Introduction
The hypothesis that chronic inflammation may
increase the risk of cancer development and progression has arisen
from a variety of cancer models (1). Inflammation is usually a host defense
against invading microbial pathogens, tissue destruction and injury
or cancer. A quintessential signaling mechanism of inflammation
uses the Toll-like receptor (TLR) family, which is a highly
conserved family of transmembrane proteins that recognize a range
of microbial agents as well as endogenous macromolecules released
by injured tissue (2). TLRs play a
crucial role in the innate immune response and the subsequent
induction of the adaptive immune response (3,4). TLRs
are expressed by immune cells and also on cancer cells. Activation
of TLRs in cancer cells may promote cancer progression,
anti-apoptotic activity and resistance to host immune responses
(2,5). To date, considerable efforts have been
directed towards developing immunochemotherapeutic regimens based
on natural or synthetic TLR agonists (6).
Head and neck squamous cell carcinoma (HNSCC) is the
sixth most common cancer worldwide, affecting 600,000 new patients
each year. In the United States, 50,000 new cases are diagnosed,
and nearly 10,000 deaths are attributable to this disease, annually
(7). Despite advances in
multimodality therapy, the overall 5-year survival rate is 40–50%,
and has increased only incrementally in the past two decades
(8,9). It has been reported that TLR4
expression is associated with progression in a variety of head and
neck cancers, and that stimulation of HNSCC cells with
lipopolysaccharide (LPS), a TLR4 ligand, potently induces
interleukin-6 (IL-6), IL-8, vascular endothelial growth factor
(VEGF) and granulocyte macrophage colony-stimulating factor
(GM-CSF) expression (10–12).
Rapamycin, which specifically inhibits the mammalian
target of rapamycin (mTOR), has shown promising results in
preclinical and clinical trials in a variety of solid tumors
(13–16). Rapamycin exerts a potent antitumor
effect, leading to a rapid decrease in tumor vascularity and
increased cell death, thus provoking tumor regression (17). Rapamycin derivatives also diminish
microscopic residual disease and enhance the effectiveness of
epidermal growth factor receptor (EGFR) inhibitors in experimental
models of squamous cell carcinoma (18–20).
Notably, rapamycin does not exert significant growth suppressive or
proapoptotic activities in HNSCC cells in vitro; however,
in vivo rapamycin inhibits hypoxia inducible factor (HIF-1α)
expression and vascular endothelial growth factor (VEGF) production
(10), suggesting that the
antitumor effects of rapamycin may result from an effect on
TLR4-induced tumor promotion or protection. It had been reported
that rapamycin significantly inhibits TLR4-triggered NF-κB
activation, IL-6 and prostaglandin E2 (PGE2) production and
invasion of colon cancer cells (21). The starting part of the digestive
tract in head and neck (i.e. oral cavity and pharyngeal cavity) and
the ending part of the digestive tract in colon were believed to
share some similarity of features concerning the diversity and
temporal stability of selected bacterial groups (22). It was reasonable to hypothesize that
TLR4 plays similar roles in the development and progression of the
head neck cancer and colon cancer in the similar bacterial
environments. In this study, focusing on TLR4-induced pro-oncogenic
effects, we investigated whether rapamycin exerts efforts
regulating TLR4-induced pro-oncogenic effects on HNSCC, and our
results demonstrated inhibitory effects of rapamycin on
TLR4-induced HNSCC proliferation, migration, invasion, tumor
necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced
apoptosis resistance and signal transduction using the TLR4 ligand
LPS, suggesting that rapamycin has the ability to attenuate
TLR4-induced pro-oncogenic effects on HNSCC.
Materials and methods
Reagents
LPS and rapamycin were obtained from Sigma (St.
Louis, MO, USA). The M-PER® Mammalian Protein Extraction
Reagent, NE-PER Nuclear and Cytoplasmic Extraction Reagents, BCATM
Protein Assay Kit and SuperSignal West Femto Maximum Sensitivity
Substrate were purchased from Pierce (Rockford, IL, USA). Lamin A
polyclonal antibody, NF-κB p65, COX-2, TLR4, phosphor-extracellular
signal regulated kinase p38 (Thr180/Tyr182), I-κB (Ser32) and
IKKα/β monoclonal antibodies were obtained from Cell Signaling
(Beverly, MA, USA). ELISA kits for IL-6, PGE2, VEGF and TGFβ were
purchased from R&D Systems (Minneapolis, MN, USA).
Cell culture
The human HNSCC lines SCC4 and CAL27 (ATCC,
Manassas, VA, USA) were cultured as previously described (23). Both cell lines were obtained from
SCC of the tongue. CAL27 was established in 1982 from tissue taken
prior to treatment from a 56-year-old male. SCC4 was first reported
in 1981 from tissue of a 55-year-old male. Briefly, the cells were
maintained in RPMI-1640 media containing 10% fetal calf serum (FCS;
Gibco, Grand Island, NY, USA), 0.29 mg/ml glutamine, 100 IU/ml
penicillin and 100 IU/ml streptomycin at 37°C in a humidified
atmosphere containing 5% CO2.
MTT assay
For the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay, HNSCC cells were seeded at 2,000 cells/well in 96-well
plates, cultured for 12 h, serum starved overnight, pretreated with
2, 5, 10 ng/ml rapamycin or PBS, then stimulated with 1 μg/ml LPS
or PBS for 48 h. Cell growth was assessed using the MTT assay
(Promega, Madison, WI, USA) following the manufacturer’s
instructions. Each experiment consisted of three to four replicate
wells for each dose, and each experiment was performed at least in
triplicate.
TRAIL-induced apoptosis assay
For TRAIL-induced apoptosis assay, HNSCC cells were
cultured in 24-well plates, pretreated with 2, 5, 10 ng/ml
rapamycin or PBS for 4 h, stimulated with 1 μg/ml LPS for 12 h,
followed by treatment with 20 μg/ml TRAIL for 20 h. The cells were
stained with FITC-Annexin V/PI and assayed by fluorescence
activated cell sorting (FACS). The results represent the percentage
of Annexin V positive cells in the total cell number.
Cell migration and invasion assays
In vitro migration and invasion assays were
performed using Matrigel-coated (for invasion) or uncoated (for
migration) 5-μm pore size polycarbonate filters in 24-well
chambers, according to the manufacturer’s instructions (BD
Biosciences, Bedford, MA, USA). Briefly, after rehydration of the
chambers, 1×105 HNSCC cells were seeded onto the upper
chamber in 200 μl RPMI-1640 media containing 5% FBS. The lower
chamber was filled with 600 μl RPMI-1640 media containing 10% FBS
and the cells were stimulated with 1 μg/ml LPS in the presence of
2, 5, 10 ng/ml of rapamycin or absence of rapamycin. After 24 h,
HNSCC cells which had migrated through the membrane were stained
using leucocrystal violet and the number of cells on the membrane
was counted in 20 randomly selected ×100 fields of view.
siRNA
siRNA targeting TLR4 and duplex control were from
Santa Cruz Inc. (Santa Cruz, CA, USA). The sense and antisense
strands of siRNA were: 5′-GUCUAGUGGCUAAUUCCUA-3′ and
5′-UAGGAAUUAGCCACUAGAC-3′. Transfection was performed as
recommended by the siRNA manufacturer. The negative control
consisted of siRNA with no homology to known sequences from humans.
Cells were incubated in complete DMEM medium at 37°C in an
atmosphere of 5% CO2. For transient transfection of
siRNA in CAL27 and SCC4 cells, the INTERFERin® reagent
was used (Polyplus-transfection Co., Illkirch, France). CAL27 and
SCC4 cells were pre-transfected with TLR4-specific siRNA or
negative control (Si-NC) for 36 h before further experiments were
carried out.
Western blotting
Cells were cultured in 6-well plates, pretreated
with 10 ng/ml rapamycin or PBS for 4 h, then stimulated with 1
μg/ml LPS for the indicated time and cytosolic and nuclear extracts
were prepared and subjected to western blotting. Cytosolic and
nuclear extracts were subjected to western blotting for IκBα,
IKKβ/α, COX2 and HIF-1α, and nuclear extracts were subjected to
western blotting for p65. For caspase 8 (p20) activation, cells
were pretreated with 10 ng/ml rapamycin or PBS for 4 h and then
stimulated with 1 μg/ml LPS for 12 h, followed by treatment with 20
ng/ml TRAIL for 20 h.
Cells were lysed in lysis buffer, separated using
SDS-PAGE gels and transferred to nitrocellulose membranes (Amersham
Biosciences, Piscataway, NJ, USA) using standard protocols. The
membranes were blocked and probed with primary antibodies, then
incubated with HRP-labeled secondary antibody (DAKO, Carpinteria,
CA, USA) and color was visualized using the TMB Membrane Peroxidase
Substrate (KPL, Gaithersburg, MD, USA).
Statistical analysis
Data are represented as the mean ± standard
deviation (SD) of more than three independent experiments.
Statistical analysis was performed using Student’s t-test and
P-values <0.05 were considered to indicate statistically
significant differences.
Results
Rapamycin inhibits LPS-induced CAL27 and
SCC4 cell proliferation and resistance to apoptosis
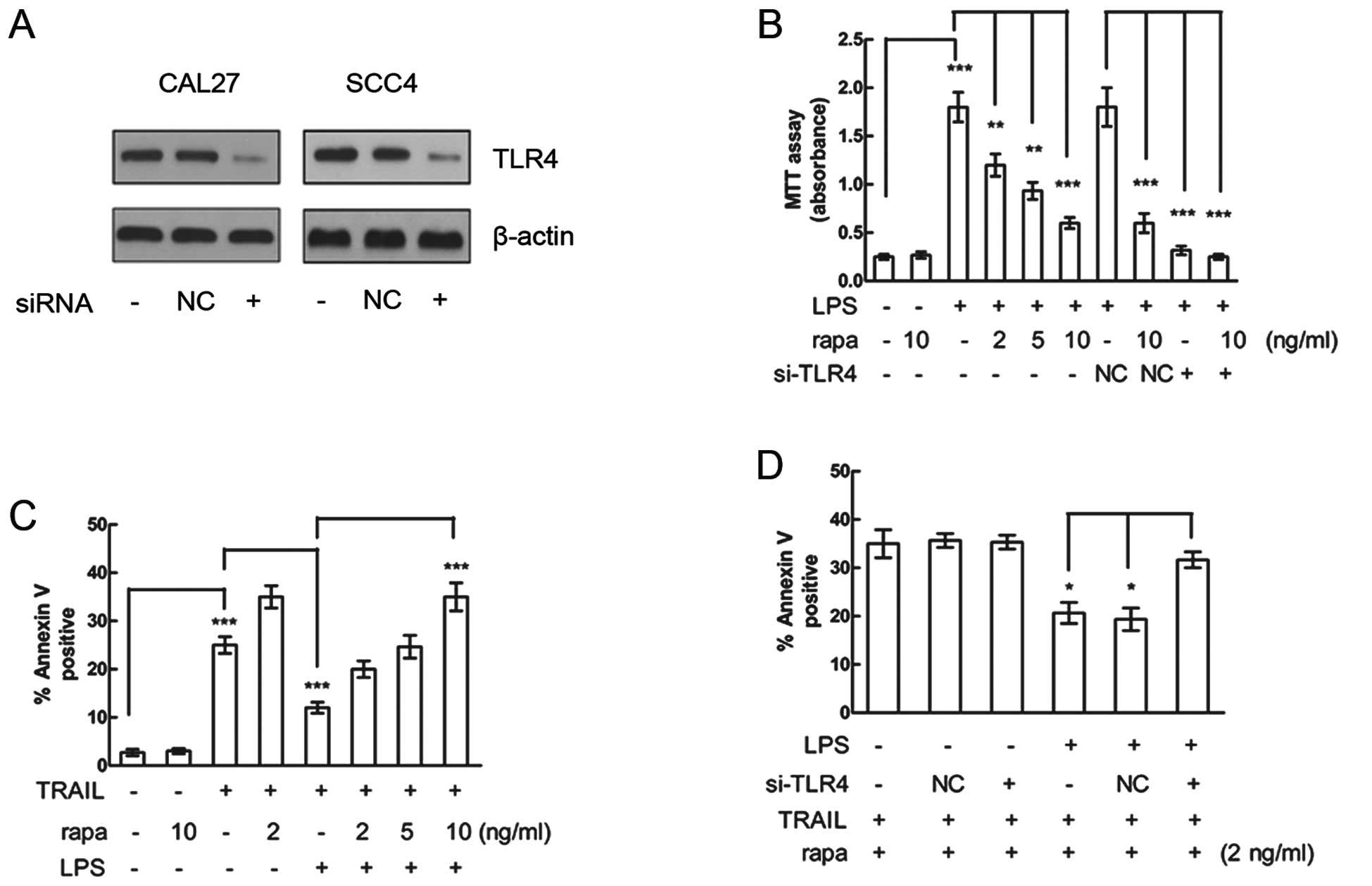
The effect of TLR4 knockdown was determined by
western blot analysis in both HNSCC cell lines treated with LPS
(Fig. 1A). Although rapamycin
exerts a potent antitumor effect in a variety of HNSCC xenograft
models, the results of in vitro studies using the same HNSCC
cell lines do not reflect the effect of rapamycin in vivo
(10). To address this discrepancy,
we pretreated the HNSCC cell lines CAL27 and SCC4 with various
concentrations of rapamycin for 4 h, then stimulated the cells with
the TLR4 ligand LPS for 48 h and measured proliferation using the
MTT assay. While LPS significantly promoted both CAL27 (Fig. 1B) and SCC4 (data not shown) cell
proliferation, rapamycin significantly inhibited LPS-induced
proliferation of both cell lines in a dose-dependent manner.
Moreover, LPS-induced proliferation was decreased by TLR4 siRNA
compared with that of the NC siRNA in CAL27 cells (Fig. 1B), whereas rapamycin alone had no
effect on the proliferation of cells not induced by LPS or treated
by TLR4 siRNA (Fig. 1B). These
results suggest that rapamycin can inhibit LPS-induced
proliferation and the effect was mediated by TLR4.
Resistance to apoptosis is an important feature of
HNSCC. Next, we measured TRAIL-induced apoptosis in HNSCC cell line
to investigate the effect of LPS and rapamycin on apoptosis in
HNSCC. CAL27 cells were pretreated with rapamycin, treated with LPS
and then stimulated with TRAIL to induce apoptosis. LPS treatment
significantly inhibited TRAIL-induced apoptosis in CAL27 cells
(Fig. 1C), while rapamycin
attenuated the LPS-mediated resistance to TRAIL-induced apoptosis
in a dose-dependent manner.
To examine whether the rapamycin-attenuated
resistance to TRAIL-induced apoptosis was mediated by TLR4, CAL27
cells were pretreated by TLR4 siRNA. The siRNA knockdown of TLR4
decreased the effect of LPS on resistance to TRAIL-induced
apoptosis. Rapamycin alone exerted no effect of attenuation of
resistance to TRAIL-induced apoptosis on cells which had not been
induced by LPS or had been treated with TLR4 siRNA (Fig. 1D). These results suggest that the
attenuation effect of rapamycin on the resistance to TRAIL-induced
apoptosis was mediated by TLR4.
Rapamycin attenuates LPS-induced CAL27
and SCC4 cell invasion and migration
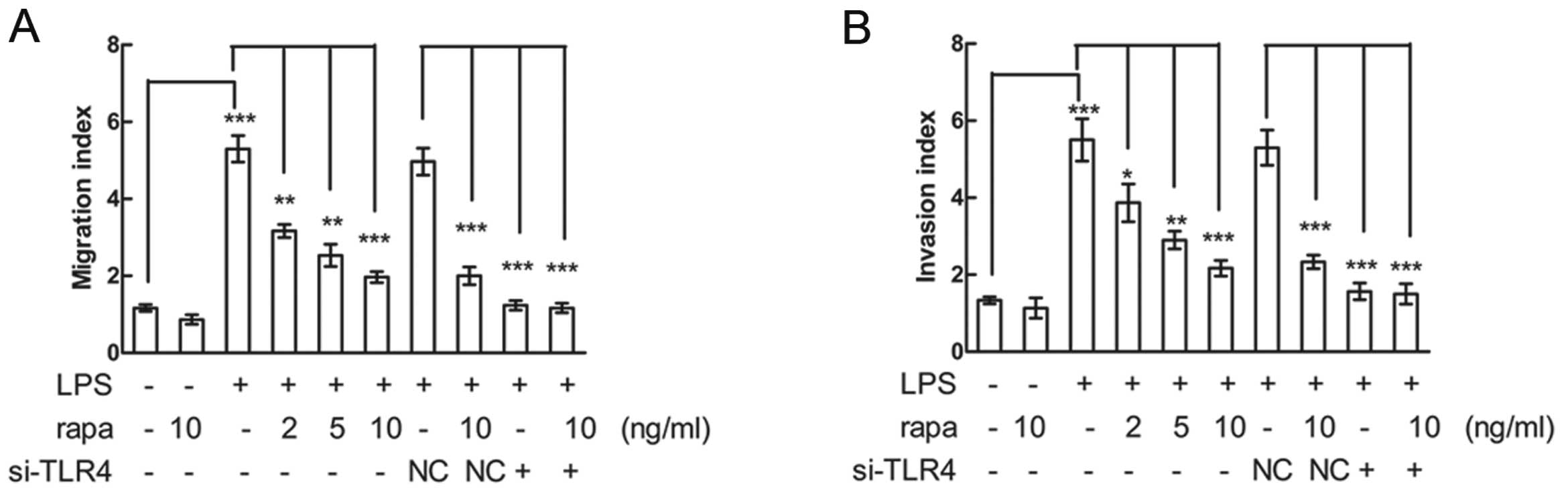
To investigate whether LPS promotes HNSCC migration
and invasion, and to determine whether rapamycin affects
LPS-induced migration and invasion, CAL27 and SCC4 cells were
pretreated with various concentrations of rapamycin, stimulated
with LPS and then cell migration and invasion were assayed. LPS
promoted CAL27 and SCC4 migration and invasion, while rapamycin
significantly inhibited LPS-induced migration and invasion in a
dose-dependent manner. However, siRNA knockdown of TLR4 decreased
the LPS-induced migration and invasion, and rapamycin alone had no
effects of inhibition on cell migration or invasion in both CAL27
(Fig. 2A and B) and SCC4 (data not
shown) cells. These results suggest that rapamycin attenuated the
LPS-induced CAL27 and SCC4 cell migration and invasion and the
effects were mediated by TLR4.
Rapamycin reduces LPS-induced cytokine
production
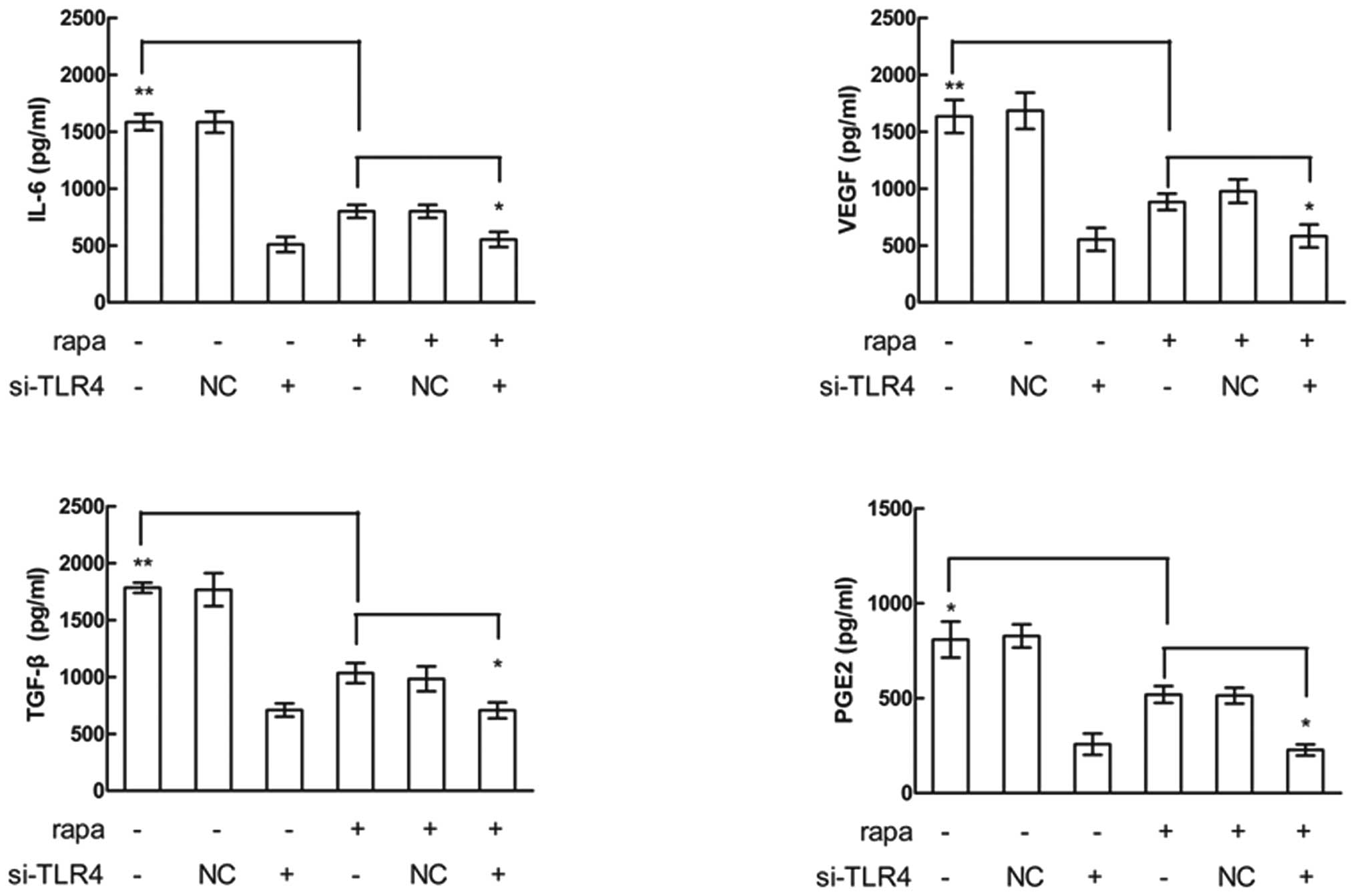
Cytokines secreted by tumor cells may contribute to
immune suppression and angiogenesis, which may promote tumor
survival and metastasis. LPS stimulates the expression of factors
related to inflammation and tumor immune escape, including IL-6,
IL-8, VEGF and prostaglandin E2 (PGE2) in a variety of cancer cells
(10–12). To investigate whether rapamycin has
an effect on LPS-induced cytokine production in HNSCC, CAL27
(Fig. 3) and SCC4 (data not shown)
cells were pretreated with rapamycin, stimulated with LPS and
cytokine production was measured using ELISAs. LPS-induced
expression of IL-6, VEGF, transformation growth factor-β (TGF-β)
and PGE2, and pre-treatment with rapamycin inhibited the
LPS-induced upregulation of these cytokines. However, siRNA
knockdown of TLR4 decreased the LPS-induced upregulation of the
cytokines, and rapamycin alone had no effects on cytokine
expression inhibition after the cells had been pretreated by TLR4
siRNA. These results suggested that rapamycin reduced LPS-induced
cytokine production and the effect was mediated by TLR4.
Rapamycin inhibits LPS-induced NF-κB
activation
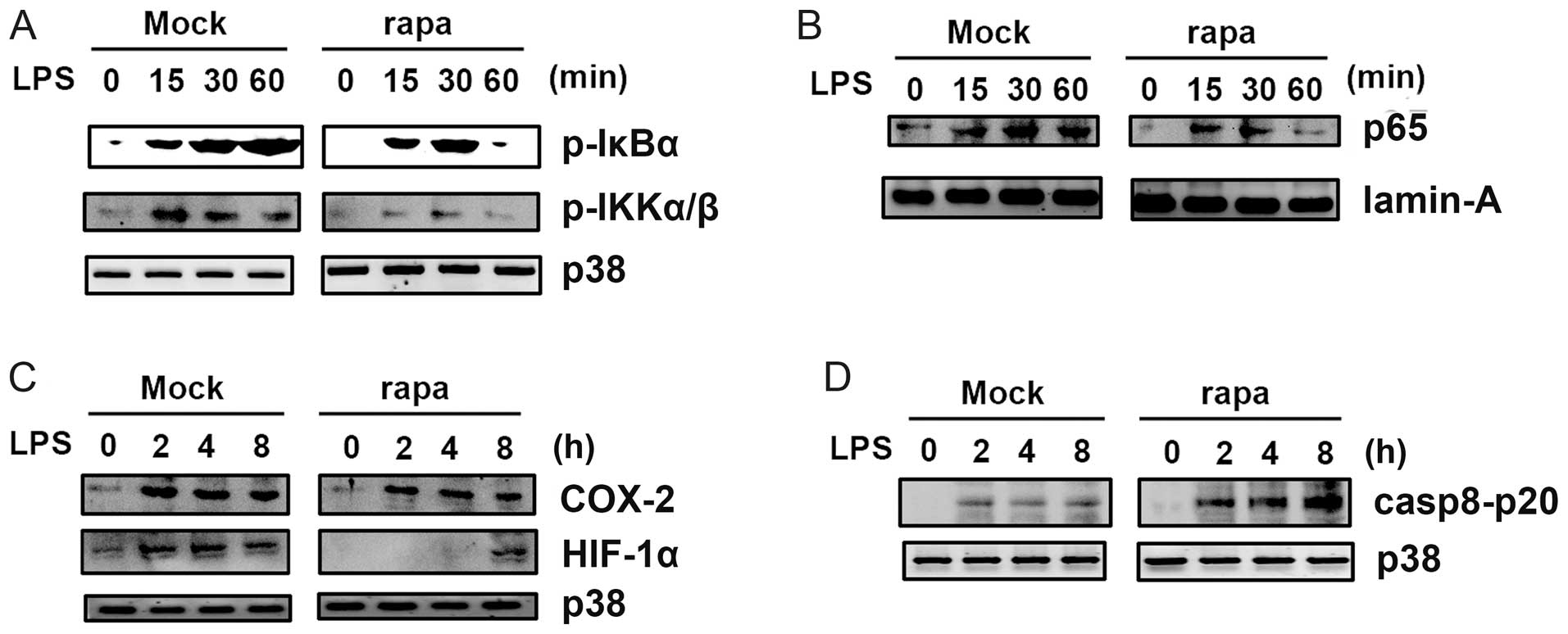
To further investigate the mechanism by which
rapamycin inhibited LPS-induced effects, we analyzed LPS-induced
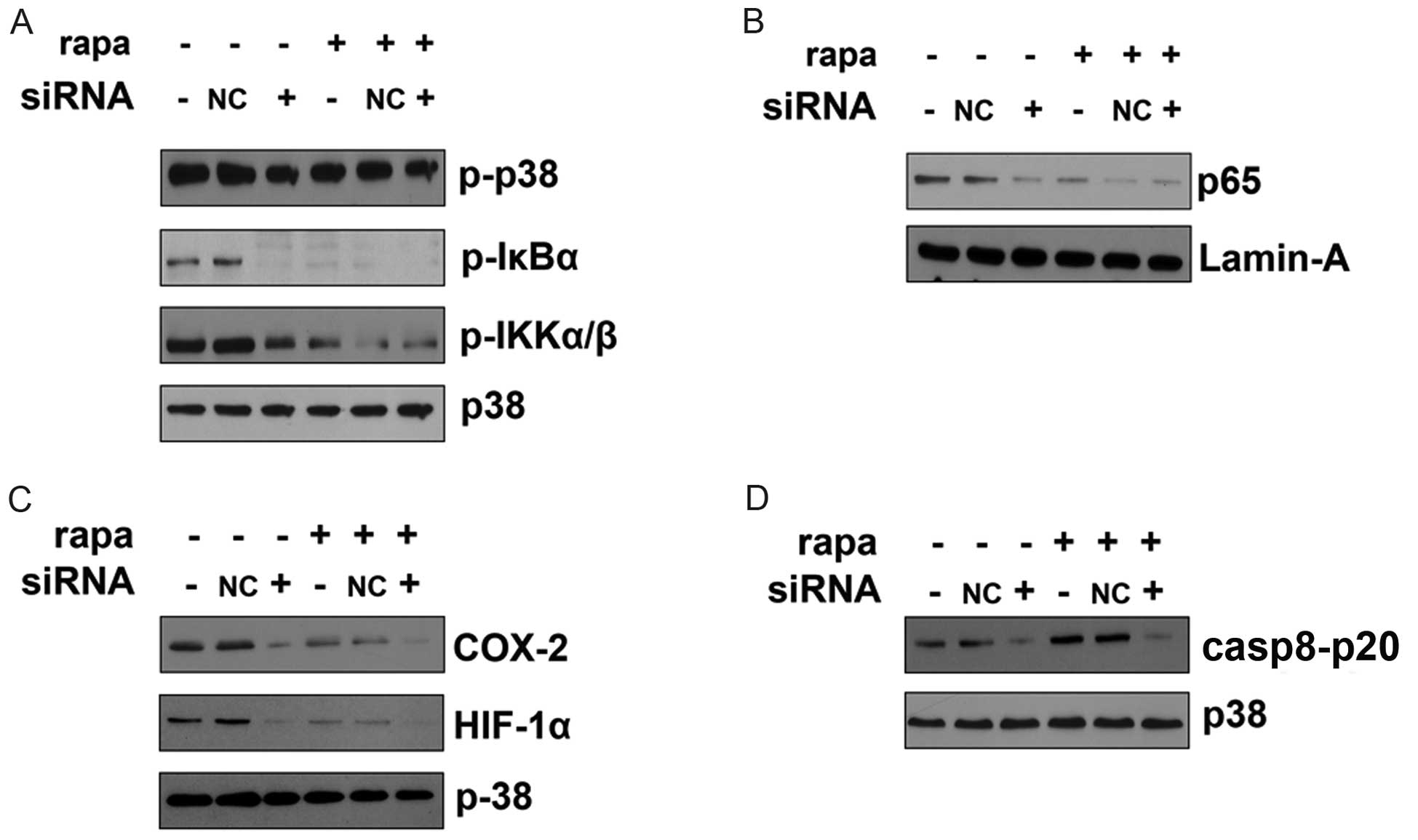
signal transduction in CAL27 cells (Fig. 4). Pretreatment with rapamycin
significantly inhibited LPS-induced activation of NF-κB IKKα/β and
IκBα (Fig. 4A). Rapamycin also
reduced nuclear translocation of the p65 subunit of NF-κB (Fig. 4B), which may account for the
decreased levels of cytokines induced by LPS in the presence of
rapamycin. LPS-induced upregulation of cyclooxygenase 2 (COX2) and
HIF-1α was also inhibited by rapamycin (Fig. 4C), which may lead to decreased
activation of NF-κB (21).
Rapamycin increased caspase 8 activation, as the p20 form of
caspase 8 was upregulated in cells pretreated with rapamycin in the
TRAIL-induced apoptosis model (Fig.
4D). Collectively, these results indicate that rapamycin
inhibits LPS-induced NF-κB activation; rapamycin attenuates
LPS-induced COX2 and HIF-1α upregulation; rapamycin also increases
LPS-induced caspase 8 activation in the TRAIL-induced apoptosis
model.
Rapamycin inhibits LPS-induced signal
transduction via TLR4
In order to further examine whether
rapamycin-inhibited LPS-induced NF-κB activation was mediated by
TLR4, CAL27 cells were transfected with TLR4-specific siRNA or a
control siRNA (NC siRNA). TLR4 siRNA decreased LPS-induced
activation of NF-κB pathways compared with the negative control
siRNA (NC siRNA). Rapamycin alone did not affect the expressions of
NF-κB pathway-related downstream proteins (Fig. 5A and B). The LPS-induced COX2 and
HIF-1α upregulation were also decreased by TLR4-specific silencing.
Rapamycin alone did not affect the protein expression of COX2 and
HIF-1α when TLR4 was silenced (Fig.
5C). The activation of p20 form of caspase 8 by LPS was
decreased in CAL27 cells when TLR4 was silenced. Rapamycin alone
did not affect expression of the p20 form of caspase 8 in CAL27
cells in the TRAIL-induced apoptosis model when TLR4 was silenced
(Fig. 5D). Hence, rapamycin
inhibits LPS-induced signal transduction via TLR4.
Discussion
TLR4 is expressed in HNSCC cells and the expression
level is correlated with tumor progression and tumor grade
(10,11,24).
The mRNA and protein of TLR4 were highly expressed in HNSCC cell
lines, while the expression of TLR4 in a human immortalized oral
epithelial cell line (HIOEC) (25),
which had been obtained from normal oral mucosa immortalized by
transfection of HPV16 E6/E7 gene, was very low (24). Previous studies of TLR4 and HNSCC
suggested that TLR4 may be functionally important in HNSCC
cells.
In contrast to the protective role of the TLRs in
pathogen infection, our study suggests that TLR4 expression in
HNSCC cells contributes to tumor growth and progression in
vitro. Consistent with other tumor types (11), we demonstrated that rapamycin
inhibits oncogenic effects induced by the TLR4 ligand LPS in
vitro, indicating that TLR4 signaling may the target of
rapamycin in HNSCC in vivo xenograft models.
Rapamycin inhibits FKBP12 from binding to the
atypical serine/threonine kinase mTOR, leading to inhibition of
mTOR and reducing the translation of many mRNA transcripts whose
protein products drive cell growth and cell cycle progression
(17). Despite marked pharmacologic
activity of rapamycin in a variety of experimental cancer models,
its anti-proliferative effect in cancer cells in vitro is
often limited. For example, rapamycin does not significantly affect
survival or growth in HNSCC cells in vitro (10). This raises the possibility that
rapamycin may not target proliferation directly and may instead
interact with other signaling pathways such as the TLRs in HNSCC
cells, as reported in colon and lung cancer cells in vitro
(21,26). Our in vitro results indicate
that rapamycin inhibits LPS-induced HNSCC cell proliferation,
apoptotic resistance, migration, invasion, cytokine production and
signal transduction. As LPS is a ligand of the TLR4 receptor, these
results indicate that antitumor effects of rapamycin may be
mediated via TLR4-signaling in HNSCC cells. This hypothesis was
further supported by our results of siRNA knockdown of TLR4.
The effects of LPS on tumor cell survival and
proliferation have been reported previously, although the results
depend on the tumor type tested (27,28).
Stimulation of TLR4 by LPS was shown to induce tumor progression,
by increasing proliferation and activating NF-κB, p65 nuclear
translocation, upregulating COX2 and HIF-1α and increasing
production of the proinflammatory cytokines IL-6, VEGF, TGFβ and
PGE2 in HNSCC or other types of cancer cells (11,29),
and we observed similar effects in LPS-treated HNSCC cell lines in
this study. NF-κB, p65, COX2, HIF-1α and proinflammatory cytokines
promote tumor progression and the development of myeloid-derived
suppressor cells (MDSC), and, in turn, MDSC may induce chronic
inflammation and lead to immune suppression through activation of
regulatory T cells.
Our observations in HNSCC cells show that
LPS-induced cell proliferation, migration, invasion and resistance
to TRAIL-mediated apoptosis was correlated with increased nuclear
translocation of the NF-κB p65 subunit. Activated NF-κB has
anti-apoptotic properties, and high levels of NF-κB activation in
tumor cells are associated with tumor progression and induction of
chronic inflammation in the tumor microenvironment. Apart from LPS,
there were also many types of endogenous ligands for TLR4, such as
S100A and HMGB1 (2,30). The other endogenous ligands could
promote tumor growth and progression via activation of TLR4 in
situ, and the manner may be similar to LPS.
In accordance with previous reports (21,31),
our study demonstrates that rapamycin can inhibit LPS-induced NF-κB
activation and TLR4 plays a pivotal role. However, the underlying
mechanism requires further examination.
In summary, this study indicates that rapamycin can
inhibit TLR4-signaling-induced proliferation, invasion, migration,
TRAIL-induced apoptosis resistance, NF-κB activation in HNSCC
cells. Our findings suggest that rapamycin may be efficient in the
treatment of HNSCC by attenuation of TLR4-induced pro-oncogenic
effects.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (No. 31140007, 81302351),
Shanghai Leading Academic Discipline Project (No. S30206) and
Jiangsu Provincial Natural Science Foundation (No. BK2012075, BK
20131080)
References
|
1
|
Elinav E, Nowarski R, Thaiss CA, et al:
Inflammation-induced cancer: crosstalk between tumours, immune
cells and microorganisms. Nat Rev Cancer. 13:759–771. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ridnour LA, Cheng RY, Switzer CH, et al:
Molecular pathways: toll-like receptors in the tumor
microenvironment - poor prognosis or new therapeutic opportunity.
Clin Cancer Res. 19:1340–1346. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Akira S and Takeda K: Toll-like receptor
signalling. Nat Rev Immunol. 4:499–511. 2004. View Article : Google Scholar
|
|
4
|
Karin M, Lawrence T and Nizet V: Innate
immunity gone awry: linking microbial infections to chronic
inflammation and cancer. Cell. 124:823–835. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Belmont L, Rabbe N, Antoine M, et al:
Expression of TLR9 in tumor-infiltrating mononuclear cells enhances
angiogenesis and is associated with a worse survival in lung
cancer. Int J Cancer. 765–777. 2013.PubMed/NCBI
|
|
6
|
Vacchelli E, Eggermont A, Sautes-Fridman
C, et al: Trial Watch: Toll-like receptor agonists for cancer
therapy. Oncoimmunology. 2:e252382013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rothenberg SM and Ellisen LW: The
molecular pathogenesis of head and neck squamous cell carcinoma. J
Clin Invest. 122:1951–1957. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bonner JA, Harari PM, Giralt J, et al:
Radiotherapy plus cetuximab for squamous-cell carcinoma of the head
and neck. N Engl J Med. 354:567–578. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Posner MR, Hershock DM, Blajman CR, et al:
Cisplatin and fluorouracil alone or with docetaxel in head and neck
cancer. N Engl J Med. 357:1705–1715. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Amornphimoltham P, Patel V,
Leelahavanichkul K, et al: A retroinhibition approach reveals a
tumor cell-autonomous response to rapamycin in head and neck
cancer. Cancer Res. 68:1144–1153. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Szczepanski MJ, Czystowska M, Szajnik M,
et al: Triggering of Toll-like receptor 4 expressed on human head
and neck squamous cell carcinoma promotes tumor development and
protects the tumor from immune attack. Cancer Res. 69:3105–3113.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mao L, Hong WK and Papadimitrakopoulou VA:
Focus on head and neck cancer. Cancer Cell. 5:311–316. 2004.
View Article : Google Scholar
|
|
13
|
Easton JB and Houghton PJ: mTOR and cancer
therapy. Oncogene. 25:6436–6446. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Faivre S, Kroemer G and Raymond E: Current
development of mTOR inhibitors as anticancer agents. Nat Rev Drug
Discov. 5:671–688. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jimeno A, Rudek MA, Kulesza P, et al:
Pharmacodynamic-guided modified continuous reassessment
method-based, dose-finding study of rapamycin in adult patients
with solid tumors. J Clin Oncol. 26:4172–4179. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Decaens T, Luciani A, Itti E, et al: Phase
II study of sirolimus in treatment-naive patients with advanced
hepatocellular carcinoma. Dig Liver Dis. 44:610–616. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang S, Bjornsti MA and Houghton PJ:
Rapamycins: mechanism of action and cellular resistance. Cancer
Biol Ther. 2:222–232. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Molinolo AA, Hewitt SM, Amornphimoltham P,
et al: Dissecting the Akt/mammalian target of rapamycin signaling
network: emerging results from the head and neck cancer tissue
array initiative. Clin Cancer Res. 13:4964–4973. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Le Tourneau C, Faivre S and Siu LL:
Molecular targeted therapy of head and neck cancer: review and
clinical development challenges. Eur J Cancer. 43:2457–2466.
2007.PubMed/NCBI
|
|
20
|
Dobashi Y, Suzuki S, Matsubara H, et al:
Critical and diverse involvement of Akt/mammalian target of
rapamycin signaling in human lung carcinomas. Cancer. 115:107–118.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun Q, Liu Q, Zheng Y, et al: Rapamycin
suppresses TLR4-triggered IL-6 and PGE(2) production of colon
cancer cells by inhibiting TLR4 expression and NF-kappaB
activation. Mol Immunol. 45:2929–2936. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maukonen J, Mätto J, Suihko ML, et al:
Intra-individual diversity and similarity of salivary and faecal
microbiota. J Med Microbiol. 57:1560–1568. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wong TS, Liu XB, Wong BY, et al: Mature
miR-184 as potential oncogenic microRNA of squamous cell carcinoma
of tongue. Clin Cancer Res. 14:2588–2592. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sun Z, Luo Q, Ye D, et al: Role of
toll-like receptor 4 on the immune escape of human oral squamous
cell carcinoma and resistance of cisplatin-induced apoptosis. Mol
Cancer. 11:332012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhong LP, Yang X, Zhang L, et al:
Overexpression of insulin-like growth factor binding protein 3 in
oral squamous cell carcinoma. Oncol Rep. 20:1441–1447.
2008.PubMed/NCBI
|
|
26
|
He W, Liu Q, Wang L, et al: TLR4 signaling
promotes immune escape of human lung cancer cells by inducing
immunosuppressive cytokines and apoptosis resistance. Mol Immunol.
44:2850–2859. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li X, Jiang S and Tapping RI: Toll-like
receptor signaling in cell proliferation and survival. Cytokine.
49:1–9. 2009. View Article : Google Scholar
|
|
28
|
Yu L and Chen S: Toll-like receptors
expressed in tumor cells: targets for therapy. Cancer Immunol
Immunother. 57:1271–1278. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen R, Alvero AB, Silasi DA and Mor G:
Inflammation, cancer and chemoresistance: taking advantage of the
toll-like receptor signaling pathway. Am J Reprod Immunol.
57:93–107. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Erridge C: Endogenous ligands of TLR2 and
TLR4: agonists or assistants? J Leukoc Biol. 87:989–999. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sun Q, Zheng Y, Liu Q and Kao X: Rapamycin
reverses TLR4 signaling-triggered tumor apoptosis resistance by
disrupting Akt-mediated Bcl-xL upregulation. Int Immunopharmacol.
8:1854–1858. 2008. View Article : Google Scholar : PubMed/NCBI
|