Introduction
Anthocyanins (ATCs) are natural phytochemicals which
are produced by fruits and vegetables. ATCs have noteworthy
biological functions, including anti-inflammatory and
apoptosis-inducing effects. It has been reported that ATCs induce
apoptosis in various cancer cell lines including hepatoma,
colorectal carcinoma, gastric adenocarcinoma, leukemia and prostate
cancer (1–5). The ATC-induced apoptosis occurs via
both caspase-dependent and -independent mechanisms. It has been
demonstrated that ATCs initially induced an alteration of protein
kinase B (AKT) and mitogen-activated protein kinases (MAPKs) such
as p38 MAPK, extracellular signal-regulated kinase (ERK)1/2 and
c-Jun N-terminal kinase (JNK), which led to the upregulation of the
BAX/BCL2 ratio and hence the activation of intrinsic apoptosis in
cancer cells (1–4). However, in prostate cancer PC3 cells,
ATCs induced caspase-independent apoptosis via the nuclear
translocation of endonuclease G and AIF (5). Regarding their anti-inflammatory
activity, ATCs prevent the LPS- and UVB-induced expression of
inflammatory mediators such as cyclooxygenase (COX)-2 and
prostaglandin E2 (PGE2) by suppressing the nuclear
factor of κ light polypeptide gene enhancer in activated B cells
(NF-κB) and phosphoinositide 3-kinase (PI3K)/AKT pathways in human
keratinocytes and macrophage cell lines (6,7). This
anti-inflammatory activity of ATCs was shown to be effective in
vivo to subside circulatory and multiple organ failures due to
endotoxemia (8). Based on these
apoptosis-inducing and anti-inflammatory as well as anti-invasive
and anti-angiogenic effects (9,10),
attempts have been made to develop ATCs as a chemopreventive agent,
and promising results of pilot studies in rodent models of
esophageal and colorectal cancers and human colorectal cancer
patients have provided support for this hypothesis (11,12).
In addition to inducing apoptosis, it has been
suggested that ATCs induce autophagy in cancer and glial cells
(13,14), although the mechanism for this
remains to be clarified. Autophagy is considered to be a cellular
defense system that can protect cancer cells from anticancer
therapeutics. Accordingly, autophagy inhibition augments
ATC-induced apoptosis (13). A more
detailed understanding of the mechanism underlying ATC-induced
autophagy could contribute to the development of effective
strategies for the use of ATCs as a chemopreventive or
chemotherapeutic agent against cancer in the future.
AMPK, which plays a central role in energy balance
and metabolism, is one of the critical regulators of autophagy
induction. AMPK becomes activated once it is phosphorylated by
several kinases (15,16). Under metabolic stresses that
restrain ATP synthesis, elevated AMP binds to the γ subunit of AMPK
for both allosteric activation of AMPK and prevention of the
dephosphorylation of AMPK, leading to a sustained activity of AMPK.
Allosterically activated AMPK is phosphorylated by liver kinase B1
(LKB1) at Thr172 of the α subunit to become fully activated. In
response to elevated intracellular calcium levels and cytokine
signaling, calmodulin-dependent protein kinase kinase (CaMKK) β and
transforming growth factor-β-activated kinase 1 (TAK1),
respectively, phosphorylate AMPK. Activated AMPK phosphorylates
mTORC1, an inhibitor of autophagy, and a serine/threonine kinase
with homology to yeast atg1 (ULK1), an initiator of autophagy,
thereby leading to autophagy induction (17,18).
P27KIP1, a CDK inhibitor, is directly phosphorylated at
Thr198 and stabilized by AMPK for the induction of autophagy and,
notably, the knockdown of p27KIP1 in the presence of
activated AMPK induced apoptosis instead of autophagy, an
observation that explains the inhibitory effect of apoptosis by
autophagy and the possibility that p27KIP1 may mediate
the decision between apoptosis and autophagy (19). Forkhead box O3a (FOXO3a), an
important regulator of glucose metabolism and tumor suppression,
was reported to be phosphorylated by AMPK (20). Recently, FOXO3a was shown to
upregulate p27KIP1 to induce cell cycle arrest in glioma
cells (21,22). Therefore, it can be argued that
under metabolic stress, autophagy formation can be induced through
interactions among AMPK, FOXO3a, and p27KIP1.
ATC was recently reported to suppress IGF-I-induced
mammalian target of rapamycin (mTOR) phosphorylation by AMPK
activation in colon cancer cells, providing further support for its
anticancer activity (23). However,
the relationship between autophagy and AMPK in ATC-treated cells is
not yet fully understood. To address this issue, we used ATCs
extracted from black soybeans (cv Cheongja 3). These ATC extracts
were shown to promote apoptosis in a model of prostate hyperplasia
in vivo and to induce autophagy in glial cells, which proved
to contain biologically active components of autophagy induction as
well as apoptosis (14,24). In the present study, we observed the
induction of autophagy and AMPK activation by ATCs and analyzed the
role of AMPK in this autophagy.
Materials and methods
Chemicals
AICAR, compound C (CC), SP600125, SB203580, U0126,
and triciribine were obtained from Sigma-Aldrich Corp. (St. Louis,
MO, USA) or Merck (Darmstadt, Germany). All chemicals used are of
molecular biology or cell culture grade.
Extraction of ATCs
ATCs were extracted from black seed coated soybean,
Cheongja 3, developed by the National Institute of Crop Science,
Rural Development Administration at Miryang, Korea, as previously
described, with minor modifications (25). Briefly, hand-peeled seed coats were
extracted twice with 80% ethanol containing 0.1% acetic acid for 2
days at 4°C. These ethanol extracts were filtered through a 0.45-μm
filter and concentrated in a rotary evaporator to obtain crude ATC
extracts. The crude extracts were freeze-dried and kept below
−70°C.
Establishment of U2OS-GFP-LC3 cells
U2OS cells were cultured in high glucose DMEM
(Invitrogen, Carlsbad, CA, USA) supplemented with 10%
heat-inactivated fetal bovine serum (FBS; Hyclone Laboratories
Inc., Logan, UT, USA), 100 U/ml penicillin/streptomycin (Hyclone).
Plasmid EGFP-LC3 was generously donated by Professor S.H. Lee at
the Department of Pathology, the Catholic University of Korea. To
establish stable cell lines expressing GFP-LC3, pEGFP-empty vector
or pEGFP-LC3 was transfected into U2OS cells. Following the
selection of transfected cells using G418 for 14 days, viable cells
were pooled, and GFP expression was observed under a fluorescence
microscope. Total cellular extracts were prepared and subjected to
immunoblot analysis against GFP or LC3.
Immunoblot analysis
U2OS cells treated with ATCs with or without other
chemicals as indicated were lysed with RIPA buffer (50 mM Tris-HCl,
pH 7.4, 150 mM NaCl, 0.25% Na-deoxycholate, 1% NP-40, 1 mM EDTA)
containing protease inhibitors (Roche Applied Sciences,
Indianapolis, IN, USA). Total cellular proteins were separated by
SDS-polyacrylamide gels and transferred to nitrocellulose membranes
(Millipore, Billerica, MA, USA), which were hybridized with primary
and then secondary antibodies. Finally, protein bands were detected
by enhanced chemiluminescence (ECL; GE Healthcare, UK). Antibodies
against phospho-AMPK-Thr172, phospho-p38 MAPK (T180/Y182), p38
MAPK, JNK, Erk1/2, phospho-Erk1/2 (T202/Y204), mTOR, and
phospho-mTOR (S2448) were obtained from Cell Signaling Technology,
Inc. (Danvers, MA, USA) and antibodies against phospho-JNK
(Y185/Y223), AKT, phospho-AKT (S473), ACC, phospho-ACC (S79), and
cleaved PARP were from Epitomics, Inc. (Burlingame, CA, USA).
Antibodies against actin, α-tubulin, and GAPDH from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA) and LC3 from
Sigma-Aldrich Corp. were used.
Transfection of small interfering
RNA
Small interfering RNA against AMPKα1
(siAMPK) was purchased from Sigma-Aldrich Corp., and small
interfering RNAs against p27KIP1 and
FOXO3a were from Bioneer (Daejeon, Korea). Each siRNA was
transfected into U2OS cells or U2OS-GFP-LC3 cells by the reverse
transfection method using RNAiMAX reagent (Invitrogen) according to
the manufacturer's protocol. The sequences of duplex siRNAs are as
follows: siAMPK,
5′-CCCAUAUUAUUUGCGUGUA(dTdT)::5′-UACACGCAAAUAAUAUGGG(dTdT);
sip27KIP1, 5′-CGA
CGAUUCUUCUACUCAA(dTdT)::5′-UUGAGUAGAAGA AUCGUCG(dTdT);
siFOXO3a, 5′-GACGAUGAUGCGCCU
CUCU(dTdT)::5′-AGAGAGGCGCAUCAUCGUC(dTdT).
Assessment of cell death
Cell death was examined by measuring sub-G1 cells
and phosphatidylserine translocation. For measurement of sub-G1
cells, cells were fixed in ice-cold 70% ethanol for 2 h at 4°C and
stained with propidium iodide (PI; 0.2 mg/ml), followed by flow
cytometric analysis (FACSCalibur, BD Bioscience, San Jose, CA,
USA). To determine phosphatidylserine translocation, the cells
stained with PI and annexin V using ApoScan Kit (BioBud,
Gyunggi-do, Korea) were analyzed using a previously described
method (26).
Measurement of intracellular ATP
levels
Intracellular ATP levels were measured using a
CellTiter Glo Kit (Promega, Madison, WI, USA) following the
manufacturer's instructions.
Statistical analysis
Statistical significances were evaluated using the
one-way ANOVA test. The data represent three independent
experiments performed in triplicate as the mean ± SD.
Results
ATCs induce autophagy in U2OS cells
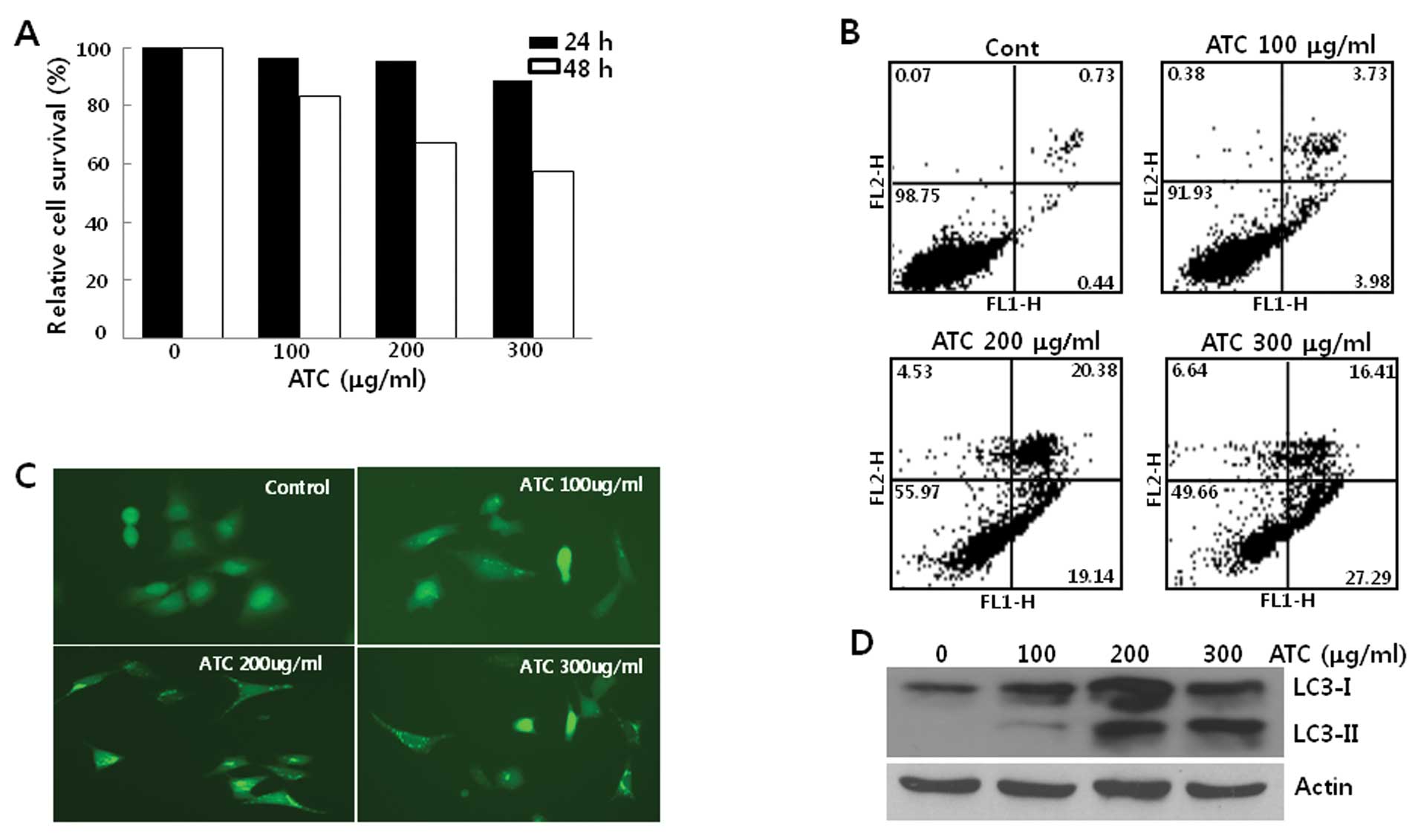
When human osteosarcoma U2OS cells were treated with
ATCs, their growth was not inhibited in the first 24 h. However,
after 24 h, ATCs suppressed the growth of U2OS cells associated
with apoptosis (Fig. 1A and B),
suggesting that ATCs activate the cell death program after 24 h in
this cell line and autophagy may be induced before 24 h. To
investigate this further, we examined the autophagy by ATCs before
24 h. For this end, we generated U2OS cells stably expressing
GFP-LC3 (U2OS-GFP-LC3) and first observed LC3 puncta formation in
these cells. When U2OS-GFP-LC3 cells were treated with ATCs for 6
h, it appeared that GFP-LC3 puncta were formed (Fig. 1C). Consistent with LC3 foci
formation, the conversion of LC3-I to LC3-II occurred in both U2OS
(Fig. 1D) and U2OS-GFP-LC3 cells
(data not shown) in a dose-dependent manner, reaching a plateau
level at concentrations of ATCs above 200 μg/ml of ATCs. These
findings demonstrated that ATCs induced autophagy in U2OS cells
prior to the activation of apoptosis.
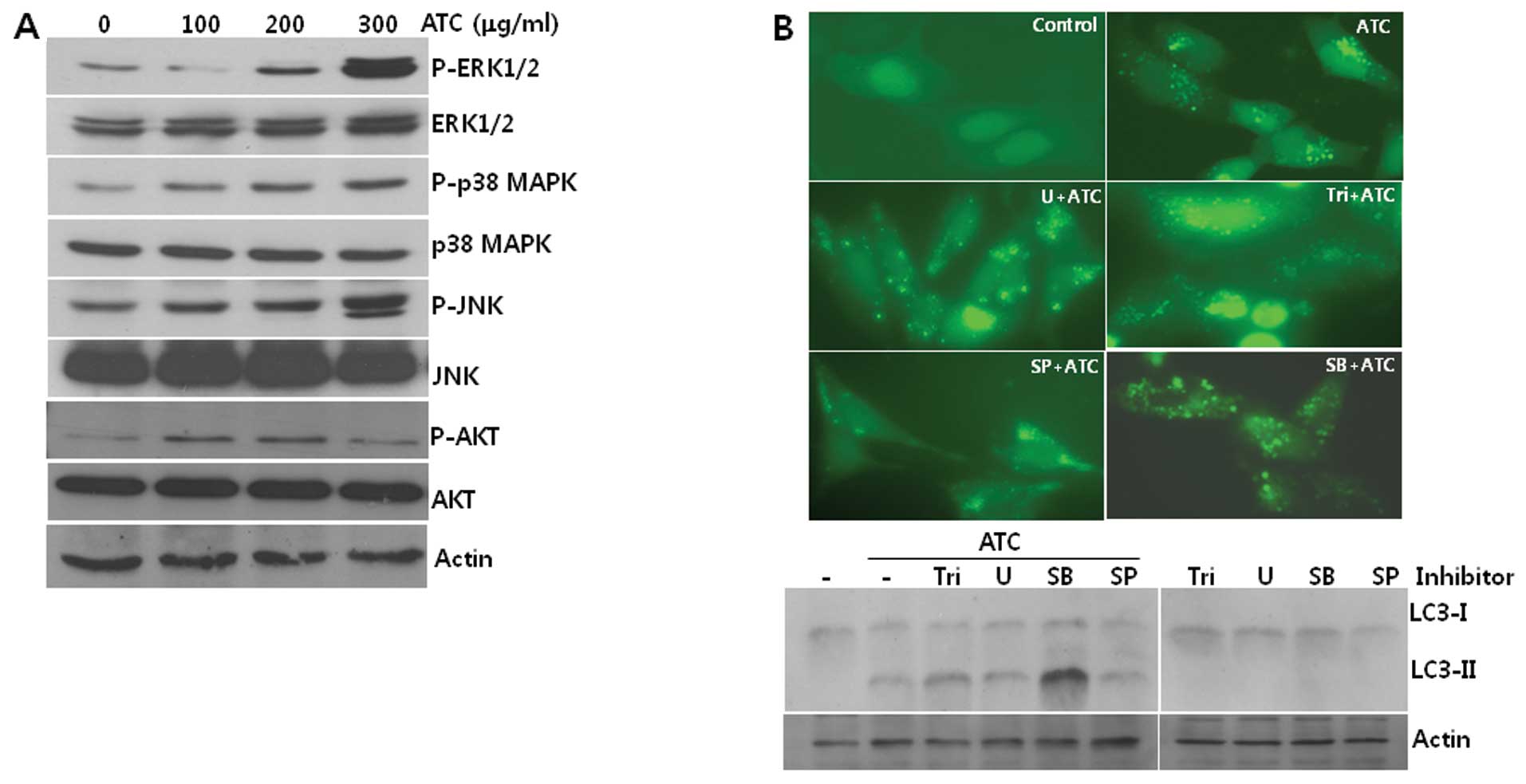
ATC-induced autophagy is not prevented by
inhibitors of MAPKs or AKT
To identify the signal transduction pathway leading
to autophagy in ATC-treated cells, we investigated the
phosphorylation of signal transduction proteins which have been
reported to be involved in ATC-induced apoptosis. As shown in
Fig. 2A, the phosphorylation of
ERK1/2, p38 MAPK, JNK, and AKT was increased by ATC treatment,
suggesting that they are involved in this autophagy. However,
neither the formation of GFP-LC3 puncta nor LC3-I conversion to
LC3-II was prevented by any of the chemical inhibitors of AKT
(Triciribine, Tri), MEK (U0126, U), p38 MAPK (SB203580, SB), and
JNK (SP600125, SP) (Fig. 2B). These
findings led us to speculate that pathways other than AKT and MAPKs
including p38 MAPK, ERK, and JNK may contribute to the induction of
autophagy by ATCs.
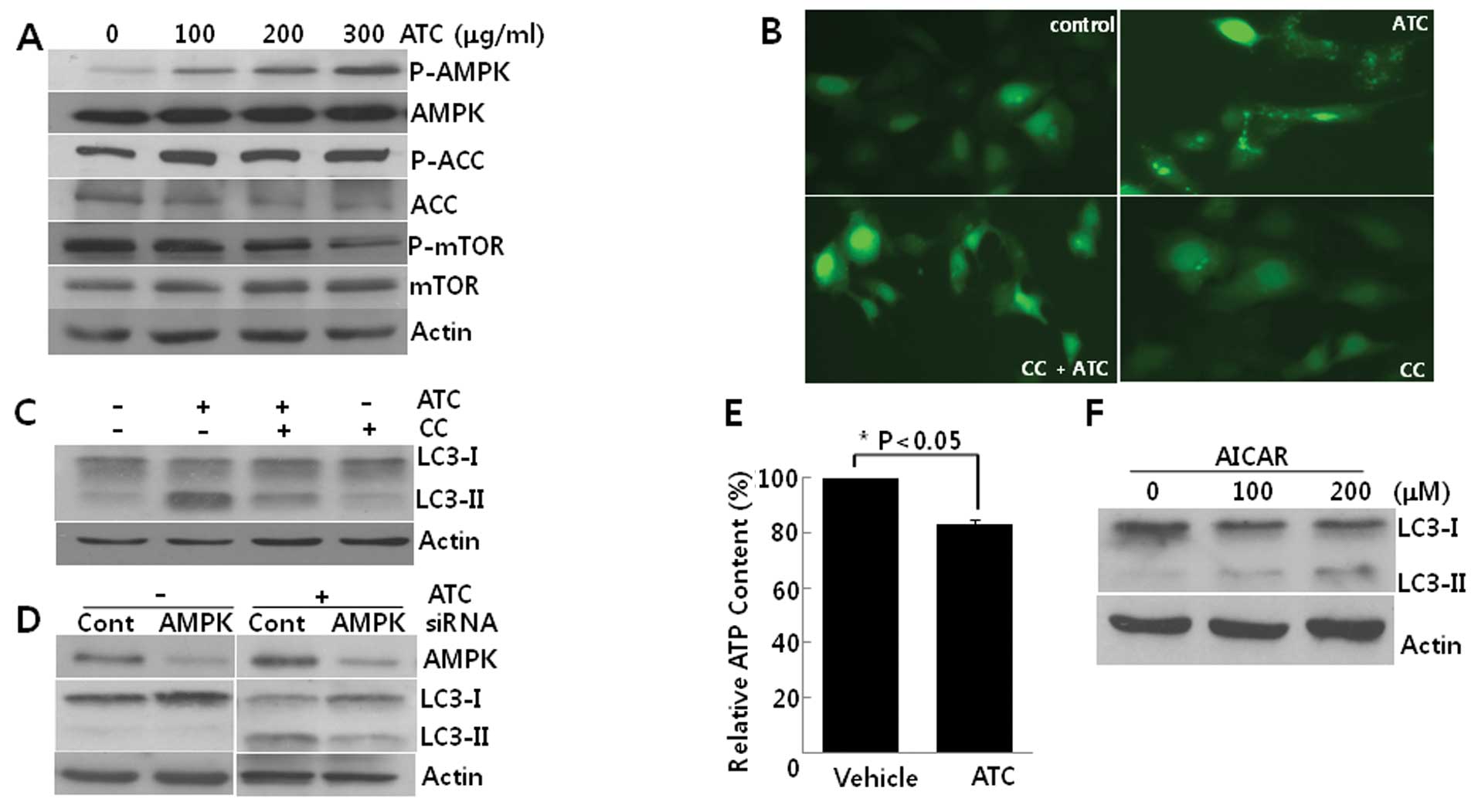
ATC-induced autophagy is prevented by
AMPK inhibition
We then explored the activation of AMPK in
ATC-treated cells. While the phosphorylation of mTOR was decreased,
the phosphorylation of both AMPKα1 and its substrate, acetyl CoA
carboxylase (ACC) increased according to concentrations of ATCs,
indicating that AMPK is activated in this autophagy (Fig. 3A). Pretreatment with CC, an
inhibitor of AMPK, reduced both the GFP-LC3 puncta formation
(Fig. 3B) and the conversion of
LC3-I to LC3-II (Fig. 3C).
Consistent with the effect of CC, the knockdown of AMPKα1 using
siRNA against AMPKα1 also suppressed the conversion of LC3-I
to LC3-II (Fig. 3D). Furthermore,
ATCs diminished intracellular ATP levels (Fig. 3E) and AICAR, an analog of AMP, also
induced autophagy in U2OS cells (Fig.
3F). Collectively, these findings suggest that ATC-induced
autophagy is dependent on AMPK activation which may be due to the
depletion of ATP.
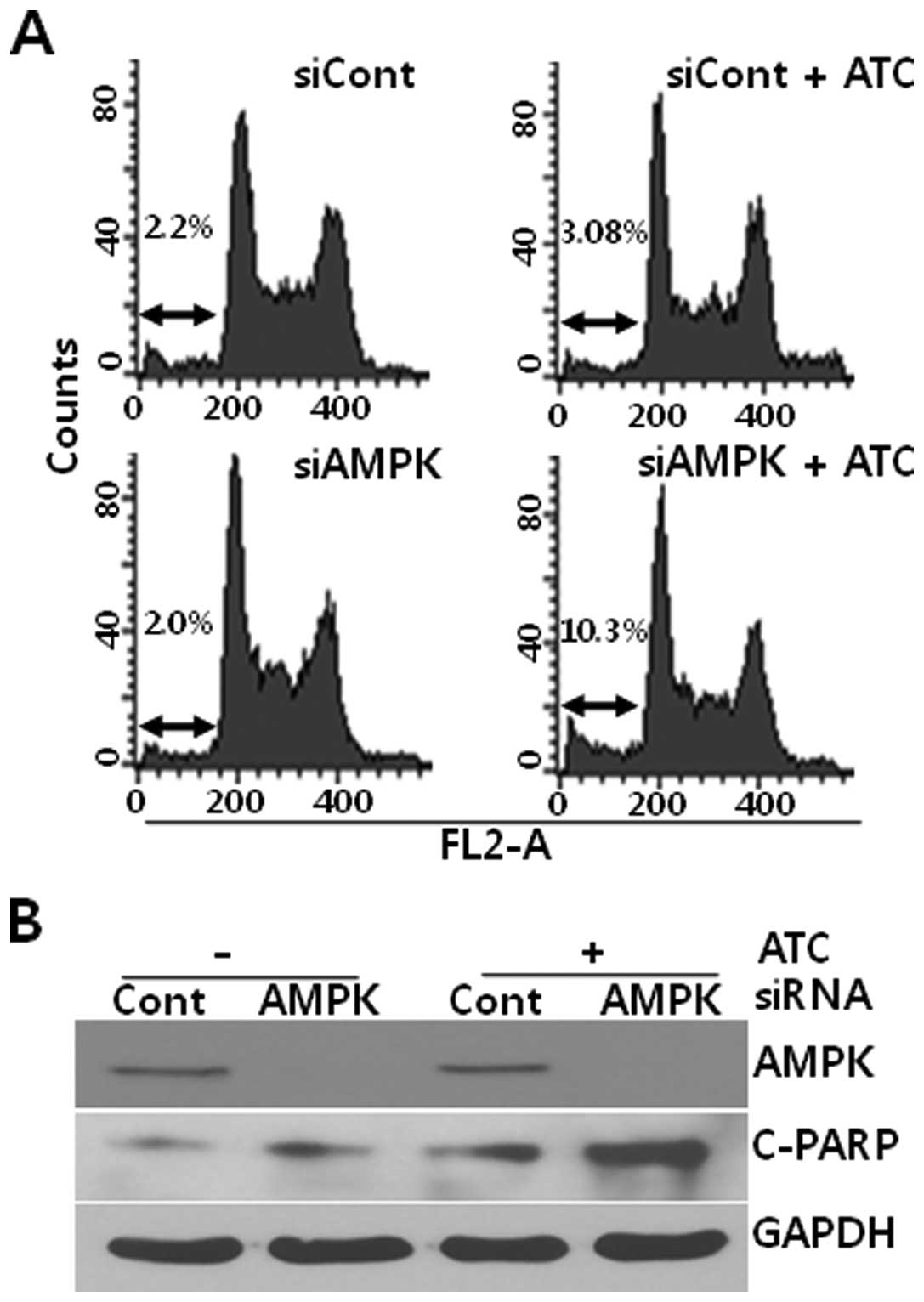
AMPK inhibition enhances ATC-induced
apoptosis
Based on the report that autophagy inhibition
augmented the ATC-induced apoptosis in hepatocellular carcinoma
cells (13), we analyzed the effect
of AMPK on apoptosis in ATC-treated U2OS cells. As shown in
Fig. 4, while ATCs induced a subtle
apoptosis at 6 h in U2OS cells that had been transfected with
control siRNA, ATCs dramatically induced apoptotic features such as
accumulation of hypo-diploidic cells (Fig. 4A) and poly(ADP-ribose) polymerase-1
(PARP) cleavage (Fig. 4B) in
AMPKα1 siRNA-transfected U2OS cells. Thus, these data
suggest that ATC-activated AMPK protects U2OS cells from
apoptosis.
Differential effects of
p27KIP1 and FOXO3a on ATC-induced autophagy
To investigate the downstream targets of AMPK that
play a role in the induction of autophagy, we attempted the
transfection of siRNAs against ULK1, p27KIP1, and
FOXO3a which have been demonstrated to be autophagy
modulators under AMPK. The knockdown of ULK1 by ULK1 siRNA
did not exert an inhibitory effect on either ATC-induced autophagy
or apoptosis, ruling out the involvement of ULK1 in this autophagy
(data not shown). As shown in Fig.
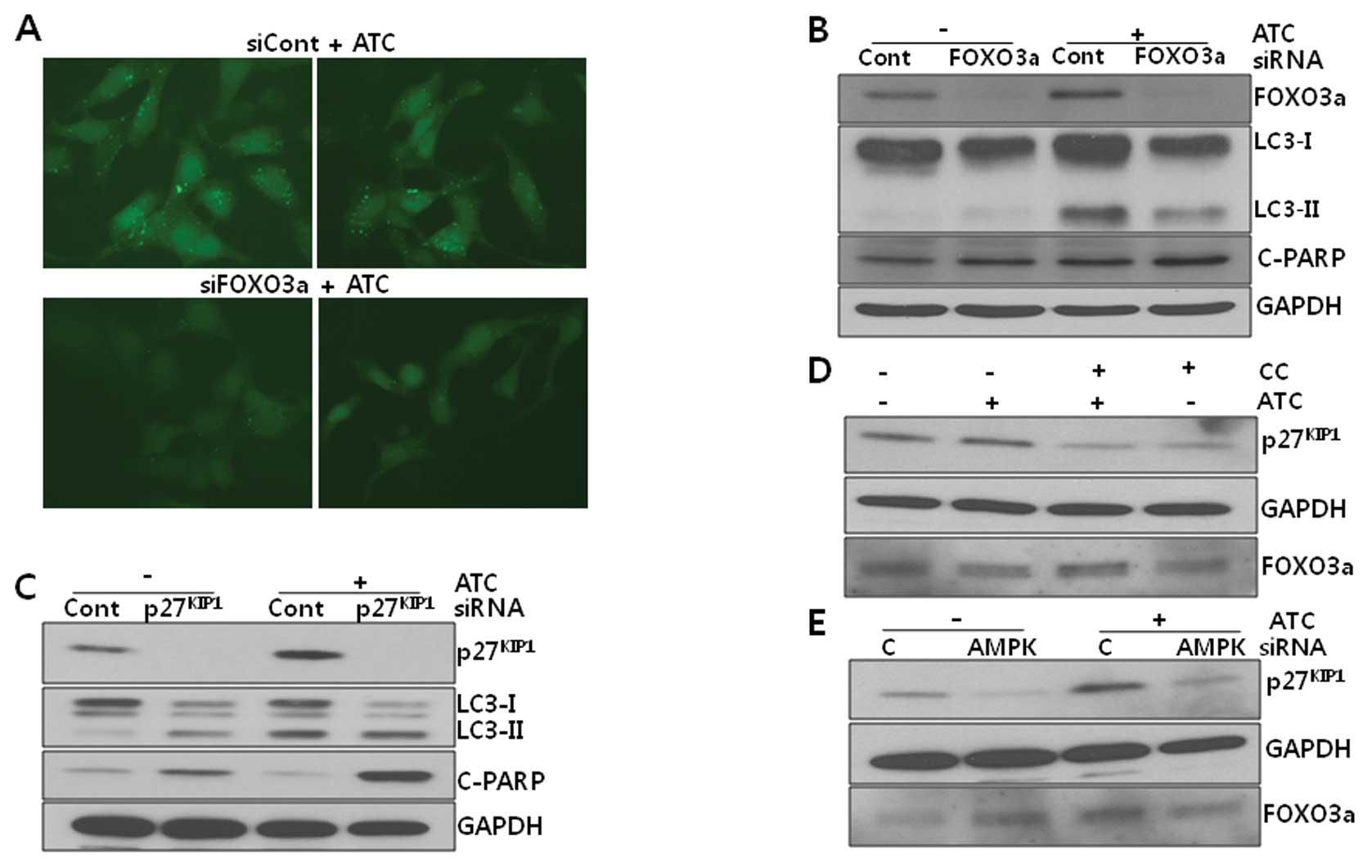
5, both ATC-induced LC3 puncta formation (Fig. 5A) and the conversion of LC3-I to
LC3-II (Fig. 5B) were reduced, but
ATC-induced PARP cleavage was not altered by FOXO3a knockdown,
suggesting that FOXO3a may be responsible for the induction of
autophagy induction under active AMPK. In contrast to the effect of
FOXO3a siRNA, p27KIP1 siRNA
potentiated ATC-induced PARP cleavage but had no effect on LC3-II
levels (Fig. 5C), suggesting that
p27KIP1 may inhibit ATC-induced apoptosis, but has no
effect on autophagy. Thus, it can be suggested that AMPK activated
by ATCs may induce autophagy via FOXO3a and inhibit apoptosis via
p27KIP1.
Discussion
The findings reported herein indicate that ATCs
induce autophagy in human osteosarcoma U2OS cells, accompanied by
the activation of MAPKs and AMPK. By analyzing the effects of
chemical inhibitors and siRNAs against MAPKs and AMPK on
ATC-induced autophagy, the findings suggest that AMPK plays a
critical role in this autophagy. However, how ATCs specifically
activate AMPK, remains unknown. It has been reported that
phytochemicals such as galegine, berberine, as well as resveratrol
activate AMPK in an AMP-dependent manner (27). ATCs also reduced ATP levels in this
cell line and AICAR, an analog of AMP, induced autophagy (Fig. 3E and F), indicating that
AMP/LKB1-dependent AMPK activation results from ATC treatment.
However, LKB1 was not phosphorylated by ATCs and AMPK was still
phosphorylated in U2OS cells that had been transfected with LKB1
siRNA (data not shown). In addition, ATC-induced AMPK
phosphorylation was not altered by BAPTA-AM, a calcium chelator,
CaMKK siRNA, TAK1 siRNA or ROS scavengers (data not
shown). Collectively, it can be assumed that ATCs may enrich
intracellular AMP levels which induce the allosteric activation of
AMPK without the action of LKB1 or inhibit the dephosphorylation of
AMPK that was phosphorylated by unidentified enzymes other than
LKB1. However, as it is known that the allosteric activation of
AMPK by AMP is induced in AMPK phosphorylated by basal LKB1
activity, this assumption leaves many questions to be resolved.
Considering the numerous studies reporting that AMPK-activating
compounds including phytochemicals inhibit mitochondrial
respiratory complexes to reduce synthesis of ATP, we speculate that
ATCs might contain two major features for activating AMPK: one is
to reduce ATP synthesis possibly via the inhibition of
mitochondrial respiratory complexes and the other is to induce the
basal phosphorylation of AMPK or the allosteric activation of AMPK
in conjunction with AMP even in the absence of LKB1 activity.
A recent study showed that AMPK phosphorylates ULK1
to initiate autophagy (18). In the
present model, ATC-induced AMPK phosphorylation was accompanied by
the dephosphorylation of mTOR (Fig.
3) and rapamycin, an inhibitor of mTOR, induced autophagy (data
not shown). Moreover, ATCs induced autophagy in ULK1-knocked down
cells to a comparable extent as in control siRNA-transfected U2OS
cells (data not shown), suggesting that AMPK-induced mTORC1
inhibition may play a more important role in ATC-induced autophagy
than does ULK1 activation. In addition to the mTOR and ULK1
pathways, AMPK modulates downstream target proteins such as
p27KIP1 and FOXO3a to induce autophagy.
P27KIP1, an inhibitor of CDK1, which can be
phosphorylated by AMPK and can be induced transcriptionally by
FOXO3a as well, was reported to induce autophagy but inhibit
apoptosis (19), suggesting that it
is a strong molecular target of ATC-activated AMPK. Different from
these reports, in our model, it appeared that p27KIP1
inhibited apoptosis but was not involved in autophagy, whereas
FOXO3a contributed to the induction of autophagy but not apoptosis.
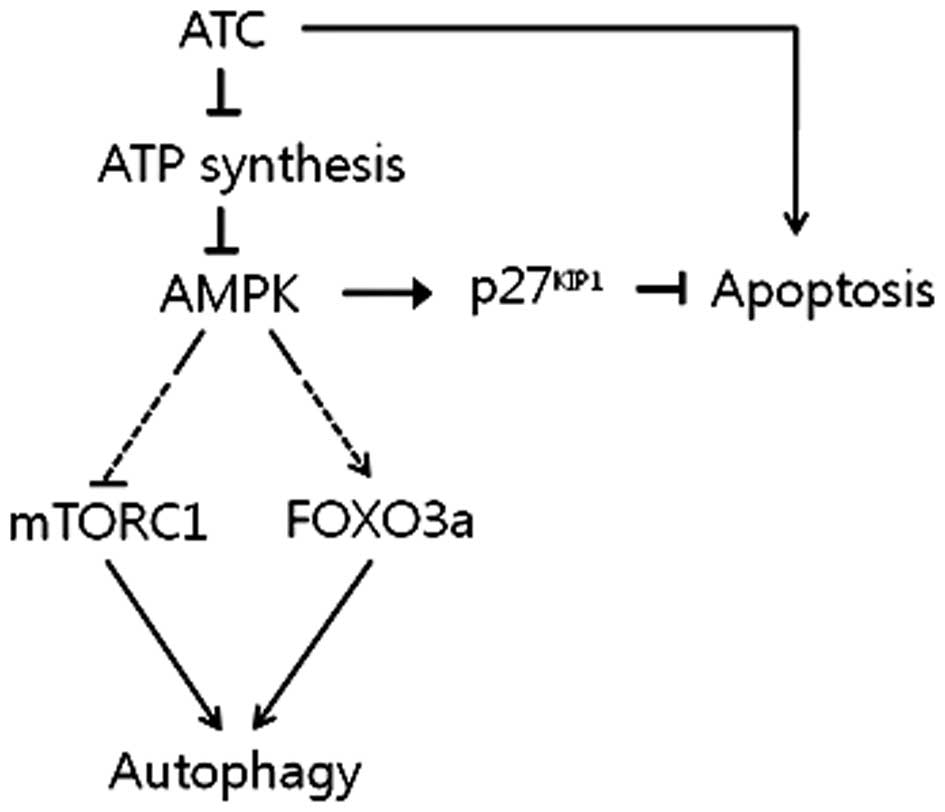
Therefore, it can be concluded that AMPK activated by ATC-inhibited
ATP synthesis induces autophagy by activating FOXO3a while
suppressing mTORC1, and inhibits ATC-induced apoptosis via the
upregulation of p27KIP1, as summarized in Fig. 6. To verify this, however, the
mechanism by which p27KIP1 and FOXO3a are regulated by
ATCs remains to be clarified. As shown in Fig. 5C-E, p27KIP1 protein
levels were upregulated by ATC treatment, which returned to basal
levels in U2OS cells that had been pretreated with compound C (CC)
or transfected with AMPKα1 siRNA before ATC treatment,
suggesting that p27KIP1 may be stabilized by active
AMPK. In contrast to p27KIP1, FOXO3a protein levels were
not consistently altered by ATCs and the phosphorylation of
FOXO3a-Ser413, a site known to be phosphorylated by AMPK, was not
detected either (data not shown), suggesting that the
phosphorylation of Ser or Thr residues other than Ser413 by AMPK or
an indirect effect of AMPK on FOXO3a induces autophagy.
The role of AMPK in cancer cells is known to be
varied. AMPK can activate p53 by phosphorylating p53-Ser15 to
induce cell cycle arrest and apoptosis (28,29).
For example, a combined treatment with metformin and
2-deoxyguanosine, as well as vincristine, an anticancer agent,
induced apoptosis in prostate and melanoma cells, respectively, by
activating AMPK (30,31). Contrary to these reports, AMPK
inhibition resulted in the sensitization of cancer cells to
apoptosis by anticancer drugs such as cisplatin as well as
metabolic stress such as hypoxia (32,33).
This variable effect of AMPK on cell death may be due to both types
of stresses and cellular context. It was recently reported that
autophagy inhibition can enhance the cytotoxicity of ATCs in
hepatoma cells (13), which may be
consistent with the data in our study showing that AMPK and
p27KIP1 siRNAs enhanced ATC-induced apoptosis (Figs. 4 and 5C). Therefore, AMPK activation by ATCs may
contribute to the cancer cell survival via the induction of
autophagy.
In non-transformed cells, autophagy serves to
preserve genomic integrity, thereby preventing malignant
transformations (34). This
autophagy-inducing activity of ATCs could explain the
chemopreventive effect of ATCs reported in the esophagus and the
colon (11,12), although the autophagy-inducing
activity of ATCs in non-transformed cells remains to be determined.
A metabolic shift to aerobic glycolysis has generally been regarded
as a driving force for carcinogenesis (35,36),
and dysregulated metabolic homeostasis and obesity have been
reported to increase the risk of certain types of cancer, such as
breast cancer (37). Since AMPK is
a central regulator of metabolism and preserves metabolic
homeostasis, AMPK could prevent cancer formation under conditions
of metabolic distress. Consistent with this assumption, metformin
was reported to inhibit the formation of pre-neoplastic lesions in
the colon and showed chemopreventive activity against breast cancer
associated with obesity (38).
Therefore, even if ATCs do not induce autophagy in non-transformed
cells, only the AMPK-activating effect of ATCs could contribute to
chemopreventive effects against cancer.
Autophagy can prevent the induction of apoptosis
and, instead, initiate necrotic cell death in cancer cells in
vivo, accompanied by an inflammatory response, which can
contribute to the invasion and metastasis of cancer cells (39,40).
This autophagy-inducing activity of ATCs could dampen the
anticancer effect of ATCs, thereby constituting a negative feedback
loop of ATC-induced apoptosis.
Collectively, it could be concluded that while ATCs
may activate AMPK and, thus, prevent carcinogenesis in
non-transformed cells by activating autophagy and preserving
metabolic homeostasis, autophagy in cancer cells may diminish the
apoptosis-inducing activity of ATCs. Therefore, to develop ATCs for
use as efficient anticancer therapy or as a chemopreventive agent
against cancer, the mechanism underlying the autophagy-inducing
activity of ATCs as well as ATC-induced apoptosis needs to be
thoroughly examined to determine whether it is possible to modulate
genes to enhance the anticancer effects of ATCs.
Acknowledgements
This study was supported by the Biogreen 21 Program
(code no. PJ007186), Rural Development Administration Korea. Part
of this study was also supported by the Basic Science Research
Program through the National Research Foundation of Korea (NRF)
funded by the Ministry of Education, Science and Technology
(2011–0027115).
Abbreviations:
|
AKT
|
protein kinase B
|
|
AMPK
|
adenosyl monophosphate-dependent
protein kinase
|
|
ATCs
|
anthocyanins
|
|
CaMKK
|
calmodulin-dependent protein kinase
kinase
|
|
CC
|
compound C
|
|
COX
|
cyclooxygenase
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
FOXO3a
|
forkhead box O3A
|
|
JNK
|
c-Jun N-terminal kinase
|
|
LC3
|
light chain 3
|
|
LKB1
|
liver kinase B1
|
|
MAPK
|
mitogen-activated protein kinase
|
|
mTOR
|
mammalian target of rapamycin
|
|
NF-κB
|
nuclear factor of κ light polypeptide
gene enhancer in activated B cells
|
|
PGE2
|
prostaglandin E2
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
TAK1
|
transforming growth factor-β-activated
kinase 1
|
|
ULK1
|
a serine/threonine kinase with
homology to yeast atg1
|
References
|
1
|
Yeh CT and Yen GC: Induction of apoptosis
by the anthocyanidins through regulation of Bcl-2 gene and
activation of c-Jun N-terminal kinase cascade in hepatoma cells. J
Agric Food Chem. 53:1740–1749. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shin DY, Lee WS, Lu JN, et al: Induction
of apoptosis in human colon cancer HCT-116 cells by anthocyanins
through suppression of Akt and activation of p38-MAPK. Int J Oncol.
35:1499–1504. 2009.PubMed/NCBI
|
|
3
|
Shih PH, Yeh CT and Yen GC: Effects of
anthocyanidin on the inhibition of proliferation and induction of
apoptosis in human gastric adenocarcinoma cells. Food Chem Toxicol.
43:1557–1566. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee SH and Park SM and Park SM: Induction
of apoptosis in human leukemia U937 cells by anthocyanins through
down-regulation of Bcl-2 and activation of caspases. Int J Oncol.
34:1077–1083. 2009.PubMed/NCBI
|
|
5
|
Reddivari L, Vanamala J, Chintharlapalli
S, et al: Anthocyanin fraction from potato extracts is cytotoxic to
prostate cancer cells through activation of caspase-dependent and
caspase-independent pathways. Carcinogenesis. 28:2227–2235. 2007.
View Article : Google Scholar
|
|
6
|
Hou DX, Fujii M, Terahara N and Yoshimoto
M: Molecular mechanisms behind the chemopreventive effects of
anthocyanins. J Biomed Biotechnol. 5:321–325. 2004.PubMed/NCBI
|
|
7
|
Tsoyi K, Park HB, Kim YM, et al:
Anthocyanins from black soybean seed coats inhibit UVB-induced
inflammatory cylooxygenase-2 gene expression and PGE2
production through regulation of the nuclear factor-κB and
phosphatidylinositol 3-kinase/Akt pathway. J Agric Food Chem.
56:8969–8974. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sautebin L, Rossi A, Serraino I, et al:
Effect of anthocyanins contained in a blackberry extract on the
circulatory failure and multiple organ dysfunction caused by
endotoxin in the rat. Planta Med. 70:745–752. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shin DY, Lu JN, Kim GY, et al:
Anti-invasive activities of anthocyanins through modulation of
tight junctions and suppression of matrix metalloproteinase
activities in HCT-116 human colon carcinoma cells. Oncol Rep.
25:567–572. 2011.
|
|
10
|
Matsubara K, Kaneyuki T, Miyake T and Mori
M: Antiangiogenic activity of nasunin, an antioxidant anthocyanin,
in eggplant peels. J Agric Food Chem. 53:6272–6275. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang LS, Hecht SS, Carmella SG, et al:
Anthocyanins in black raspberries prevent esophageal tumors in
rats. Cancer Prev Res. 2:84–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thomasset S, Berry DP, Cai H, et al: Pilot
study of oral anthocyanins for colorectal cancer chemoprevention.
Cancer Prev Res. 2:625–633. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Longo L, Platini F, Scardino A, Alabiso O,
Vasapollo G and Tessitore L: Autophagy inhibition enhances
anthocyanin-induced apoptosis in hepatocellular carcinoma. Mol
Cancer Ther. 7:2476–2485. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim YK, Yoon HH, Lee YD, et al:
Anthocyanin extracts from black soybean (Glycine max L.)
protect human glial cells against oxygen-glucose deprivation by
promoting autophagy. Biomol Ther. 20:68–74. 2012.
|
|
15
|
Hardie DG: AMP-activated protein kinase:
an energy sensor that regulates all aspects of cell function. Genes
Dev. 25:1895–1908. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yun H and Ha J: AMP-activated protein
kinase modulators: a patent review (2006–2010). Expert Opin Ther
Pat. 21:983–1005. 2011.PubMed/NCBI
|
|
17
|
Alers S, Löffler AS, Wesselborg S and
Stork B: Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy:
cross talk, shortcuts, and feedbacks. Mol Cell Biol. 32:2–11. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Egan DF, Shackelford DB, Mihaylova MM, et
al: Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase
connects energy sensing to mitophagy. Science. 331:456–461. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liang J, Shao SH, Xu ZX, et al: The energy
sensing LKB1-AMPK pathway regulates p27KIP1
phosphorylation mediating the decision to enter autophagy or
apoptosis. Nat Cell Biol. 9:218–224. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Greer EL, Oskoui PR, Banko MR, et al: The
energy sensor AMP-activated protein kinase directly regulates the
mammalian FOXO3 transcription factor. J Biol Chem. 282:30107–30119.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Shi J, Zhang L, Shen A, et al: Clinical
and biological significance of forkhead class box O 3a expression
in glioma: mediation of glioma malignancy by transcriptional
regulation of p27KIP1. J Neurooncol. 98:57–69. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gopinath S, Malla RR, Gondi CS, et al:
Co-depletion of cathepsin B and uPAR induces G0/G1 arrest in glioma
via FOXO3a mediated p27 upregulation. PLoS One. 5:e116682010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee YK, Lee WS, Kim GS and Park OJ:
Anthocyanins are novel AMPKα1 stimulators that suppress tumor
growth by inhibiting mTOR phosphorylation. Oncol Rep. 24:1471–1477.
2010.
|
|
24
|
Jang H, Ha US, Kim SJ, Yoon BI, Han DS,
Yuk SM and Kim SW: Anthocyanin extracted from black soybean reduces
prostate weight and promotes apoptosis in the prostatic
hyperplasia-induced rat model. J Agric Food Chem. 58:12686–12691.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee JH, Kang NS, Shin SO, et al:
Characterization of anthocyanins in the black soybean (Glycine
max L.) by HPLC-DAD-ESI/MS analysis. Food Chem. 112:226–231.
2009. View Article : Google Scholar
|
|
26
|
Jang JY, Kim MK, Jeon YK, Joung YK, Park
KD and Kim CW: Adenovirus adenine nucleotide translocator-2 shRNA
effectively induces apoptosis and enhances chemosensitivity by the
down-regulation of ABCG2 in breast cancer stem-like cells. Exp Mol
Med. 44:251–259. 2012. View Article : Google Scholar
|
|
27
|
Hawley Simon A, Ross Fiona A, Chevtzoff
Cyrille, et al: Use of cells expressing γ subunit variants to
identify diverse mechanisms of AMPK activation. Cell Metab.
11:554–565. 2010.
|
|
28
|
Jones RG, Plas DR, Kubek S, et al:
AMP-activated protein kinase induces a p53-dependent metabolic
checkpoint. Mol Cell. 18:283–293. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Imamura K, Ogura T, Kishimoto A, Kaminishi
M and Esumi H: Cell cycle regulation via p53 phosphorylation by a
5′-AMP activated protein kinase activator,
5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside, in a human
hepatocellular carcinoma cell line. Biochem Biophys Res Commun.
287:562–567. 2001.
|
|
30
|
Ben Sahra I, Laurent K, Giuliano S, et al:
Targeting cancer cell metabolism: The combination of metformin and
2-deoxyglucose induces p53-dependent apoptosis in prostate cancer
cells. Cancer Res. 70:2465–2475. 2010.
|
|
31
|
Chen MB, Shen WX, Yang Y, Wu XY, Gu JH and
Lu PH: Activation of AMP-activated protein kinase is involved in
vincristine-induced cell apoptosis in B16 melanoma cell. J Cell
Physiol. 226:1915–1925. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim HS, Hwang JT, Yun H, et al: Inhibition
of AMP-activated protein kinase sensitizes cancer cells to
cisplatin-induced apoptosis via hyper-induction of p53. J Biol
Chem. 283:3731–3742. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chhipa RR, Wu Y and Ip C: AMPK-mediated
autophagy is a survival mechanism in androgen-dependent prostate
cancer cells subjected to androgen deprivation and hypoxia. Cell
Signal. 23:1466–1472. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Karantza-Wadsworth V, Patel S, Kravchuk O,
et al: Autophagy mitigates metabolic stress and genome damage in
mammary tumorigenesis. Genes Dev. 21:1621–1635. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim JW and Dang CV: Cancer's molecular
sweet tooth and the Warburg effect. Cancer Res. 66:8927–8930.
2006.
|
|
36
|
Levine AJ and Puzio-Kuter AM: The control
of the metabolic switch in cancers by oncogenes and tumor
suppressor genes. Science. 330:1340–1344. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cleary MP and Grossman ME: Minireview:
obesity and breast cancer - the estrogen connection. Endocrinology.
150:2537–2542. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Goodwin PJ and Stambolic V: Obesity and
insulin resistance in breast cancer - Chemoprevention strategies
with a focus on metformin. Breast. S3:S31–S35. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar
|
|
40
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: apoptosis, autophagy and the
cross-talk between them. Cell Death Diff. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|