Introduction
Lung cancer continues to be the leading cause of
death due to cancer in Western countries. In 2012, lung cancer was
estimated to account for approximately 26% of female and 29% of
male deaths due to cancer (1).
Among them, the formation of metastasis is the most common cause of
death.
The process of epithelial-mesenchymal transition
(EMT) plays a fundamental role in tumor progression and formation
of metastasis. In EMT, epithelial tumor cells with a cobblestone
phenotype acquire mesenchymal cell characteristics with a
spindle/fibroblast-like morphology (2). This process involves the loss or
downregulation of epithelial markers, including E-cadherin and
upregulation of mesenchymal molecular markers such as vimentin and
α-smooth muscle actin (αSMA). During the acquisition of EMT, loss
of epithelial markers, especially E-cadherin, is a critical process
that is regulated by several important transcriptional repressors.
The processes of EMT may be triggered by many growth factors,
including transforming growth factor β1 (TGF-β1), which is the most
important stimulus that can be influenced by the tumor
microenvironment.
In recent years, the role of the renin-angiotensin
system (RAS) in tumor progression or metastasis has been
extensively characterized (3–8).
Changes in the expression of RAS components, particularly in local
tumor tissue, appear to correlate with tumor grade and clinical
outcome (9). We previously reported
that the low expression of angiotensin-converting enzyme 2 (ACE2)
is associated with tumor grade in lung cancer (10). Lower levels of ACE2 expression
co-localized with areas of malignant tumors. Moreover, recent
studies suggest that angiotensin II (Ang II), which is the key
effector of the RAS system, was found to be enhanced in the EMT
process of intrahepatic cholangiocarcinoma at a local tissue level
(11). It is of note that marked
progress continues to be made in unraveling the contribution of the
RAS system, particularly the role of the vasodepressor pathway
composed by ACE2.
As we previously reported, decreased local
expression of ACE2 was shown to correlate with the progression of
lung cancer while transcriptional overexpression of ACE2, using an
adenoviral-mediated plasmid, reduced invasion and angiogenesis in
A549 cells in vitro and inhibited tumor growth in a mouse
model (10). In our present study,
we sought to investigate whether ACE2 inhibits metastasis in lung
cancer and whether it influences the EMT process in lung cancer
cells using the A549 cell line as a model.
Materials and methods
Materials and reagents
F-12K nutrient mixture (F-12K) and fetal bovine
serum (FBS) were obtained from Gibco (Grand Island, NY, USA).
Antibiotics (100 U/ml penicillin and 100 mg/ml streptomycin) were
purchased from Invitrogen Life Technologies (Carlsbad, CA, USA).
Recombinant human TGF-β1 was purchased from R&D Systems
(Minneapolis, MN, USA). Mouse anti-β-actin antibody was from Santa
Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Mouse monoclonal
anti-E-cadherin and mouse monoclonal anti-vimentin were from BD
Biosciences (San Diego, CA, USA). Goat polyclonal antibody to ACE2
was from R&D Systems. Rabbit polyclonal antibody to αSMA was
from Abcam (Cambridge, MA, USA). DX600 was from AnaSpec (San Jose,
CA, USA).
Cell lines and culture conditions
A549 cells were purchased from the Cytology Center
of the Chinese Academy of Sciences (Shanghai, China). Cells were
maintained in F-12K nutrient mixture (F-12K; Gibco, Carlsbad, CA,
USA) containing 10% FBS and 100 U/ml each of
penicillin/streptomycin at 37°C in 5% CO2. The media
were replaced every 48 h. In order to examine cell survival, cells
were cultured up to 70% confluence and serum-starved overnight.
They were then treated with TGF-β1 in culture medium containing 1%
FBS for 72 h. DX600, which is a specific inhibitor of ACE2, was
added to the culture 30 min before the addition of TGF-β1.
Gene transfection
To analyze the function of ACE2, we established
stable clones of A549 cells overexpressing ACE2 (A549-ACE2). The
transfection procedure was performed as previously reported
(12). Briefly, infection of A549
cells with MSCV-ACE2 resulted in robust ACE2 expression compared
with A549 cells infected with the vector alone. Overexpression of
ACE2 was confirmed by western blot analysis as described below.
RT-PCR
The mRNA levels of Twist, Snail2, ZEB1 and
E-cadherin were examined by RT-PCR. The procedure of RT-PCR was
performed as previously reported (13). Total RNA was extracted from cells or
tumor tissues using TRIzol reagent (Invitrogen, Karlsruhe, Germany)
according to the manufacturer’s protocol. To determine the mRNA
transcript level from cDNA, PCR was carried out using the following
primers: E-cadherin forward, 5′-CCACCAAAGTCACGCTGAAT AC-3′ and
reverse, 5′-GGAGTTGGGAAATGTGAGCAA-3′; Twist forward,
5′-TCTCGGTCTGGAGGATGGAG-3′ and reverse, 5′-GTTATCCAGCTCCAGAGTCT-3′;
Snail2 forward, 5′-GAGCATTTGCAGACAGGTCA-3′ and reverse, 5′-CCTCA
TGTTTGTGCAGGAGA-3′; ZEB1 forward, 5′-GCACAACC AAGTGCAGAAGA-3′ and
reverse, 5′-GCCTGGTTCAGGA GAAGATG-3′; and β-actin, which served as
an internal control, forward, 5′-AAGATGACGCAGATCATGTTTGAG-3′ and
reverse, 5′-AGGAGGAGCAATGATCTTGATCTT-3′.
SDS-PAGE and western blot analysis
Cultured A549 cells (106–107)
were washed with cold phosphate-buffered saline (PBS; Mediatech)
three times and disrupted in 0.2 ml ice-cold cell lysis buffer
containing 10% phenylmethanesulfonyl fluoride (PMSF). After
incubation for 5 min at room temperature, cells were scraped from
the 6-well plates. Total cell lysates were sonicated, and insoluble
materials were removed by centrifugation at 13,000 rpm for 15 min
at 4°C. Protein concentrations were determined by the Bradford
method (Bio-Rad, Herts, UK). Equal amounts of protein (10 μg) were
separated on an 8% SDS-polyacrylamide gel (Bio-Rad). After
electrophoresis, separated proteins were transferred onto
immunoblot polyvinylidene difluoride (PVDF) membranes (Merck
Millipore, USA). Membranes were then blocked with 5% non-fat dried
milk in Tris-buffered saline with Tween-20 (TBST) for 1 h at room
temperature. Primary antibodies were added to TBST, and membranes
were incubated overnight at 4°C on a rocking platform. Primary
antibodies used were; mouse anti-β-actin (1:3,000) from Santa Cruz
Biotechnology Inc., mouse monoclonal anti-E-cadherin (1:1,500) and
mouse anti-vimentin (1:1,500) from BD Biosciences. Rabbit
polyclonal to αSMA (1:500) was from Abcam. After three washing
steps with TBST (15 min each), membranes were probed with the
corresponding anti-rabbit horseradish peroxidase (HRP)-conjugated
secondary antibody for 2 h at room temperature. Membranes went to a
second stage of washing in TBST, three times each for 15 min.
Immunoblots were visualized by enhanced chemiluminescence (GE
Healthcare, Chalfont St. Giles, UK). β-actin band density was used
as a loading control, and results were digitalized and quantified
using ImageJ software.
Animal experiments
Six-week-old male athymic nu/nu mice were purchased
from the Shanghai Laboratory Animal Center of Chinese Academy of
Sciences (Shanghai, China) and maintained under specific
pathogen-free conditions. Animals were maintained in a
temperature-controlled room (at 22°C) and supplied with food and
water. A single-cell suspension containing 2×106 cells
in 0.1 ml phosphate-buffered saline was injected into the lateral
tail veins of nu/nu mice. There were six mice per group. Three
weeks after treatment, mice were sacrificed and then lung, liver
and brain tissues were extracted, fixed with 4% formaldehyde, and
the number of metastatic colonies was counted under a dissecting
microscope. For the lung cancer xenograft model, wild-type or ACE2
overexpressing A549 cells (2×107 in 0.1 ml PBS) were
transplanted subcutaneously. Four weeks later, mice were
sacrificed, and tumor tissues were harvested and fixed with 4%
formaldehyde. Tumor and lung tissue extracts were analyzed by
hematoxylin and eosin (H&E) staining and immunohistochemical
analysis.
Immunohistochemical analysis
Immunohistochemical analyses were performed as
previously described. Briefly, all tissue samples were fixed in
phosphate-buffered neutral formalin, embedded in paraffin and cut
into 5-μm serial sections. After being deparaffinized in xylene,
tissue sections were rehydrated in graded ethanol solutions,
permeabilized in 0.1% Triton X-100 and 0.1% sodium citrate and
incubated overnight with primary antibodies. Immunohistochemical
staining with antibodies to E-cadherin (1:50), vimentin (1:25, both
from BD Biosciences) and ACE2 (1:50, R&D Systems) was performed
according to standard procedures. Results were observed and
photographed under a fluorescence microscope (Leica, Germany) with
Image-Pro Plus 6.0 software (Media Cybernetics). Specimens were
classified as positive when >10% of cancer cells were stained.
The intensity of each type of staining was graded as negative or
positive microscopically as previously reported (12).
Statistical analysis
Data are presented as averages and their respective
standard deviation (means ± SEM). All statistical analyses were
performed with the SPSS Statistical Program (version 17.0; SPSS,
Chicago, IL, USA). Comparisons of data between two groups were
conducted with the Student’s t-test. P-values of <0.05 were
considered to indicate statistically significant results.
Results
Overexpression of ACE2 decreases
metastasis of lung cancer in a mouse model
Our previous study showed that the overexpression of
ACE2 inhibited the proliferation of lung cancer cells in
vitro. It was also demonstrated that ACE2 overexpression
reduced tumor growth in a mouse lung xenograft model (10). However, it is still unknown whether
ACE2 decreases metastasis formation of lung cancer. Consequently, a
cell line stably overexpressing ACE2 (A549-ACE2) was constructed in
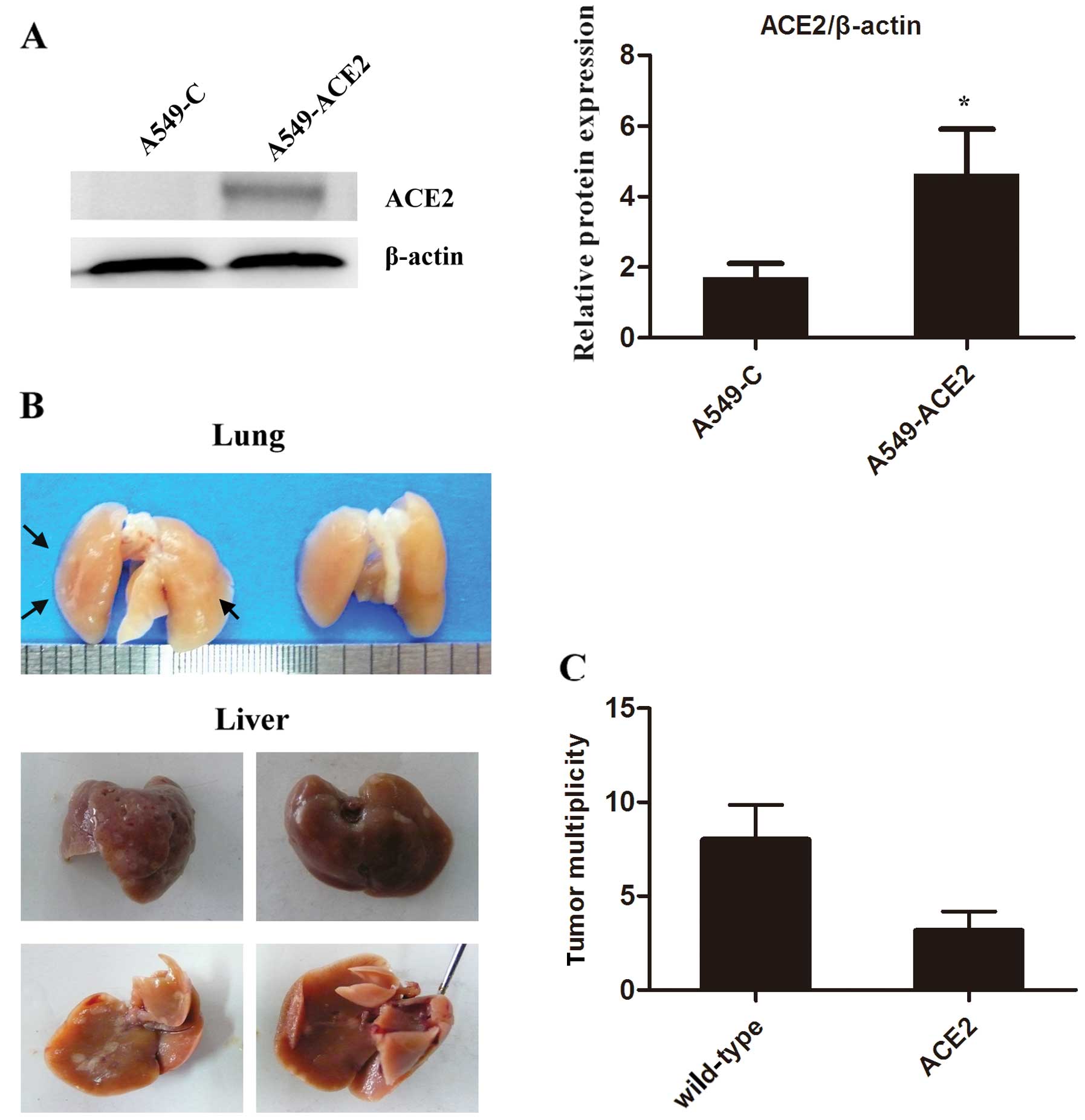
the present study. As shown in Fig.
1A, the increased expression of ACE2 in A549-ACE2 cells was
confirmed by western blot analysis compared to the barely
detectable levels in the wild-type cells (A549-C). In our model of
lung cancer metastasis, gross examination of the lungs and livers
extracted from nu/nu mice revealed numerous tumors (Fig. 1B). Age-matched nu/nu mice
transplanted with A549 cells overexpressing ACE2 developed
significantly fewer tumors (P<0.05) (Fig. 1C). These data revealed that the
overexpression of ACE2 reduced the metastatic potential of lung
cancer cells in vivo. Therefore, the regulation of ACE2
expression could prove a novel strategy for anticancer therapy.
Overexpression of ACE2 upregulates
expression of E-cadherin in vitro
EMT has been recently found to play a critical role
in tumor development, particularly during the induction of
metastasis (14). Decrease or loss
of E-cadherin expression is a key event during EMT. We previously
showed that ACE2 overexpression inhibited lung cancer metastasis
in vivo, therefore we hypothesized that ACE2 decreases lung
cancer metastasis through inhibiting EMT. To test this hypothesis,
we utilized the previous cell lines to study the role of ACE2 on
the expression of E-cadherin in vitro. To examine the
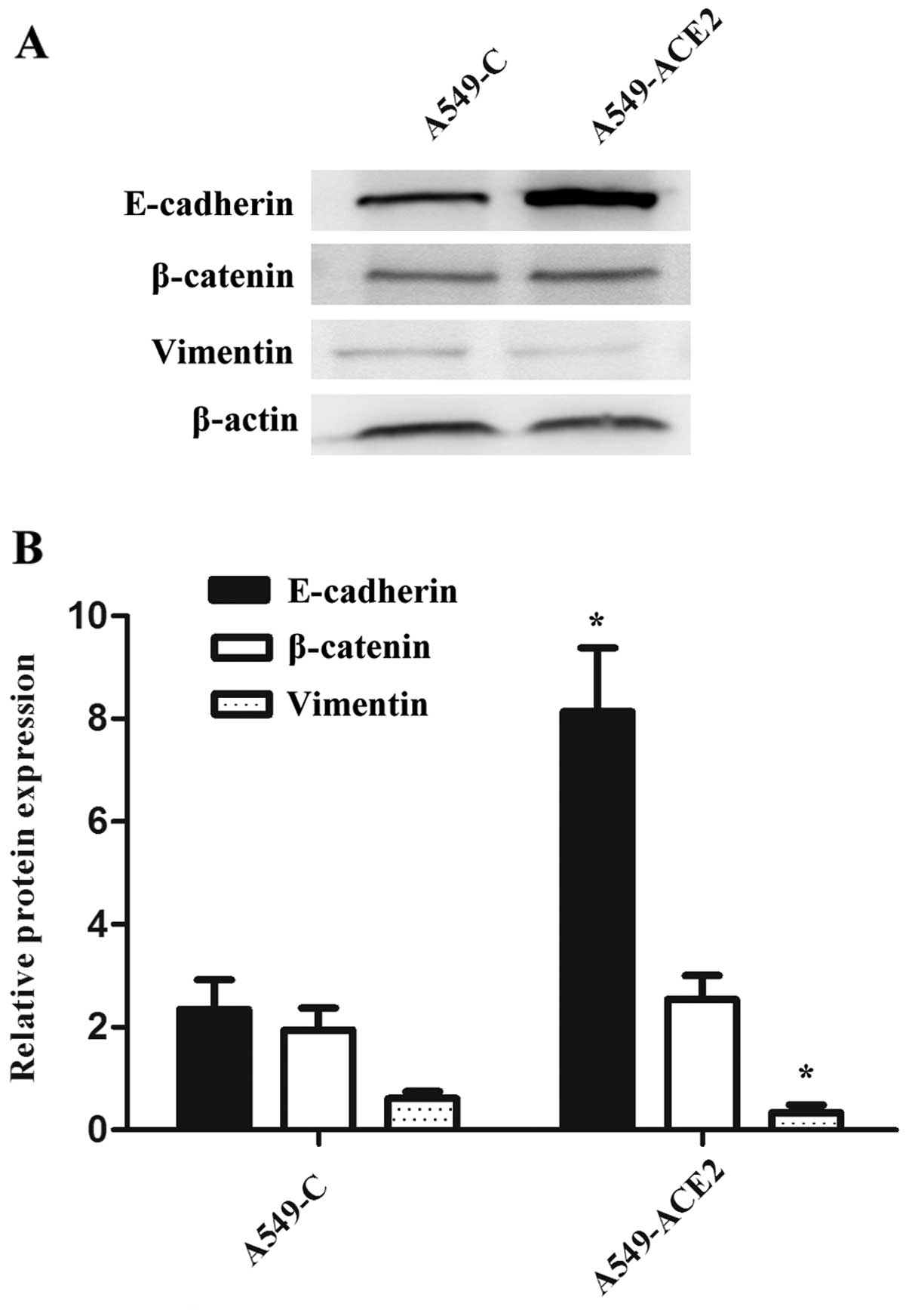
influence of ACE2 on epithelial markers, including E-cadherin and
β-catenin, we performed western blot analysis using anti-E-cadherin
and anti-β-catenin antibodies (Fig.
2A). ACE2 overexpression significantly increased the expression
of E-cadherin when compared to wild-type cells. This was determined
by densitometric analysis of the western blot bands (Fig. 2B). In contrast, mesenchymal markers,
particularly vimentin, were decreased in A549-ACE2 cells when
compared to A549-C cells.
ACE2 influences the expression of EMT
markers in a lung cancer xenograft model
We then evaluated the doubling times of A549-C and
A549-ACE2 cells in vivo. A549-C or A549-ACE2 cells
(2×107) were injected subcutaneously in mice; 21 days
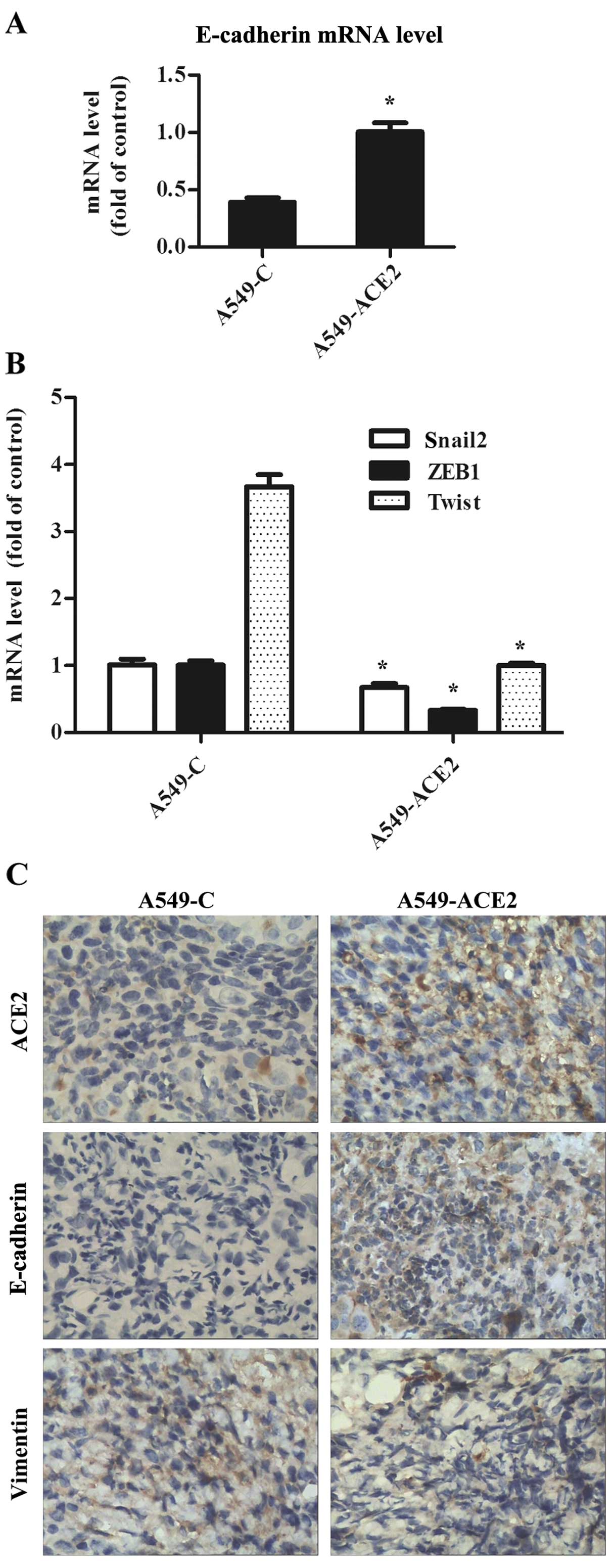
later, the resulting tumors were extracted. Immunohistochemical
staining showed that the expression of E-cadherin was significantly
higher and was located mainly on the cell membrane at the cell-cell
junctions in all tumor specimens of the A549-ACE2 mouse group
(Fig. 3C). Similarly, we found that
ACE2 was localized on the cell membrane of A549 cells
overexpressing ACE2 (4/6 positive in the A549-ACE2 group vs. 2/6
positive in the control group). In addition, the patterns of
expression and distribution of mesenchymal markers were different
between the two groups. Notably, the transfection of A549 cells
with ACE2 led to a decreased vimentin expression (Fig. 4). Vimentin expression has been shown
to positively correlate with EMT. In the A549-ACE2 mouse group,
vimentin was expressed in only one specimen while it was expressed
in 4 specimens extracted from the control group. In addition, we
found that overexpression of ACE2 in A549 cells significantly
upregulated the expression of epithelial-specific genes such as
E-cadherin; E-cadherin is associated with decreased expression of
mesenchymal markers (Fig. 3A).
ACE2 decreases mRNA levels of
transcriptional repressors which are associated with EMT
We aimed to ascertain whether A549 cells
overexpressing ACE2 had a differential expression pattern of genes
involved in the induction of EMT. ACE2 overexpression was
correlated with decreased mRNA levels of transcripts such as
Snail2, ZEB1 and Twist, which are causally involved in the EMT
process (Fig. 3B). In contrast, the
mRNA level of E-cadherin was restored in the tumor tissues of the
A549-ACE2 mouse group compared to the A549-C group (Fig. 3A). These data are consistent with
the notion that ACE2 inhibits a set of genes and markers involved
in EMT in a lung cancer xenograft model.
ACE2 attenuates EMT of A549 cells induced
by TGF-β1 in vitro
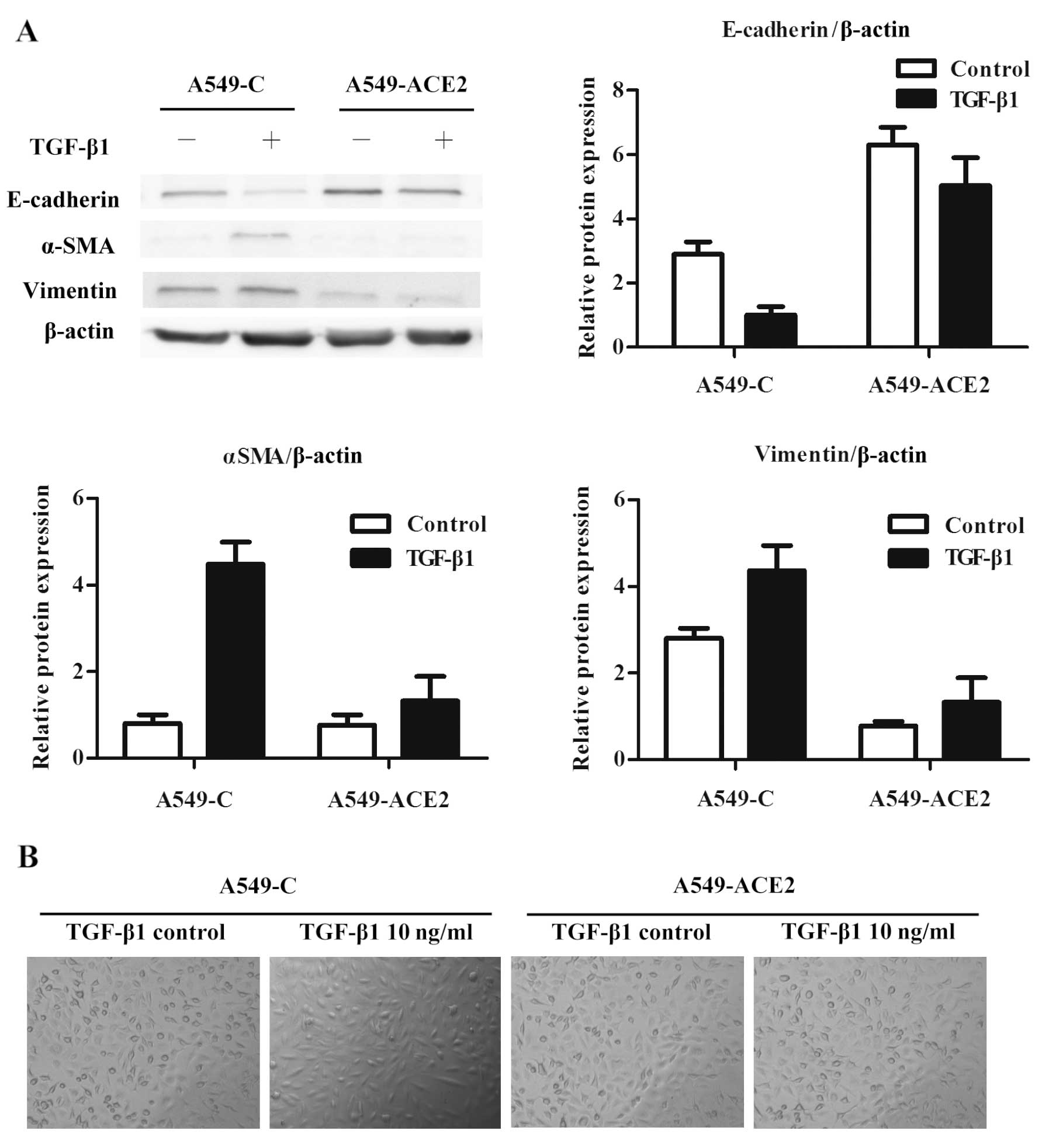
The upregulatory effect of ACE2 on E-cadherin
expression in lung cancer cells led us to explore the direct impact
of ACE2 on EMT. To further investigate whether ACE2 suppresses the
EMT process, a classical EMT model was developed using A549 cells
stimulated with TGF-β1. The presence of ACE2 in A549 cells
attenuated the decrease in the levels of E-cadherin due to TGF-β1
treatment. In addition, ACE2 significantly abrogated the
upregulation of mesenchymal markers, including vimentin and αSMA
(Fig. 4). The cell morphologic
phenotypes of A549-C and A549-ACE2 cells were examined using a
phase contrast microscope. TGF-β1-treated A549-C cells showed a
spindle-like shape and a loss of cell-to-cell attachments.
Untreated A549-C and A549-ACE2 cells with or without TGF-β1
treatment retained the morphological appearance of epithelial cells
(Fig. 4B).
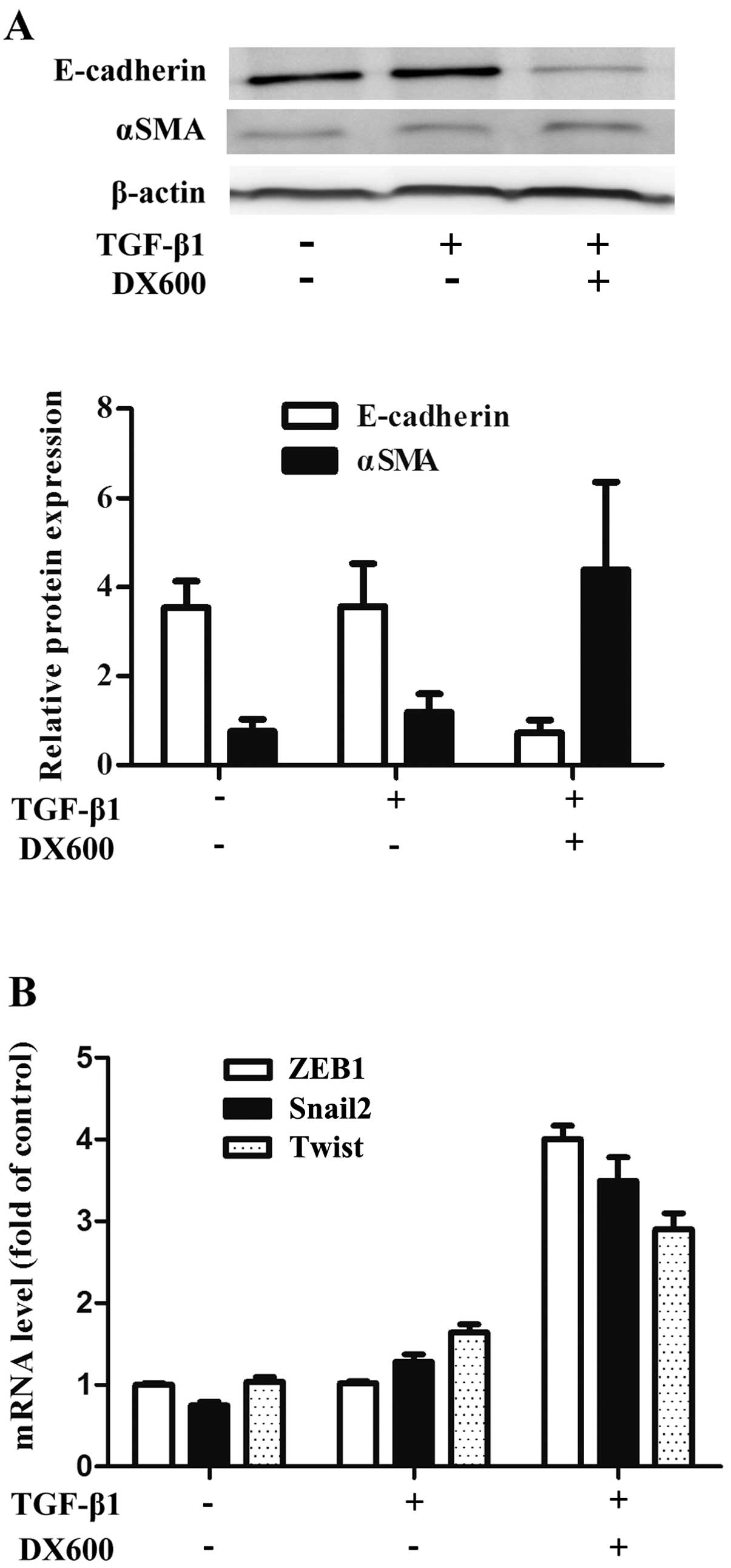
Effect of ACE2 inhibition on the
expression of EMT markers
An ACE2 antagonist was used to further confirm the
effect of ACE2 on EMT. Pretreatment of A549 cells with DX600 (at
106 M) abolished the increase in E-cadherin expression
caused by ACE2. Furthermore, the upregulation of αSMA caused by
TGF-β1 treatment was recovered in A549-ACE2 cells following
treatment with DX600 (Fig. 5A).
A549-ACE2 cells treated with TGFβ-1 followed by treatment with
DX600, lost the effect of ACE2-mediated restoration of E-cadherin
expression to control levels. These results demonstrate that the
lack of ACE2 increases the sensitivity of A549 cells to
TGF-β1-induced EMT.
In order to mechanistically investigate the role of
ACE2 in the regulation of transcriptional repressors, including
Snail2 and ZEB1 during the process of EMT, we determined the
transcriptional repertoire of A549 cells following treatment with
or without TGF-β1. We found that the expression levels of ZEB1,
Snail2 and Twist were markedly higher in A549-ACE2 cells treated
with TGF-β1 and DX600 when compared to TGF-β1 treatment alone. This
was consistent with the increased expression of mesenchymal markers
(Fig. 5B).
Discussion
In the present study we investigated the role of
ACE2 in lung cancer metastasis and EMT. To the best of our
knowledge, this is the first demonstration of how ACE2, in a A549
cell lung cancer model, decreases metastasis in vivo. ACE2
upregulates the expression of E-cadherin both in vitro and
in vivo as well as downregulates vimentin. These proteins
are representative markers of the EMT process. Furthermore, western
blot analysis indicated that ACE2 attenuated TGF-β1-mediated EMT of
A549 cells. ACE2 decreased the transcriptional levels of genes
associated with EMT in vitro. Exposing cells to DX600, an
inhibitor of ACE2, recovered the sensitivity of lung cancer cells
to TGF-β1.
Components of the RAS system have been found to
influence tumor growth and development (15–17).
ACE2 is a critical enzyme of the RAS system, which plays a
counterbalancing role by degrading Ang II to Ang 1–7 (18). Although numerous large clinical
trials have been carried out, the contribution of the RAS system to
tumor development remains controversial (19,20).
One study showed a negative association of losartan and a positive
association of candesartan and telmisartan with the overall
occurrence of cancer (21). Despite
the different origins of cancer, the results still remain distinct.
Bhaskaran et al(22) found a
decreased risk of lung cancer but increased risks for breast and
prostate cancers. The contradictory findings among the two methods
of angiotensin receptor blockade (ARB) suggest that a more
complicated regulatory mechanism exists in the RAS system. As the
RAS system is not linear, we hypothesized that the imbalance of the
different pathways of RAS plays a critical role in tumor growth and
development, and not Ang II and its receptors alone. Among these,
ACE2 may be the key component to link and regulate opposing Ang II
and Ang 1–7 pathways (23). Several
components of RAS may be expressed in the lung (24). The local concentration of these
cytokines is also altered in different lung diseases. The
beneficial effect of ACE2 has been studied in pulmonary
hypertension and pulmonary fibrosis (25).
In lung cancer, we found that decreased expression
of ACE2 correlates with poor clinical outcomes, which suggested
that ACE2 acts as a suppressor of lung cancer progression. It has
also been reported that the overexpression of ACE2 in human
pancreatic carcinoma cells decreased tumor growth, both in
vitro and in vivo. To examine the influence of ACE2 on
lung cancer metastasis, we evaluated in two xenograft experiments
the doubling times of A549-C and A549-ACE2 cancer cells. Here, ACE2
significantly attenuated the metastasis of lung cancer in
vivo.
Several key factors have been noted to be involved
in the malignant behavior of cancer cells. For example, loss of
E-cadherin has been considered an early event in cancer
development, which is well known to promote cancer cell invasion
and lymph node metastasis. Recently, EMT has been reported to play
a role in tumor progression (14,26).
The results of our study showed that ACE2 overexpression markedly
attenuated the effect of TGF-β1-induced EMT. Notably, TGF-β1 is a
key mediator of the EMT process (27,28).
Ang II, which is known as a key molecule of the RAS system, shares
many cellular responses with TGF-β1 (29). In renal epithelial cells, treatment
with Ang II resulted in a transition of cellular morphology from a
‘cobblestone’ epithelial to a mesenchymal phenotype. Altered
expression of the biomarkers for EMT were also demonstrated
(30). Ang II-induced key events of
the EMT process in intrahepatic cholangiocarcinoma were found to
include the downregulation of epithelial adherins and upregulation
of vimentin. Ang II was found to enhance cell invasiveness and
migration. Ang II may also serve as a growth factor in tumor
development and facilitate tumor metastasis of cancer cells
(31). However, it is still unknown
whether Ang II is a stimulus of EMT in lung cancer.
In the present study, we found that ACE2, which is a
newly identified component of the RAS system, attenuated lung
cancer metastasis through its inhibition of the EMT process. This
finding suggests that ACE2 may be a potential therapeutic target of
lung cancer where EMT contributes to the development of tumor
metastasis.
Acknowledgements
This study was supported by a grant from the
National Natural Science Foundation of China (81071925).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
2
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhou L, Zhang R, Zhang L, Yao W, Li J and
Yuan Y: Angiotensin-converting enzyme 2 acts as a potential
molecular target for pancreatic cancer therapy. Cancer Lett.
307:18–25. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pickel L, Matsuzuka T, Doi C, Ayuzawa R,
Maurya DK, Xie S-X, Berkland C and Tamura M: Overexpression of
angiotensin II type 2 receptor gene induces cell death in lung
adenocarcinoma cells. Cancer Biol Ther. 9:277–285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
George AJ, Thomas WG and Hannan RD: The
renin-angiotensin system and cancer: old dog, new tricks. Nat Rev
Cancer. 10:745–759. 2010.PubMed/NCBI
|
|
6
|
Wilop S, von Hobe S, Crysandt M, Esser A,
Osieka R and Jost E: Impact of angiotensin I converting enzyme
inhibitors and angiotensin II type 1 receptor blockers on survival
in patients with advanced non-small-cell lung cancer undergoing
first-line platinum-based chemotherapy. J Cancer Res Clin Oncol.
135:1429–1435. 2009. View Article : Google Scholar
|
|
7
|
Li H, Qi Y, Li C, Braseth LN, Gao Y,
Shabashvili AE, Katovich MJ and Sumners C: Angiotensin type 2
receptor-mediated apoptosis of human prostate cancer cells. Mol
Cancer Ther. 8:3255–3265. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Redondo-Müller MA, Stevanovic-Walker M,
Barker S, Puddefoot JR and Vinson GP: Anti-cancer actions of a
recombinant antibody (R6313/G2) against the angiotensin II AT1
receptor. Endocr Relat Cancer. 15:277–288. 2008.PubMed/NCBI
|
|
9
|
Nakai Y, Isayama H, Ijichi H, Sasaki T,
Sasahira N, Hirano K, Kogure H, Kawakubo K, Yagioka H, Yashima Y,
et al: Inhibition of renin-angiotensin system affects prognosis of
advanced pancreatic cancer receiving gemcitabine. Br J Cancer.
103:1644–1648. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Feng Y, Ni L, Wan H, Fan L, Fei X, Ma Q,
Gao B, Xiang Y, Che J and Li Q: Overexpression of ACE2 produces
antitumor effects via inhibition of angiogenesis and tumor cell
invasion in vivo and in vitro. Oncol Rep.
26:1157–1164. 2011.PubMed/NCBI
|
|
11
|
Okamoto K, Tajima H, Nakanuma S, Sakai S,
Makino I, Kinoshita J, Hayashi H, Nakamura K, Oyama K, Nakagawara
H, et al: Angiotensin II enhances epithelial-to-mesenchymal
transition through the interaction between activated hepatic
stellate cells and the stromal cell-derived factor-1/CXCR4 axis in
intrahepatic cholangiocarcinoma. Int J Oncol. 41:573–582. 2012.
|
|
12
|
Feng Y, Wan H, Liu J, Zhang R, Ma Q, Han
B, Xiang Y, Che J, Cao H, Fei X, et al: The angiotensin-converting
enzyme 2 in tumor growth and tumor-associated angiogenesis in
non-small cell lung cancer. Oncol Rep. 23:941–948. 2010.PubMed/NCBI
|
|
13
|
Ni L, Feng Y, Wan H, Ma Q, Fan L, Qian Y,
Li Q, Xiang Y and Gao B: Angiotensin-(1–7) inhibits the migration
and invasion of A549 human lung adenocarcinoma cells through
inactivation of the PI3K/Akt and MAPK signaling pathways. Oncol
Rep. 27:783–790. 2012.
|
|
14
|
Iwatsuki M, Mimori K, Yokobori T, Ishi H,
Beppu T, Nakamori S, Baba H and Mori M: Epithelial-mesenchymal
transition in cancer development and its clinical significance.
Cancer Sci. 101:293–299. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Puddefoot JR, Udeozo UK, Barker S and
Vinson GP: The role of angiotensin II in the regulation of breast
cancer cell adhesion and invasion. Endocr Relat Cancer. 13:895–903.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Uemura H, Ishiguro H, Nagashima Y, Sasaki
T, Nakaigawa N, Hasumi H, Kato S and Kubota Y: Antiproliferative
activity of angiotensin II receptor blocker through cross-talk
between stromal and epithelial prostate cancer cells. Mol Cancer
Ther. 4:1699–1709. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kanehira T, Tani T, Takagi T, Nakano Y,
Howard EF and Tamura M: Angiotensin II type 2 receptor gene
deficiency attenuates susceptibility to tobacco-specific
nitrosamine-induced lung tumorigenesis: involvement of transforming
growth factor-beta-dependent cell growth attenuation. Cancer Res.
65:7660–7665. 2005.
|
|
18
|
Tikellis C, Bernardi S and Burns WC:
Angiotensin-converting enzyme 2 is a key modulator of the
renin-angiotensin system in cardiovascular and renal disease. Curr
Opin Nephrol Hypertens. 20:62–68. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bangalore S, Kumar S, Kjeldsen SE, Makani
H, Grossman E, Wetterslev J, Gupta AK, Sever PS, Gluud C and
Messerli FH: Antihypertensive drugs and risk of cancer: network
meta-analyses and trial sequential analyses of 324,168 participants
from randomised trials. Lancet Oncol. 12:65–82. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pasternak B, Svanström H, Callréus T,
Melbye M and Hviid A: Use of angiotensin receptor blockers and the
risk of cancer. Circulation. 123:1729–1736. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chang CH, Lin JW, Wu LC and Lai MS:
Angiotensin receptor blockade and risk of cancer in type 2 diabetes
mellitus: a nationwide case-control study. J Clin Oncol.
29:3001–3007. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bhaskaran K, Douglas I, Evans S, van Staa
T and Smeeth L: Angiotensin receptor blockers and risk of cancer:
cohort study among people receiving antihypertensive drugs in UK
General Practice Research Database. BMJ. 344:e26972012. View Article : Google Scholar
|
|
23
|
Ferrario CM: ACE2: more of Ang-(1–7) or
less Ang II? Curr Opin Nephrol Hypertens. 20:1–6. 2011.
|
|
24
|
Gembardt F, Sterner-Kock A, Imboden H,
Spalteholz M, Reibitz F, Schultheiss HP, Siems WE and Walther T:
Organ-specific distribution of ACE2 mRNA and correlating peptidase
activity in rodents. Peptides. 26:1270–1277. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shenoy V, Ferreira AJ, Qi Y, Fraga-Silva
RA, Diez-Freire C, Dooies A, Jun JY, Sriramula S, Mariappan N,
Pourang D, et al: The angiotensin-converting enzyme
2/angiogenesis-(1–7)/Mas axis confers cardiopulmonary protection
against lung fibrosis and pulmonary hypertension. Am J Respir Crit
Care Med. 182:1065–1072. 2010.
|
|
26
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Moustakas A and Heldin CH: Induction of
epithelial-mesenchymal transition by transforming growth factor β.
Semin Cancer Biol. 22:446–454. 2012.
|
|
28
|
Perez RE, Navarro A, Rezaiekhaligh MH,
Mabry SM and Ekekezie II: TRIP-1 regulates TGF-β1-induced
epithelial-mesenchymal transition of human lung epithelial cell
line A549. Am J Physiol Lung Cell Mol Physiol. 300:L799–L807.
2011.PubMed/NCBI
|
|
29
|
Arnold SA, Rivera LB, Carbon JG, Toombs
JE, Chang CL, Bradshaw AD and Brekken RA: Losartan slows pancreatic
tumor progression and extends survival of SPARC-null mice by
abrogating aberrant TGFβ activation. PLoS One.
7:e313842012.PubMed/NCBI
|
|
30
|
Burns WC, Velkoska E, Dean R, Burrell LM
and Thomas MC: Angiotensin II mediates epithelial-to-mesenchymal
transformation in tubular cells by ANG 1-7/MAS-1-dependent
pathways. Am J Physiol Renal Physiol. 299:F585–F593. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rodrigues-Ferreira S, Abdelkarim M,
Dillenburg-Pilla P, Luissint AC, di-Tommaso A, Deshayes F, Pontes
CL, Molina A, Cagnard N, Letourneur F, et al: Angiotensin II
facilitates breast cancer cell migration and metastasis. PLoS One.
7:e356672012. View Article : Google Scholar : PubMed/NCBI
|