Introduction
Polycomb group (PcG) proteins, which are known to
maintain the silenced state of homeotic genes, are important for
constituting a cellular memory system responsible for maintaining
the epigenetic status of target genes throughout the lifetime of
the organism (1,2). PcG proteins play a crucial role in
many physiological processes, such as germline development, cell
differentiation, pluripotency and stem cell self-renewal (3,4). PcG
proteins form transcriptional repressor modules that functionally
can be divided into at least three distinct classes of complexes:
polycomb repression complex 1 (PRC1), including RING1A, HPC1-3,
HPH1-3, and Bmi-1, is to maintain repression; PRC2, with the core
proteins EZH2, EED, and SUZ12, is to inhibit gene expression
directly. Both PRC1 and PRC2 members have been found involved in
malignant transformation and tumor development in various
hematological and epithelial cancers (5).
B-cell-specific moloney murine leukemia virus
integration site 1 (Bmi-1) is a member of PRC1 that was initially
identified as an oncogene involved in the development of mouse pre
B-cell lymphoma cooperating with c-Myc (6,7). Many
studies have demonstrated that Bmi-1 protein regulates the
INK4a/ARF locus, which encodes the two tumor suppressors,
p16INK4a and p14ARF (p19ARF in
mouse), which act in pRb and p53 cell cycle control pathways,
respectively (8,9). Bmi-1 promotes cellular proliferation
by repression the expression of the INK4a/ARF locus (9). Moreover, overexpression of Bmi-1 in
epithelial cells could induce human telomerase reverse
transcriptase activity, which is associated with cell
immortalization (10,11). It has also been shown that Bmi-1
overexpression together with H-Ras promotes human mammary
epithelial cell (HMEC) transformation and breast oncogenesis
(12). Interestingly, Bmi-1 has
been recently shown to play a crucial role in self renewal of
hematopoietic and neural stem cells and leukemic stem cells
(13–15). Previous studies also showed that
Bmi-1 plays important roles in regulating self-renewal of normal
and tumorigenic human mammary stem cells (16).
In clinical study, overexpression of Bmi-1 has been
correlated with cancer susceptibility and poor prognosis in several
human cancers, including non-small cell lung cancer (17), gastric carcinoma (18), hepatocellular carcinoma (19), acute myeloid leukemia (20), breast cancer (21), nasopharyngeal carcinoma (22), and bladder cancer (23). Furthermore, Bmi-1-associated gene
expression pathway, which is 11 gene Bmi-1 stem cell signature, is
a powerful predictor of a short interval to distant metastasis,
highly malignant clinical course of disease progression, and high
likehood of therapy failure in multiple types of human cancer
(24). Epithelial-mesenchymal
transition (EMT), epithelial cells acquire mesenchymal-like
properties, which increase cell motility, and EMT generates cells
with properties of stem cells (25). Bmi-1 is essential in EMT process
(22,26,27),
and EMT also mediates radioresistance in human cancer cells
(28,29). Our previous study demonstrated that
Bmi-1 promotes the invasion and metastasis of human breast cancer
and predicts poor survival, the inhibition of Bmi-1 reverses the
expression of EMT markers and inhibits the Akt/GSK3β/Snail pathway
(30). These observations led us to
hypothesize that abrogation of Bmi-1 expression could be a
potential therapeutic strategy against human cancers. We
hypothesized that Bmi-1 inhibition combined radiotherapy could
induce synergistic effect on MCF-7 tumor cells. We tested this
hypothesis by evaluating the effects of Bmi-1 inhibition by shRNA
on DNA damage, apoptosis, mitochondrial membrane potential (ΔΨm)
and apoptosis related protein for the purpose of a more improved
cancer therapy.
Materials and methods
Cell culture
MCF-7 (human breast cancer) cell line was obtained
from the American Type Culture Collection (ATCC, Manassas, VA) and
incubated in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10%
fetal bovine serum (FBS; Hyclone, Logan, UT), penicillin (100
units/ml), and streptomycin (100 units/ml) at 37°C in humidified 5%
CO2 incubator.
Generation of stable Bmi-1 overexpression
and Bmi-1 knockdown (KD) cell lines
Retroviral vector pMSCV-Bmi-1 and Bmi-1 short
hairpin RNA (shRNA) was previously described (22, 28).
Retroviruses were generated by transients transfection as described
(31). Bmi-1 gene was introduced
into MCF-7 cells by infecting cells with a retroviral vector
pMSCV-Bmi-1 or pMSCV-Bmi-1-shRNA (knockdown, KD). Control cells
were infected with the empty retroviral vector pMSCV.
Retrovirus-infected were selected and maintained in 0.5 μg/ml
puromycin for 7 days and used as stable cells.
Radiation clonogenic survival assay
Cells were seeded in triplicate into 60-mm culture
dishes in a range of 100–10,000 cells per dish, depending on the
radiation dose that the cells received and the test condition, so
as to yield 0–100 colonies per dish. Cells were then irradiated
with a single dose of X ionizing radiation (irradiation rate of
104.93 cGy/min at 210 kV and 12 mA) (32), including 0, 1, 2, 4 and 6 Gy.
Immediately after irradiation, the treated cells were cultured in a
37°C, 5% CO2 incubator for 10–14 days. Individual
colonies (>50 cells per colony) were fixed with methanol and
stained with crystal violet. Plating efficiency (PE) and survival
fractions (SF) were calculated. Survival curves were fitted and
analyzed using linear-quadratic model [S=exp(−αD−βD2)]
by GraphPad Prism software (version 4.0, GraphPad Prism software,
San Diego, CA). The radiation sensitizing enhancement ratio (SER)
by Bmi-1 inhibition was calculated using the following formula:
SER=(SF2 of MCF-7 infected by control
vector)/(SF2 of Bmi-1-overexpressing MCF-7 or
Bmi-1-silencing MCF-7). SER=1 suggests an additive radiation effect
and SER >1, a supra-additive effect as against a sub-additive
effect in the case of SER <1.
Immunofluorescence staining
Cells were seeded on coverslips in 6-well plates and
allowed to grow overnight. Cells were irradiated with a single dose
of 0, 1, 2, 4 and 6 Gy. After 15 min, cells was washed and fixed
with 4% paraformaldehyde. Or cells were fixed at designed measuring
time (0, 15, 30 min, 1, 3, 6 and 20 h) after irradiated with a
single dose of 5 Gy. Then, cells were rinsed in phosphate-buffered
saline (PBS). After blocking in 10% normal blocking serum at room
temperature for 10 min, slides were incubated with γH2AX antibody
(Cell Signaling Technology, Beverly, MA) at 4°C overnight and then
incubated with goat anti-rabbit IgG conjugated with FITC (Molecular
Probe, Carlsbad, CA). Slides mounted with Prolong Gold antifade
reagent with DAPI (Molecular Probe) and examined by fluorescence
microscopy (Carl Zeiss Axioskop 2, Thornwood, NY). Cells were
judged as ‘positive’ for γH2AX foci when they displayed 10 or more
discrete dots of brightness. For quantitation of foci, a minimum of
100 cells were analyzed for each time point. All data points
represent mean ± SD of three experiments.
FACS analysis
After 8 Gy irradiation, cells were harvested at
designed measuring time (0, 3, 5 and 7 days) and fixed with 70%
ice-cold ethanol. Cells were treated with RNase A (50 μg/ml,
Sigma-Aldrich, St. Louis, MO) and stained with propidiumiodide (PI,
50 μg/ml, Sigma-Aldrich). The fluorescence of DNA-bound PI in cells
was measured with FACSCalibur Flow Cytometer (Becton-Dickinson,
Franklin Lakes, CA).
Measurement of mitochondrial membrane
potential (ΔΨ m
Mitochondrial membrane potential was measured using
the mitochondrial-specific dual-fluorescence probe, JC-1, based on
the method previously described (33). Cells were irradiated with 0 or 5 Gy.
After 24 h, cells were incubated with the 10 μg/ml JC-1 for 10 min
followed by two wash with PBS. A CytoFluor plate reader (excitation
wavelength 485 nm, slit width 20 nm) was used to monitor the
fluorescence intensities for the monomer and the aggregated JC-1
molecules (emission wavelength 520 nm, slit width of 25 nm, and 580
nm, slit width of 30 nm, respectively). Results were expressed as
fluorescence ratio (580/530 nm).
Western blot analysis
Cells were washed in cold phosphate-buffered saline
(PBS) and lysed in a RIPA buffer containing 50 mM Tris-HCl pH 7.4,
150 mM NaCl, 1 mM PMSF, 1 mM EDTA, 5 μg/ml aprotinin, 5 μg/ml
leupeptin, 1% Triton X-100, 1% sodium deoxycholate and 0.1% SDS.
The protein concentration was determined by the Bradford dye method
(Bio-Rad Laboratories, Hercules, CA). Equal amounts of cell extract
were subjected to electrophoresis in 10% or 15% SDS-PAGE and
transferred to polyvinylidene difluoride membranes (Amersham
Pharmacia Biotech, Piscataway, NJ). The membrane was probed with
primary antibody overnight, subsequently incubated with horseradish
peroxidase-conjugated anti-rabbit or anti-mouse immunoglobulin G
(1:2000; Santa Cruz Biotechnology, Santa Cruz, CA) and detected by
enhanced chemiluminescence (Amersham Pharmacia Biotech) according
to the manufacturer’s suggested protocols. Mouse anti-Bmi-1 (F6)
antibody was from Upstate Biotechnical (Lake Placid, NY), rabbit
anti-phosphorylated Akt (Ser473) antibody were from Cell Signaling,
and mouse anti-p53 (DO-1), mouse anti-p21 (F-5), mouse anti-Bax
(6A7), Bcl-2 (C-2), mouse anti-α-tubulin antibodies were from Santa
Cruz Biotechnology.
Statistical analysis
Statistical analysis was performed using Student’s
t-test or ANOVA test by SPSS 12.0 software (Abbott Laboratories,
North Chicago, IL). Differences were considered statistically
significant at P<0.05. We performed each study at least three
times under identical conditions.
Results
Bmi-1 expression is associated with
radiation response of MCF-7 cells
We first studied whether Bmi-1 overexpression was
correlated with radiosensitivity of MCF-7 cells. For this purpose,
we infected MCF-7 cells with a retroviral vector expressing Bmi-1
or pMSCV control vector. After puromycin selection, overexpression
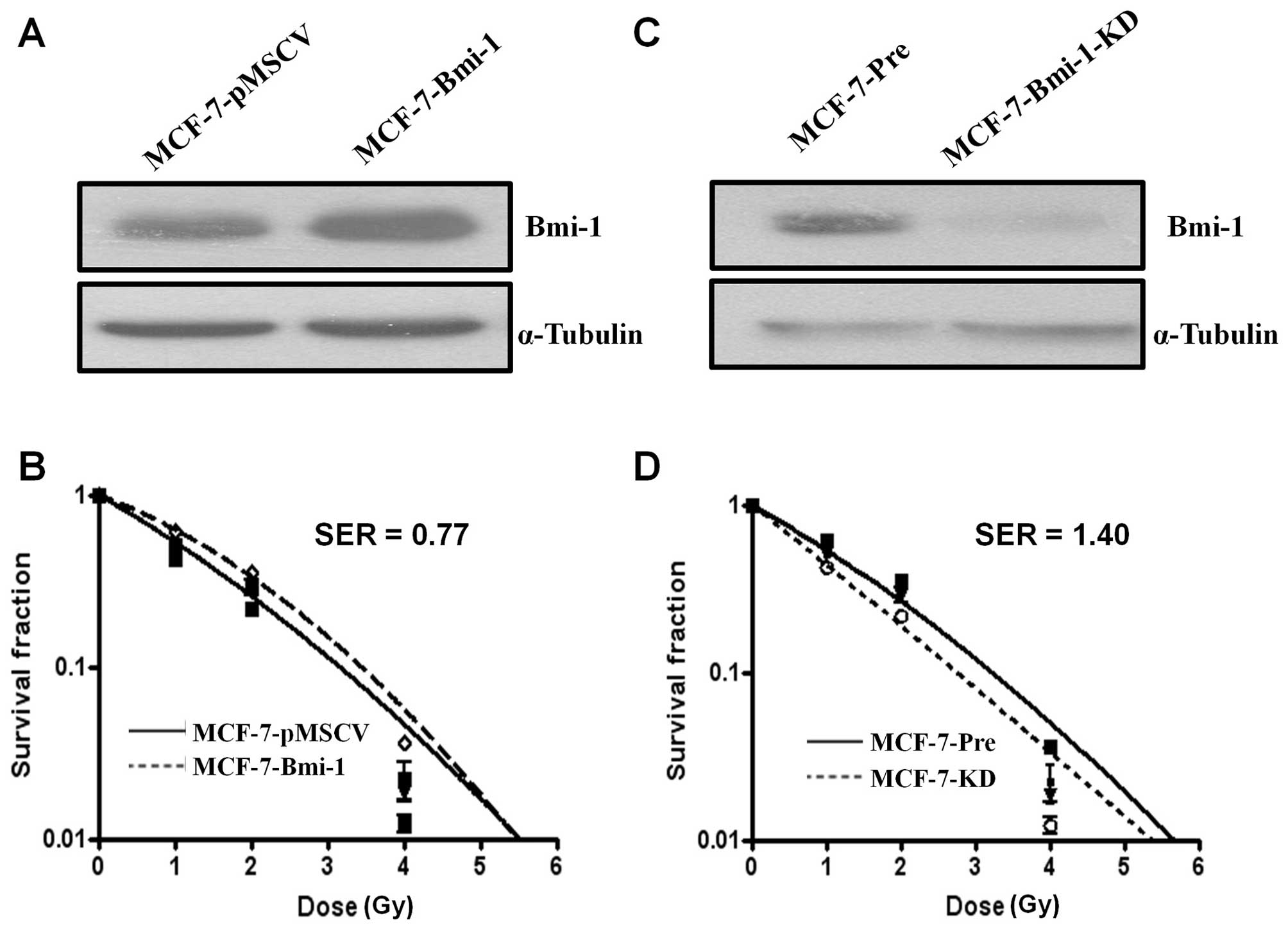
of Bmi-1 was confirmed by Western blot analysis (Fig. 1A). By classical radiation clonogenic
survival assay, we found that Bmi-1-overexpressing cells
(MCF-7-Bmi-1) had more radioresistance and higher survival than
pMSCV vector infected (MCF-7-pMSCV) cells (F=4.183, P=0.007; ANOVA
test) (Fig. 1B). Statistical
analysis of the survival parameters calculated using the
linear-quadratic model revealed that there was a significant
difference in the α value, characterizing the initial slope of the
curving radiation survival curve fitted to all the data from the
three repeat experiments as a function of dose, between MCF-7-Bmi-1
cells (α=0.380±0.039 Gy−1) and MCF-7-pmscv cells
(α=0.588±0.054 Gy−1) (P<0.05). Bmi-1 overexpression
enhanced radiation resistance by SER=0.77 (SF2=0.257 for
MCF-7-pMSCV cells; SF2=0.333 for MCF-7-Bmi-1 cells).
Next, we asked whether silencing of Bmi-1 expression
by RNA interference (RNAi) was able to increase the sensitivity of
MCF-7 cells to irradiation. As expected, Bmi-1 shRNA-expressing
cells (MCF-7-KD cells) exhibited significantly enhanced
radiosensitivity compared with control vector infected cells
(MCF-7-Pre cells) (F=4.183, P<0.001; ANOVA test) (Fig. 1C and D). Statistical analysis of the
survival parameters calculated using the linear-quadratic model
also revealed a significant sensitization effect. The α value for
MCF-7-KD and MCF-7-Pre cells was 0.814±0.056 and 0.580±0.041
Gy−1, respectively. Bmi-1 inhibition enhanced
radiosensitivity by SER=1.40 (SF2=0.264 for MCF-7-Pre
cells; SF2=0.189 for MCF-7-KD cells). Thus, these data
suggested that Bmi-1 might be a critical regulator in radiation
response in MCF-7 cells.
Increased DNA double strand break (DSB)
and decreased DSB repair caused by Bmi-1 silencing
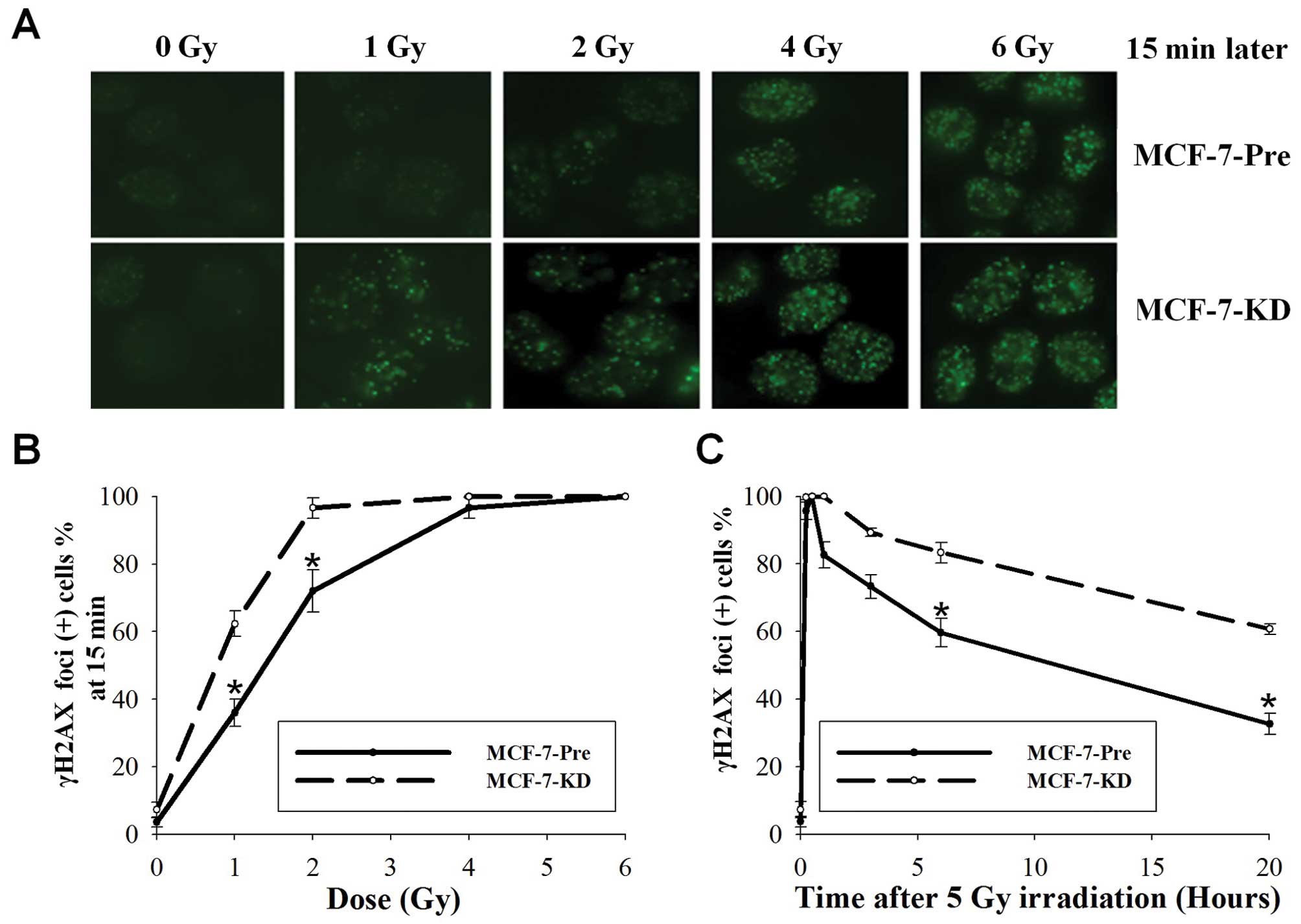
Phosphorylated histone H2AX (γH2AX) foci
measurement, a more sensitive detecting method of DNA DSBs, was
used to verify further this increase DNA DSBs after Bmi-1
silencing. As shown in Fig. 2A and
B, by 15 min after 1 Gy, 36.0% of MCF-7-Pre cells demonstrated
γH2AX foci compared with 62.3% of MCF-7-KD cells (P=0.001; t-test).
At 2 Gy, 72.0% of MCF-7-Pre cells and 94.7% of MCF-7-KD cells
displayed γH2AX foci (P=0.005; t-test). Foci were no difference in
4 Gy and observed in 100% of both cells at 6 Gy. These data further
demonstrated that Bmi-1 knockdown enhanced DNA DSBs.
We next examined the kinetics of γH2AX in MCF-7-Pre
and MCF-7-KD cells after irradiation. Fifteen minutes after 5 Gy,
95.7% of MCF-7-Pre cells and 100% of MCF-7-KD cells retained γH2AX
foci (Fig. 2C). At 1 h, foci
remained in 82% of MCF-7-Pre cells and 100% of MCF-7-KD cells. At 6
h, foci persisted in 59.7% of MCF-7-Pre cells and in 83.3% of
MCF-7-KD cells (P=0.001; t-test). After 20 h, the number of foci
for MCF-7-Pre and MCF-7-KD cells was 32.7 and 60.7%, respectively
(P<0.001; t-test). The results showed that the residual number
of γH2AX foci in MCF-7-KD cells was significantly higher than that
of MCF-7-Pre cells after irradiation. Thus, DSB repair capacity in
MCF-7-Pre and MCF-7-KD cells was significantly different,
indicating that Bmi-1 silencing deceased DSB repair capacity.
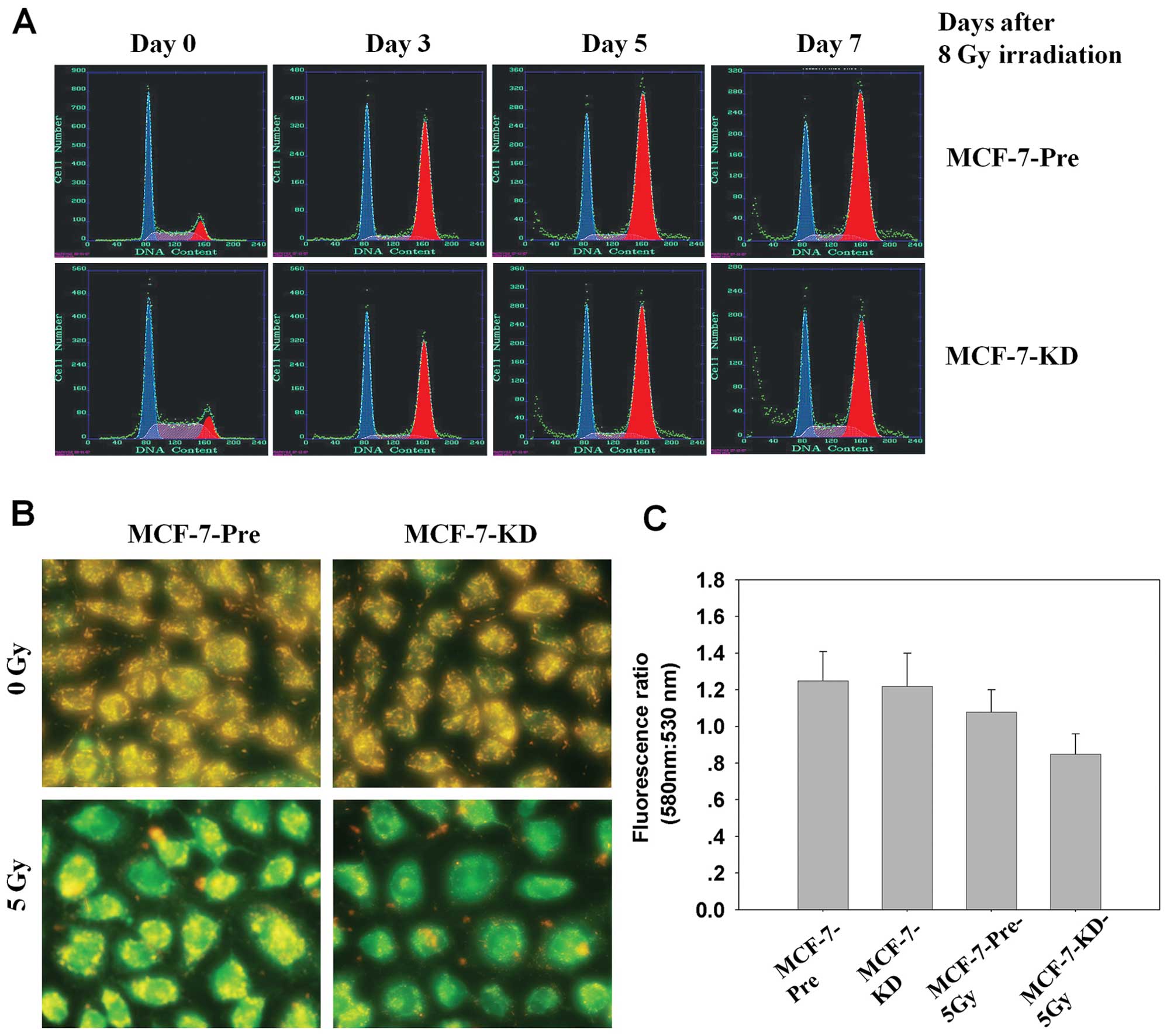
Bmi-1 knockdown promotes sub-G1
population and induces loss of mitochondrial membrane potential in
irradiated MCF-7 cells
It has reported that Bmi-1 enhanced cell survival by
altering cell cycle regulatory protein expression and inhibiting
apoptosis (34). Thus, we
investigate cell cycle distribution in MCF-7-Pre and MCF-7-KD cells
after 8 Gy irradiation in different time points by FACS. Radiation
induced G1-phase and G2-phase cell cycle arrest in both MCF-7-Pre
and MCF-7-KD cells, but there was no significant difference between
them. However, the percentage of the sub-G1 population, which is
indicative of apoptosis, significant increased in MCF-7-KD cells
(11.3±1.1%) compared with MCF-7-Pre cells (6.6±0.8%, P<0.001;
ANOVA) (Fig. 3A). The data
indicated that Bmi-1 knockdown was able to induce apoptosis in
MCF-7 cells.
Next, we examined whether Bmi-1 inhibtion induced
apoptosis in MCF-7 cells could be coincident with changes in
mitochondrial membrane potential (ΔΨm). We used the fluorescent
dye, JC-1, as an indicator of the energy state of the mitochondria.
A loss in mitochondria membrane potential is indicated by a
decrease in red/green fluorescence intensity ratio. As shown in
Fig. 3B and C, Bmi-1 inhibition led
to a rapid drop in mitochondrial energy, as reflected by a
fluorescence shift from green (emission 530 nm) to red (emission
580 nm) by 24 h after 5 Gy irradiation. These data demonstrated
that apoptosis induced by Bmi-1 inhibition combined radiation may
be closely related to mitochondrial function.
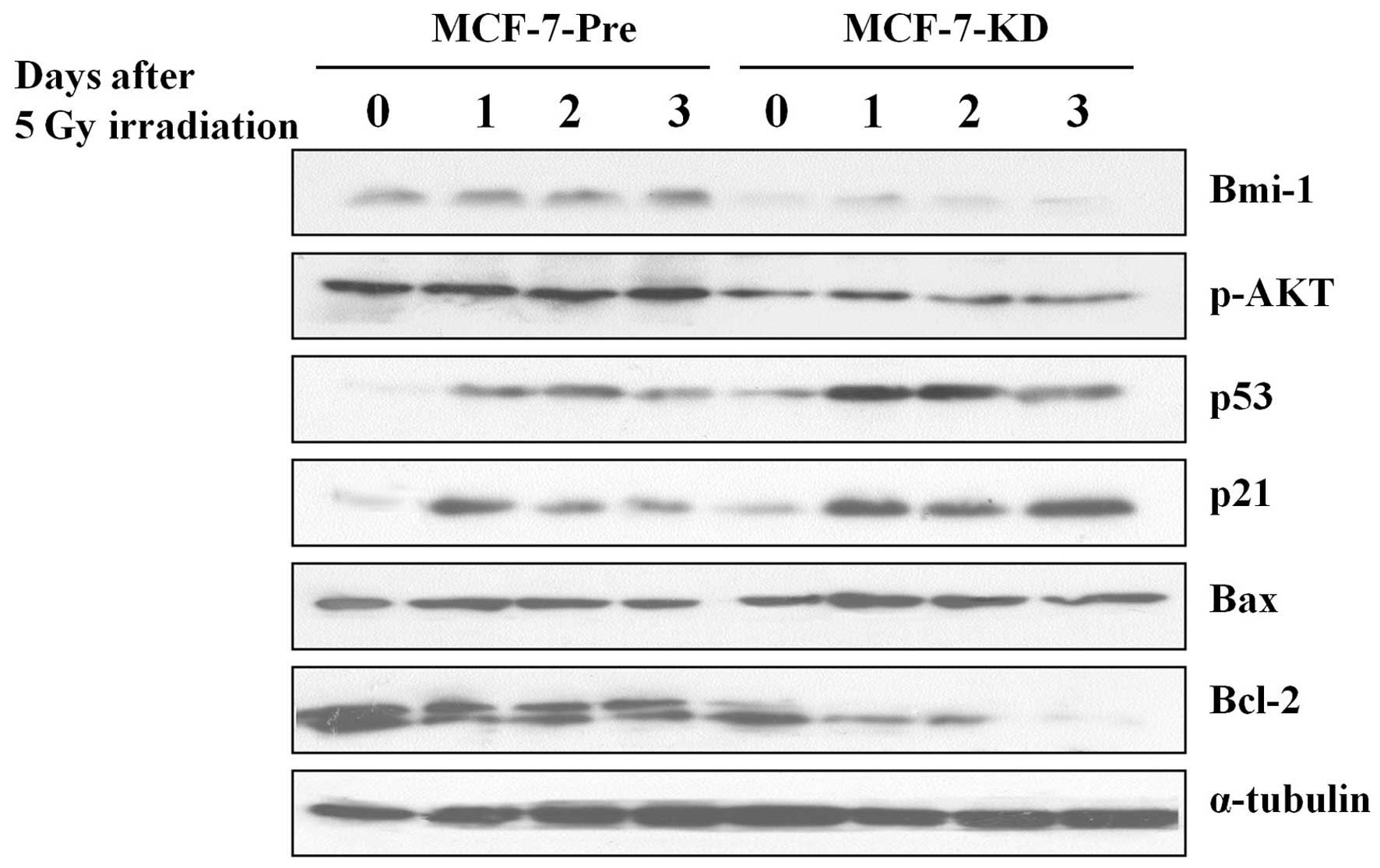
Bmi-1 knockdown changes the signal
pathway involved in apoptosis in MCF-7 cells
Then, we examined the levels of several apoptosis
related proteins in MCF-7-Pre and MCF-7-KD cells. Western blot
analysis showed that dramatically increased expression of p53, p21
and Bax protein, and decreased expression of p-AKT and Bcl-2 in
irradiated MCF-7-KD cells compared with those in irradiated
MCF-7-Pre cells (Fig. 4). This data
indicated that Bmi-1 depletion sensitizes radiotherapy through
activating apoptosis pathway.
Discussion
In this report, we present the first evidence that
Bmi-1 was correlated with radiation response in MCF-7 breast cancer
cells. We showed that Bmi-1 overexpression developed radiation
resistance, whereas Bmi-1 inhibition significantly increased
radiation sensitivity with a DER of 1.40 by radiation clonogenic
survival assay. It suggested that Bmi-1 might play an important
role in the regulation of cellular response to radiation in MCF-7
cells. Thus, the results underscore the importance of Bmi-1
targeting in combination with irradiation in tumor therapy. In our
previous study, we showed that Bmi-1 could enhance the invasion and
metastasis of human breast cancer and predicts poor survival, Bmi-1
silencing reverses the expression of EMT markers and inhibits the
Akt/GSK3β/Snail pathway (30). EMT
promotes radioresistance in human tumor cells (28,29),
down-regulation of Bmi-1 could be a novel strategy to sensitize
radiotherapy by reversing EMT in human breast cancer.
It is well known that DSBs are suggestive of
critical lesions in DNA caused by ionizing radiation. DSB is the
main mechanism of tumor cell death after irradiation, and some
protein involve in DSB could be used as a prognostic predictor for
cancer (35). The major cause of
radiotherapy failure is the success of DSB repair in tumor cells,
leading to prolonged tumor cell survival. Molecules that are
involved in DSB repair may be potential prognostic markers for the
prediction of radiotherapy outcome, and hence, for optimization of
treatment. We found that Bmi-1 inhibition dramatically increased
DSB and significantly decreased the rate of DNA DSB recovery
induced by irradiation, suggesting a participation of Bmi-1 in DNA
strand damage and repair. Studies have showed that increased p-Akt
had been linked to decreased radiation responsiveness and
inhibition of p-Akt have radiosensitizing effects in various
malignancies (36–39). Our study showed that p-Akt decreased
after Bmi-1 inhibition, this might be one of reasons resulting in
increased DSBs and decreased DSB repair in Bmi-1 knockdown
cells.
If complete DNA damage repair fails, apoptosis is
triggered for the elimination of damaged cells. We noticed that the
number of apoptotic cells in Bmi-1 knockdown cells was much higher
compared with that in the controls using FACS analysis. The
mechanism of Bmi-1 inhibition combined radiation induced apoptosis
is complex. A possible reason is that mitochondria-dependent
pathway for apoptosis was activated in response to combined Bmi-1
inhibition with ionizing radiation. In many tumor models of
apoptosis, oxidative stress induced by ROS is a frequent mediator
of apoptosis and produced massive cellular damage associated with
lipid peroxidation, loss of mitochondrial membrane potential (ΔΨm),
deletion of cellular antioxidants and Bax translocation to
mitochondrial (40–43). In this study, we found MCF-7 cell
silencing of Bmi-1 seems to be more susceptible to cell death with
loss of ΔΨm induced by irradiation (Fig. 3B and C). The underlying mechanism by
which Bmi-1 inhibition induces loss of ΔΨm in MCF-7 cells remains
undefined. Several mitochondrial proteins, including Bcl-2 and Bax,
possible play important roles in the process. During apoptosis, the
imbalance between the expression of anti- and pro-apoptotic
proteins is important. Our data showed that Bmi-1 inhibition
enhanced proapototic p53, p21 and Bax expression but decreased
levels of the antiapoptotic proteins p-AKT and Bcl-2 expression
compared with control cells following irradiation.
In conclusion, we report the enhanced
radiosensitivity of a breast cancer cell line after Bmi-1
inhibition using shRNA. The increased sensitivity was associated
with increased DSBs and decreased DSB repair. We also showed that
combination Bmi-1 inhibition with irradiation could induce
apoptosis, collapse of mitochondrial membrane potential, elevated
p53, p21, Bax expresseion, and deceased Bcl-2 expression. Our
results suggest that Bmi-1 inhibition play an additive effect to
radiation therapy in MCF-7 mammary carcinoma cells providing a
novel target for radio-sensitizing breast cancer.
Abbreviations:
|
Bmi-1
|
B-cell-specific moloney murine
leukemia virus integration site 1
|
|
EMT
|
epithelial-mesenchymal transition
|
|
DSB
|
DNA double strand break
|
|
KD
|
knock down
|
|
GSK3β
|
glycogen synthase kinase 3β
|
|
Akt
|
protein kinase B
|
References
|
1
|
Jacobs JJ and van Lohuizen M: Cellular
memory of transcriptional states by Polycomb-group proteins. Semin
Cell Dev Biol. 10:227–235. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schwartz YB and Pirrotta V: Polycomb
silencing mechanisms and the management of genomic programmes. Nat
Rev Genet. 8:9–22. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schuettengruber B, Chourrout D, Vervoort
M, Leblanc B and Cavalli G: Genome regulation by polycomb and
trithorax proteins. Cell. 128:735–745. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Spivakov M and Fisher AG: Epigenetic
signatures of stem-cell identity. Nat Rev Genet. 8:263–271. 2007.
View Article : Google Scholar
|
|
5
|
Rajasekhar VK and Begemann M: Concise
review: roles of polycomb group proteins in development and
disease: a stem cell perspective. Stem Cells. 25:2498–2510. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Haupt Y, Alexander WS, Barri G, Klinken SP
and Adams JM: Novel zinc finger gene implicated as myc collaborator
by retrovirally accelerated lymphomagenesis in E mu-myc transgenic
mice. Cell. 65:753–763. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
van Lohuizen M, Verbeek S, Scheijen B,
Wientjens E, van der Gulden H and Berns A: Identification of
cooperating oncogenes in E mu-myc transgenic mice by provirus
tagging. Cell. 65:737–752. 1991.PubMed/NCBI
|
|
8
|
Jacobs JJ, Scheijen B, Voncken JW, Kieboom
K, Berns A and van Lohuizen M: Bmi-1 collaborates with c-Myc in
tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF.
Genes Dev. 13:2678–2690. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jacobs JJ, Kieboom K, Marino S, DePinho RA
and van Lohuizen M: The oncogene and Polycomb-group gene Bmi-1
regulates cell proliferation and senescence through the ink4a
locus. Nature. 397:164–168. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dimri GP, Martinez JL, Jacobs JJ, et al:
The Bmi-1 oncogene induces telomerase activity and immortalizes
human mammary epithelial cells. Cancer Res. 62:4736–4745.
2002.PubMed/NCBI
|
|
11
|
Guney I and Sedivy JM: Cellular
senescence, epigenetic switches and c-Myc. Cell Cycle. 5:2319–2323.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Datta S, Hoenerhoff MJ, Bommi P, et al:
Bmi-1 cooperates with H-Ras to transform human mammary epithelial
cells via dysregulation of multiple growth-regulatory pathways.
Cancer Res. 67:10286–10295. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Molofsky AV, Pardal R, Iwashita T, Park
IK, Clarke MF and Morrison SJ: Bmi-1 dependence distinguishes
neural stem cell self-renewal from progenitor proliferation.
Nature. 425:962–967. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Raaphorst FM: Self-renewal of
hematopoietic and leukemic stem cells: a central role for the
Polycomb-group gene Bmi-1. Trends Immunol. 24:522–524. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Grinstein E and Wernet P: Cellular
signaling in normal and cancerous stem cells. Cell Signal.
19:2428–2433. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu S, Dontu G, Mantle ID, et al: Hedgehog
signaling and Bmi-1 regulate self-renewal of normal and malignant
human mammary stem cells. Cancer Res. 66:6063–6071. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vrzalikova K, Skarda J, Ehrmann J, et al:
Prognostic value of Bmi-1 oncoprotein expression in NSCLC patients:
a tissue microarray study. J Cancer Res Clin Oncol. 134:1037–1042.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu JH, Song LB, Zhang X, et al: Bmi-1
expression predicts prognosis for patients with gastric carcinoma.
J Surg Oncol. 97:267–272. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang H, Pan K, Zhang HK, et al: Increased
polycomb-group oncogene Bmi-1 expression correlates with poor
prognosis in hepatocellular carcinoma. J Cancer Res Clin Oncol.
134:535–541. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chowdhury M, Mihara K, Yasunaga S, Ohtaki
M, Takihara Y and Kimura A: Expression of Polycomb-group (PcG)
protein BMI-1 predicts prognosis in patients with acute myeloid
leukemia. Leukemia. 21:1116–1122. 2007.PubMed/NCBI
|
|
21
|
Arnes JB, Collett K and Akslen LA:
Independent prognostic value of the basal-like phenotype of breast
cancer and associations with EGFR and candidate stem cell marker
BMI-1. Histopathology. 52:370–380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Song LB, Zeng MS, Liao WT, et al: Bmi-1 is
a novel molecular marker of nasopharyngeal carcinoma progression
and immortalizes primary human nasopharyngeal epithelial cells.
Cancer Res. 66:6225–6232. 2006. View Article : Google Scholar
|
|
23
|
Qin ZK, Yang JA, Zeng MS, et al:
Expression and clinical significance of Bmi-1 protein in bladder
cancer. Ai Zheng. 27:1327–1330. 2008.(In Chinese).
|
|
24
|
Glinsky GV, Berezovska O and Glinskii AB:
Microarray analysis identifies a death-from-cancer signature
predicting therapy failure in patients with multiple types of
cancer. J Clin Invest. 115:1503–1521. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mani SA, Guo W, Liao MJ, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang MH, Hsu DS, Wang HW, et al: Bmi1 is
essential in Twist1-induced epithelial-mesenchymal transition. Nat
Cell Biol. 12:982–992. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Song LB, Li J, Liao WT, et al: The
polycomb group protein Bmi-1 represses the tumor suppressor PTEN
and induces epithelial-mesenchymal transition in human
nasopharyngeal epithelial cells. J Clin Invest. 119:3626–3636.
2009. View
Article : Google Scholar
|
|
28
|
Theys J, Jutten B, Habets R, et al:
E-Cadherin loss associated with EMT promotes radioresistance in
human tumor cells. Radiother Oncol. 99:392–397. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kurrey NK, Jalgaonkar SP, Joglekar AV, et
al: Snail and slug mediate radioresistance and chemoresistance by
antagonizing p53-mediated apoptosis and acquiring a stem-like
phenotype in ovarian cancer cells. Stem Cells. 27:2059–2068. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guo BH, Feng Y, Zhang R, et al: Bmi-1
promotes invasion and metastasis, and its elevated expression is
correlated with an advanced stage of breast cancer. Mol Cancer.
10:102011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dimri GP, Itahana K, Acosta M and Campisi
J: Regulation of a senescence checkpoint response by the E2F1
transcription factor and p14(ARF) tumor suppressor. Mol Cell Biol.
20:273–285. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu ZG, Chen HY, Cheng JJ, Chen ZP, Li XN
and Xia YF: Relationship between methylation status of ERCC1
promoter and radiosensitivity in glioma cell lines. Cell Biol Int.
33:1111–1117. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Reers M, Smiley ST, Mottola-Hartshorn C,
Chen A, Lin M and Chen LB: Mitochondrial membrane potential
monitored by JC-1 dye. Methods Enzymol. 260:406–417. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lee K, Adhikary G, Balasubramanian S, et
al: Expression of Bmi-1 in epidermis enhances cell survival by
altering cell cycle regulatory protein expression and inhibiting
apoptosis. J Invest Dermatol. 128:9–17. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yan SS, Liu L, Liu ZG, Zeng MS, Song LB
and Xia YF: Expression and clinical significance of DNA-PKcs in
nasopharyngeal carcinoma. Ai Zheng. 27:979–983. 2008.(In
Chinese).
|
|
36
|
Kraus AC, Ferber I, Bachmann SO, et al: In
vitro chemo- and radio-resistance in small cell lung cancer
correlates with cell adhesion and constitutive activation of AKT
and MAP kinase pathways. Oncogene. 21:8683–8695. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liang K, Jin W, Knuefermann C, et al:
Targeting the phosphatidylinositol 3-kinase/Akt pathway for
enhancing breast cancer cells to radiotherapy. Mol Cancer Ther.
2:353–360. 2003.PubMed/NCBI
|
|
38
|
Soderlund K, Perez-Tenorio G and Stal O:
Activation of the phosphatidylinositol 3-kinase/Akt pathway
prevents radiation-induced apoptosis in breast cancer cells. Int J
Oncol. 26:25–32. 2005.PubMed/NCBI
|
|
39
|
LoPiccolo J, Granville CA, Gills JJ and
Dennis PA: Targeting Akt in cancer therapy. Anticancer Drugs.
18:861–874. 2007.
|
|
40
|
Lemasters JJ, Qian T, Trost LC, et al:
Confocal microscopy of the mitochondrial permeability transition in
necrotic and apoptotic cell death. Biochem Soc Symp. 66:205–222.
1999.PubMed/NCBI
|
|
41
|
Kang YH, Yi MJ, Kim MJ, et al:
Caspase-independent cell death by arsenic trioxide in human
cervical cancer cells: reactive oxygen species-mediated
poly(ADP-ribose) polymerase-1 activation signals apoptosis-inducing
factor release from mitochondria. Cancer Res. 64:8960–8967. 2004.
View Article : Google Scholar
|
|
42
|
Kim WH, Park WB, Gao B and Jung MH:
Critical role of reactive oxygen species and mitochondrial membrane
potential in Korean mistletoe lectin-induced apoptosis in human
hepatocarcinoma cells. Mol Pharmacol. 66:1383–1396. 2004.
View Article : Google Scholar
|
|
43
|
Park MT, Kim MJ, Kang YH, et al:
Phytosphingosine in combination with ionizing radiation enhances
apoptotic cell death in radiation-resistant cancer cells through
ROS-dependent and -independent AIF release. Blood. 105:1724–1733.
2005. View Article : Google Scholar : PubMed/NCBI
|