated with different
concentrations of VPA (0.5, 1 and 5 mM for UKF-NB-3 and 1, 5 and 10
mM for SK-N-AS) for 24, 48 and 72 h (Fig. 4A). We observed a time- and
dose-dependent cleavage of BID in the UKF-NB-3 cell line under
normoxic conditions. Whereas under hypoxic conditions BID was
cleaved only when treatment with a relatively high concentration of
VPA (5 mM). In the case of the SK-N-AS line, corresponding
concentrations of VPA also led to a decrease of full-length BID
albeit only marginally (Fig. 4B).
This is in concert with the lower overall sensitivity of this cell
line to VPA. We used 20 mM of VPA to confirm the dose-dependent
manner of BID cleavage in SK-N-AS. This enormous concentration of
VPA, exceeding IC50 values of SK-N-AS, decreased
full-length BID, confirmed that BID cleavage caused by VPA was
really dose-dependent and also demonstrated the poor sensitivity of
this cell line to VPA. Together these data indicate that BID is
cleaved upon VPA treatment and can subsequently transfer the
apoptotic signal from the receptor-mediated to the intrinsic
apoptotic pathway.
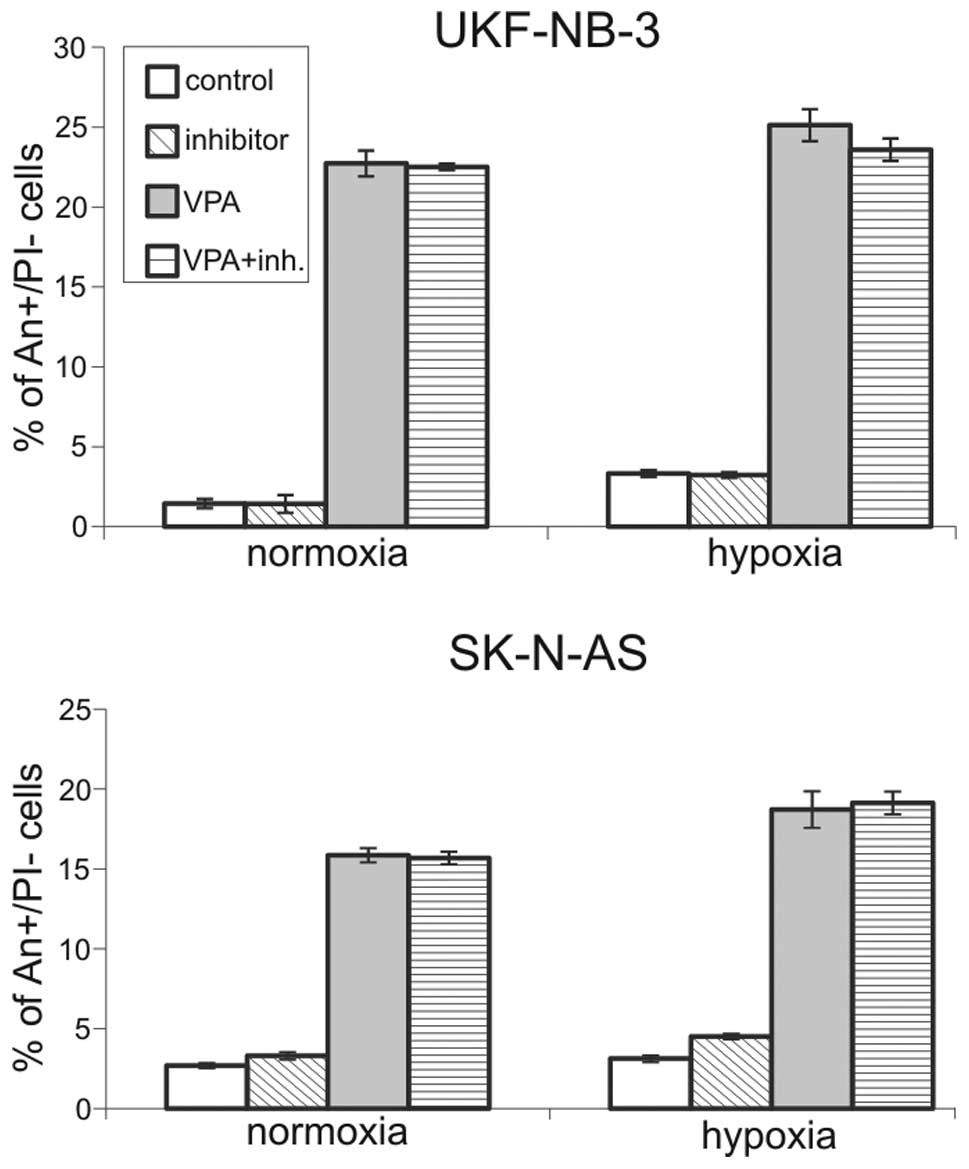
Inhibition of caspase-8 does not
influence VPA-induced apoptosis
Caspases-8 has been reported as the main effector
responsible for BID cleavage (22).
We therefore inhibited caspase-8 using a specific inhibitor,
z-IETD-fmk, to determine whether its inhibition was capable of
blocking apoptosis induced by VPA. Cells were treated with 2 μM
z-IETD-fmk for 15 min preceding VPA addition. Cell cultures were
then incubated together with caspase-8 inhibitor and VPA for 48 h.
We employed Annexin V/PI labeling to detect apoptotic changes.
Surprisingly, overall viability measured as Annexin V/PI double
negative cells was not increased in samples treated with the
caspase-8 inhibitor. This inhibition did not influence the
percentage of early apoptotic cells (An+/PI−)
(Fig. 5) nor the percentage of
necrotic/late apoptotic cells (An+/PI+). We
did not observe a shift of Annexin V/propidium iodide double
positive cells to the Annexin V single positive population, which
would have signaled that caspase-8 inhibition only delayed
apoptotic progress. Moreover, there were no differences between
normoxic and hypoxic conditions. WB analysis showed that BID was
cleaved regardless of caspase-8 inhibition (Fig. 6), which further points to a
non-essential role for caspase-8 in apoptosis induction.
The effectivity of caspase-8 inhibition was also
determined by measuring its activity after treatment with
z-IETD-fmk. It was found that it was decreased to the level of
untreated samples (data not shown); this confirmed that the
concentration of z-IEDT-fmk used was sufficient. It is therefore
evident that inhibition of caspase-8 has no significant effect on
apoptosis and BID cleavage in NBL cell lines.
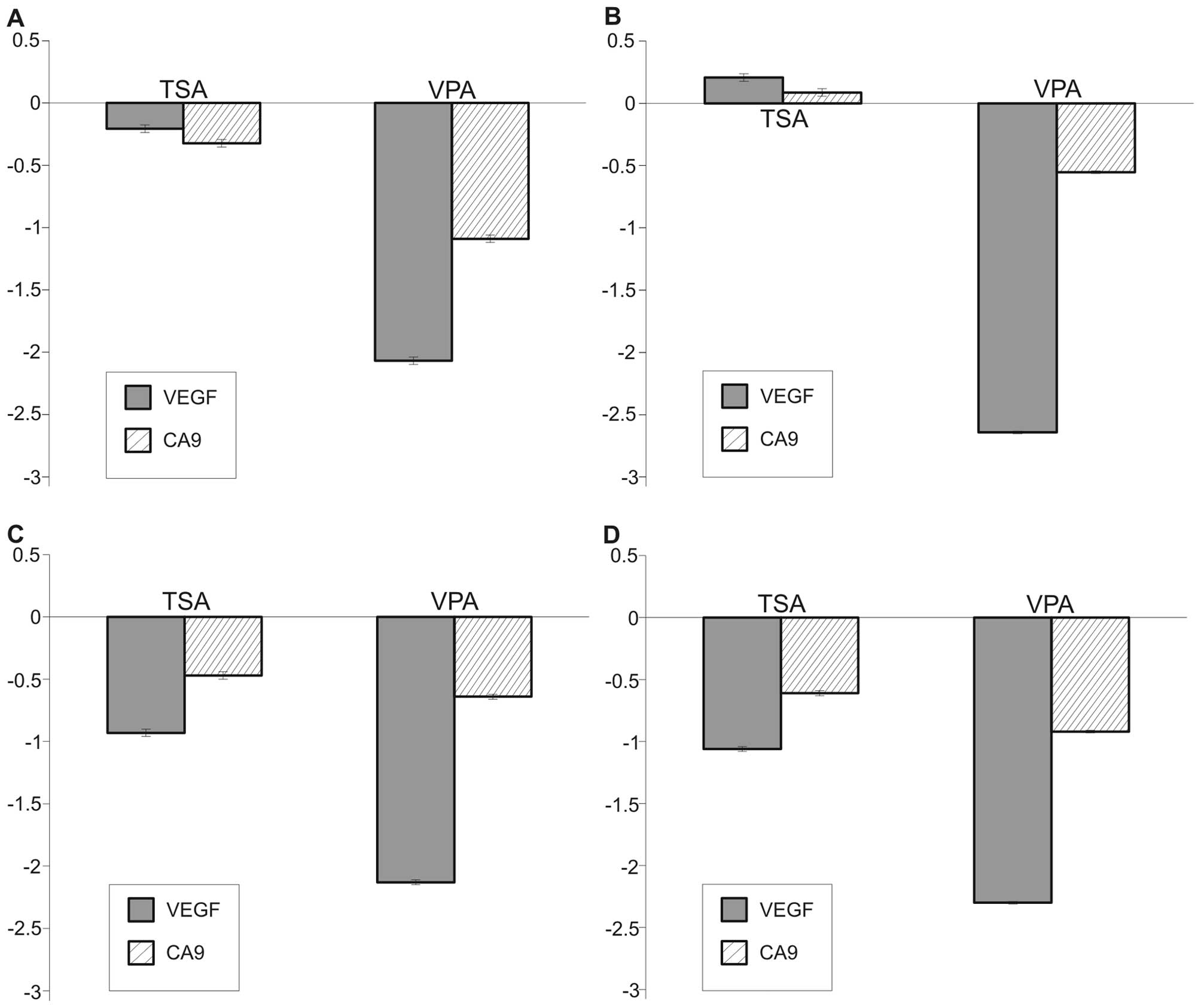
VPA decreases transcriptional activity of
HIF-1
Hypoxia inducible factor 1 (HIF-1) influences the
expression of many genes which can directly or indirectly inhibit
apoptosis (23,24). HDACi have been described to
attenuate stability of HIF-1 hence re-establishing sensitivity to
apoptosis. We employed real-time PCR techniques for determination
of mRNA levels of two well-described (25,26)
HIF-1 target genes, VEGF and carbonic-anhydrase 9 (CA9) in order to
assess whether VPA diminish HIF-1 transcriptional activity in NBL
cells. Cells were preincubated with 2 mM VPA or 100 μM trichostatin
A for 24 h and then placed into a hypoxia chamber for 3 and 8 h,
respectively. Expression of both genes was significantly
(P<0.01) decreased, in a time-dependent manner in both SK-N-AS
and UKF-NB-3 cell lines (Fig. 7).
VPA attenuated expression of VEGF 2.2-fold and CA9 4.2-fold
compared with untreated samples of UKF-NB-3 after 3 h of hypoxia.
Similar results were obtained for SK-N-AS cells. These results
indicate that inhibition of HIF-1 by VPA participates with higher
efficiency of VPA under hypoxic conditions by sensitizing NBL cells
to apoptosis as discussed below.
Discussion
Hypoxia is regarded as a negative prognostic factor
for solid tumors. It correlates with higher risk of cancer
malignancy, resistance to radio- and chemotherapy and poorer
patient outcomes (27,28). Hence, agents capable of overcoming
hypoxia resistance would be beneficial for cancer treatment. We
found that VPA was able to induce apoptosis under hypoxic
conditions and moreover, was even more efficient than under
normoxic conditions. To our knowledge this is the first observation
of increased VPA efficacy under hypoxic conditions. Moderate
hypoxia (1% O2) caused apoptosis resistance in hypoxic
cells (29,30). Resistance can be caused by both
HIF-1-dependent and -independent mechanisms. The role of HIF-1 as
an anti- or pro-apoptotic transcription factor is still
controversial (31). It is
dependent on the severity and duration of hypoxia, HIF-1
phosphorylation status and cell type (32). HDACi have been previously reported
to attenuate HIF-1 transcription activity (33). In concert with this observation, we
showed that two HDACi (VPA and TSA) down-regulate expression of
HIF-1 target genes VEGF and CA9 in hypoxic NBL cells. Several
mechanisms can be proposed by which inhibition of HIF-1 by VPA
promotes apoptosis under hypoxic conditions via attenuation of
HIF-1 transcriptional activity.
p53 is usually said to be stabilized by HIF-1
(34) hence promoting apoptosis.
However, it has been recently shown that HIF-1 can also antagonize
p53 pro-apoptotic function through several mechanisms. First, HIF-1
increases expression of tyrosinase-related protein 2 (TRP2; also
called DCT) which then down-regulates p53, thereby impeding
apoptosis (35). Second,
homeodomain-interacting protein kinase-2 (HIPK2) is an important
co-activator of p53. HIF-1 increases proteasomal degradation of
HIPK2 under hypoxic conditions, which eventually attenuates p53
pro-apoptotic function (36). Taken
together, inhibition of HIF-1 by VPA can promote apoptosis by both
re-establishing HIPK2 levels and attenuation of TRP2
expression.
AP-1 is another transcription factor induced by
hypoxia. Recent studies showed that induction of AP-1 is also
involved in hypoxia induced resistance to apoptosis (37–39).
On the other hand, we do not suspect a role for AP-1 regarding the
higher efficacy of VPA during hypoxic conditions, since it has been
shown that VPA enhances AP-1 mediated gene expression in the
SH-SY5Y NBL cell line (40).
Therefore, VPA acts, most likely, as an inductor of AP-1 rather
than a suppressor. Additionally, lithium chloride (LiCl) also
increases transcription activity of AP-1, however, we did not
observe higher efficacy of LiCl during hypoxia (data not shown). It
is therefore probable that AP-1 has no significant role in VPA
induced apoptosis during hypoxia. The actual contribution of
different transcription factors to hypoxia-induced apoptosis
resistance depends on several things (e.g. cell type, severity and
length of hypoxia and/or type of pro-apoptotic stimuli); therefore
a substantial role for HIF-1 is very likely in NBL cell lines.
Two points concerning the question of whether VPA
should be used as monotherapy or in a combination regimen need to
be addressed. First, despite the ability of VPA to overwhelm
hypoxia resistance, sensitivity of some NBL cell lines, e.g.
SK-N-AS in this study and UKF-NB-4, reported in our previous study
(41) is quite low. For example,
there was only a 20% induction of apoptotic cells by 5 mM VPA in
SK-N-AS after 72 h, whereas 1 mM VPA has been reported to induce
apoptosis in >50% of cells in some hematological malignancies
(5). Second, plasma levels of VPA,
in patients treated for epilepsy, usually do not exceed 0.7 mM and
have minimal or no side effects in such concentrations. Serious
adverse reactions are seen when the concentration exceeds 3.1 mM
(42). It can be argued that unlike
epilectic patients, where very long-term therapy is necessary,
cancer patients could tolerate short-term application of higher
doses of VPA. Our measurement of apoptosis when cells were treated
with VPA and CDDP together demonstrated that low concentration of
VPA (1 mM) were enough to overcome hypoxia induced apoptosis
resistance to CDDP while still maintaining low VPA toxicity. Based
on this we see VPA, in NBL treatment, mainly used in combination
regimens in which its low concentration would have minimal side
effects, yet it would be able to synergize with other agents even
in the hypoxic areas of a tumor.
BID is thought to be cleaved by caspase-8 upon
activation of receptor mediated apoptosis. Truncated BID (tBID)
then translocates from the cytosol to mitochondria where it
promotes release of cytochrome c and caspase-9 which, in turn,
forms apoptosome and activates executive caspase-3. However, BID
can also be cleaved by caspase-3 and served as a self-amplification
loop (22). We suspect that BID
cleavage, during VPA treatment, is mediated by caspase-3, since
inhibition of caspase-8 neither prevented BID cleavage nor
influenced the number of apoptotic cells.
Although HDACi have been described to trigger
apoptosis through both receptor mediated (43) and intrinsic pathways, the latter was
shown to be dominant in NBL cells during VPA treatment (44). We also demonstrated this in our
experimental setting. We further showed that mitochondrial
activation is the first event in apoptosis induction and BID
cleavage and caspase-8 activation were a consequence of the
progressing apoptotic cascade. Notably, there was no difference in
the pathway through which apoptosis proceeded relative to normoxic
or hypoxic conditions and VPA treatment.
To conclude, we showed that VPA is effective in both
normoxic and hypoxic conditions and can overcome hypoxia induced
resistance to CDDP-induced apoptosis. Considering all its
advantages (i.e. orally applicable, low toxicity, an already
approved drug), VPA alone might be beneficial in NBL treatment
keeping in mind that VPA alone failed to induce significant
apoptosis in some NBL cell lines. However, VPA combined with
conventional chemotherapeutic drugs should be much more effective
and is worthy of consideration. Additionally, VPA seems to be a
very suitable compound for continued research regarding
hypoxia-induced resistance. We also presented a possible role for
HIF-1 as it relates to the VPA mode of action, but the direct
mechanisms by which it acts are unknown and need further
elucidation.
Acknowledgements
This study was supported by grants GAUK 72208/2008
and GACR P301/10/0356. We thank Professor Jindrich Cinatl who
kindly provided cell lines and Dr Michael Pfaffl for developing and
freely distributing REST-MCS software for calculating relative
expression in real-time PCR techniques.
References
|
1
|
Brodeur GM: Neuroblastoma: biological
insights into a clinical enigma. Nat Rev Cancer. 3:203–216. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Furchert SE, Lanvers-Kaminsky C, Juurgens
H, Jung M, Loidl A and Frühwald MC: Inhibitors of histone
deacetylases as potential therapeutic tools for high-risk embryonal
tumors of the nervous system of childhood. Int J Cancer.
120:1787–1794. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mossman D and Scott RJ: Long term
transcriptional reactivation of epigenetically silenced genes in
colorectal cancer cells requires DNA hypomethylation and histone
acetylation. PloS One. 6:e231272011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Santini V, Gozzini A and Ferrari G:
Histone deacetylase inhibitors: molecular and biological activity
as a premise to clinical application. Curr Drug Metab. 8:383–393.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bokelmann I and Mahlknecht U: Valproic
acid sensitizes chronic lymphocytic leukemia cells to apoptosis and
restores the balance between pro- and antiapoptotic proteins. Mol
Med. 14:20–27. 2007.PubMed/NCBI
|
|
6
|
Fullgrabe J, Hajji N and Joseph B:
Cracking the death code: apoptosis-related histone modifications.
Cell Death Differ. 1:1238–1243. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hrebackova J, Poljakova J, Eckschlager T,
et al: Histone deacetylase inhibitors valproate and trichostatin A
are toxic to neuroblastoma cells and modulate cytochrome P450 1A1,
1B1 and 3A4 expression in these cells. Interdiscip Toxicol.
2:205–210. 2009. View Article : Google Scholar
|
|
8
|
Cinatl J, Scholz M, Driever PH, et al:
Antitumor activity of sodium valproate in cultures of human
neuroblastoma cells. Anticancer Drugs. 7:766–773. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Michaelis M, Suhan T and Cinatl J, Driever
PH and Cinatl J: Valproic acid and interferon-α synergistically
inhibit neuroblastoma cell growth in vitro and in
vivo. Int J Oncol. 25:1795–1799. 2004.
|
|
10
|
Harris AL: Hypoxia - a key regulatory
factor in tumour growth. Nat Rev Cancer. 2:38–47. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vaupel P, Kallinowski F and Okunieff P:
Blood flow, oxygen and nutrient supply, and metabolic
microenvironment of human tumors: a review. Cancer Res.
49:6449–6465. 1989.PubMed/NCBI
|
|
12
|
Hussein D, Estlin EJ, Dive C and Makin GW:
Chronic hypoxia promotes hypoxia-inducible factor-1alpha-dependent
resistance to etoposide and vincristine in neuroblastoma cells. Mol
Cancer Ther. 5:2241–2250. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lara PC, Lloret M, Clavo B, et al: Severe
hypoxia induces chemo-resistance in clinical cervical tumors
through MVP over-expression. Radiat Oncol. 4:292009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Song X, Liu X, Chi W, et al:
Hypoxia-induced resistance to cisplatin and doxorubicin in
non-small cell lung cancer is inhibited by silencing of HIF-1alpha
gene. Cancer Chemother Pharmacol. 58:776–784. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Giuntoli S, Rovida E, Barbetti V,
Cipolleschi MG, Olivotto M and Dello Sbarba P: Hypoxia suppresses
BCR/Abl and selects imatinib-insensitive progenitors within clonal
CML populations. Leukemia. 20:1291–1293. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang LE, Bindra RS, Glazer PM and Harris
AL: Hypoxia-induced genetic instability - a calculated mechanism
underlying tumor progression. J Mol Med (Berl). 85:139–148. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brizel DM, Scully SP, Harrelson JM, et al:
Tumor oxygenation predicts for the likelihood of distant metastases
in human soft tissue sarcoma. Cancer Res. 56:941–943.
1996.PubMed/NCBI
|
|
18
|
Hockel M, Schlenger K, Aral B, et al:
Association between tumor hypoxia and malignant progression in
advanced cancer of the uterine cervix. Cancer Res. 56:4509–4515.
1996.PubMed/NCBI
|
|
19
|
Hockel M, Schlenger K, Hockel S and Vaupel
P: Hypoxic cervical cancers with low apoptotic index are highly
aggressive. Cancer Res. 59:4525–4528. 1999.PubMed/NCBI
|
|
20
|
Bossy-Wetzel E and Green DR: Detection of
apoptosis by annexin V labeling. Methods Enzymol. 322:15–18. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pfaffl MW, Horgan GW and Dempfle L:
Relative expression software tool (REST) for group-wise comparison
and statistical analysis of relative expression results in
real-time PCR. Nucleic Acids Res. 30:e362002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yin X-M: Bid, a BH3-only multi-functional
molecule, is at the cross road of life and death. Gene. 369:7–19.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Baek JH, Jang JE, Kang CM, Chung HY, Kim
ND and Kim KW: Hypoxia-induced VEGF enhances tumor survivability
via suppression of serum deprivation-induced apoptosis. Oncogene.
19:4621–4631. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu X-H, Yu EZ, Li Y-Y and Kagan E:
HIF-1alpha has an anti-apoptotic effect in human airway epithelium
that is mediated via Mcl-1 gene expression. J Cell Biochem.
97:755–765. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee J-W, Bae S-H, Jeong J-W, Kim S-H and
Kim K-W: Hypoxia-inducible factor (HIF-1)alpha: its protein
stability and biological functions. Exp Mol Med. 36:1–12. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Semenza GL: HIF-1: mediator of
physiological and pathophysiological responses to hypoxia. J Appl
Physiol. 88:1474–1480. 2000.PubMed/NCBI
|
|
27
|
Shannon AM, Bouchier-Hayes DJ, Condron CM
and Toomey D: Tumour hypoxia, chemotherapeutic resistance and
hypoxia-related therapies. Cancer Treat Rev. 29:297–307. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Um JH, Kang CHD, Bae JH, et al:
Association of DNA-dependent protein kinase with hypoxia inducible
factor-1 and its implication in resistance to anticancer drugs in
hypoxic tumor cells. Exp Mol Med. 36:233–242. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Graeber TG, Osmanian C, Jacks T, et al:
Hypoxia-mediated selection of cells with diminished apoptotic
potential in solid tumours. Nature. 379:88–91. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu L, Ning X, Sun L, et al:
Hypoxia-inducible factor-1 alpha contributes to hypoxia-induced
chemoresistance in gastric cancer. Cancer Sci. 99:121–128.
2008.PubMed/NCBI
|
|
31
|
Piret J-P, Mottet D, Raes M and Michielis
C: Is HIF-1alpha a pro- or an anti-apoptotic protein? Biochem
Pharmacol. 64:889–892. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Suzuki H, Tomida a and Tsuruo T:
Dephosphorylated hypoxia-inducible factor 1alpha as a mediator of
p53-dependent apoptosis during hypoxia. Oncogene. 20:5779–5788.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim SH, Jeong JW, Park JA, et al:
Regulation of the HIF-1α stability by histone deacetylases. Oncol
Rep. 17:647–651. 2007.
|
|
34
|
An WG, Kanekal M, Simon MC, Maltepe E,
Blagosklonny MV and Neckers LM: Stabilization of wild-type p53 by
hypoxia-inducible factor 1alpha. Nature. 392:405–408. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sendoel A, Kohler I, Fellmann C, Lowe WS
and Hengartner OM: HIF-1 antagonizes p53-mediated apoptosis through
a secreted neuronal tyrosinase. Nature. 465:577–585. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nardinocchi L, Puca R and Orazi GD: HIF-1α
antagonizes p53-mediated apoptosis by triggering HIPK2 degradation.
Aging. 3:33–43. 2011.
|
|
37
|
Flamant L, Notte A, Ninane N, Raes M and
Michiels C: Anti-apoptotic role of HIF-1 and AP-1 in paclitaxel
exposed breast cancer cells under hypoxia. Mol Cancer. 9:1912010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dong Z, Venkatachalam Ma, Wang J, et al:
Up-regulation of apoptosis inhibitory protein IAP-2 by hypoxia.
Hif-1-independent mechanisms. J Biol Chem. 276:18702–18709. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Piret J-P, Cosse J-P, Ninane N, Raes M and
Michiels C: Hypoxia protects HepG2 cells against etoposide-induced
apoptosis via a HIF-1-independent pathway. Exp Cell Res.
312:2908–2920. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen G, Yuan PX, Jiang YM, Huang LD and
Manji HK: Valproate robustly enhances AP-1 mediated gene
expression. Brain Res Mol Brain Res. 64:52–58. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hrebackova J, Hrabeta J and Eckschlager T:
Valproic acid in the complex therapy of malignant tumors. Curr Drug
Targets. 11:361–379. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Catalano MG, Fortunati N, Pugliese M, et
al: Valproic acid induces apoptosis and cell cycle arrest in poorly
differentiated thyroid cancer cells. J Clin Endocrinol Metab.
90:1383–1389. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nebbioso A, Clarke N, Voltz E, et al:
Tumor-selective action of HDAC inhibitors involves TRAIL induction
in acute myeloid leukemia cells. Nat Med. 11:77–84. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Muhlethaler-Mottet A, Meier R, Flahaut M,
et al: Complex molecular mechanisms cooperate to mediate histone
deacetylase inhibitors anti-tumour activity in neuroblastoma cells.
Mol Cancer. 7:552008. View Article : Google Scholar
|