Introduction
RAS proteins are molecular switches for signaling
cascades that modulate many aspects of cell biology (1,2). They
have two distinct conformations, including an inactive GDP-bound
form and an active GTP-bound form, which are regulated by RAS
guanine nucleotide exchange factors (GEFs) and RAS
GTPase-activating proteins (GAPs) (3). Approximately 30% of human tumors
express an oncogenic form of RAS (i.e., Ha-, K-, or N-RAS), which,
in its active form, is insensitive to Ras GAPs (3). RASAL1, which is a member of the Ras
GAPs family, has been shown to be downregulated in several solid
tumors, including brain, skin, bladder, head, neck, lung, liver,
esophageal, and multiple cell lines. Loss of RASAL1 activity has
been correlated with hyperactive Ras in the colon and
hepatocellular carcinoma lacking oncogenic Ras (4,5).
Matkar et al have reported that systemic K-ras
activation in mice leads to rapid changes in gastric cellular
homeostasis, and conditional K-ras activation results in
MAPK pathway activation and the hyperproliferation of the squamous
epithelium in the forestomach and metaplasia in the glandular
stomach. Parietal cells almost completely disappear from the upper
portion of the stomach that is adjacent to forestomach in
K-ras activated mice (6).
The activated embryonic oncogene ERas may be associated with the
tumorigenic growth of somatic cells and may be a putative molecule
responsible for cancer stem cell-like characteristics in gastric
cancer (7). Liu et al
(8) found that different types of
K-ras mutations may play a role in gastric cancer
development at different stages in the Chinese population. However,
Ras mutations are rare, and normally, wild-type Ras is found in
gastric cancer. Given that Ras GAPs including RASAL1 suppression
may lead to aberrant Ras activation, which promotes tumorigenesis
(9), in this study, we investigated
RASAL1 expression patterns in gastric cancer tissues and cell
lines, exploring the potential epigenetic mechanism of RASAL1
inactivation in gastric cancer cell lines. Enforced expression of
RASAL1 is to evaluate its biological function in gastric cancer
cells. These findings may be beneficial in exploring the mechanism
and treatment options for gastric cancer.
Materials and methods
Tissue samples and cell lines
Gastric cancer and adjacent non-cancerous tissue
specimens were obtained from the First Hospital of Nanjing. The
samples and our study were approved by the Committees for Ethical
Review of Research at the first hospital of Nanjing in China and
the patients signed informed consent forms. The clinicopathological
features are shown in Table I. Four
human gastric cancer cell lines AGS, MCG-803, SGC-7901 and BGC-823
were obtained from the Chinese Academy of Science cell bank.
 | Table IThe association of RASAL1 expression
with clinicopathological features in 50 GC cases. |
Table I
The association of RASAL1 expression
with clinicopathological features in 50 GC cases.
| Parameters | T<N | T≥N | P-value |
|---|
| Age | | | 1 |
| ≤60 | 12 | 8 | |
| >60 | 18 | 12 | |
| Gender | | | 0.626 |
| Male | 19 | 14 | |
| Female | 11 | 6 | |
| Differentiation | | | 1 |
| Poor | 16 | 11 | |
| Moderate | 13 | 9 | |
| Invasive degree | | | 0.936 |
| Early stage | 3 | 2 | |
| Progression | 25 | 18 | |
| Lymph node
metastasis | | | 0.742 |
| Yes | 19 | 14 | |
| No | 10 | 6 | |
Western blot analysis
Western blot analysis was performed to detect RASAL1
protein expression in gastric cancer specimens and cell lines. The
protein concentration of each extract was standardized using the
BCA assay (Pierce, USA). The RASAL1 primary antibody (1:1500,
Abcam) and the mouse monoclonal anti-β-actin antibody (1:8000,
Sigma, USA) were used to detect RASAL1 protein levels. The
intensities of specific protein bands were quantified with Gel Pro
3.2 (UVP, CLL, USA), corrected for the intensity of the respective
β-actin band.
Reverse-transcription (RT)-PCR and
quantitative real-time polymerase chain reaction (qPCR)
Total RNA from the cases and cell lines was isolated
using TRIzol reagent (Invitrogen, USA), and first-strand cDNA was
prepared from the total RNA with an oligo(dT)18 primer
and AMV reverse transcriptase (BioFlux, Japan) according to the
manufacturer’s instructions. Rasal1 gene expression was
examined using a SYBR-Green PCR kit (Takara, Japan). The
Rasal1 gene RT-PCR and qPCR primers, resulting in a 265-bp
DNA product, were as follows: 5′-GCAGGGAGGCGATTACAGCCGACCCCCGAG-3′
(sense) and 5′-GGGAAGCGAGTCTTCTTGATGGTTGAGGTC TCC-3′ (antisense)
(10).
5′-AZA and TSA treatment
A total of 1.5×105 AGS, BGC-823, MCG-803
and SGC-7901 cells were plated in 6-well plates. The cells were
cultured in medium containing 0, 5, 10 or 50 μmol/l of the DNA
methyltransferase inhibitor 5′-AZA (Sigma) for 72 h. BGC-823 cells
were also treated with 0, 0.1, 0.2, 0.3, 0.4 or 0.5 μmol/l TSA and
MCG-803 cells were treated with 0, 0.1, 0.2, or 0.3 μmol/l TSA
(Sigma), a histone deacetylase inhibitor, for 24 h.
Transfection of RASAL1 into BGC-823 and
MCG-803 cells
BGC-823 and MCG-803 cells were transfected with a
RASAL1 construct or pEGFP-N1 as a control (a gift from Q. Tao,
Chinese University of Hong Kong) using the FuGene HD transfection
reagent (Roche, Switzerland) according to the manufacturer’s
instructions. The cells were grown and selectively cultured in 0.4
mg/ml G418 (Life Technologies, USA) for 2 months after the initial
transfection. BGC-823 and MCG-803 RASAL1-transfected cells were
named BGC-RASAL1 and MCG-RASAL1, respectively, and those
transfected with pEGFP-N1 were referred to as the control.
Cell growth and apoptosis assay
The Cell Counting kit-8 (CCK-8) (Dojindo
Laboratories, Kumamoto, Japan) was used to measure the cellular
growth of BGC-RASAL1, MCG-RASAL1 and the control cells. The 450-nm
absorbance was measured to determine the cell viability. All of the
experiments were independently repeated at least three times.
Apoptosis was detected by PI and Annexin V-FITC staining. The cells
were seeded at 3×105 cells/well in 6-well plates and
incubated for 24 h and 48 h. Trypsinized cells were washed three
times with PBS. The cells were then conjugated with Annexin V-FITC
using the PI/Annexin V-FITC kit (Biouniquer, USA), according to the
manufacturer’s protocol, and analyzed by flow cytometry (Olympus,
Japan).
In vitro tumorigenicity assays and
migration detection
For soft agar colony formation, 1×103
cells were seeded in 6-well plates with RPMI-1640 containing 10%
FBS and incubated at 37°C in 5% CO2. Colonies consisting
of >80 cells were counted after 2 weeks, and the data are
expressed as the mean ± SD. of triplicate wells within the same
experiment. The foci formation assay was performed by seeding
1×103 cells in a 6-well plate. After 12 days, the
surviving colonies (450 cells per colony) were counted following
crystal violet (Invitrogen) staining. Triplicate independent
experiments were performed.
Cell migration was assessed by measuring the
movement of cells into a scraped area created by a 200-μl pipette
tip, and the spread of the wound closure was observed after 36 h.
The cells were photographed under a microscope.
Statistical analysis
Correlations between the RASAL1 expression levels
and pathological features were analyzed with the χ2 test
using SPSS 13.0 software for Windows. Differences were analyzed by
Fisher’s exact test. The independent Student’s t-test was used to
compare the results, which were expressed as the mean ± SD between
any two preselected groups. Results with a P<0.05 were
considered statistically significant.
Results
Decreased RASAL1 expression in gastric
cancer cases and cell lines
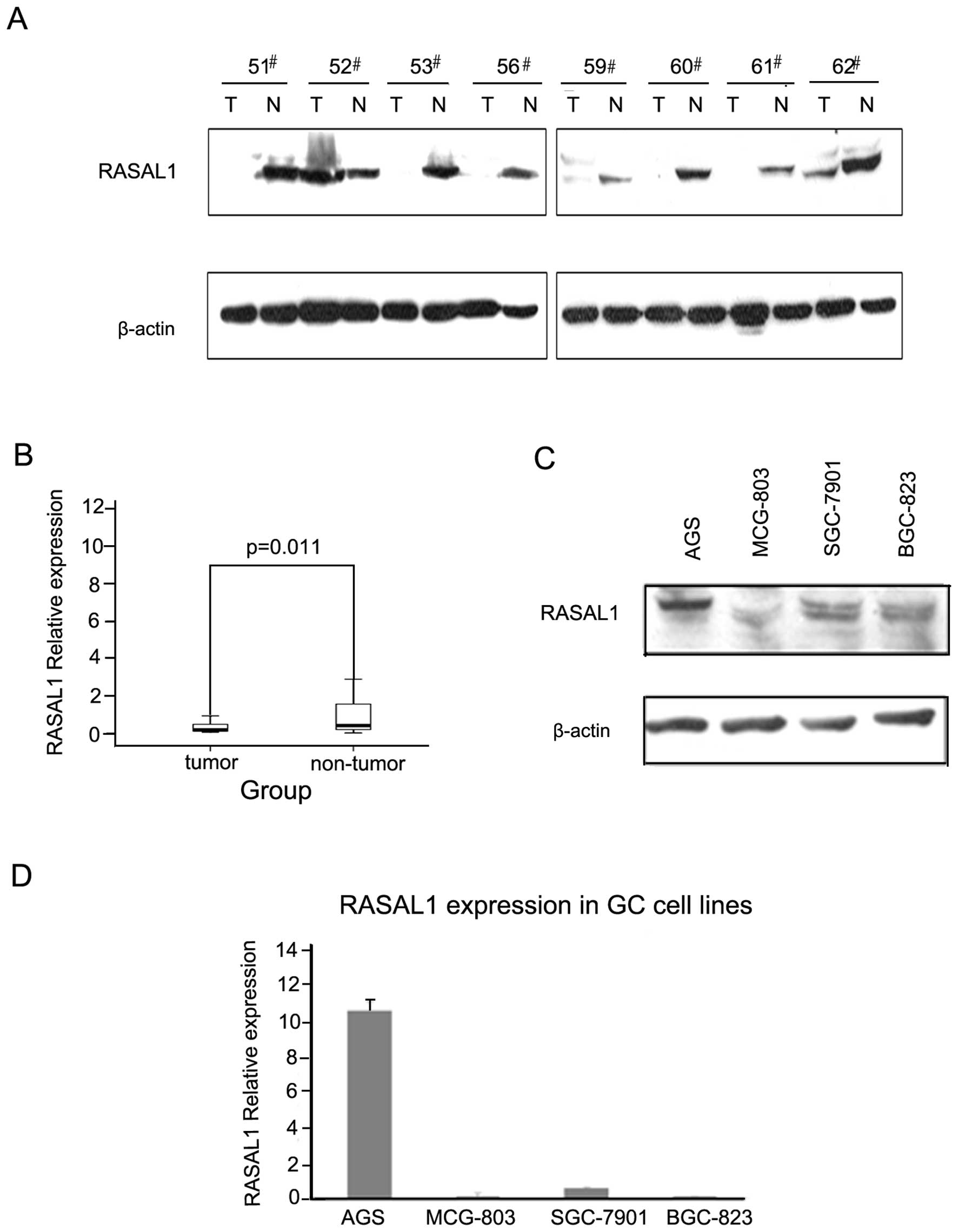
The RASAL1 protein levels were detected by western
blotting in 50 pairs of tumor and matched non-tumor tissue
specimens. The frequency of RASAL1 downregulation was 64% (32/50)
(Fig. 1A). As detected by qPCR, the
RASAL1 mRNA expression level was 58.8% (20/34), which is consistent
with the protein expression level (Fig.
1B). Western blotting and qPCR demonstrated that RASAL1
expression was reduced markedly in gastric cancer cells BGC-823,
7901 and MCG-803 compared to AGS (Fig.
1C and 1D). These data indicated that decreased RASAL1
expression may play an important role in gastric tumorigenesis.
Clinicopathology of decreased RASAL1
expression in gastric cancer
To explore the clinicopathological significance of
the RASAL1 expression pattern in gastric cancer tumorigenesis, the
clinical features of patients with GCs were analyzed. A total of 34
patients with gastric cancer were categorized into two groups
according to their RASAL1 expression level. However, there was no
statistically significant association between RASAL1 and
clinicopathological features, including age, gender,
differentiation, invasion degree and lymph node metastasis
(Table I).
Correlation between RASAL1 expression and
environmental factors (H. pylori and Epstein-Barr virus
infection)
The presence of H. pylori and EB virus
infection was determined by PCR for H. pylori 16S rDNA and
the EBNA-1 region, respectively, as previously described (11). The relationships between the H.
pylori and Epstein-Barr virus (EBV) infection status of the
patient and the RASAL1 expression level were analyzed. Of the 50 GC
cases, 22 samples were EBV-positive, and 28 samples were
EBV-negative; 22 samples were H. pylori-positive, and 28
samples were H. pylori-negative (Table II). No significant correlation
between the RASAL1 expression and environmental factors was
observed.
| Table IIAssociation of RASAL1 expression with
H. pylori and EBV infection in 50 GC cases. |
Table II
Association of RASAL1 expression with
H. pylori and EBV infection in 50 GC cases.
| Cases | T<N | T≥N | P-value |
|---|
| H. pylori and
EBV infection | | | | 0.554 |
| Both | 9 | 5 | 4 | |
| Either | 26 | 15 | 11 | |
| Neither | 15 | 11 | 4 | |
| H. pylori or
EBV infection | | | | 0.280 |
| (+) | 35 | 20 | 15 | |
| (−) | 15 | 11 | 4 | |
Silenced RASAL1 was restored by either
5′-AZA or TSA depending on the drug concentration
To investigate whether the RASAL1 downregulation
observed in gastric cancer tissues and cell lines is due to
epigenetic inactivation, gastric cancer cell lines AGS, BGC-823,
MCG-803 and SGC-7901 were treated with DNA methylation inhibitor
5-aza-2′-deoxycytidine (5′-AZA) or HDAC inhibitor trichostatin A
(TSA). RASAL1 expression was restored after treatment with 5′-AZA
for 72 h in the three gastric cell lines, depending on the drug
concentration (MCG-803 and BGC-823: 0–50 μmol/l; SGC-7901: 5–50
μmol/l) (Fig. 2A). Moreover, RASAL1
expression was also restored depending on the TSA concentration in
the BGC-823 (0–0.5 μmol/l) and MCG-803 (0–0.3 μmol/l) cell lines
(Fig. 2B). These findings
demonstrate the expression of RASAL1 is affected by epigenetic
modification in gastric cancer cells.
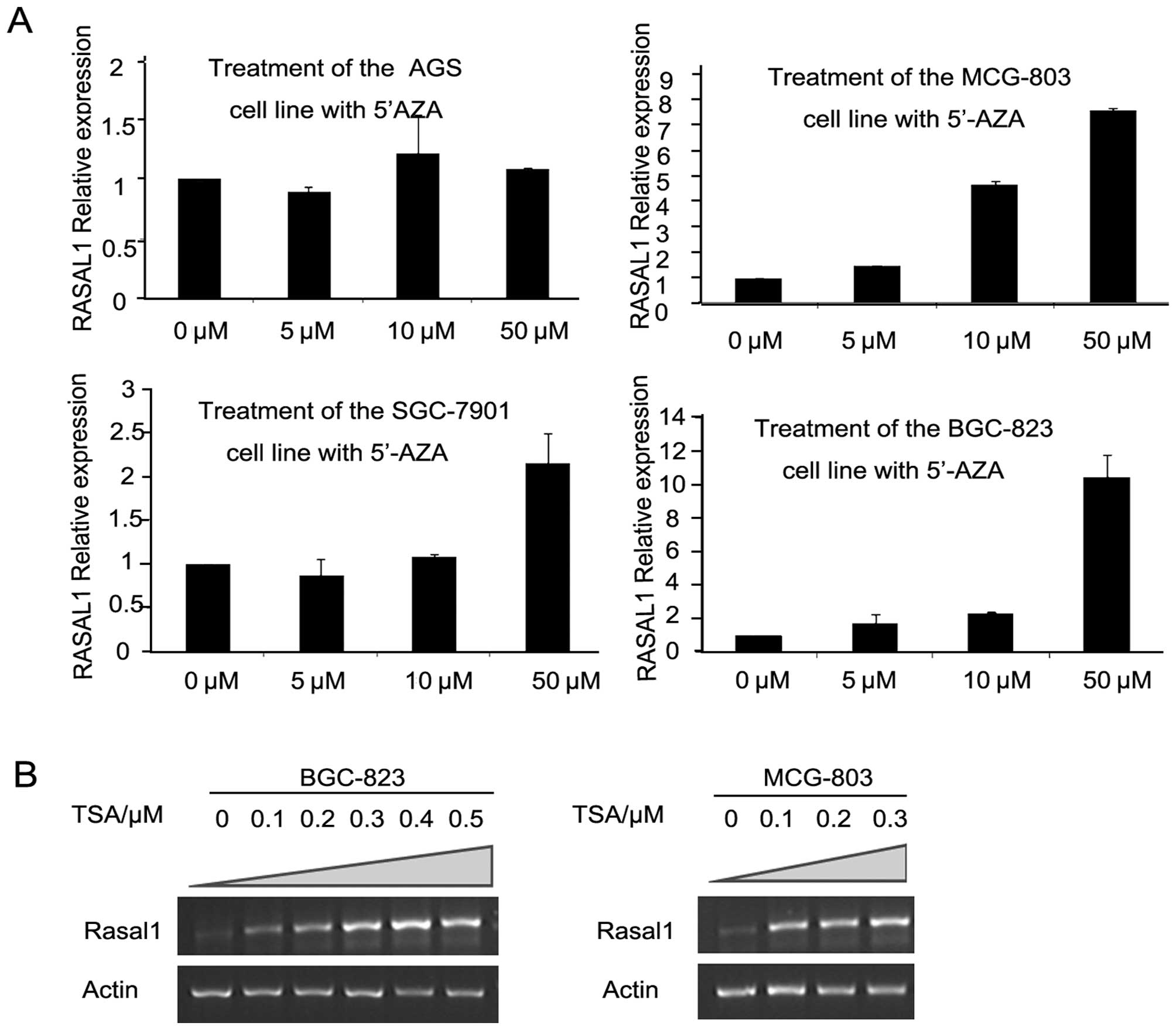 | Figure 2RASAL1 expression was restored by
epigenetic modification drugs. (A) RASAL1 expression was detected
in gastric cancer cell lines (AGS, MCG-803, SGC-7901 and BGC-823)
after 5′-AZA treatment (0, 5, 10 and 50 μM) for 72 h by qPCR. (B)
RT-PCR showed RASAL1 expression after induced for 24 h by 0, 0.1,
0.2, 0.3, 0.4 and 0.5 μM TSA in the BGC-823 cell line and by 0,
0.1, 0.2 and 0.3 μM in the MCG-803 cell line, respectively. |
Restoration of RASAL1 expression
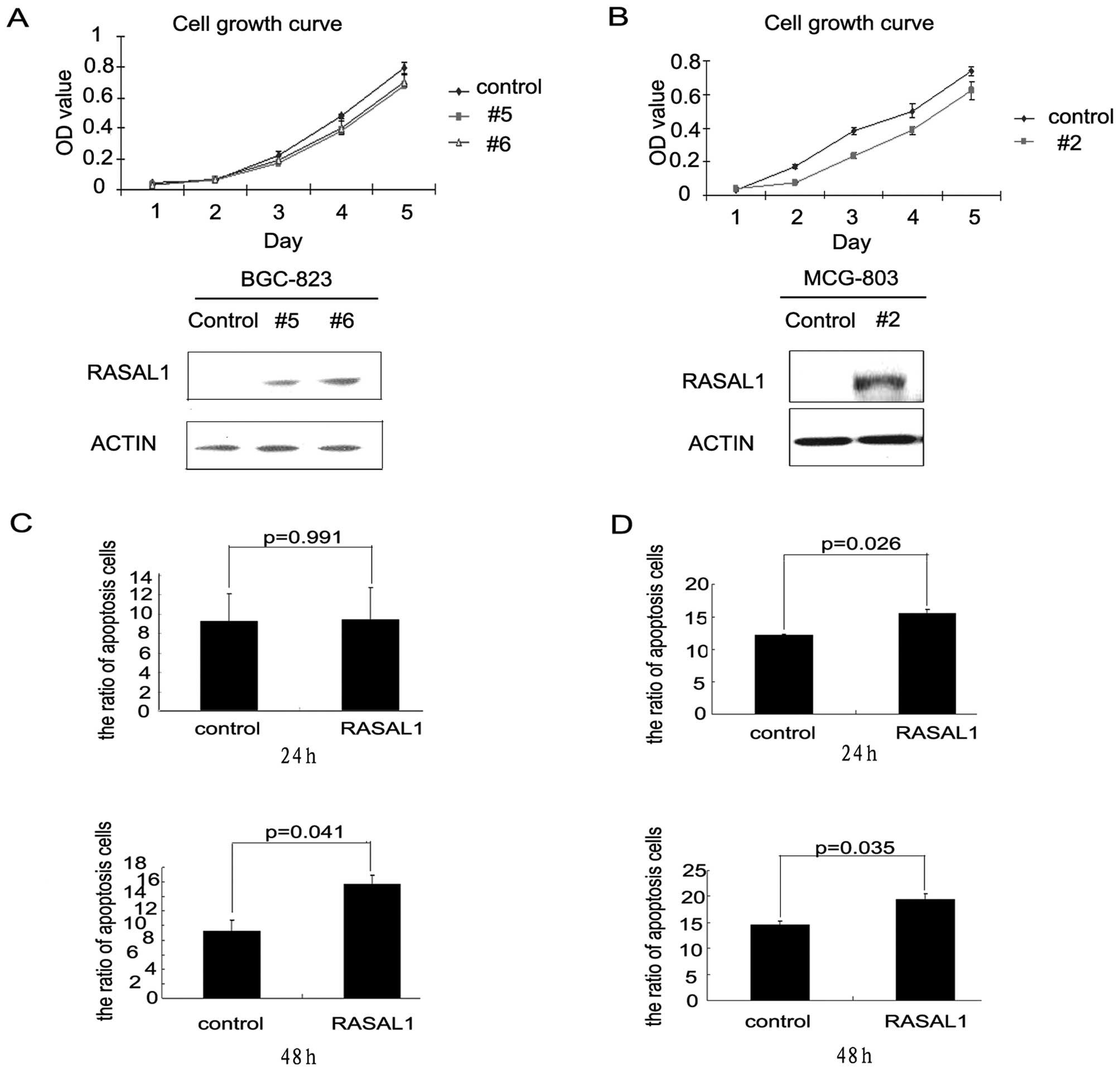
inhibited tumor cell proliferation by inducing apoptosis
BGC-823 and MCG-803 cells were stably transfected
with a pEGFP-RASAL1 expression construct or a pEGFP control
construct. RASAL1 expression increased in the RASAL1-transfected
cells (BGC-RASAL1 #5 and #6 and MCG-RASAL1 #2) as compared with the
pEGFP-transfectants. RASAL1 overexpression decreased the cell
growth rates of the BGC-RASAL1 #5 and 6 cells as compared with the
BGC-pEGFP control, especially from the third to the fifth day
(Fig. 3A). The effect of RASAL1
overexpression on cell growth in the MCG-RASAL1 #2 cells was more
significant beginning on the second day (Fig. 3B). Flow cytometric analysis showed
that RASAL1 overexpression increased apoptosis in the BGC-RASAL1 #6
cells at 48 h. (15.7 vs 9.29%, *P=0.041; Fig. 3C). The MCG-RASAL1 #2 cells showed
significantly induced apoptosis as compared to the MCG-pEGFP
control at 24 h (15.48 vs 12.18%, *P=0.026) and 48 h
(19.45 vs. 14.54%, *P=0.035) (Fig. 3D). These data implied that RASAL1
inhibited gastric cancer cell line growth by inducing cell
apoptosis.
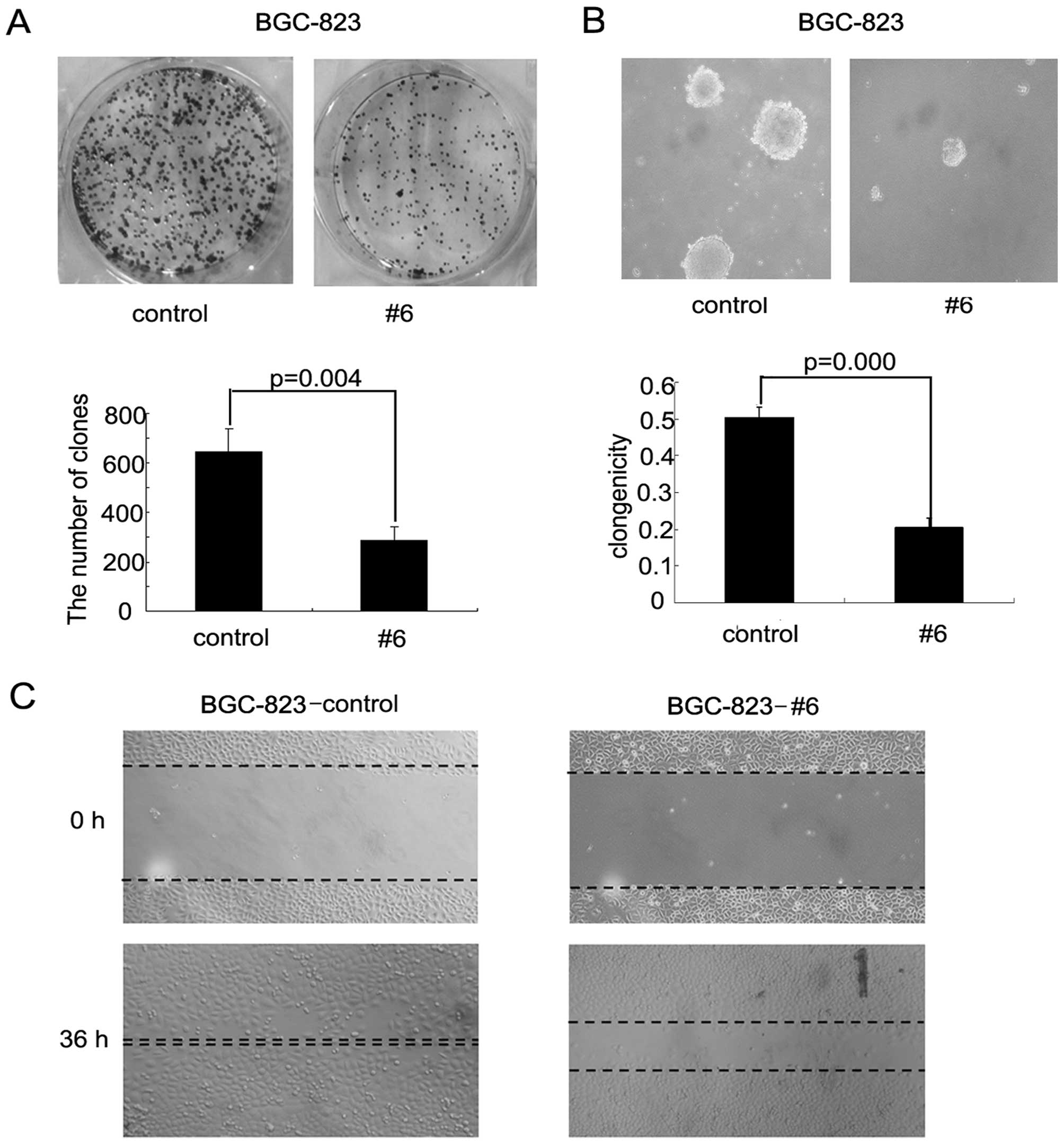
Enforced expression of RASAL1 suppressed
gastric cancer cells transformation and migration ability
RASAL1 tumor-suppressive effects were assessed by
colony and foci formation assays in soft agar and wound healing
assays. The frequency of foci formation was significantly lower in
the BGC-RASAL1 #6 cells compared to the BGC-pEGFP control cells
(P=0.004; Fig. 4A). Colony
formation in soft agar was significantly decreased in the
BGC-RASAL1 #6 cells compared to the BGC-pEGFP cells (P=0.000;
Fig. 4B). Additionally, wound
healing assay showed that RASAL1 inhibited cell migration ability
in BGC cells (Fig. 4C). Together,
these data provide evidence that RASAL1 has significant
tumor-suppressive effects in gastric cancer cells.
Discussion
Ras GAPs play an important role in regulating Ras
activation, which is an alternative mechanism to Ras gene mutation.
RASAL1 is a member of the Ras GAPs family, and it is a
Ca2+-regulated Ras GAPs that decodes the frequency of
Ca2+ oscillations (10).
Other family members include p120GAP, neurofibromin (NF1), the GAP1
family (GAP1IP4BP, Ca2+-promoted Ras
inactivator (CAPRI), and Ras GTPase activating-like protein
RASAL1), and the SynGAP family (DAB2IP, nGAP, SynGAP) (9,12–18).
The expression profiles and cellular localizations of the Ras GAP
family members differ. Previous studies have shown a reduction in
RASAL1 expression in nasopharyngeal carcinoma, breast, lung, liver,
and esophageal squamous cell carcinoma cell lines (10) as well as in colon (5).
Our findings showed that RASAL1 is downregulated in
gastric cancer tissues. Although most gastric cancers are
considered to be derived from chronic inflammatory mucosa induced
by H. pylori and Epstein-Barr virus (EBV) infection,
decreased expression of RASAL1 was not observed relationships
between the RASAL1 expression level and H. pylori and
Epstein-Barr virus (EBV) infection, Seto et al (19) also reported that reduced expression
of RASAL1 was not observed in any inflammatory mucosa or intestinal
metaplasia. Therefore, RASAL1 possibly contributes to gastric
carcinogenesis as a tumor suppressor gene (TSG), but is not
effected by environment factors in gastric cancer. However, the
potential role of RASAL1 in gastric progression and its ability to
inhibit gastric tumorigenicity is less well known. In order to
explore the biological role of RASAL1 in gastric tumorigenesis, we
established RASAL1 overexpression cell models and evaluated whether
RASAL1 restoration affects the malignant phenotype of tumor cells.
The present study found that RASAL1 overexpression inhibited cell
proliferation induced by cell apoptosis. RASAL1 enforced expression
significantly inhibited foci formation and cell migration, which
implied RASAL1 is associated with the epithelial-mesenchymal
transition (EMT) process, and EMT-related markers should be
identified.
A previous report has found that RASAL1
downregulation resulted from an underexpressed transcription factor
PITX1 in certain tumors (20).
However, decreased expression of RASAL1 was not observed related
with the expression of PITX1 (data not shown) in our study. Jin
et al (10) have reported an
epigenetic silencing mechanism of RASAL1 in a variety of
carcinomas. In this study, silenced RASAL1 was restored after
treatment with 5′-AZA or TSA depending on the drug dose. Together
with previous studies, our findings supported that DNA methylation
and histone deacetylation contribute, at least in part, to reduced
RASAL1 expression in gastric carcinogenesis.
In conclusion, the present study provide the first
insight into the biological function of RASAL1 and implied its
potential role in gastric progression and its ability to inhibit
gastric tumorigenicity. Our findings are useful in understanding if
RASAL1 may be beneficial for the development of new treatment
options for gastric cancer. However, the underlying molecular
mechanism by which RASAL1 promotes apoptosis and controls gastric
carcinogenesis remains to be determined.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China, grant no. 81171915. This study was
also supported by Nanjing Medical Key Scientific Foundation, (no.
ZKX08012). We are grateful to Professor Qian Tao for providing the
pEGFP-RASAL1 plasmid.
References
|
1
|
Hancock JF: Ras proteins: different
signals from different locations. Nat Rev Mol Cell Biol. 4:373–384.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mitin N, Rossman KL and Der CJ: Signaling
interplay in Ras superfamily function. Curr Biol. 15:R563–R574.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Downward J: Role of receptor tyrosine
kinases in G-protein-coupled receptor regulation of Ras:
transactivation or parallel pathways? Biochem J. 376:e9–10. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Calvisi DF, Ladu S, Conner EA, et al:
Inactivation of Ras GTPase-activating proteins promotes
unrestrained activity of wild-type Ras in human liver cancer. J
Hepatol. 54:311–319. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ohta M, Seto M, Ijichi H, et al: Decreased
expression of the RAS-GTPase activating protein RASAL1 is
associated with colorectal tumor progression. Gastroenterology.
136:206–216. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Matkar SS, Durham A, Brice A, Wang TC,
Rustgi AK and Hua X: Systemic activation of K-ras rapidly induces
gastric hyperplasia and metaplasia in mice. Am J Cancer Res.
1:432–445. 2011.PubMed/NCBI
|
|
7
|
Yashiro M, Yasuda K, Nishii T, et al:
Epigenetic regulation of the embryonic oncogene ERas in gastric
cancer cells. Int J Oncol. 35:997–1003. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu ZM, Liu LN, Li M, Zhang QP, Cheng SH
and Lu S: Mutation detection of KRAS by high-resolution melting
analysis in Chinese with gastric cancer. Oncol Rep. 22:515–520.
2009.PubMed/NCBI
|
|
9
|
Bernards A and Settleman J: GAPs in growth
factor signalling. Growth Factors. 23:143–149. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jin H, Wang X, Ying J, et al: Epigenetic
silencing of a Ca(2+)-regulated Ras GTPase-activating protein RASAL
defines a new mechanism of Ras activation in human cancers. Proc
Natl Acad Sci USA. 104:12353–12358. 2007.
|
|
11
|
Su X, Lv C, Qiao F, et al: Expression
pattern and clinical significance of DNA methyltransferase 3B
variants in gastric carcinoma. Oncol Rep. 23:819–826.
2010.PubMed/NCBI
|
|
12
|
Yarwood S, Bouyoucef-Cherchalli D, Cullen
PJ and Kupzig S: The GAP1 family of GTPase-activating proteins:
spatial and temporal regulators of small GTPase signalling. Biochem
Soc Trans. 34:846–850. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cullen PJ and Lockyer PJ: Integration of
calcium and Ras signalling. Nat Rev Mol Cell Biol. 3:339–348. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Donovan S, Shannon KM and Bollag G: GTPase
activating proteins: critical regulators of intracellular
signaling. Biochim Biophys Acta. 1602:23–45. 2002.PubMed/NCBI
|
|
15
|
Le LQ and Parada LF: Tumor
microenvironment and neurofibromatosis type I: connecting the GAPs.
Oncogene. 26:4609–4616. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dasgupta B and Gutmann DH:
Neurofibromatosis 1: closing the GAP between mice and men. Curr
Opin Genet Dev. 13:20–27. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pamonsinlapatham P, Hadj-Slimane R,
Lepelletier Y, et al: p120-Ras GTPase activating protein (RasGAP):
a multi-interacting protein in downstream signaling. Biochimie.
91:320–328. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Grewal T and Enrich C: Molecular
mechanisms involved in Ras inactivation: the annexin A6-p120GAP
complex. Bioessays. 28:1211–1220. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Seto M, Ohta M, Ikenoue T, et al: Reduced
expression of RAS protein activator like-1 in gastric cancer. Int J
Cancer. 128:1293–1302. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kolfschoten IG, van Leeuwen B, Berns K, et
al: A genetic screen identifies PITX1 as a suppressor of RAS
activity and tumorigenicity. Cell. 121:849–858. 2005. View Article : Google Scholar : PubMed/NCBI
|