Introduction
Human hepatocellular carcinoma (HCC) is the fifth
most common malignancy in the world (1), and has a marked increase in
younger-aged groups (2). The
occurrence of tumor is induced by the imbalance between cell
proliferation and apoptosis. Apoptosis is a fundamental cellular
event during development and is critical for the cytotoxicity
induced by anticancer drugs (3); it
can be initiated by extracellular and intracellular signals that
trigger a complex mechanism of pro-apoptotic proteases and
mitochondrial changes and is the integration of multiple survival
and death signals that determine whether a cell is to survive or
undergo apoptosis. Previously, it was reported that compound K
could induce apoptosis in cancer cell lines (4,5).
However, the detailed molecular mechanism and crosstalk between
these apoptosis-related signals remains largely unknown.
Ginseng radix, the root of Panax ginseng C.A.
Meyer, is frequently used as a traditional medicine in Asian
countries. It is used worldwide for preventive and therapeutic
purposes (6). In recent years,
ginsenosides, extracted from ginseng radix, have become a research
hotspot for their wide range of biological and pharmacological
activities (7). However,
ginsenosides are considered a prodrug, and are rarely absorbed into
the blood from the gastrointestinal tract (8). The really active components is
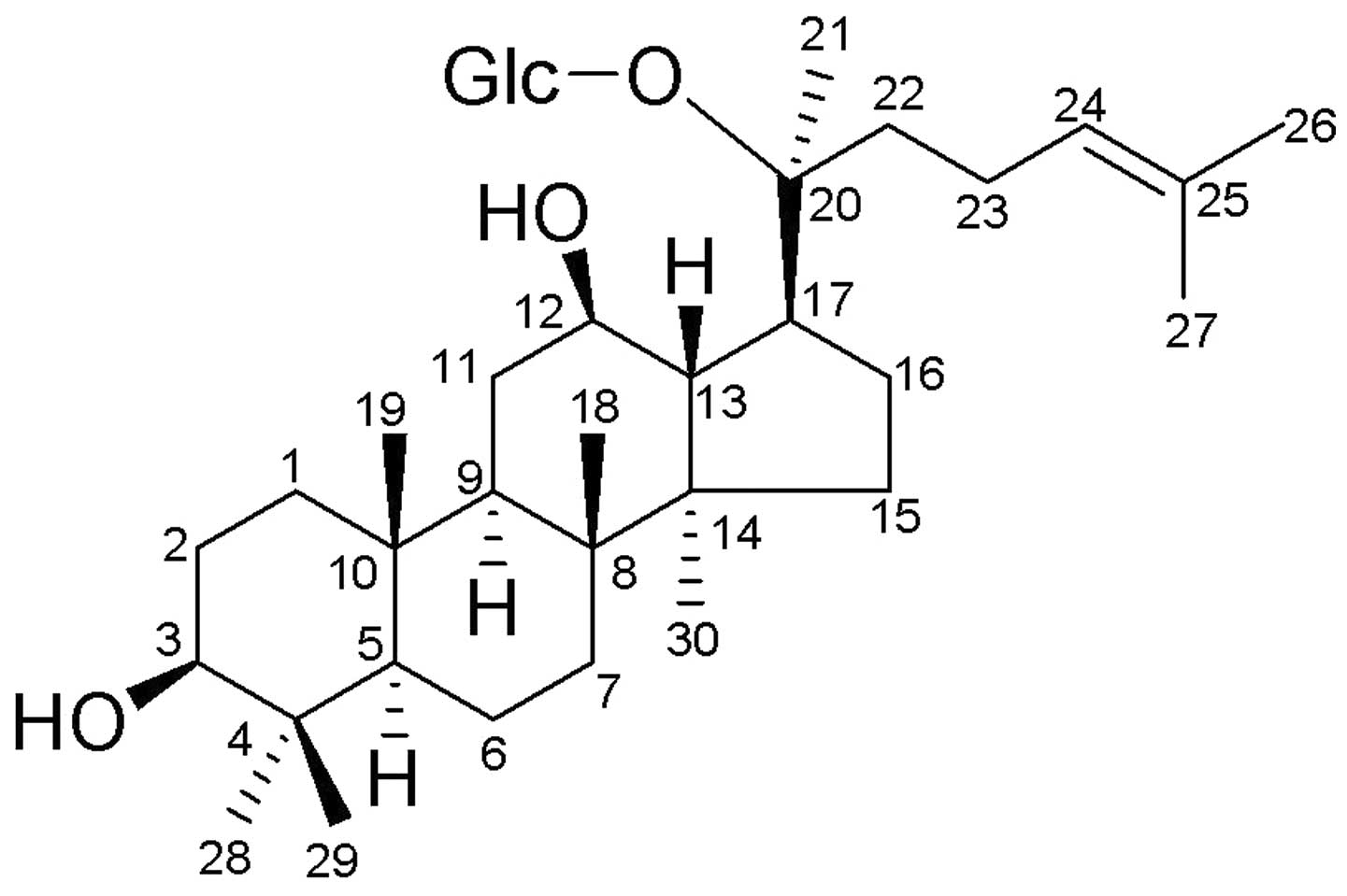
20-O-(β-D-glucopyranosyl)-20(S)-protopanaxadiol (compound K or M1;
Fig. 1).
Compound K is a novel ginseng saponin metabolite,
formed from ginsenosides Rb1, Rb2 and Rc by the human intestinal
bacteria deglycosylation (9,10). It
has been identified and purified after giving ginseng extract in
humans and rats, and detected as one of the major metabolites after
oral administration (11,12). Thus, compound K was speculated to be
the major form of protopanaxadiol saponin absorbed by the
intestine. Moreover, compound K has previously been found to
possess chemopreventive and chemotherapeutic potential, including
cardiac protection (13),
antimetastasis effect (14),
attenuating hepatic lipid accumulation (15), antigenotoxic and anticlastogenic
activity induced by benzopyrene (16), antitumor activity in
cisplatin-resistant pulmonary adenocarcinoma cells (17) and reversing multidrug resistance in
tumor cells (18).
In the present study, we specifically selected
MHCC97-H cell line as the experiment model, as it is a highly
metastatic HCC cell line, and this may provide more insight into
some cases than normal HCC. The main focus of this study was to
test the hypothesis that compound K inhibits the growth of MHCC97-H
cells through induction of apoptosis and possible apoptosis signal
pathways.
Materials and methods
Materials
Compound K (purity >98%) was prepared and
identified as previously described (19). HCC cell line MHCC97-H and normal
human hepatocyte cell line chang-liver were purchased from the Cell
Bank of the Chinese Academy of Sciences (Shanghai, China).
Propidium iodide (PI) was obtained from Bio Basic Canada Inc.
(Toronto, Canada). Bisbenzimide (Hoechst 33258), acridine orange
(AO), ethidium bromide (EB), methyl thiazolyl tetrazolium (MTT) and
monoclonal mouse anti-β-actin antibody were purchased from
Sigma-Aldrich Co. (St. Louis, MO, USA). Mitochondrial membrane
potential (Δψm) detection kit was purchased from Nanjing KeyGen
Biotech, Co., Ltd. (Nanjing, China). Antibodies against
pro-caspase-9, pro-caspase-3, cleaved-caspase-8, Fas, FasL, p-Akt,
Akt, Bax, Bcl-2, horseradish peroxidase-conjugated goat anti-rabbit
and goat anti-mouse antibodies were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA).
Cell culture and treatment
MHCC97-H cell line was cultured in Dulbecco’s
modified Eagle’s medium (Gibco, Grand Island, NY, USA),
supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT,
USA), 100 U/ml penicillin and 100 μg/ml streptomycin, at 37°C in a
water-saturated atmosphere of 5% CO2 in air. Compound K
was prepared in dimethyl sulphoxide (DMSO) at a concentration of 50
mM as a stock solution. Prior to use, the stock solution was
diluted to the required concentration immediately. Control cells
were treated with equivalent amount of DMSO without compound K.
Cell viability assay
Cell viability was examined by MTT assay. Briefly,
cells were plated in 96-well plates at a density of
5×103 cells/well in a 200 μl medium, and then treated
with compound K or DMSO for a predetermined period. Then, 200 μl of
MTT (0.5 mg/ml) was added to each well and the plates were
incubated for an additional 4 h at 37°C. The medium was replaced
with DMSO to dissolve the formazan produced from MTT by viable
cells. The optical absorbance at 570 nm was proportional to the
percentage of cell viability. Fifty percent inhibitory
concentration value (IC50) was calculated using
Log-Probit regression analysis in SPSS 13.0.
Morphological study with fluorescence
microscope
Exponential growth phase MHCC97-H cells seeded in
6-well plates were treated with 50 μM compound K or 0.1% DMSO for
48 h, harvested with 0.25% trypsin and resuspended in DMEM medium.
Cells (1×106) were washed and resuspended in PBS,
followed by observation of the morphological changes. In addition,
some cells were incubated with 10 μg/ml Hoechst 33258 for 10 min in
the dark. Furthermore, 25 μl of cell suspension was mixed with 1 μl
of dye mixture containing 100 μg/ml AO and EB in PBS. Thereafter,
cell morphology was visualized immediately under a fluorescence
microscope (Leica DM IRB).
Cell cycle analysis
Cells were treated with compound K for 24 h and were
harvested and suspended in 1:1 (v/v) mixture of PBS and 0.2 M
Na2HPO4-0.1 M citric acid (pH 7.5). Then,
cells were fixed with 100% ice-cooled ethanol at 4°C for 4 h,
centrifuged at 800 × g for 5 min and washed twice, resuspended with
PBS (1×106 cells/ml). Subsequently, 1×106
cells were stained with 10 μg/ml PI containing 100 μg/ml DNase-free
RNase A in the dark for 1 h at 37°C. Then 20,000 cells were
measured using a FACSCalibur flow cytometer (Becton-Dickinson, San
Jose, CA, USA). Data were analyzed with ModFit LT 3.2 software
(Becton-Dickinson).
Mitochondrial membrane potential
assay
Cells treated with 50 μM compound K for 48 h and
control cells were incubated with
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolocarbocyanine
iodide (JC-1) (0.1 μg/ml) for 15 min. After washing with PBS, the
cells were suspended with PBS and observed with fluorescence
microscope as previously described (20). The green fluorescence from JC-1
monomer and the red fluorescence from the aggregated form of JC-1
were visualized.
Single-cell gel electrophoresis
(SCGE)
The comet assay was performed as previously
described (21,22). Briefly, cells were suspended in 1%
(w/v) low-melting point agarose and pipetted on to slides which
were precoated with a layer of 1% (w/v) normal melting-point
agarose (warmed to 37°C before use). The agarose was allowed to set
at 4°C for 10 min, and the slides were then immersed for 1 h at 4°C
in a lysis solution. Slides were placed in single rows in a 30 cm
wide horizontal electrophoresis tank containing 0.3 M NaOH and 1 mM
EDTA, pH 13.0 (unwinding solution) and kept at 4°C for 40 min.
Electrophoresis was performed for 30 min in unwinding solution at
30 V (1 V/cm) and 300 mA. Finally, the slides were washed 5 min for
3 times in 0.4 M Tris (pH 7.5, 4°C) and stained with EB.
Western blot analysis
Cells were washed and suspended in lysis buffer on
ice for 10 min. Lysates were cleared by centrifugation at 12,000 ×
g for 10 min at 4°C. The total protein, as determined by Bio-Rad
protein assay (Bio-Rad Laboratories, Hercules, CA, USA), was mixed
with 4X loading buffer and pre-heated. Equal amounts of cell
extracts were resolved by SDS-PAGE, and transferred onto a PVDF
membrane (Millipore, Billerica, MA, USA). The membrane was blocked
and incubated 1 h with appropriate primary antibody. Horseradish
peroxidase (HRP)-conjugated anti-rabbit or anti-mouse IgG was used
as the secondary antibody. Final detection was performed with ECL™
western blotting reagents (Amersham, Livingston, NJ, USA).
Statistical analysis
Data are provided as the mean ± standard deviation
and intergroup differences were analyzed using the Student’s
t-test. Statistical analysis was conducted by SPSS 13.0.
Results
Compound K inhibits MHCC97-H cell
proliferation
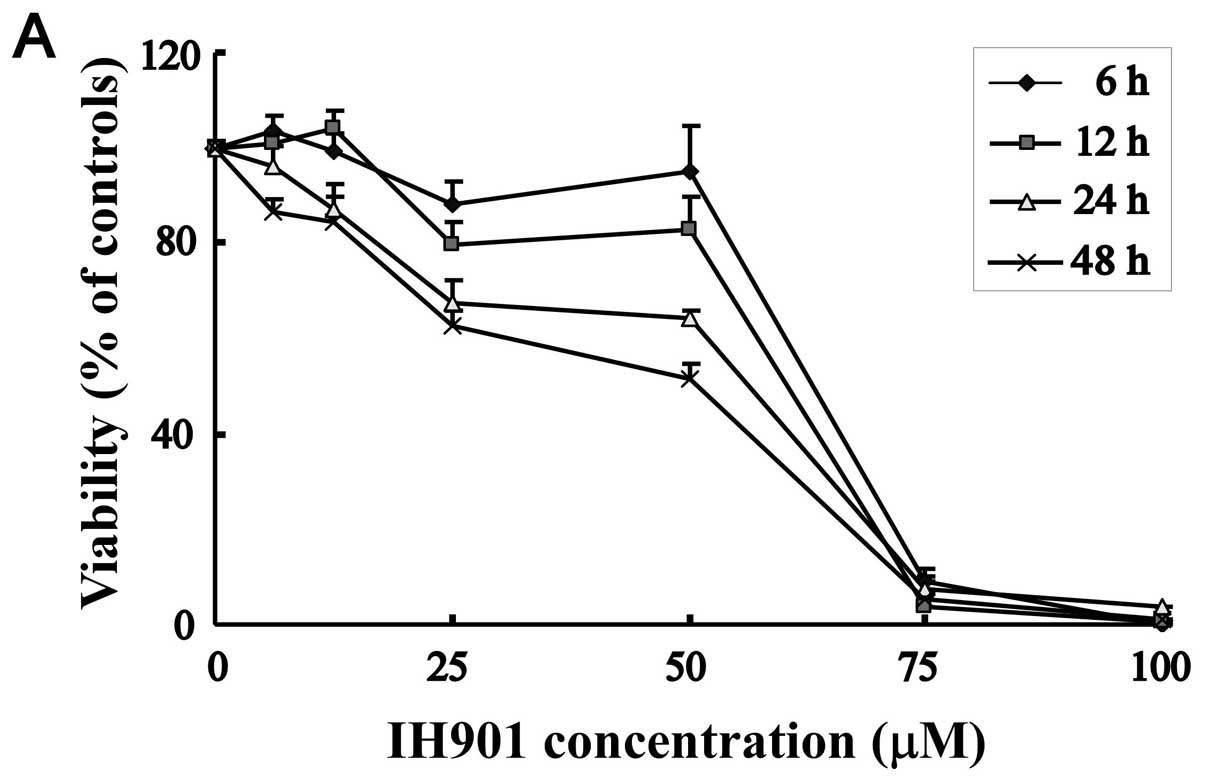
To evaluate the cytotoxicity of compound K by MTT
assay, MHCC97-H cells were treated with increasing concentrations
of compound K for 6, 12, 24 and 48 h. As shown in Fig. 2A, cell viability was significantly
reduced from 25–100 μM. In the same concentration, cell inhibitory
rate is proportioned to the exposure time. Thus, compound K
exhibited a dose and time-dependent decrease in MHCC97-H
proliferation. In contrast, normal hepatocyte chang-liver showed
relatively strong resistance to compound K, with 50% inhibitory
concentration (IC50) of 71.3±3.7 μM, whereas the
IC50 against MHCC97-H was 49.8±2.5 μM for 48 h.
Morphological changes of MHCC97-H cells
after exposure to compound K
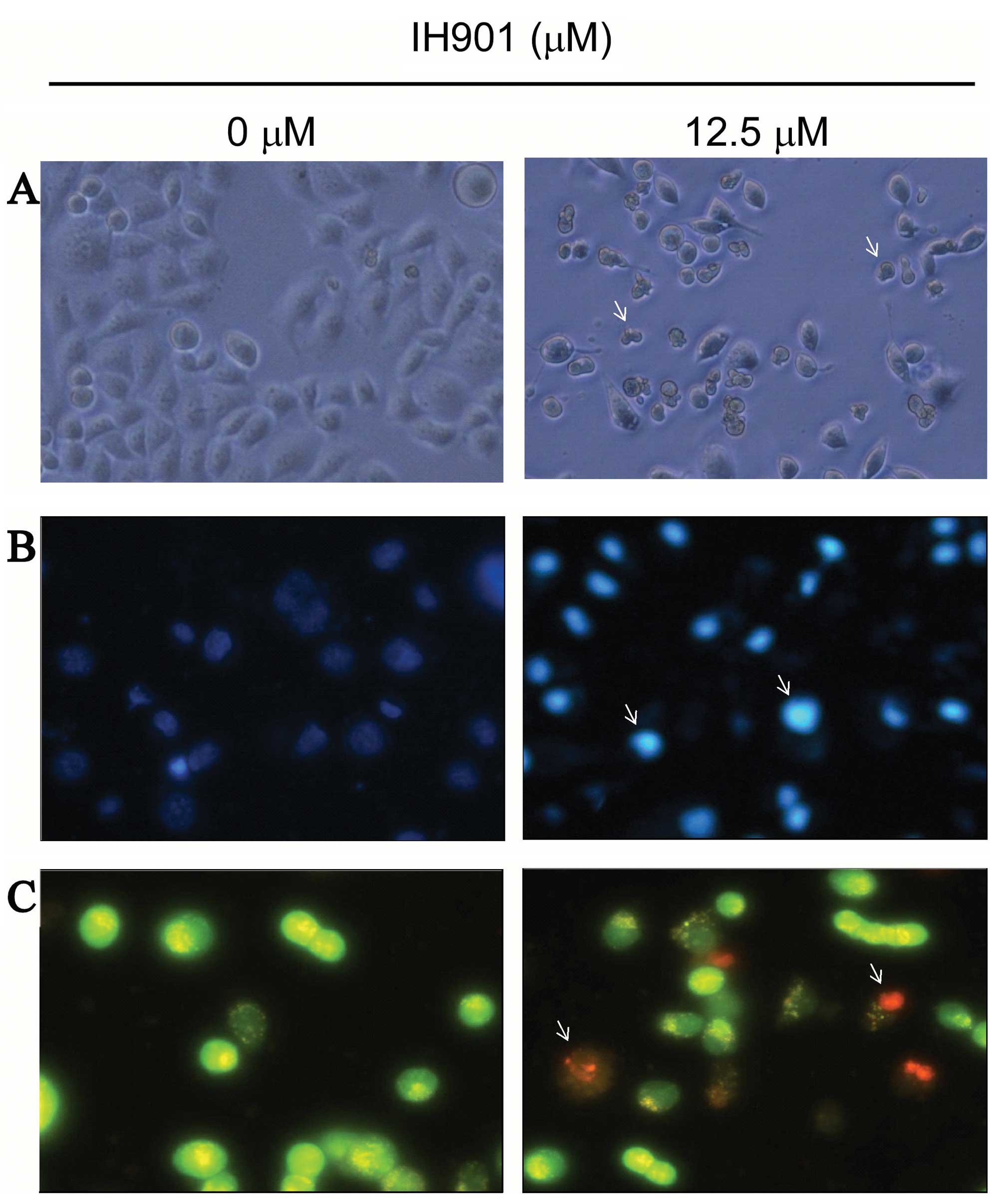
In order to verify compound K-induced inhibition, we
examined the changes of cell morphology after compound K exposure.
As shown in Fig. 3A, cells treated
with 50 μM compound K for 48 h showed the typical appearance of
apoptotic cells, such as nuclear fragmentation, cell shrinkage and
apoptotic bodies under the phase-contrast microscope. In addition,
with Hoechst 33258 staining, the blue emission light in apoptotic
cells was much brighter than in the control cells (Fig. 3B). By comparison, the control cells
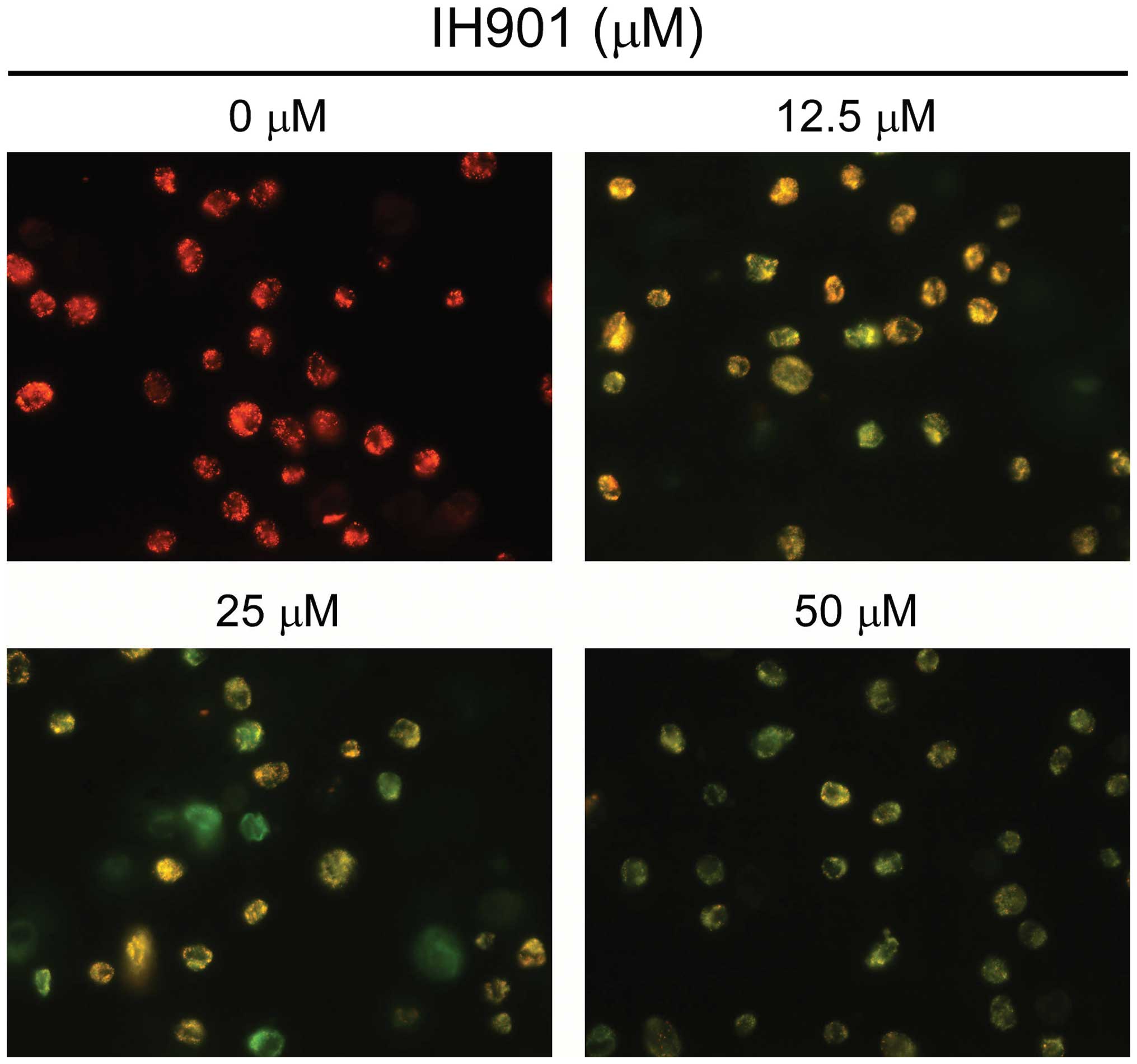
were less bright blue and more homogeneous. With AO/EB staining,
different cells (alive, apoptotic or necrotic) were clearly
differentiated by different colors (Fig. 3C). These results showed that the
compound K-treated cells exhibited morphological changes indicating
apoptosis, including chromatin condensation and nuclear
fragmentation.
Effect of compound K on the proportion of
apoptotic MHCC97-H cells
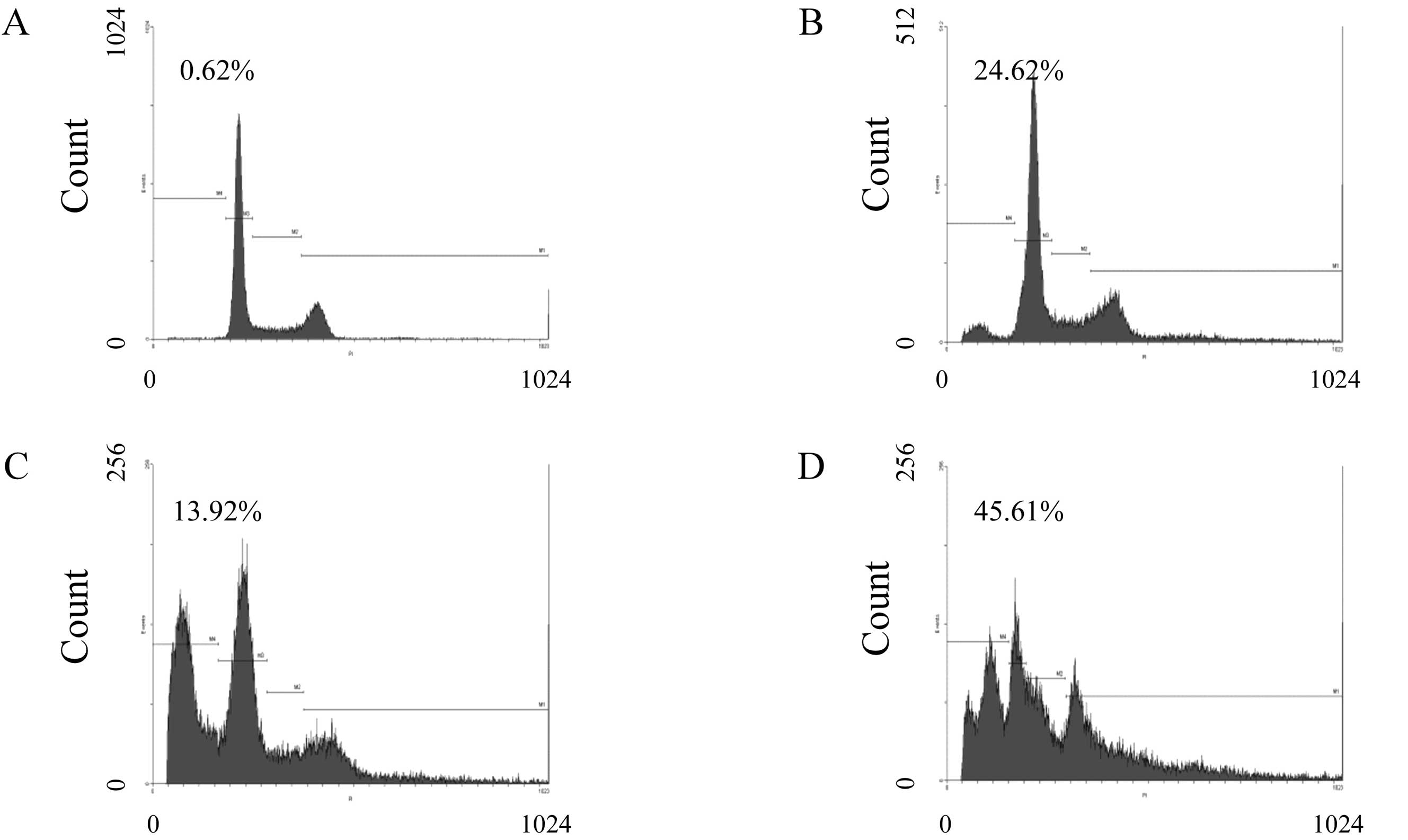
To determine the effect of compound K on the
proportion of MHCC97-H cells, apoptosis and cell cycle distribution
were evaluated by flow cytometer. As shown in Fig. 4, MHCC97-H cells were treated with
compound K at the concentrations of 25 μM (Fig. 4B), 50 μM (Fig. 4C), and 75 μM (Fig. 4D) for 24 h, and then analyzed for
cell cycle progression by flow cytometer. Sub-G1 fraction was
detected from 13.92±0.14 to 45.61±7.78% in MHCC97-H cells, while
only 0.62±2.12% in the control group (P<0.05; Fig. 4A and E). As shown in Fig. 4E, compound K induced cell cycle
arrest at the G0/G1 phase in MHCC97-H cells as well as a
progressive decline of S phase. These results revealed that
compound K induced apoptotic cell death concurrent with cell cycle
arrest in MHCC97-H cells.
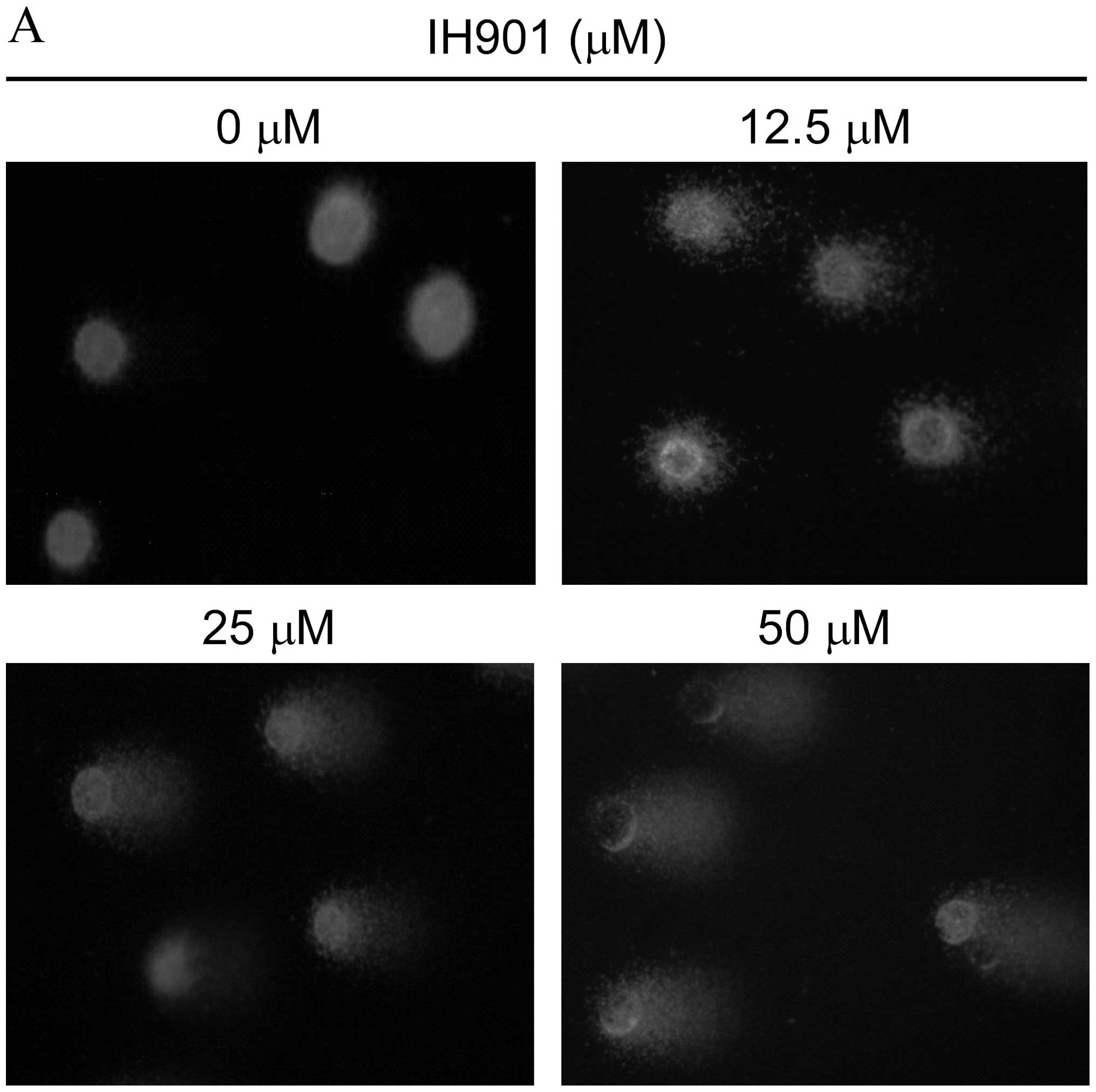
Disruption of Δψm by compound K
To investigate the loss of Δψm during apoptosis
induced by compound K, cells were stained with JC-1 and monitored
with a fluorescence microscope. JC-1 forms monomer and emits green
fluorescence when Δψm is depolarized (common in apoptosis), while
JC-1 aggregates and emits red fluorescence at a highly polarized
Δψm. As shown in Fig. 5, JC-1 was
accumulated in intact cells where it displayed red fluorescence
indicating a high potential. In contrast, JC-1 was poorly
accumulated in compound K-treated cells, which displayed only green
or weak red fluorescence, indicating low membrane potential. These
results strongly support the hypothesis that, after exposure to
compound K, an initial interaction with redox-active iron takes
place in the mitochondrial membrane compartments, resulting in
destabilization of their membranes and Δψm is disrupted.
Tail-DNA is induced by compound K in
SCGE
SCGE was employed to investigate the DNA damage
induced by compound K in MHCC97-H cells. As shown in Fig. 6A, the comets resulting from exposure
to compound K differed from control. Relatively undamaged cells
gave comets consisting of a compact head without tail, indicating
double-stranded DNA, while the comets originating from cells
exposed to compound K (lower panel) had a distinct head with a
tail, indicating the induction of DNA damage by compound K.
As compared with appropriate control, the mean
tail-DNA is presented in Fig. 6B.
Compound K evoked more increase in tail-DNA at concentrations as
compared with the control. The increase of tail-DNA is positively
proportioned to the concentration of compound K. These results
indicate that DNA induced by compound K is closely associated with
apoptosis.
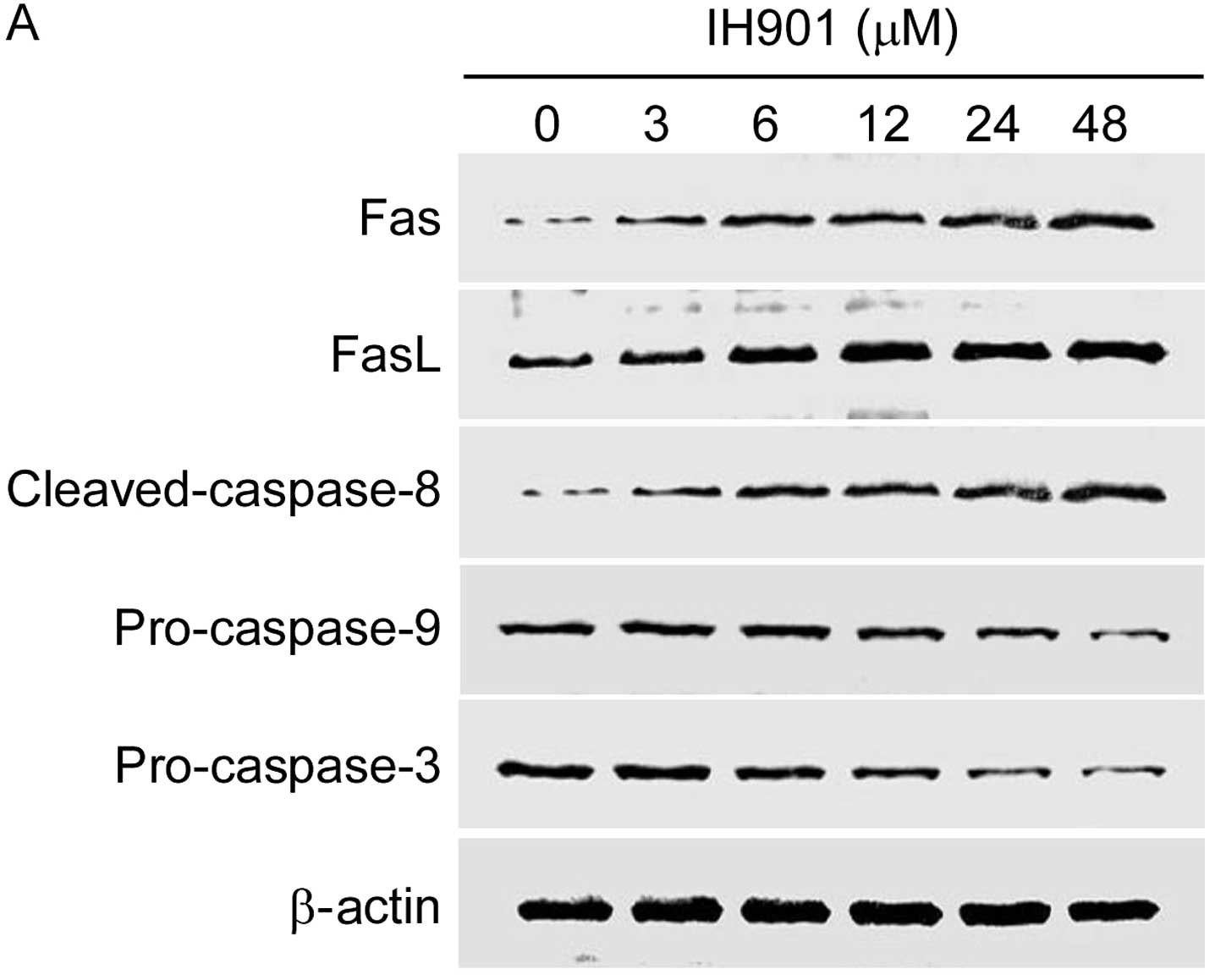
Effect of compound K on the expression of
apoptosis-related proteins
Apoptosis is characterized by a well-organized
sequence of cellular events, resulting in the activation of the
Fas/FasL and caspases cascade. In the group of apoptosis-related
proteins, Fas/FasL, PI3K/Akt, caspase-8 and Bax/Bcl-2 are very
important molecules for their regulatory role during apoptosis. We
examined these proteins in MHCC97-H with compound K for 48 h.
As shown in Fig. 7,
compound K not only significantly upregulated the expression of
Fas/FasL and cleaved-caspase-8, but also decreased downstream
proteins pro-caspase-9, pro-caspase-3 in a dose-dependent manner.
In addition, compound K gradually inhibited Akt phosphorylation and
increased Bax/Bcl-2 ratio. These results indicated that compound
K-induced apoptosis may occur through Fas- and
mitochondria-mediated caspase-dependent pathways.
Discussion
Apoptosis is required for proper tissue homeostasis.
Defects in apoptosis signaling pathways contribute to
carcinogenesis. Previous studies demonstrated that compound K may
be the active metabolite responsible for the anticarcinogenic
effects of ginseng saponin. This prompted us to investigate the
effects of compound K in more detail. Although previous studies
have reported that compound K exhibits a broad range of important
pharmacological effects, including anticancer activities (23,24),
the precise mechanisms induced by compound K remain unclear.
Our previous study revealed that compound K can
induce apoptosis in HCC (25). In
the present study, we sought to demonstrate the mechanisms of
apoptosis induced by compound K. We found that compound K could
inhibit the cell proliferation of MHCC97-H in a dose- and
time-dependent manner with a relatively low cytotoxicity to normal
hepatocytes, and increased sub-G1 phase in MHCC97-H cells. In
addition, both phase contrast microscopy and fluorescence staining
showed the typical appearance of apoptosis in compound K-treated
cells. The results of SCGE showed the DNA damage in compound K is
positively correlated with the drug concentration. These results
suggest that compound K induces apoptosis in MHCC97-H cells.
Moreover, the western blot analysis and Δψm of mitochondrial
membrane results revealed apoptosis induced by compound K is
through the caspase-dependent extrinsic and intrinsic pathways
(26).
In the extrinsic pathway, Fas and FasL are widely
recognized as key regulators of the apoptosis signal transduction
pathway. Fas is a type I transmembrane receptor protein and Fas
ligand is a type II transmembrane protein. The ligation of Fas and
FasL results in receptor trimerization followed by the binding of
the adaptor molecule, Fas-associated death domain (FADD) to the
cytoplasmic domain of the receptor (27,28).
Then, FADD activates caspase-8, which triggers the caspase cascade
through tbid and cytochrome c (29). Finally, caspase-3 and several other
effector pro-caspases were considerably activated (30) and caused DNA damage. In the present
study, we found that compound K can upregulate the expression level
of Fas and FasL in a dose-dependent manner. These results indicated
that Fas-mediated caspase-dependent pathway is involved in the
compound K-induced apoptosis in MHCC97-H cells.
In the intrinsic pathway, mitochondria play a
central role in the commitment of cells to chemical-induced
apoptosis (31). Cytochrome
c normally resides in the mitochondrial intermembrane space,
where it serves as a transducer of electrons in the respiratory
chain. Compound K can disrupt the Δψm of mitochondrial membrane,
leading to the release of cytochrome c into the cytosol
(32). After release from
mitochondria, cytochrome c binds to apoptosis protease
activating factor 1 (Apaf-1), which activates caspase-9 and
downstream caspase-3. Then, caspase-9 and caspase-3 act together to
destroy the death signal and finally lead to a unilateral process
to apoptosis (33,34) by DNA damage. In our experiments, we
found that compound K induced the loss of Δψm, downregulated
pro-caspase-9 and pro-caspase-3, in a dose-dependent manner. These
data indicated that the mitochondria-mediated caspase-dependent
signal pathway is involved in the compound K-induced apoptosis in
MHCC97-H cells.
The Akt kinase regulates the balance between
survival and apoptosis factors. It is activated by binding with
phospholipids. Activated Akt (p-Akt) promotes cell survival by
inhibiting apoptosis through phospho-inactivation of several
targets, including Bad and caspase-9 (35). In the present study, we found that
phosphorylation of Akt at Ser 473 was suppressed by compound K
(Fig. 8A). The inhibition of Akt is
likely to be involved in compound K-mediated growth inhibition of
MHCC97-H cells. In addition, compound K promoted pro-apoptotic
protein Bax expression, and decreased anti-apoptotic protein Bcl-2
expression in MHCC97-H cells, leading to a decrease of Bax/Bcl-2
ratio. The ratio between pro- and anti-apoptotic proteins
determines the susceptibility of cells to an apoptotic death signal
(36). Thus, p-Akt, Bax and Bcl-2
were also involved in compound K-induced apoptosis in MHCC97-H
cells.
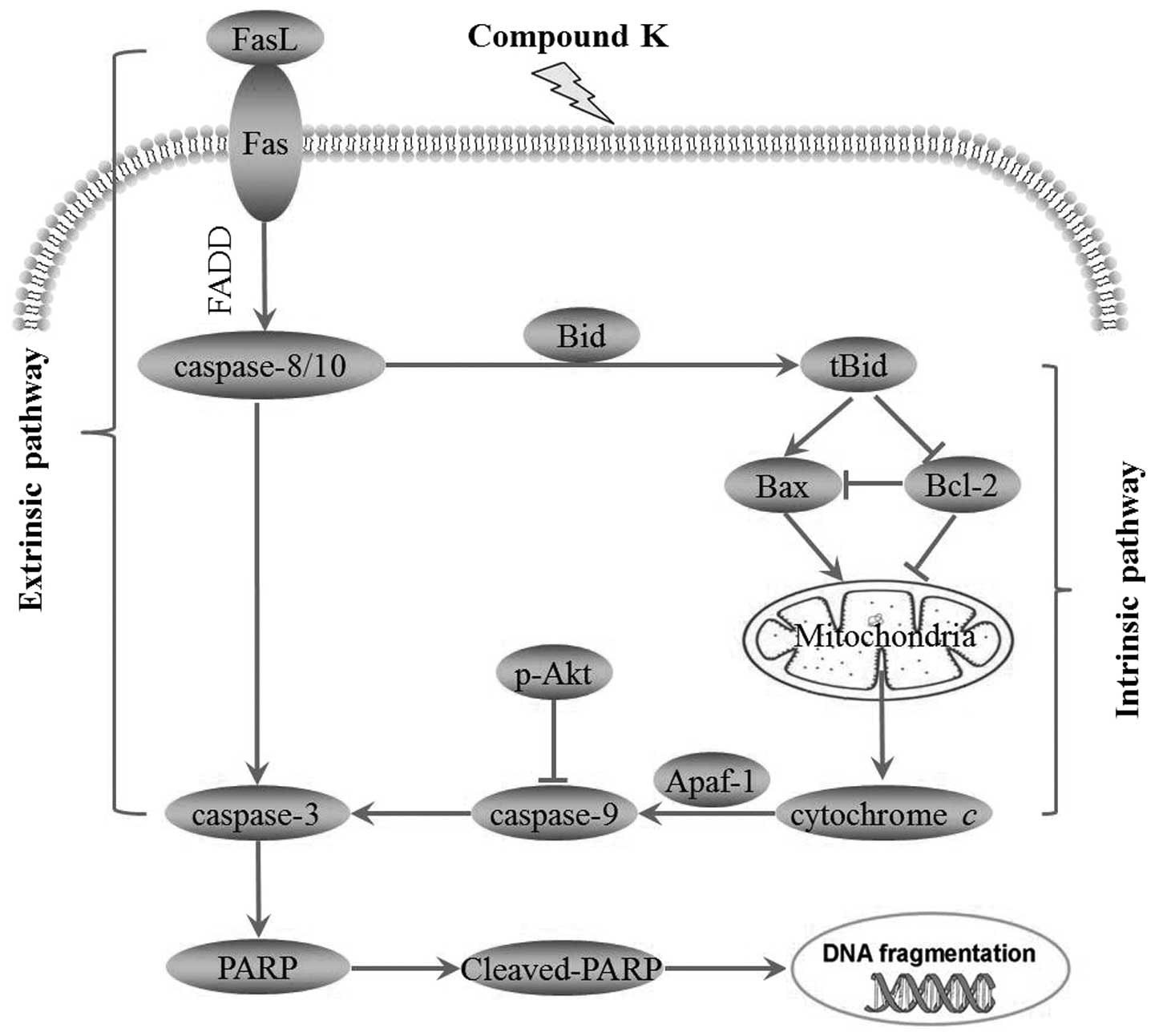
Taken together, the present study demonstrated that
compound K, a ginseng saponin metabolite, significantly inhibited
cell proliferation and induced apoptosis in MHCC97-H via Fas- and
mitochondria-mediated caspase-dependent pathways (Fig. 8). The relatively low toxicity in
normal hepatocytes and high activity in HCC MHCC97-H cells suggest
that compound K might be a promising experimental cancer
chemotherapeutic and chemopreventive agent for human HCC (37).
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81274149) and the
Natural Science Foundation of Fujian Province, China (grant no.
2010D012).
References
|
1
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Estimating the world cancer burden: Globocan 2000. Int J Cancer.
94:153–156. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ogunbiyi JO: Hepatocellular carcinoma in
the developing world. Semin Oncol. 28:179–187. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cotter TG: Apoptosis and cancer: the
genesis of a research field. Nat Rev Cancer. 9:501–507. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Choi HH, Jong HS, Park JH, et al: A novel
ginseng saponin metabolite induces apoptosis and down-regulates
fibroblast growth factor receptor 3 in myeloma cells. Int J Oncol.
23:1087–1093. 2003.PubMed/NCBI
|
|
5
|
Hu C, Song G, Zhang B, Liu Z, Chen R,
Zhang H and Hu T: Intestinal metabolite compound K of panaxoside
inhibits the growth of gastric carcinoma by augmenting apoptosis
via Bid-mediated mitochondrial pathway. J Cell Mol Med. 16:96–106.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Attele AS, Wu JA and Yuan CS: Ginseng
pharmacology: multiple constituents and multiple actions. Biochem
Pharmacol. 58:1685–1693. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Joh EH, Lee IA, Jung IH and Kim DH:
Ginsenoside Rb1 and its metabolite compound K inhibit IRAK-1
activation - the key step of inflammation. Biochem Pharmacol.
82:278–286. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kobashi K and Akao T: Relation of
intestinal bacteria to pharmacological effects of glycosides.
Bioscience Microflora. 16:1–7. 1997. View Article : Google Scholar
|
|
9
|
Wakabayashi C, Hasegawa H, Murata J and
Saiki I: The expression of in vivo antimetastatic effect of ginseng
protopanaxatriol saponins is mediated by their intestinal bacterial
metabolites after oral administration. J Traditional Med.
14:180–185. 1997.
|
|
10
|
Wakabayashi C, Hasegawa H, Murata J and
Saiki I: In vivo antimetastatic action of ginseng
protopanaxadiol saponins is based on their intestinal bacterial
metabolites after oral administration. Oncol Res. 9:411–417.
1997.
|
|
11
|
Akao T, Kanaoka M and Kobashi K:
Appearance of compound K, a major metabolite of ginsenoside Rb1 by
intestinal bacteria, in rat plasma after oral administration -
measurement of compound K by enzyme immunoassay. Biol Pharm Bull.
21:245–249. 1998. View Article : Google Scholar
|
|
12
|
Wang CZ, Kim KE, Du GJ, et al:
Ultra-performance liquid chromatography and time-of-flight mass
spectrometry analysis of ginsenoside metabolites in human plasma.
Am J Chin Med. 39:1161–1171. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tsutsumi YM, Tsutsumi R, Mawatari K,
Nakaya Y, Kinoshita M, Tanaka K and Oshita S: Compound K, a
metabolite of ginsenosides, induces cardiac protection mediated
nitric oxide via Akt/PI3K pathway. Life Sci. 88:725–729. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Choo MK, Sakurai H, Kim DH and Saiki I: A
ginseng saponin metabolite suppresses tumor necrosis
factor-α-promoted metastasis by suppressing nuclear factor-κB
signaling in murine colon cancer cells. Oncol Rep. 19:595–600.
2008.PubMed/NCBI
|
|
15
|
Kim DY, Yuan HD, Chung IK and Chung SH:
Compound K, intestinal metabolite of ginsenoside, attenuates
hepatic lipid accumulation via AMPK activation in human hepatoma
cells. J Agric Food Chem. 57:1532–1537. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee BH, Lee SJ, Hur JH, Lee S, Sung JH,
Huh JD and Moon CK: In vitro antigenotoxic activity of novel
ginseng saponin metabolites formed by intestinal bacteria. Planta
Med. 64:500–503. 1998. View Article : Google Scholar
|
|
17
|
Lee SJ, Sung JH, Lee SJ, Moon CK and Lee
BH: Antitumor activity of a novel ginseng saponin metabolite in
human pulmonary adenocarcinoma cells resistant to cisplatin. Cancer
Lett. 144:39–43. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hasegawa H, Sung JH, Matsumiya S, Uchiyama
M, Inouye Y, Kasai R and Yamasaki K: Reversal of daunomycin and
vinblastine resistance in multidrug-resistant P388 leukemia in
vitro through enhanced cytotoxicity by triterpenoids. Planta
Med. 61:409–413. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ming YL, Song G, Chen LH, et al:
Anti-proliferation and apoptosis induced by a novel intestinal
metabolite of ginseng saponin in human hepatocellular carcinoma
cells. Cell Biol Int. 31:1265–1273. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cossarizza A, Baccarani-Contri M,
Kalashnikova G and Franceschi C: A new method for the
cytofluorimetric analysis of mitochondrial membrane potential using
the J-aggregate forming lipophilic cation
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine
iodide (JC-1). Biochem Biophys Res Commun. 197:40–45.
1993.PubMed/NCBI
|
|
21
|
Collins AR, Dobson VL, Dusinska M, Kennedy
G and Stetina R: The comet assay: what can it really tell us? Mutat
Res. 375:183–193. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Panayiotidis M, Tsolas O and Galaris D:
Glucose oxidase-produced H2O2 induces
Ca2+-dependent DNA damage in human peripheral blood
lymphocytes. Free Radic Biol Med. 26:548–556. 1999.
|
|
23
|
Hasegawa H: Proof of the mysterious
efficacy of ginseng: basic and clinical trials: metabolic
activation of ginsenoside: deglycosylation by intestinal bacteria
and esterification with fatty acid. J Pharmacol Sci. 95:153–157.
2004. View Article : Google Scholar
|
|
24
|
Lee JY, Shin JW, Chun KS, et al: Antitumor
promotional effects of a novel intestinal bacterial metabolite
(IH-901) derived from the protopanaxadiol-type ginsenosides in
mouse skin. Carcinogenesis. 26:359–367. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ming YL, Zheng ZZ, Chen LH and Tong QX:
Apoptosis induced by a novel intestinal metabolite of ginseng
saponin in human hepatocellular carcinoma BEL-7402 cells. Chin Trad
Herbal Drugs. 38:1511–1514. 2007.
|
|
26
|
Qi F, Li A, Inagaki Y, et al: Induction of
apoptosis by cinobufacini preparation through mitochondria- and
Fas-mediated caspase-dependent pathways in human hepatocellular
carcinoma cells. Food Chem Toxicol. 50:295–302. 2012. View Article : Google Scholar
|
|
27
|
Vaithinathan S, Saradha B and Mathur PP:
Methoxychlor induces apoptosis via mitochondria- and FasL-mediated
pathways in adult rat testis. Chem Biol Interact. 185:110–118.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nagata S: Fas ligand-induced apoptosis.
Annu Rev Genet. 33:29–55. 1999. View Article : Google Scholar
|
|
29
|
Schug ZT, Gonzalvez F, Houtkooper RH, Vaz
FM and Gottlieb E: BID is cleaved by caspase-8 within a native
complex on the mitochondrial membrane. Cell Death Differ.
18:538–548. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Medema JP, Scaffidi C, Kischkel FC,
Shevchenko A, Mann M, Krammer PH and Peter ME: FLICE is activated
by association with the CD95 death-inducing signaling complex
(DISC). EMBO J. 16:2794–2804. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Herr I and Debatin KM: Cellular stress
response and apoptosis in cancer therapy. Blood. 98:2603–2614.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu X, Kim CN, Yang J, Jemmerson R and
Wang X: Induction of apoptotic program in cell-free extracts:
requirement for dATP and cytochrome c. Cell. 86:147–157. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Srinivasula SM, Ahmad M, Fernandes-Alnemri
T and Alnemri ES: Autoactivation of procaspase-9 by Apaf-1-mediated
oligomerization. Mol Cell. 1:949–957. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Slee EA, Harte MT, Kluck RM, et al:
Ordering the cytochrome c-initiated caspase cascade: hierarchical
activation of caspases-2, -3, -6, -7, -8, and -10 in a
caspase-9-dependent manner. J Cell Biol. 144:281–292. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jeong SJ, Dasgupta A, Jung KJ, Um JH,
Burke A, Park HU and Brady JN: PI3K/AKT inhibition induces
caspase-dependent apoptosis in HTLV-1-transformed cells. Virology.
370:264–272. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Moon DO, Park SY, Choi YH, Kim ND, Lee C
and Kim GY: Melittin induces Bcl-2 and caspase-3-dependent
apoptosis through downregulation of Akt phosphorylation in human
leukemic U937 cells. Toxicon. 51:112–120. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Oh SH, Yin HQ and Lee BH: Role of the
Fas/Fas ligand death receptor pathway in ginseng saponin
metabolite-induced apoptosis in HepG2 cells. Arch Pharm Res.
27:402–406. 2004. View Article : Google Scholar : PubMed/NCBI
|