Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common cancer in the world (1).
Although numerous anticancer drugs have been used in the routine
clinical treatment of HCC and result in a reduction in tumor volume
at early stages, recurrence, the development of multidrug
resistance, toxicity and side-effects are unfortunately common in
patients. Therefore, there is a pressing need for new therapeutic
drugs with increased efficacy and decreased toxicity.
Cell cycle deregulation is a hallmark of tumor cells
and targeting the proteins that mediate critical cell cycle
processes is an emerging strategy for the treatment of cancer
(2). The G2/M checkpoint is the
most conspicuous target for several anticancer drugs (3,4).
CDK1/cyclin B1 and CDK1/cyclin A complexes play a key role in
promoting the G2/M phase transition. A number of proteins are known
to regulate the stepwise activation of CDK1, which controls the G2
to M transition. This process involves additional proteins,
including Wee1 (5), Myt1 (6) and Cdc25C (7). The phosphatase activity of Cdc25C is
inactivated by Chk1/Chk2, which are activated by ATM/ATR in
response to DNA damage (8,9). Activation of ATM/ATR initiates the
subsequent protein kinase cascade through both p53 dependent and
independent pathways. In the p53 dependent pathways, p53 is
phosphorylated on Ser15 and Ser20 and then activated downstream
target genes, such as p21 (10) and
14-3-3 (7), which play an important
role in G2/M checkpoint through inhibition of cyclin B1/Cdk1
(11–16). In p53 independent pathways, Chk1 and
Chk2 phosphorylate Cdc25c at Ser216, which downregulate Cdc25c
activity by promoting 14-3-3 protein and nuclear export. Chk/12
also phosphorylates wee1 and increases wee1 activity (8,12,17–22).
Dihydromyricetin (DHM) also known as Ampelopsin,
isolated from the tender stem and leaves of the plant species
Ampelopsis grossedentata, is one of the most common
flavonoids found in grapes, berries, fruits, vegetables, herbs and
other plants with certain anticancer activities. As the major
bioactive constituent of Ampelopsis grossedentata, DHM was
reported to possess numerous pharmacological activities, such as
anti-inflammatory (23),
antimicrobial activity, relieving cough, anti-oxidation (24), antihypertension as well as
hepatoprotective (25) and
anticarcinogenic effects. DHM was shown to possess certain
anticancer activities. It has been reported that DHM inhibits the
growth and metastasis in prostate cancer (26), lung cancer (27) and melanoma tumor (28,29).
DHM also possesses anti-angiogenesis activity by inhibiting the
secretion of vascular endothelial growth factor (VEGF) and basic
fibroblast growth factor (bFGF) from human HCC cells in
vitro and in mice (30). DHM
also reversed multidrug resistance in leukemia cells in
vitro in part via decreasing the expression of p-glycoprotein
(31). On the other hand, the
effect of DHM on the growth and progression of HCC is rarely
studied.
The objectives of the present study were to
systematically evaluate DHM as a potential chemopreventive and
therapeutic candidate against HCC progression, and to elucidate the
underlying cellular and molecular mechanisms of DHM actions. Our
results provided experimental evidence to support the future
development of DHM as an effective and safe candidate agent for the
prevention and/or therapy of HCC.
Materials and methods
Cell lines and cell culture
The human HCC cell lines HepG2, Hep3B and
immortalized human liver cell line L02 were provided by the Cell
Bank of the Institute of Biochemistry and Cell Biology at the China
Academy of Sciences (Shanghai, China). All cell lines were cultured
in RPMI-1640 medium (HyClone, Logan, UT, USA) containing 10%
heat-inactivated fetal bovine serum (FBS) (HyClone) and
supplemented with 100 IU/ml penicillin G and 100 μg/ml streptomycin
(HyClone). All cell lines were incubated at 37°C in a humidified
atmosphere with 5% CO2.
Drug stocks
DHM was purchased from Sigma-Aldrich and prepared at
a stock concentration of 50 mM in dimethyl sulfoxide (DMSO).
MTT assay
Cell toxicity and proliferation after DHM treatment
were determined using the MTT
[3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]
assay. Briefly, 5,000 cells/well were plated in triplicate in
96-well plates, and the cells were exposed to 2, 10, 50, 100 and
200 μM DHM for 48 h. The MTT reagent (Sigma-Aldrich) was prepared
at 5 mg/ml in PBS. This MTT stock solution was then added to each
well at a 1:10 dilution. Cells were incubated for 4 h and the
resulting crystals were dissolved in 100 μl DMSO (Sigma-Aldrich).
The absorbance at 492 nm was measured using a multiwell plate
reader. The inhibition rate was calculated as follows: Inhibition
rate = 1−A492 of treated cells/A492 of control cells.
Colony formation
To determine the frequency of colony formation,
HepG2 and Hep3B cells were plated in 6-well plates at
concentrations of 5×103 cells/ml in DMEM media
containing 2, 10, 50, 100 and 200 μM DHM and colonies were stained
with crystal violet and counted in triplicate wells after growth
for a further 2–3 weeks. DMSO was used as a negative control.
Cell cycle analysis
For cell cycle analysis, 2×105 cells were
plated in a 6-well culture plate and grown for 24 h. The cells were
then incubated with 1 mM thymidine (Sigma-Aldrich) for 24 h to
synchronize cells at the G1/S boundary. The cells were then treated
with fresh media containing 2, 10, 50, 100 and 200 μM DHM for 48 h.
Next, the cells were trypsinized, washed twice with cold PBS and
fixed with cold 70% ethanol at −4°C overnight. The cells were then
washed twice with PBS and incubated with 10 mg/ml RNase A, 400
mg/ml propidium iodide (Sigma-Aldrich) and 0.1% Triton-X in PBS at
room temperature (RT) for 30 min. Cells were subsequently analyzed
by flow cytometry.
Western blotting
At the end of the treatments, the HCC cells were
harvested and lysed with ice-cold cell lysis solution and the
homogenate was centrifuged at 10,000 × g for 15 min at 4°C. Total
protein in the supernatant was quantified using a BCA protein assay
kit. Total protein (30 μg) from each sample was separated by 12%
SDS-PAGE and transferred to a PVDF membrane which was placed in
washing buffer containing skimmed milk powder at room temperature,
blocked for 2 h, and washed 3 times. The indicated primary
antibodies, listed in Table I, were
added, respectively, and incubated at 4°C overnight. Then,
horseradish peroxidase-conjugated secondary antibody was added to
incubate for 1 h. X-ray film exposure was performed and AlphaImager
HP fluorescence/visible light gel imaging analyzer processing and
image analysis software were used to analyze gray value.
 | Table IAntibodies used in the present
study. |
Table I
Antibodies used in the present
study.
| Antibody | Cat. no., host | Dilution | Company |
|---|
| p-CDK1 | 4539, rabbit
polyclonal | 1:300 for WB | Cell Signaling |
| CDK1 | 9116, mouse mAb
IgG1 | 1:300 for WB | Cell Signaling |
| Cyclin antibody
sampler kit | 9869, rabbit
polyclonal | 1:300 for WB | Cell Signaling |
| Wee1 | 4936, abbit
polyclonal | 1:300 for WB | Cell Signaling |
| p53 | 9282, rabbit
polyclonal | 1:300 for WB | Cell Signaling |
| MDM2 | Ab137413, rabbit
polyclonal | 1:500 for WB | Abcam |
| p-MDM2 | 3521, rabbit
polyclonal | 1:300 for WB | Cell Signaling |
| Cdc25C | 4688, rabbit
polyclonal | 1:300 for WB | Cell Signaling |
| p-Cdc25C | 9528, rabbit
polyclonal | 1:300 for WB | Cell Signaling |
| Myt1 | 4282, rabbit
polyclonal | 1:300 for WB | Cell Signaling |
| p-Myt1 | 4281, rabbit
polyclonal | 1:300 for WB | Cell Signaling |
| p-Chk2 (ser
33/35) | 2665, rabbit
polyclonal | 1:300 for WB | Cell Signaling |
| p-Chk2 (Thr
68) | 2197, rabbit
polyclonal | 1:300 for WB | Cell Signaling |
| p-Chk2 (ser
19) | 2666, rabbit
polyclonal | 1:300 for WB | Cell Signaling |
| Chk2 | 3440, rabbit
polyclonal | 1:300 for WB | Cell Signaling |
| p-Chk1 (ser
296) | 2349, rabbit
polyclonal | 1:300 for WB | Cell Signaling |
| p-Chk1 (ser
317) | 8191, rabbit
polyclonal | 1:300 for WB | Cell Signaling |
| p-Chk1 (ser
345) | 2348, rabbit
polyclonal | 1:300 for WB | Cell Signaling |
| Chk1 | 2360, mouse
mAb | 1:300 for WB | Cell Signaling |
| β-actin | 8457, rabbit
polyclonal | 1:1,000 for WB | Cell Signaling |
RNA interference
Small-interfering RNA (siRNA) oligos for p53, Chk1,
Chk2 and general negative control with the sequences listed in
Table II, were synthesized and
annealed by GenePharma (Shanghai, China). Each siRNA duplex was
transfected into HepG2 using Lipofectamine® 2000
(Invitrogen) following the manufacturer’s protocol. siRNA-NC,
siRNA-NC-FAM and siRNA-GAPDH respectively served as negative
control, transfecting control and siRNA positive control targeting
GAPDH gene. Cells were exposed to 50 μM DHM after transfection and
harvested for indicated analysis.
| Table IIsiRNA sequences. |
Table II
siRNA sequences.
| Target name | Sequence |
|---|
| p53 |
5′-AAGACUCCAGUGGUAAUCUACTT-3′
(sense)
5′-GUAGAUUACCACUGGAGUCUUTT-3′ (antisense) |
| Chk1 |
5′-GACUGGGACUUGGUGCAAATT-3′
(sense)
5′-UUUGCACCAAGUCCCAGUCTT-3′ (antisense) |
| Chk2 |
5′-GUAAGAAAGUAGCCAUAAATT-3′
(sense)
5′-UUUAUGGCUACUUUCUUACTT-3′ (antisense) |
| Negative
control |
5′-UCCUCCGAACGUGUCACGUTT-3′
(sense)
5′-ACGUGACACGUUCGGAGAATT-3′ (antisense) |
| GAPDH positive
control |
5′-GUAUGACAACAGCCUCAAGTT-3′
(sense)
5′-CUUGAGGCUGUUGUCAUACTT-3′ (antisense) |
Statistical analysis
The data are presented as the mean ± standard
deviation (SD). Statistical analyses (two group comparisons) were
performed using the Student’s t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
DHM suppresses proliferation and colony
formation of HepG2 and Hep3B cells
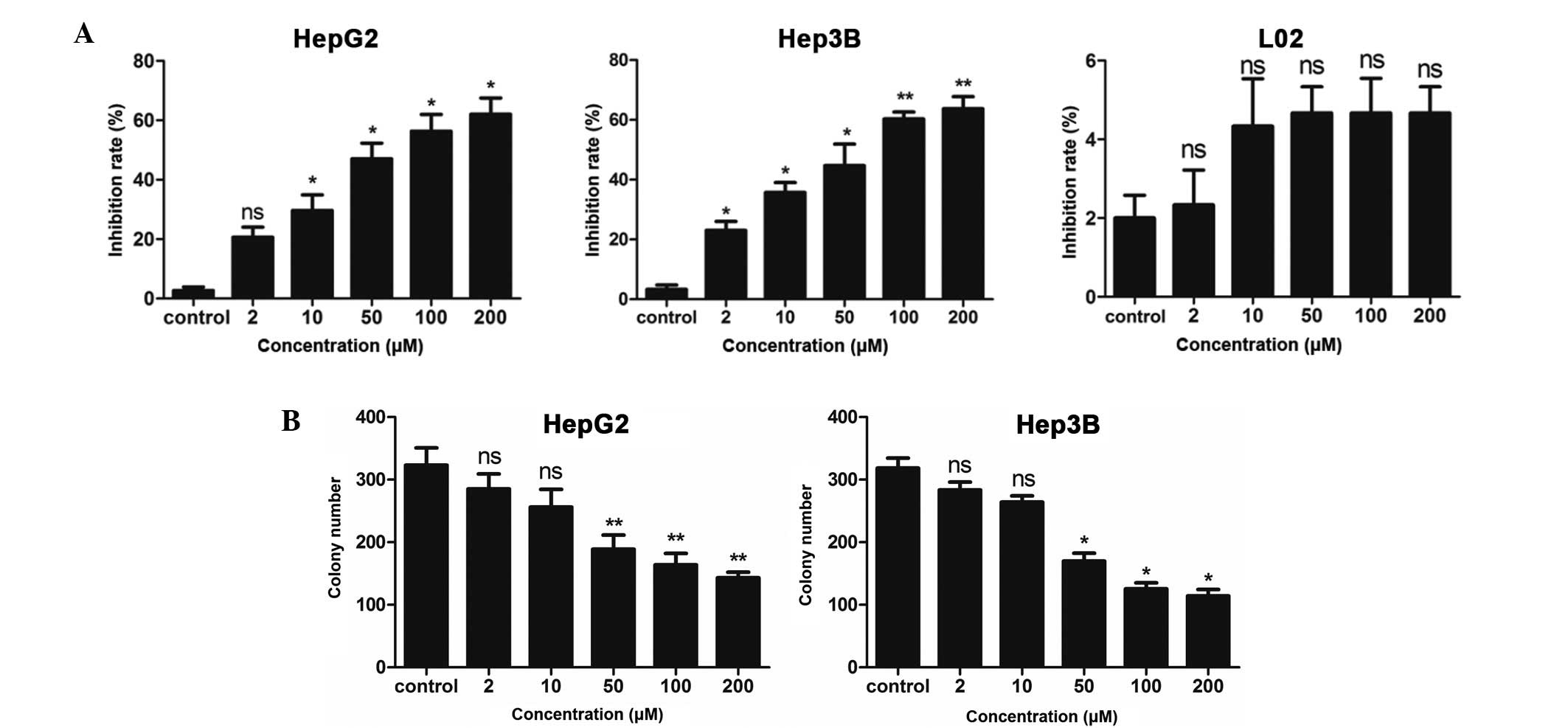
To investigate the suppressive growth effect of DHM,
the HCC cell lines HepG2 and Hep3B were incubated with 2, 10, 50,
100 and 200 μM DHM for 48 h. Cell proliferation was subsequently
measured by the MTT assay. Our results show that DHM inhibited the
growth of HepG2 and Hep3B cells in a dose-dependent manner
(Fig. 1A). To exclude the
possibility that cell death was due to drug toxicity, the effect of
DHM on the immortalized human liver cell line L02 was also
investigated. L02 cells were found to have a low sensitivity to DHM
treatment (Fig. 1A).
We also investigated whether DHM inhibited the
ability of HCC cells to initiate colonies on plastic. HepG2 and
Hep3B cells were treated with 2, 10, 50, 100 and 200 μM DHM. DMSO
was used as a negative control. HepG2 and Hep3B cells treated with
DHM showed a reduction in colony formation compared to those
treated with DMSO (Fig. 1B). These
results indicated DHM inhibited colony formation of HCC cell
lines.
DHM induces G2/M cell cycle arrest in
HepG2 and Hep3B cells
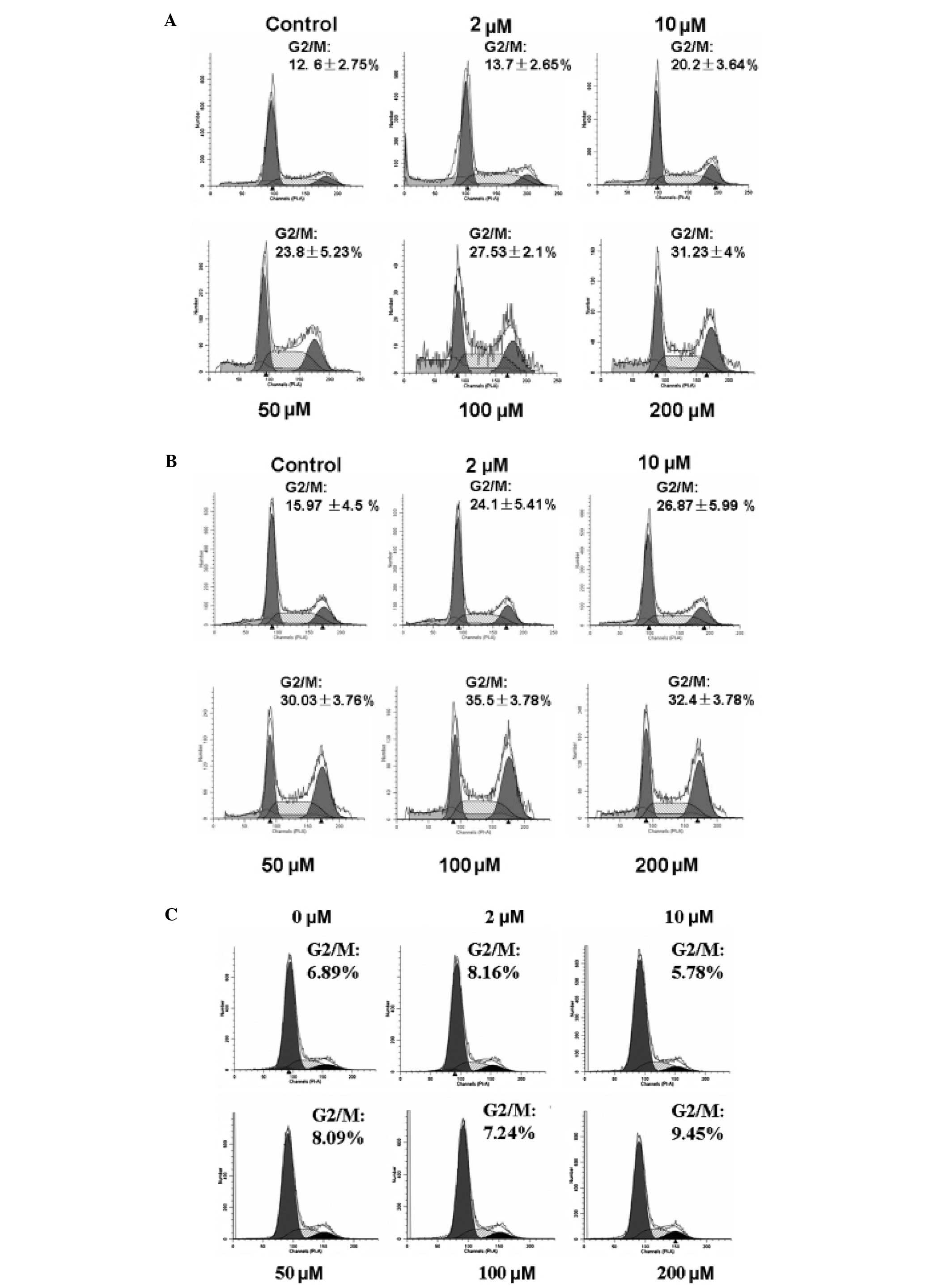
To further elucidate the inhibitory effects of DHM
on HCC cell growth, the cell cycle distributions of HepG2 and Hep3B
cells were determined by flow cytometry. Following treatment with 1
mM thymidine for 24 h to synchronize cells at the G1/S border, the
cells were incubated with 2, 10, 50, 100 and 200 μM DHM for 48 h. A
dose-dependent G2/M arrest in the cell cycle was observed in HepG2
and Hep3B cells after treatment with DHM. By contrast, G2/M arrest
was not observed in L02 cells treated with DHM for 48 h (Fig. 2).
DHM induces G2/M cell cycle arrest by
decreasing the activity of CDK1
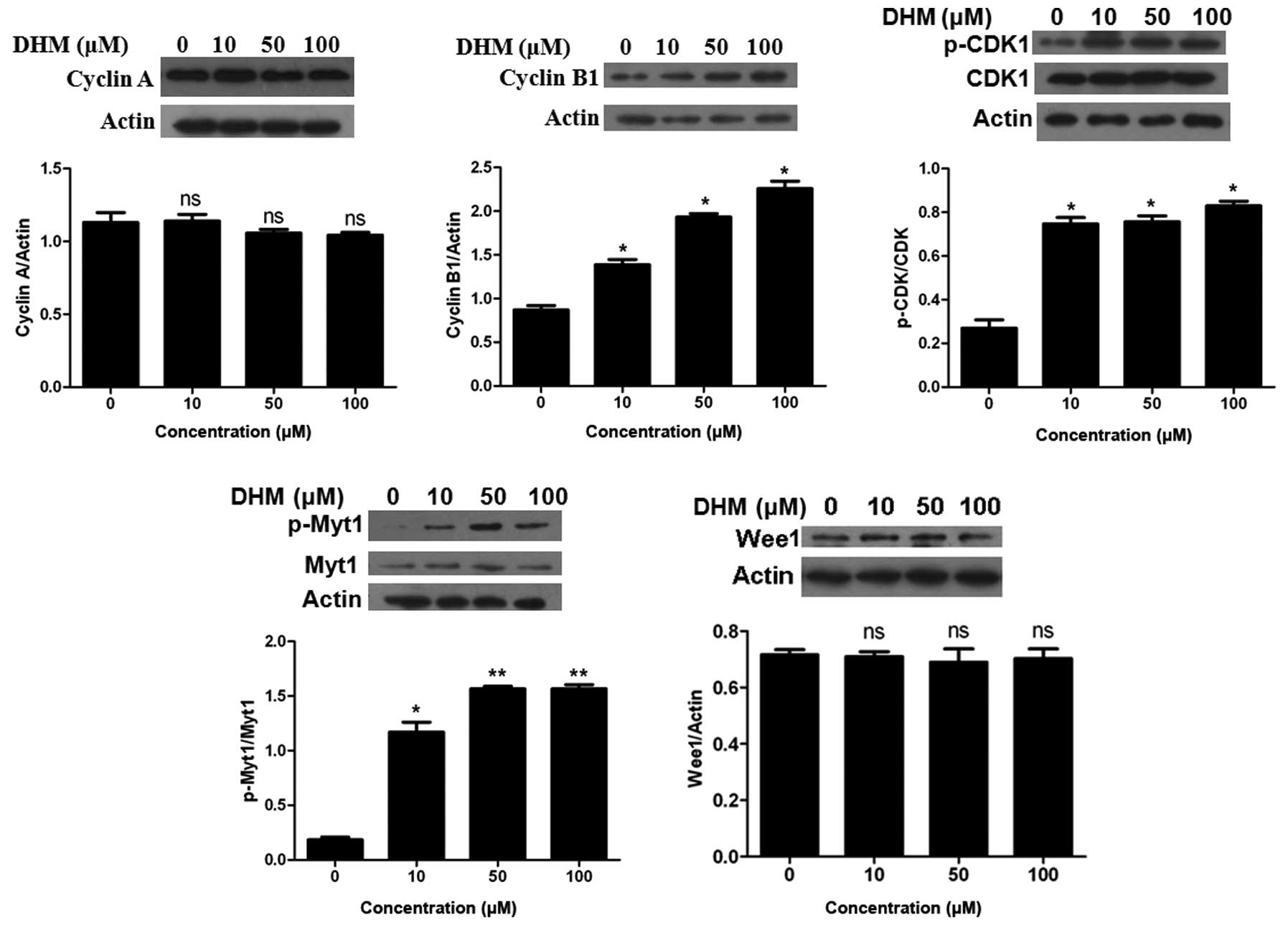
We next examined the expression of the key molecules
that promote the G2/M phase transition. The activation of CDK1 is
essential for cells to correctly enter the M phase (32). This process involves the formation
of a complex between CDK1 and cyclin B1 or cyclin A. Western blot
analysis showed that the expression level of cyclin A was not
influenced by DHM treatment. DHM increased the expression of cyclin
B1 and the inhibitory phosphorylation status of CDK1 (Tyr15) in a
dose-dependent manner (Fig. 3).
Since the accumulation of p-CDK1 (Tyr15) indicated the presence of
an inactive complex, our data suggested DHM inactivated the
CDK1/cyclin B1 complex.
Phosphorylation of CDK1 at Tyr15 and Thr14 sites is
known to be performed by the Wee1 and Myt1 protein kinases. We
observed an upregulation in the expression level of p-Myt1 protein
following DHM treatment in HepG2 cells; however, DHM did not affect
the expression level of Wee1 (Fig.
3).
DHM inactivates CDK1 independent of the
p53 pathway
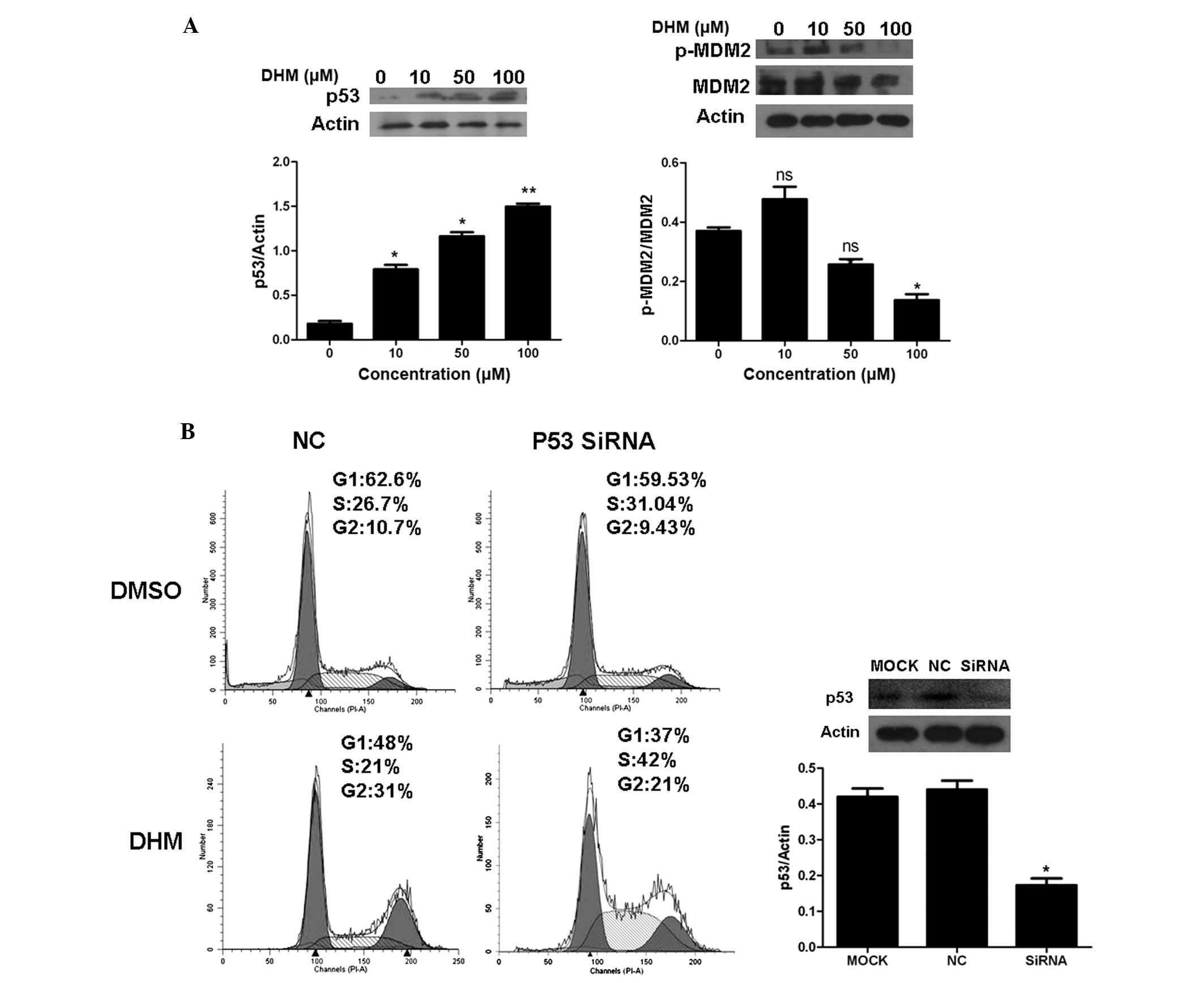
It is well known that CDK1/cyclin B1 complex can be
inactivated by the p53 pathways. Therefore, to elucidate whether
the p53 pathway is involved in the phosphorylation of CDK1 observed
in our experiments, we determined the levels of p53 by western
blotting. Our results indicated that DHM increased the protein
level of p53 and decreased p-MDM2 expression level; however, DHM
did not affect the expression level of MDM2 (Fig. 4A). To further determine the relative
contribution of p53 to DHM-induced G2/M arrest, HepG2 cells were
treated with DHM after transfection with either p53 siRNA or a
negative control. The efficiency of p53 siRNA was confirmed by
western blot analysis (Fig. 4B).
Cell cycle analysis showed that in the p53 knockdown HepG2 cells,
the G2/M percentage of negative control (NC), p53 siRNA was 31 and
21%, respectively, after DHM treatment (Fig 4B). The results suggest that p53 siRNA
does not disrupt the G2/M cell cycle arrest induced by DHM.
DHM inactivates CDK1 through the
Chk1/Chk2/Cdc25C pathway
The CDK1/cyclin B1 complex can be inactivated by the
Chk1/Chk2/Cdc25C pathways. The Cdc25C protein activates the cyclin
B1/CDK1 complex by dephosphorylating these inhibitory residues on
CDK1. Inactivated phosphatase activity of Cdc25C can contribute to
CDK inactivation. The phosphatase activity of Cdc25C is inactivated
by Chk1/Chk2, which are activated by ATM/ATR in response to DNA
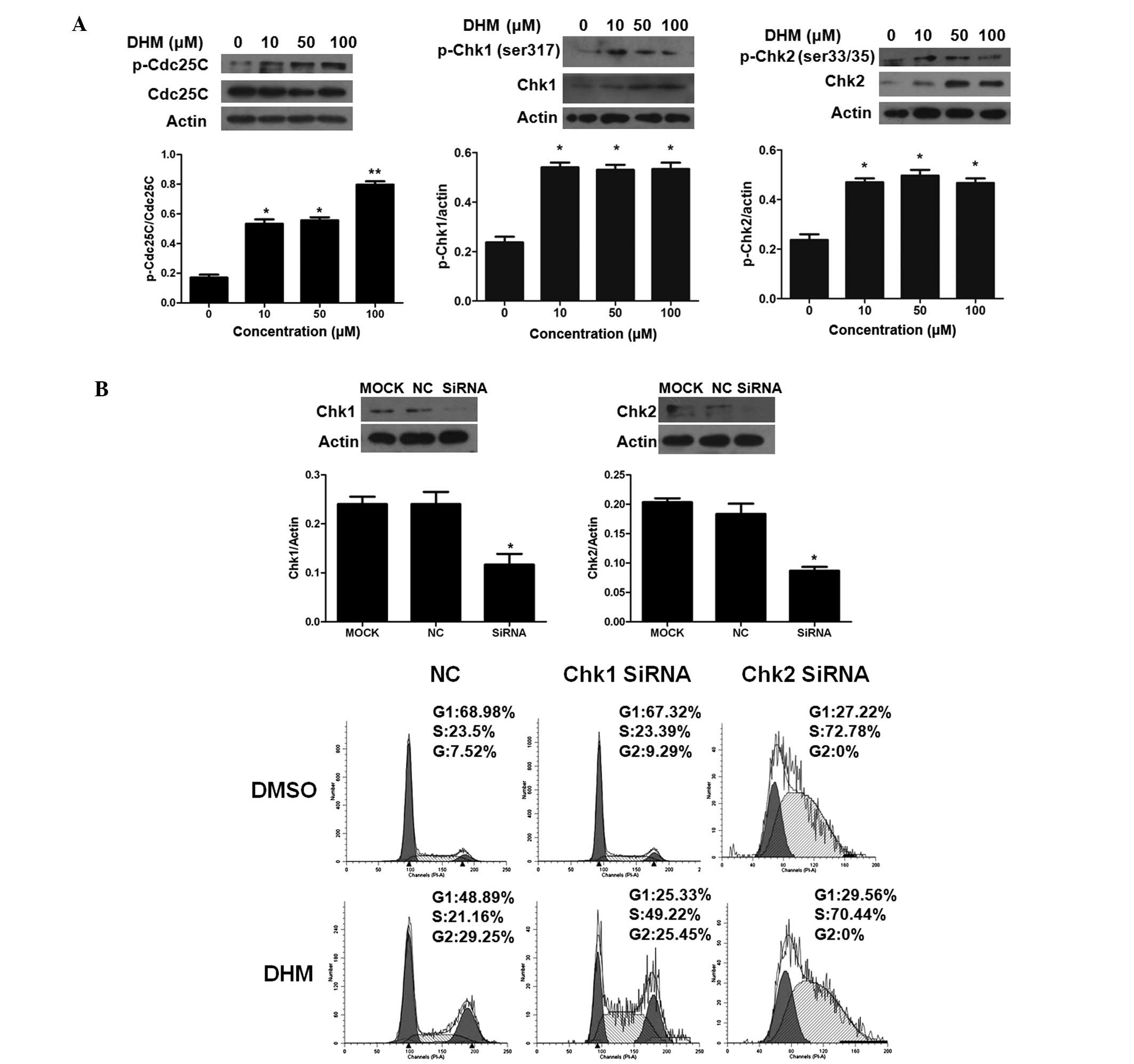
damage. We found that DHM treatment resulted in an increase in the
p-Cdc25C protein level and did not affect the total level of Cdc25C
(Fig. 5A). Therefore, these data
indicate that DHM may inactivate Cdc25C which leads to inactivation
of the CDK1/cyclin B1 complex.
In DHM-treated HepG2 cells, we observed an
upregulation of the phosphorylation of Chk1 (at Ser317) and
increased phosphorylation of Chk2 at Ser33/35, but it had no effect
on other phosphorylation sites within this protein (Fig. 5A). These results suggest that the
inactivation of CDK1 observed with DHM treatment is mainly induced
by Chk1- and Chk2-mediated phosphorylation of Cdc25C.
Since both Chk1 and Chk2 were phosphorylated after
DHM treatment, to further determine the relative contribution of
Chk1 and Chk2 to DHM-induced G2/M arrest, HepG2 cells were treated
with DHM after transfection with either Chk1/Chk2 siRNA or a
negative control. The efficiency of Chk1/Chk2 siRNA was confirmed
by western blot analysis (Fig. 5B).
Cell cycle analysis showed that in the Chk1 and Chk2 knockdown
HepG2 cells, the G2/M percentage of negative control (NC), Chk1
siRNA and Chk2 siRNA was 29.25, 25.45 and 0%, respectively, after
DHM treatment (Fig. 5B). The
results suggest that Chk2 siRNA disrupts the G2/M cell cycle
arrest, while the negative control or Chk1 do not.
Discussion
The Chinese herb Ampelopsis grossedentata is
widely distributed in South China and is used to treat cold and
tinea corporis. It contains a rich resource of phytochemicals with
dihydromyricetin (DHM), a naturally occurring flavonoid found in
grapes, berries, fruits, vegetables, herbs and other plants with
certain anticancer activities. As the major bioactive constituent
of Ampelopsis grossedentata, DHM has been shown to be mainly
responsible for the reported biological activities, including
hypoglycemic (33), anti-oxidative
(24) and hepatoprotective
activities (25). DHM also enhanced
the chemokinesis and chemotaxis effects of neutrophilic
granulocytes and monocytes (23).
To investigate the antitumor effect of DHM in
hepatocellular carcinoma (HCC), the HCC cell lines HepG2, Hep3B and
the human liver cell line L02 were exposed to DHM for 48 h. In the
present study, DHM treatment resulted in a clear inhibition of
proliferation at a relatively low concentration in HCC cell lines
(Fig. 1). By contrast, L02 cells
were found to be markedly resistant to this compound. In L02 cells,
the observed inhibitory rate was less than 5% (Fig. 1), indicating that DHM may be less
toxic to L02 cells than to cancer cells. Therefore, DHM may not
exhibit toxicity in experimental animals.
To investigate the mechanism behind the antitumoral
properties of DHM, cell cycle analysis was performed. DHM induced
G2/M phase arrest in HepG2 and Hep3B cells but not in L02 cells
(Fig. 2). Cell cycle deregulation
is an important mechanism to modulate HCC cell proliferation. Cell
cycle progression is tightly regulated by cyclin/cyclin-dependent
kinase (Cdks) complexes. For instance, cyclin D/Cdk4 and Cdk6 drive
the sequential progression from G1 to S phase (34,35);
cyclin A/Cdk2 and Cdc2 (Cdk1) complexes control the S and G2 phases
(36); and cyclin B/Cdk1 complex
drives the G2/M transition as well as processes during mitosis
(32). The G2/M checkpoint allows
the cell to repair DNA damage before entering mitosis. The stepwise
activation of CDK1 is essential for cells to correctly enter the M
phase (32). This process involves
the formation of a complex between CDK1 and cyclin B1 or cyclin A.
According to our results, DHM treatment increased expression levels
of cyclin B1 and Cdk1. The expression level of cyclin A was not
influenced by DHM treatment. These findings indicate DHM might
induce G2/M cell cycle arrest by increasing the level of inactive
cyclin B1/Cdk1 complex (Fig.
3).
Numerous proteins are known to regulate the
activation of CDK1 including Wee1 (5), Myt1 (6) and Cdc25C. CDK1 is subsequently
activated via a Cdk-activating enzyme, which phosphorylates the
activating residues on CDK1. Inhibitory phosphorylation can also be
performed at Thr160/161, Thr14 and Tyr15 by Wee1 (5) and Myt1 (6). The phosphatase Cdc25C, by contrast,
can dephosphorylate Thr14 and Tyr15 (37). According to our results, DHM
treatment induced a pronounced G2/M phase arrest by increasing the
level of cyclin B1 as well as the accumulation of
Thr14/Tyr15-phosphorylated CDK1. DHM induced Myt 1 upregulation may
contribute to the accumulation of Thr14/Tyr15-phosphorylated CDK1
(Fig. 3).
It is known that the initial activation of cyclin
B1/Cdk1 also involves Cdk1 dephosphorylation at Thr14 and Tyr15 by
Cdc25C (38). Decreased Cdc25C
phosphatase activity can lead to inactivation of cyclin B1/Cdk1
(19). In the present study, DHM
treatment led to an upregulation of Ser216-phosphorylated Cdc25C
(Ser216) which downregulates Cdc25c activity and leads to
accumulation of Thr14/Tyr15-phosphorylated CDK1. Our data,
therefore, suggest that DHM inactivates the CDK1/cyclin B1 complex
by inactivating Cdc25C.
The phosphatase activity of Cdc25C is inactivated by
Chk1/Chk2, which are activated by ATM/ATR in response to DNA damage
(13,15,16,19).
These kinases are activated upon DNA damage, which results in the
inactivation of Cdc25C. Chk1 is activated by phosphorylation at
Ser317, Ser345 and Ser296, while Chk2 is activated at Ser33/35,
Ser516, Ser296 and Thr68 (39,40).
In the present study, p-Chk1 (Ser317 and Ser345) and p-Chk2
(Ser33/35) were upregulated after DHM treatment. To determine the
relative contributions of these proteins on the DHM-induced cell
cycle arrest, we analyzed the cell cycle distribution of
siRNA-mediated Chk1 or Chk2 knockdown HepG2 cells after treatment
with DHM or DMSO (Fig. 5). The
results showed that Chk2 knockdown disrupts the G2/M arrest
compared with NC or Chk1. These results indicate that DHM treatment
activates Chk1 and Chk2, allowing these kinases to inactivate
Cdc25C. Cdc25C, Wee1 and Myt1 then decrease the activity of the
CDK1/cyclin B1 complex, resulting in an arrest of the cell cycle at
the G2/M phase.
The activation of CDK1/cyclin B1 can also be
prevented by p53. Our results indicate that DHM increased the
protein level of p53 and decreased p-MDM2 expression level;
however, DHM did not affect the expression level of MDM2 (Fig. 4). To further determine the relative
contribution of p53 to DHM-induced G2/M arrest, HepG2 cells were
treated with DHM after transfection with either p53 siRNA or a
negative control. The efficiency of p53 siRNA was confirmed by
western blot analysis (Fig. 4B).
Cell cycle analysis showed that in the p53 knockdown HepG2 cells,
the G2/M percentage of negative control (NC), p53 siRNA was 31 and
21%, respectively, after DHM treatment (Fig. 4B). The results suggest that p53
siRNA does not disrupt the G2/M cell cycle arrest induced by
DHM.
In conclusion, our present study demonstrated that
DHM inhibited HCC cell growth through G2/M phase cell cycle arrest
dependent on the Chk1/Chk2/Cdc25C pathway. Based on the present
study, DHM could be a potential agent against HCC development.
Future studies on the effects of DHM on detailed mechanisms of cell
cycle arrest and other signal pathways for in vitro cell
lines and in vivo animal models are required to further
elucidate the detailed mechanism(s) of action of DHM on HCC
chemoprevention.
Acknowledgements
The authors are grateful to Dr Qitao Yan for the
technical assistance. The present study was supported by grants
from the Science and Technology program of Guangdong Province
(2008B030301028) and the Science and Technology Innovation Fund of
the Guangdong Medical College (STIF201107).
References
|
1
|
Jemal A, Siegel R, Ward E, et al: Cancer
statistics, 2008. CA Cancer J Clin. 58:71–96. 2008. View Article : Google Scholar
|
|
2
|
Stewart ZA, Westfall MD and Pietenpol JA:
Cell-cycle dysregulation and anticancer therapy. Trends Pharmacol
Sci. 24:139–145. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huang WW, Ko SW, Tsai HY, et al:
Cantharidin induces G2/M phase arrest and apoptosis in human
colorectal cancer colo 205 cells through inhibition of CDK1
activity and caspase-dependent signaling pathways. Int J Oncol.
38:1067–1073. 2011.
|
|
4
|
Visanji JM, Thompson DG and Padfield PJ:
Induction of G2/M phase cell cycle arrest by carnosol and carnosic
acid is associated with alteration of cyclin A and cyclin B1
levels. Cancer Lett. 237:130–136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Parker LL and Piwnica-Worms H:
Inactivation of the p34cdc2-cyclin B complex by the human WEE1
tyrosine kinase. Science. 257:1955–1957. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ruiz EJ, Vilar M and Nebreda AR: A
two-step inactivation mechanism of Myt1 ensures CDK1/cyclin B
activation and meiosis I entry. Curr Biol. 20:717–723. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Peng CY, Graves PR, Thoma RS, Wu Z, Shaw
AS and Piwnica-Worms H: Mitotic and G2 checkpoint control:
regulation of 14-3-3 protein binding by phosphorylation of Cdc25C
on serine-216. Science. 277:1501–1505. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ahn J and Prives C: Checkpoint kinase 2
(Chk2) monomers or dimers phosphorylate Cdc25C after DNA damage
regardless of threonine 68 phosphorylation. J Biol Chem.
277:48418–48426. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Turowski P, Franckhauser C, Morris MC,
Vaglio P, Fernandez A and Lamb NJ: Functional cdc25C
dual-specificity phosphatase is required for S-phase entry in human
cells. Mol Biol Cell. 14:2984–2998. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Choi YH, Lee WH, Park KY and Zhang L:
p53-independent induction of p21 (WAF1/CIP1), reduction of cyclin
B1 and G2/M arrest by the isoflavone genistein in human prostate
carcinoma cells. Jpn J Cancer Res. 91:164–173. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rief N, Herges H, Prowald A, Gotz C and
Montenarh M: Binding of the growth suppressor p53 protein to the
cell cycle regulator phosphatase cdc25C. Int J Oncol. 17:189–195.
2000.PubMed/NCBI
|
|
12
|
St Clair S, Giono L, Varmeh-Ziaie S, et
al: DNA damage-induced downregulation of Cdc25C is mediated by p53
via two independent mechanisms: one involves direct binding to the
cdc25C promoter. Mol Cell. 16:725–736. 2004.PubMed/NCBI
|
|
13
|
Hirao A, Kong YY, Matsuoka S, et al: DNA
damage-induced activation of p53 by the checkpoint kinase Chk2.
Science. 287:1824–1827. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ye R, Bodero A, Zhou BB, Khanna KK, Lavin
MF and Lees-Miller SP: The plant isoflavenoid genistein activates
p53 and Chk2 in an ATM-dependent manner. J Biol Chem.
276:4828–4833. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Canman CE, Lim DS, Cimprich KA, et al:
Activation of the ATM kinase by ionizing radiation and
phosphorylation of p53. Science. 281:1677–1679. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Banin S, Moyal L, Shieh S, et al: Enhanced
phosphorylation of p53 by ATM in response to DNA damage. Science.
281:1674–1677. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dalal SN, Schweitzer CM, Gan J and
DeCaprio JA: Cytoplasmic localization of human cdc25C during
interphase requires an intact 14-3-3 binding site. Mol Cell Biol.
19:4465–4479. 1999.PubMed/NCBI
|
|
18
|
Graves PR, Lovly CM, Uy GL and
Piwnica-Worms H: Localization of human Cdc25C is regulated both by
nuclear export and 14-3-3 protein binding. Oncogene. 20:1839–1851.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Savitsky PA and Finkel T: Redox regulation
of Cdc25C. J Biol Chem. 277:20535–20540. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bulavin DV, Demidenko ZN, Phillips C,
Moody SA and Fornace AJ Jr: Phosphorylation of Xenopus Cdc25C at
Ser285 interferes with ability to activate a DNA damage replication
checkpoint in pre-midblastula embryos. Cell Cycle. 2:263–266. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Perdiguero E and Nebreda AR: Regulation of
Cdc25C activity during the meiotic G2/M transition. Cell Cycle.
3:733–737. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tyagi A, Singh RP, Agarwal C, Siriwardana
S, Sclafani RA and Agarwal R: Resveratrol causes Cdc2-tyr15
phosphorylation via ATM/ATR-Chk1/2-Cdc25C pathway as a central
mechanism for S phase arrest in human ovarian carcinoma Ovcar-3
cells. Carcinogenesis. 26:1978–1987. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zeng S, Luo GQ and Liu DY: The chemotaxis
effect of ampelopsin on the immunocytes. Zhong Yao Cai. 29:260–262.
2006.(In Chinese).
|
|
24
|
He G, Du F, Yang W, Pei G and Zhu Y:
Effects of tengcha flavonoids on scavenging oxygen free radicals
and inhibiting lipid-peroxidation. Zhong Yao Cai. 26:338–340.
2003.(In Chinese).
|
|
25
|
Murakami T, Miyakoshi M, Araho D, et al:
Hepatoprotective activity of tocha, the stems and leaves of
Ampelopsis grossedentata, and ampelopsin. Biofactors. 21:175–178.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ni F, Gong Y, Li L, Abdolmaleky HM and
Zhou JR: Flavonoid ampelopsin inhibits the growth and metastasis of
prostate cancer in vitro and in mice. PLoS One. 7:e388022012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zeng S, Liu D, Ye Y, Wang L and Wang W:
Antitumor effects of ampelopsin on human lung cancer GLC-82
implanted in nude mice. Zhong Yao Cai. 27:842–845. 2004.(In
Chinese).
|
|
28
|
Liu D and Luo M: Study on inhibitory
effect of ampelopsin on melanoma by serologic pharmacological
method. Zhong Yao Cai. 24:348–350. 2001.(In Chinese).
|
|
29
|
Liu DY, Zheng HQ and Luo GQ: Effects of
ampelopsin on invasion and metastasis of B16 mouse melanoma in vivo
and in vitro. Zhongguo Zhong Yao Za Zhi. 28:957–961. 2003.(In
Chinese).
|
|
30
|
Luo GQ, Zeng S and Liu DY: Inhibitory
effects of ampelopsin on angiogenesis. Zhong Yao Cai. 29:146–150.
2006.(In Chinese).
|
|
31
|
Ye J, Zheng Y and Liu D: Reversal effect
and its mechanism of ampelopsin on multidrug resistance in K562/ADR
cells. Zhongguo Zhong Yao Za Zhi. 34:761–765. 2009.(In
Chinese).
|
|
32
|
Nurse P: Universal control mechanism
regulating onset of M-phase. Nature. 344:503–508. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhong ZX, Qin JP, Zhou GF and Chen XF:
Experimental studies of hypoglycemic action on total flavone of
Ampelopsis grossedentata from Guangxi. Zhongguo Zhong Yao Za
Zhi. 27:687–689. 2002.(In Chinese).
|
|
34
|
Lukas J, Bartkova J, Rohde M, Strauss M
and Bartek J: Cyclin D1 is dispensable for G1 control in
retinoblastoma gene-deficient cells independently of cdk4 activity.
Mol Cell Biol. 15:2600–2611. 1995.PubMed/NCBI
|
|
35
|
Wang Z, Xie Y, Zhang L, et al: Migratory
localization of cyclin D2-Cdk4 complex suggests a spatial
regulation of the G1-S transition. Cell Struct Funct. 33:171–183.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Goldstone S, Pavey S, Forrest A, Sinnamon
J and Gabrielli B: Cdc25-dependent activation of cyclin A/cdk2 is
blocked in G2 phase arrested cells independently of ATM/ATR.
Oncogene. 20:921–932. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Galaktionov K, Lee AK, Eckstein J, et al:
CDC25 phosphatases as potential human oncogenes. Science.
269:1575–1577. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Karlsson C, Katich S, Hagting A, Hoffmann
I and Pines J: Cdc25B and Cdc25C differ markedly in their
properties as initiators of mitosis. J Cell Biol. 146:573–584.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li J and Stern DF: Regulation of CHK2 by
DNA-dependent protein kinase. J Biol Chem. 280:12041–12050. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Peng A and Chen PL: NFBD1, like 53BP1, is
an early and redundant transducer mediating Chk2 phosphorylation in
response to DNA damage. J Biol Chem. 278:8873–8876. 2003.
View Article : Google Scholar : PubMed/NCBI
|