Introduction
Gallic acid (GA) as a polyhydroxylphenolic compound
is commonly distributed in various plants, fruits and foods
(1) and is very well absorbed in
humans (2). Various biological
activities of GA have been reported, including antibacterial
(3), antiviral (4) and anti-inflammatory effects (5). GA has been shown to be selectively
cytotoxic against a variety of cancer cells such as, prostate
(6), lung (7–9),
gastric, colon, breast, cervical and esophageal cancer (10,11).
Apoptosis induced by GA is associated with oxidative stresses
derived from reactive oxygen species (ROS), mitochondrial
dysfunction and an increase in intracellular Ca2+ level
(12,13). Controversially, GA has been reported
to have both pro-oxidant and antioxidant properties depending on
iron or hydrogen peroxide (H2O2) in medium
and plasma (14,15).
ROS, such as H2O2, superoxide
anion (O2•−) and hydroxyl radical
(•OH) regulate many important cellular procedures
including transcription factor activation, gene expression,
differentiation and cell proliferation (16,17).
ROS are formed as by-products of mitochondrial respiration or by
the action of certain oxidases (18). A change in the redox state of the
tissue and cell implies an alteration in the generation or
metabolism of ROS. The main metabolic pathways include superoxide
dismutase (SOD), expressed as extracellular, cytoplasmic and
mitochondrial isoforms (19), which
metabolize O2•− to
H2O2. Further metabolism by peroxidases,
include catalase and glutathione (GSH) peroxidase, yields
O2 and H2O (20). Cells have antioxidant systems to
manage the redox state, which is important for their survival.
Excessive production of ROS leads to death in various cell types
(21,22).
Vascular endothelial cells (ECs) are implicated in
diverse regulatory responsibilities such as vascular permeability
for gases and metabolites, vascular smooth muscle tone, blood
pressure, blood coagulation, inflammation and angiogenesis
(23). The vascular endothelium can
experience widespread degrees of oxidative stress, ultimately
leading to endothelial dysfunction in cardiovascular diseases via
the initiation of EC apoptosis (24). Additionally, angiogenesis involving
configuration of new blood vessels from pre-existing vasculature is
a crucial event for the transition of tumors from a latent to a
malignant state. In spite of the vital role of vascular ECs in
tumor biogenesis and progression, the effects of GA on EC death is
not completely understood in relation to ROS and GSH. Therefore, it
is valuable to elucidate the toxicological mechanisms of GA in
human primary ECs.
Previously, we demonstrated that GA reduces the
growth of human umbilical vein endothelial cells (HUVEC) with an
IC50 of approximately 400 μM GA at 24 h and induced the
death of these cells (11). In the
present study, we assessed the effects of GA on ROS and GSH levels
in GA-treated HUVECs and investigated the effects of NAC (a well
known antioxidant) or BSO, an inhibitor of GSH synthesis (25), on GA-treated HUVEC, in relation to
cell growth, cell death, ROS and GSH levels.
Materials and methods
Cell culture
Primary HUVECs from PromoCell GmbH (Heidelberg,
Germany) were maintained in a humidified incubator containing 5%
CO2 at 37°C. HUVECs were cultured in a complete
endothelial cell growth medium (ECGM, PromoCell) with 2% fetal
bovine serum (FBS). HUVECs were grown in 100-mm plastic tissue
culture dishes (Nunc, Roskilde, Denmark). HUVECs were washed and
detached with HEPES-BSS (30 mM HEPES), trypsin-EDTA and trypsin
neutralization solution (PromoCell). HUVECs were used between
passages 4 and 8.
Reagents
GA purchased from the Sigma-Aldrich Chemical Co.
(St. Louis, MO) was dissolved in ethanol at 200 mM as a stock
solution. The pan-caspase inhibitor (Z-VAD-FMK;
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone) was obtained from
R&D Systems, Inc. (Minneapolis, MN) and was dissolved in DMSO
(Sigma) at 10 mM as a stock solution. NAC and BSO were obtained
from Sigma-Aldrich Chemical Co. NAC was dissolved in the buffer [20
mM HEPES (pH 7.0)] at 100 mM as a stock solution. BSO was dissolved
in water at 100 mM as a stock solution. Based on the previous
experiment (26), cells were
pretreated with Z-VAD, NAC or BSO for 1 h prior to treatment with
GA. Ethanol (0.2%) and DMSO (0.3%) were used as a control
vehicle.
Growth inhibition assay
The cell growth inhibition effects of drugs were
determined by measuring the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT,
Sigma-Aldrich Chemical Co.) dye absorbance by living cells as
previously described (27). In
brief, 3×104 cells/well were seeded in 96-well
microtiter plates (Nunc) for the MTT assay. After exposure to the
indicated amounts of GA with or without Z-VAD, NAC or BSO for 24 h,
20 μl of MTT solution [2 mg/ml in phosphate-buffered saline (PBS)]
were added to each well of 96-well plates. The plates were
additionally incubated for 4 h at 37°C. Media in plates were
withdrawn by pipetting, and 200 μl of DMSO was added to each well
to solubilize the formazan crystals. Optical density was measured
at 570 nm using a microplate reader (SpectraMAX 340, Molecular
Devices Co., Sunnyvale, CA).
Annexin V staining for cell death
detection
Apoptosis was determined by staining cells with
Annexin V-fluorescein isothiocyanate (FITC) (Ex/Em = 488 nm/519
nm), Pharmingen, San Diego, CA; and propidium iodide (PI;
Sigma-Aldrich; Ex/Em = 488 nm/617 nm). In brief, 1×106
cells in a 60-mm culture dish (Nunc) were incubated with the
indicated amounts of GA with or without Z-VAD, NAC or BSO for 24 h.
Cells were washed twice with cold PBS and then resuspended in 500
μl of binding buffer (10 mM HEPES/NaOH pH 7.4, 140 mM NaCl, 2.5 mM
CaCl2) at a concentration of 1×106 cells/ml.
Five microliters of Annexin V-FITC and PI (1 μg/ml) were then added
to these cells, which were analyzed with a FACStar flow cytometer
(Becton-Dickinson). Viable cells were negative for both PI and
Annexin V. Apoptotic cells were positive for Annexin V and negative
for PI, whereas late apoptotic dead cells displayed both high
Annexin V and PI labeling. Non-viable cells, which underwent
necrosis, were positive for PI and negative for Annexin V.
Measurement of MMP (Δ Ψm)
MMP (Δ Ψm) levels were measured using rhodamine 123
fluorescent dye (Sigma-Aldrich Chemical Co.; Ex/Em = 485 nm/535 nm)
as previously described (28). In
brief, 1×106 cells in a 60-mm culture dish (Nunc) were
incubated with the indicated amounts of GA with or without Z-VAD,
NAC or BSO for 24 h. Cells were washed twice with PBS and incubated
with rhodamine 123 (0.1 μg/ml) at 37°C for 30 min. Rhodamine 123
staining intensity was determined by flow cytometry
(Becton-Dickinson). An absence of rhodamine 123 from the cells
indicated the loss of MMP (Δ Ψm) in HUVEC.
Detection of intracellular ROS
levels
Intracellular ROS levels were detected by means of
an oxidation-sensitive fluorescent probe dye,
2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA,
Ex/Em = 495 nm/529 nm; Invitrogen Molecular Probes, OR; ) (29). As H2DCFDA is poorly
selective for O2•−, dihydroethidium (DHE,
Ex/Em = 518 nm/605 nm; Invitrogen Molecular Probes), which is
highly selective for O2•−, was used for its
detection. In brief, 1×106 cells in a 60-mm culture dish
(Nunc) were incubated with the indicated amounts of GA with or
without Z-VAD, NAC or BSO for 24 h. Cells were then washed in PBS
and incubated with 20 μM H2DCFDA or DHE at 37°C for 30
min. Dichlorofluorescein (DCF) and dihydroethidium (DHE)
fluorescence was detected using a FACStar flow cytometer
(Becton-Dickinson). ROS and O2•− levels were
expressed as mean fluorescence intensity (MFI), which was
calculated by the CellQuest software (Becton-Dickinson).
Detection of the intracellular
glutathione levels
Cellular glutathione (GSH) levels were analyzed
using 5-chloromethylfluorescein diacetate (CMFDA, Invitrogen
Molecular Probes; Ex/Em = 522 nm/595 nm) as previously described
(29). In brief, 1×106
cells in a 60-mm culture dish (Nunc) were incubated with the
indicated amounts of GA with or without Z-VAD, NAC or BSO for 24 h.
Cells were then washed with PBS and incubated with 5 μM CMFDA at
37°C for 30 min. Chloromethylfluorescein (CMF) fluorescence
intensity was determined using a FACStar flow cytometer
(Becton-Dickinson). Negative CMF staining (GSH depleted) cells were
expressed as the percent of (-) CMF cells.
Statistical analysis
The results represent the mean of at least three
independent experiments (mean ± SD). The data were analyzed using
Instat software (GraphPad Prism4, San Diego, CA). The Student’s
t-test or one-way analysis of variance (ANOVA) with post-hoc
analysis using the Tukey’s multiple comparison test was used for
parametric data. Statistical significance was defined as
P<0.05.
Results
Effects of Z-VAD, NAC or BSO on cell
growth in GA-treated HUVECs
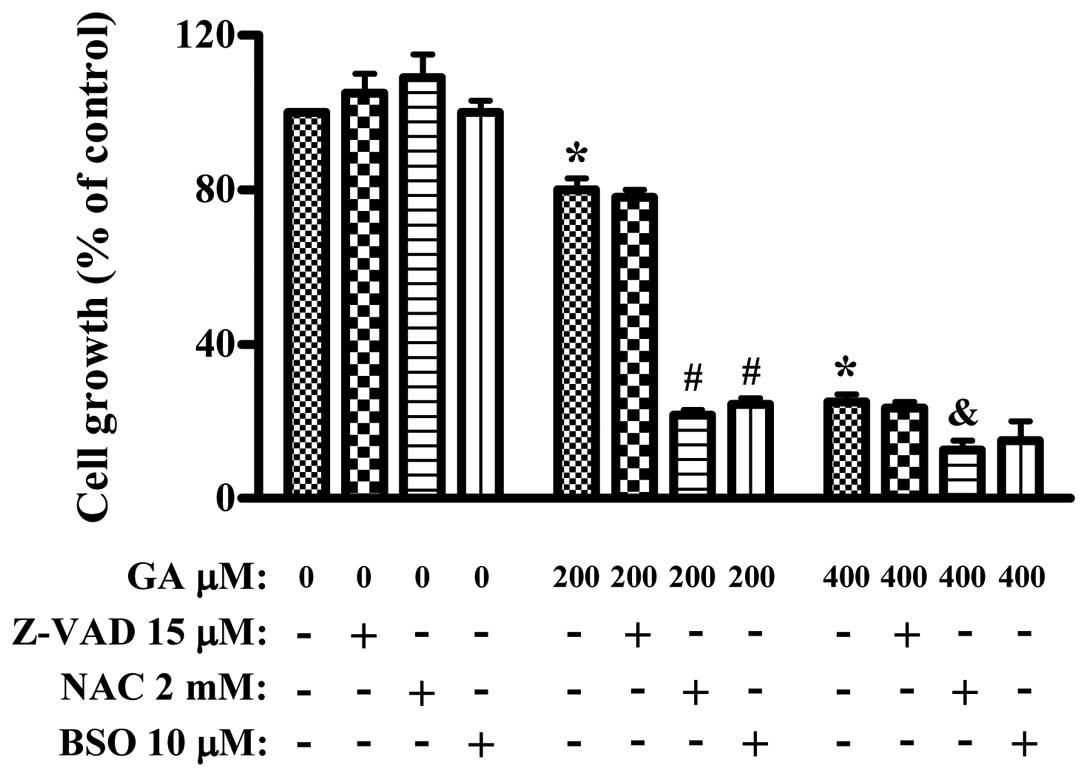
We examined the effect of Z-VAD, NAC or BSO on the
growth of GA-treated HUVEC using an MTT assay. Treatment with 200
and 400 μM GA inhibited the growth of HUVEC about 20 and 70% at 24
h, respectively (Fig. 1). Treatment
with 15 μM Z-VAD, 2 mM NAC or 10 μM BSO alone did not strongly
modify the growth of control HUVECs at 24 h (Fig. 1). In addition, Z-VAD did not affect
HUVEC growth inhibition of 200 or 400 μM GA (Fig. 1). However, both NAC and BSO
significantly enhanced the growth inhibition of HUVEC by GA
(Fig. 1).
Effects of Z-VAD, NAC or BSO on cell
death and MMP (Δ Ψm) in GA-treated HUVECs
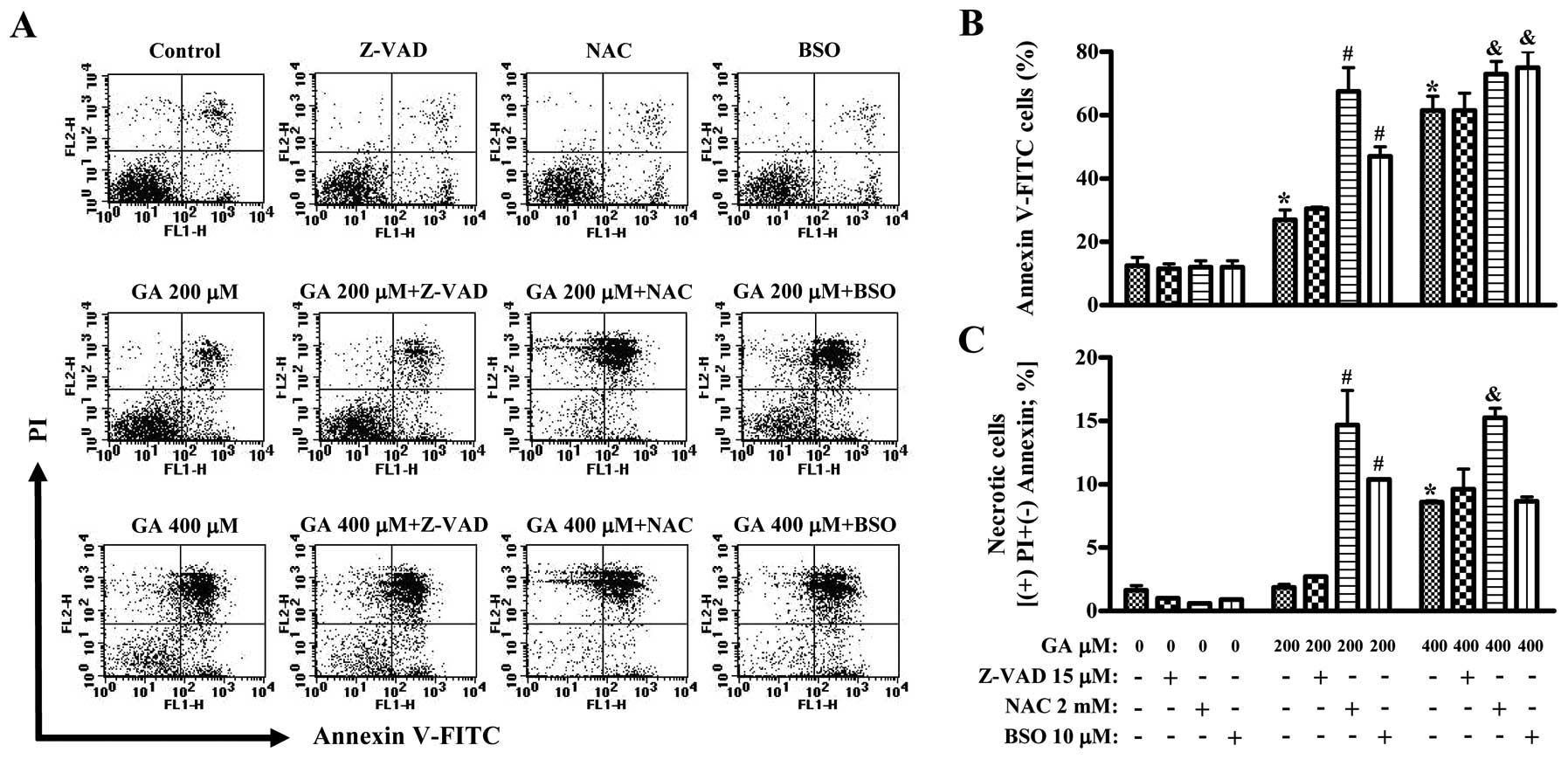
Next, we determined whether GA induces cell death in
HUVECs. As shown in Fig. 2A and B,
the numbers of Annexin V-staining cells in GA-treated HUVEC were
dose-dependently increased at 24 h. Treatment with 400 μM GA also
increased the numbers of necrotic cells (PI+ and Annexin
V-FITC− cells) (Fig. 2A and
C). Neither Z-VAD, NAC or BSO alone significantly induced
control HUVEC death (Fig. 2). Z-VAD
did not change the numbers of Annexin V-staining cells in
GA-treated HUVECs (Fig. 2A and B).
Both NAC and BSO significantly intensified the increased numbers of
Annexin V-staining cells by GA (Fig. 2A
and B). The strong effect was shown in HUVEC co-treated with
200 μM GA and NAC (Fig. 2A and B).
Interestingly, NAC and BSO induced necrotic cell death in HUVECs
treated with 200 μM GA (Fig. 2A and
C). NAC also significantly enhanced the increased numbers of
necrotic cells by 400 μM GA (Fig. 2A
and C).
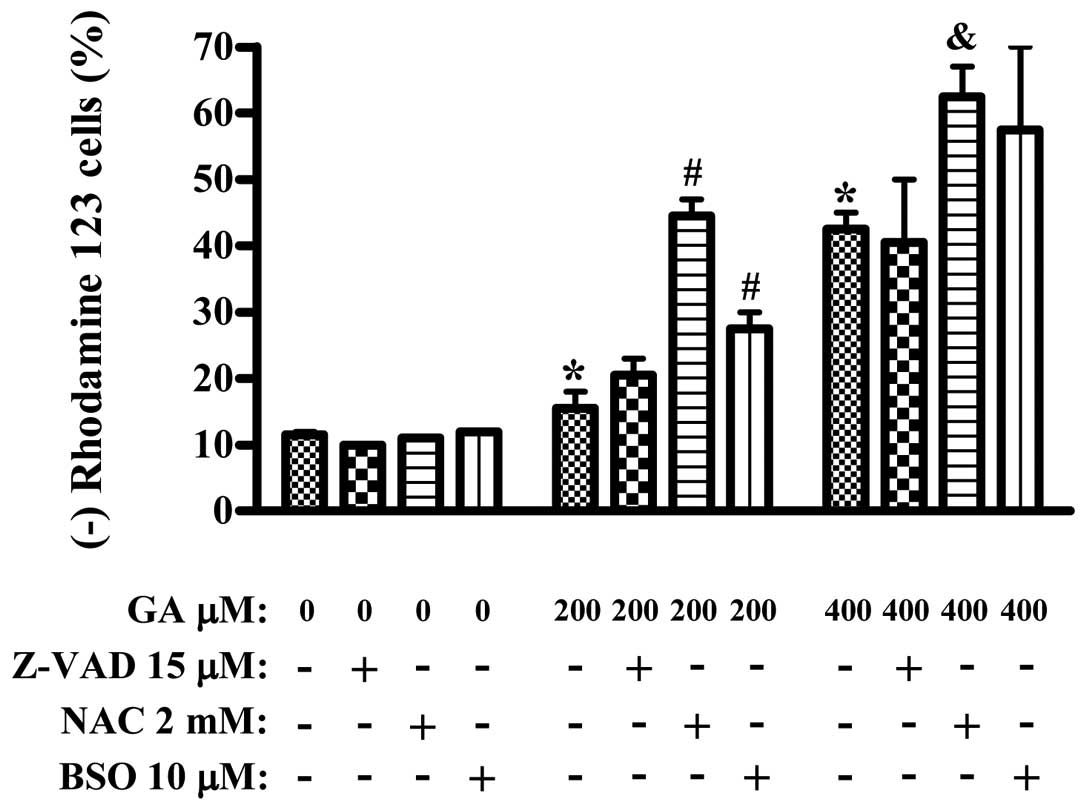
In addition, GA dose-dependently triggered the loss
of MMP (Δ Ψm) in HUVEC (Fig. 3).
Neither of Z-VAD, NAC or BSO alone induced the MMP (Δ Ψm) loss in
HUVEC, compared to the GA-untreated control HUVEC (Fig. 3). While Z-VAD did not significantly
affect the loss of MMP (Δ Ψm), both NAC and BSO enhanced this loss
in the GA-treated HUVECs (Fig.
3).
Effects of Z-VAD, NAC or BSO on ROS and
GSH levels in GA-treated HUVECs
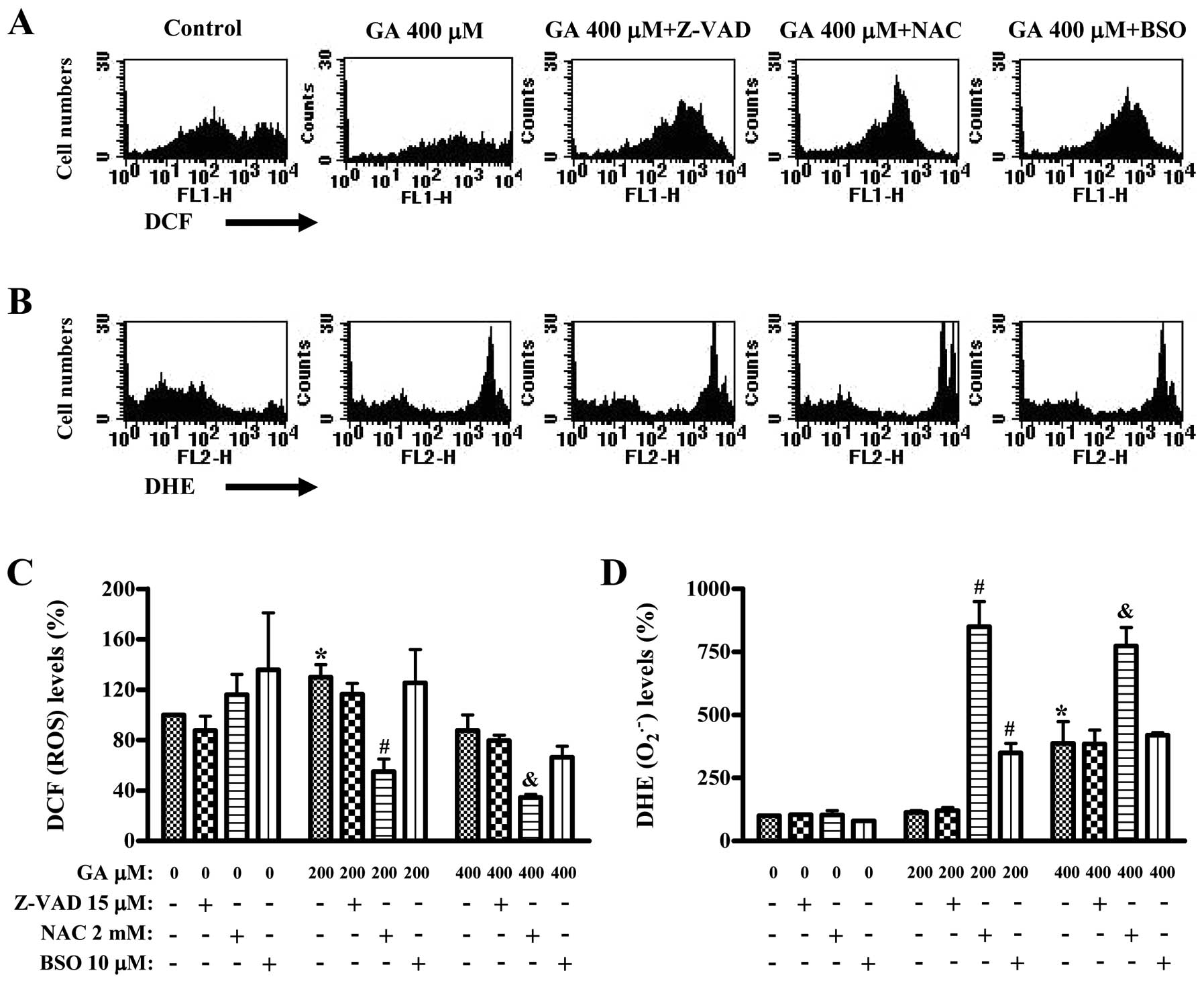
We assessed whether ROS and GSH levels in GA-treated
HUVECs were altered by Z-VAD, NAC or BSO treatment at 24 h. ROS
(DCF) levels such as H2O2 was increased in
200 μM GA-treated HUVECs, but was decreased in 400 μM GA-treated
HUVECs (Fig. 4A and C). NAC did not
reduce the basal level of ROS in HUVECs, and BSO increases the ROS
level (Fig. 4C). Both Z-VAD and BSO
did not significantly alter the ROS (DCF) levels in GA-treated
HUVECs whereas NAC strongly decreased them (Fig. 4A and C). Red fluorescence derived
from DHE reflecting intracellular O2•− levels
was not altered in the 200 μM GA-treated HUVECs but was strongly
increased in the 400 μM GA-treated HUVECs (Fig. 4B and D). Z-VAD did not alter the
O2•− levels of GA-treated HUVECs (Fig. 4B and D). Interestingly, NAC strongly
increased O2•- levels in the 200 or 400 μM
GA-treated HUVECs, and BSO also increased those in the 200 μM
GA-treated HUVECs (Fig. 4B and
D).
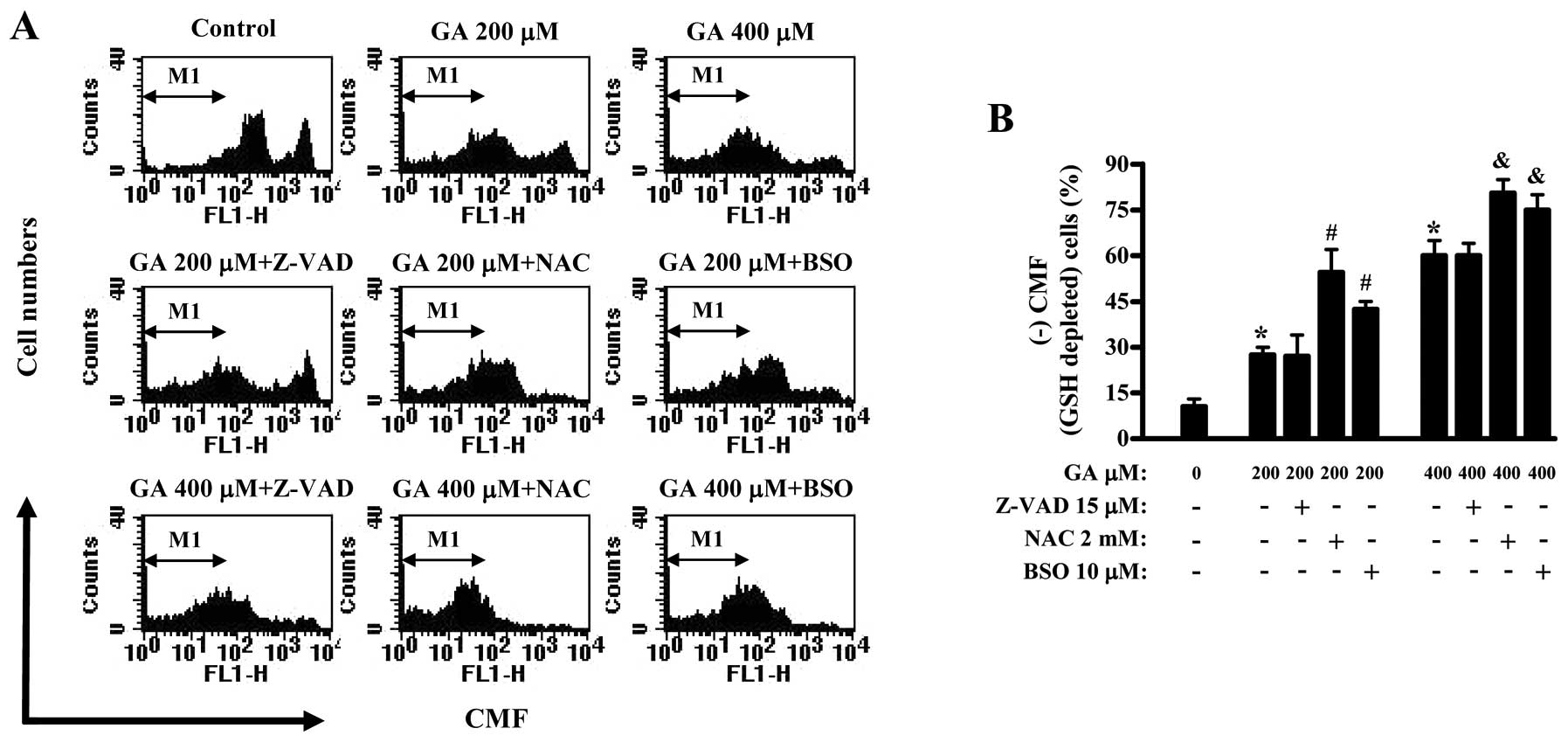
GA dose-dependently increased the number of
GSH-depleted cells in HUVECs (Fig.
5). Z-VAD did not affect GSH depletion in GA-treated HUVECs
(Fig. 5). NAC and BSO significantly
enhanced the increased numbers of GSH depleted cells by GA and a
strong effect was shown in NAC-treated HUVECs (Fig. 5).
Discussion
In the present study, we focused on evaluating the
effects of GA on the death of HUVECs in relation to ROS and GSH.
Treatment with 200 and 400 μM GA inhibited the growth of HUVECs by
about 20 and 70% at 24 h, respectively. This result was similar to
the report that GA shows lower cytotoxicity against normal
fibroblast and endothelial cells than against cancer cells
(30,31). We also observed that GA inhibited
the growth of HeLa cervical cancer cells, Calu-6 and A549 lung
cancer cells and the doses of IC50 were between 30–150
μM (9,11). Although we do not explain the reason
of this different susceptibility, Isuzugawa et al suggested
that the different susceptibilities to GA can depend on the
catalase contents of each cell (32).
GA is reported to induce apoptosis in prostate
cancer cells via mitochondrial dysfunction (13). Likewise, GA induced apoptosis in
HUVECs as evidenced by Annexin V staining, and triggered the loss
of MMP (Δ Ψm). However, Z-VAD did not affect HUVEC death and MMP (Δ
Ψm) loss of GA. In addition, 400 μM GA increased the numbers of
necrotic cells (PI+ and Annexin V-FITC−
cells). These results suggest that GA seemed to inhibit the growth
of HUVECs via caspase-independent apoptosis and/or necrosis.
GA has both pro-oxidant and antioxidant properties
(14,15). Increasing evidence suggests that
apoptosis induced by GA is associated with oxidative stresses
derived from ROS (12,13,33).
According to our results, ROS (DCF) levels such as
H2O2 were increased by 200 μM GA-treated
HUVECs. However, 400 μM GA showing an apoptotic effect decreased
the ROS levels. In contrast, 200 μM GA did not after the
O2•− levels in HUVECs, whereas 400 μM GA
significantly increased the O2•− level. These
results suggest that GA can affect different ROS levels depending
on the incubation doses. NAC showing the reduction of ROS (DCF)
level in GA-treated HUVEC intensified the growth inhibition, cell
death and MMP (Δ Ψm) loss in these cells. However, NAC strongly
increased the O2•− level in these cells.
Although NAC decreased the ROS (DCF) levels in GA-treated HUVECs,
NAC acted as a pro-oxidant in increasing the
O2•− level, thus intensifying HUVEC death.
Treatment with BSO showed enhanced effects on the growth
inhibition, cell death and MMP (Δ Ψm) loss of GA, and it did not
significantly alter the ROS (DCF) level in GA-treated HUVECs but
increased the O2•− level in HUVECs treated
with 200 μM GA rather than 400 μM GA. Similarly to the results in
O2•− levels, both NAC and BSO induced
necrotic cell death in 200 μM GA-treated HUVECs. NAC but not BSO
also enhanced the increased numbers of necrotic cells of 400 μM GA.
Taken together, these results suggest that the alteration of ROS
levels by GA is not closely related to HUVEC death, but
O2•− levels among ROS are involved in HUVEC
necrotic cell death. The exact roles of ROS in GA-induced HUVEC
death need to be further defined.
It has been reported that the intracellular GSH
content has a decisive effect on anticancer drug-induced apoptosis,
indicating that apoptotic effects are inversely comparative to GSH
content (34,35). Likewise, GA increased the number of
GSH depleted cells in HUVECs. Z-VAD did not affect GSH depletion in
GA-treated HUVECs. In addition, NAC increased cell death in
GA-treated HUVECs significantly increasing the numbers of
GSH-depleted cells. Although it is known that NAC containing a
thiol group is a GSH precursor, NAC in this study did not act as a
GSH precursor. In contrast, we recently demonstrated that NAC
significantly prevented GSH depletion in propyl gallate-treated
HeLa cervical cancer cells (26).
Therefore, NAC may be considered a GSH precursor, depending on the
co-incubated agents or cell types. In addition, BSO significantly
increased the number of GSH depleted cells in GA-treated HUVEC.
Therefore, these results supported the notion that apoptotic
effects are inversely comparative to GSH content.
In conclusion, GA inhibited the growth of HUVECs via
apoptosis and/or necrosis. The changes of ROS and GSH levels by NAC
and BSO affected cell growth and death in GA-treated HUVECs.
Acknowledgements
This study was supported by a grant from the
Ministry of Science and Technology (MoST)/Korea Science and
Engineering Foundation (KOSEF) through the Diabetes Research Center
at Chonbuk National University (2011-0028226) and the National
Research Foundation of Korea Grant funded by the Korean Government
(MEST) (2010-0021808).
Abbreviations:
|
GA
|
gallic acid
|
|
HUVEC
|
human umbilical vein endothelial
cells
|
|
ROS
|
reactive oxygen species
|
|
MMP (Δ Ψm)
|
mitochondrial membrane potential
|
|
SOD
|
superoxide dismutase
|
|
FBS
|
fetal bovine serum
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
FITC
|
fluorescein isothiocyanate
|
|
H2DCFDA
|
2′,7′-dichlorodihydrofluorescein
diacetate
|
|
DHE
|
dihydroethidium
|
|
GSH
|
glutathione
|
|
CMFDA
|
5-chloromethylfluorescein
diacetate
|
|
Z-VAD-FMK
|
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone
|
|
NAC
|
N-acetyl cysteine
|
|
BSO
|
L-buthionine sulfoximine
|
|
PI
|
propidium iodide
|
References
|
1
|
Niemetz R and Gross GG: Enzymology of
gallotannin and ellagitannin biosynthesis. Phytochemistry.
66:2001–2011. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shahrzad S, Aoyagi K, Winter A, Koyama A
and Bitsch I: Pharmacokinetics of gallic acid and its relative
bioavailability from tea in healthy humans. J Nutr. 131:1207–1210.
2001.PubMed/NCBI
|
|
3
|
Kang MS, Oh JS, Kang IC, Hong SJ and Choi
CH: Inhibitory effect of methyl gallate and gallic acid on oral
bacteria. J Microbiol. 46:744–750. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kratz JM, Andrighetti-Frohner CR, Leal PC,
Nunes RJ, Yunes RA, Trybala E, Bergstrom T, Barardi CR and Simoes
CM: Evaluation of anti-HSV-2 activity of gallic acid and pentyl
gallate. Biol Pharm Bull. 31:903–907. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim SH, Jun CD, Suk K, Choi BJ, Lim H,
Park S, Lee SH, Shin HY, Kim DK and Shin TY: Gallic acid inhibits
histamine release and pro-inflammatory cytokine production in mast
cells. Toxicol Sci. 91:123–131. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kaur M, Velmurugan B, Rajamanickam S,
Agarwal R and Agarwal C: Gallic acid, an active constituent of
grape seed extract, exhibits anti-proliferative, pro-apoptotic and
anti-tumorigenic effects against prostate carcinoma xenograft
growth in nude mice. Pharm Res. 26:2133–2140. 2009. View Article : Google Scholar
|
|
7
|
Kawada M, Ohno Y, Ri Y, Ikoma T, Yuugetu
H, Asai T, Watanabe M, Yasuda N, Akao S, Takemura G, et al:
Anti-tumor effect of gallic acid on LL-2 lung cancer cells
transplanted in mice. Anticancer Drugs. 12:847–852. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ohno Y, Fukuda K, Takemura G, Toyota M,
Watanabe M, Yasuda N, Xinbin Q, Maruyama R, Akao S, Gotou K, et al:
Induction of apoptosis by gallic acid in lung cancer cells.
Anticancer Drugs. 10:845–851. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
You BR and Park WH: Gallic acid-induced
lung cancer cell death is related to glutathione depletion as well
as reactive oxygen species increase. Toxicol In Vitro.
24:1356–1362. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Faried A, Kurnia D, Faried LS, Usman N,
Miyazaki T, Kato H and Kuwano H: Anticancer effects of gallic acid
isolated from Indonesian herbal medicine, Phaleria
macrocarpa (Scheff.) Boerl, on human cancer cell lines. Int J
Oncol. 30:605–613. 2007.PubMed/NCBI
|
|
11
|
You BR, Moon HJ, Han YH and Park WH:
Gallic acid inhibits the growth of HeLa cervical cancer cells via
apoptosis and/or necrosis. Food Chem Toxicol. 48:1334–1340. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Inoue M, Sakaguchi N, Isuzugawa K, Tani H
and Ogihara Y: Role of reactive oxygen species in gallic
acid-induced apoptosis. Biol Pharm Bull. 23:1153–1157. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen HM, Wu YC, Chia YC, Chang FR, Hsu HK,
Hsieh YC, Chen CC and Yuan SS: Gallic acid, a major component of
Toona sinensis leaf extracts, contains a ROS-mediated anti-cancer
activity in human prostate cancer cells. Cancer Lett. 286:161–171.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Strlic M, Radovic T, Kolar J and Pihlar B:
Anti- and prooxidative properties of gallic acid in fenton-type
systems. J Agric Food Chem. 50:6313–6317. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sakagami H and Satoh K: Prooxidant action
of two antioxidants: ascorbic acid and gallic acid. Anticancer Res.
17:221–224. 1997.PubMed/NCBI
|
|
16
|
Gonzalez C, Sanz-Alfayate G, Agapito MT,
Gomez-Nino A, Rocher A and Obeso A: Significance of ROS in oxygen
sensing in cell systems with sensitivity to physiological hypoxia.
Respir Physiol Neurobiol. 132:17–41. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baran CP, Zeigler MM, Tridandapani S and
Marsh CB: The role of ROS and RNS in regulating life and death of
blood monocytes. Curr Pharm Des. 10:855–866. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial ROS-induced ROS release: an update and review.
Biochim Biophys Acta. 1757:509–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zelko IN, Mariani TJ and Folz RJ:
Superoxide dismutase multigene family: a comparison of the CuZn-SOD
(SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures,
evolution, and expression. Free Radic Biol Med. 33:337–349. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wilcox CS: Reactive oxygen species: roles
in blood pressure and kidney function. Curr Hypertens Rep.
4:160–166. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wallach-Dayan SB, Izbicki G, Cohen PY,
Gerstl-Golan R, Fine A and Breuer R: Bleomycin initiates apoptosis
of lung epithelial cells by ROS but not by Fas/FasL pathway. Am J
Physiol Lung Cell Mol Physiol. 290:L790–L796. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Simon HU, Haj-Yehia A and Levi-Schaffer F:
Role of reactive oxygen species (ROS) in apoptosis induction.
Apoptosis. 5:415–418. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bassenge E: Endothelial function in
different organs. Prog Cardiovasc Dis. 39:209–228. 1996. View Article : Google Scholar
|
|
24
|
Irani K: Oxidant signaling in vascular
cell growth, death, and survival : a review of the roles of
reactive oxygen species in smooth muscle and endothelial cell
mitogenic and apoptotic signaling. Circ Res. 87:179–183. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bailey HH: L-S,R-buthionine sulfoximine:
historical development and clinical issues. Chem Biol Interact.
111–112:239–254. 1998.PubMed/NCBI
|
|
26
|
Han YH and Park WH: Propyl gallate
inhibits the growth of HeLa cells via regulating intracellular GSH
level. Food Chem Toxicol. 47:2531–2538. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Park WH, Seol JG, Kim ES, Hyun JM, Jung
CW, Lee CC, Kim BK and Lee YY: Arsenic trioxide-mediated growth
inhibition in MC/CAR myeloma cells via cell cycle arrest in
association with induction of cyclin-dependent kinase inhibitor,
p21, and apoptosis. Cancer Res. 60:3065–3071. 2000.
|
|
28
|
Han YH, Kim SZ, Kim SH and Park WH:
Arsenic trioxide inhibits growth of As4.1 juxtaglomerular cells via
cell cycle arrest and caspase-independent apoptosis. Am J Physiol
Renal Physiol. 293:F511–F520. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Han YH, Kim SH, Kim SZ and Park WH:
Caspase inhibitor decreases apoptosis in pyrogallol-treated lung
cancer Calu-6 cells via the prevention of GSH depletion. Int J
Oncol. 33:1099–1105. 2008.PubMed/NCBI
|
|
30
|
Inoue M, Suzuki R, Koide T, Sakaguchi N,
Ogihara Y and Yabu Y: Antioxidant, gallic acid, induces apoptosis
in HL-60RG cells. Biochem Biophys Res Commun. 204:898–904. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Inoue M, Suzuki R, Sakaguchi N, Li Z,
Takeda T, Ogihara Y, Jiang BY and Chen Y: Selective induction of
cell death in cancer cells by gallic acid. Biol Pharm Bull.
18:1526–1530. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Isuzugawa K, Inoue M and Ogihara Y:
Catalase contents in cells determine sensitivity to the apoptosis
inducer gallic acid. Biol Pharm Bull. 24:1022–1026. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Serrano A, Palacios C, Roy G, Cespon C,
Villar ML, Nocito M and Gonzalez-Porque P: Derivatives of gallic
acid induce apoptosis in tumoral cell lines and inhibit lymphocyte
proliferation. Arch Biochem Biophys. 350:49–54. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Poot M, Teubert H, Rabinovitch PS and
Kavanagh TJ: De novo synthesis of glutathione is required for both
entry into and progression through the cell cycle. J Cell Physiol.
163:555–560. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schnelldorfer T, Gansauge S, Gansauge F,
Schlosser S, Beger HG and Nussler AK: Glutathione depletion causes
cell growth inhibition and enhanced apoptosis in pancreatic cancer
cells. Cancer. 89:1440–1447. 2000. View Article : Google Scholar : PubMed/NCBI
|