Introduction
It is becoming increasingly apparent that
cholesterol and its metabolites contribute to the development of
breast cancer (1–3). Various mechanisms by which cholesterol
promotes the growth of breast tumors have been identified;
cholesterol is also the precursor of steroid hormones such as
estrogen and testosterone, both of which have well-recognized tumor
promoting effects (4). The
cholesterol metabolite 27-hydroxycholesterol has been shown to
exert selective estrogen receptor modulation (SERM) effects
(5,6) and also to promote the growth of
estrogen receptor (ER)-positive breast tumors (3). Current evidence, therefore, suggests
that by disrupting cholesterol biosynthesis, we may inhibit cell
cycle progression and induce cell death (7). Most previous studies have targeted
HMG-CoA reductase, a rate limiting enzyme in the cholesterol
biosynthetic pathway. However, using in silico analysis, we
recently found that oxidosqualene cyclase (OSC), which is
downstream of HMG-CoA reductase and is a critical enzyme that
catalyzes the cyclization of 2,3-oxidosqualene to lanosterol, may
also be a potential target by which to control the proliferation of
ER+ve tumors (8). On the basis of
these initial findings, and after examining the effects of
different OSC inhibitors on human breast cancer cells, we selected
RO 48-8071 (RO) as a prototype inhibitor. Our decision to use RO
was also based on the aforementioned study where we showed that the
compound is a suitable candidate for suppressing human breast
cancer cells in vitro (8).
Since there are differences in intra-tumor estrogen
levels between ER+ve and triple negative breast cancer tissue
(9), we hypothesized that the
levels of enzymes involved in the cholesterol biosynthetic pathway
may be altered in breast cancer cells. This could include
differential expression or activity of OSC. Taking this into
consideration, we measured levels of OSC in a panel of human breast
cancers and normal tissues and found no significant difference in
levels of OSC mRNA between steroid responsive and
hormone-independent tumors. Furthermore, OSC expression in tumor
tissue was not significantly different than in normal mammary
tissue, suggesting that RO must suppress breast cancer cell growth
via alternative, off-target effects. In this respect, we discovered
that RO suppresses the transcriptional activity of ERα and to some
extent that of ERβ, under conditions that preserve cell viability.
Moreover, RO also suppresses androgen receptor (AR) transcriptional
activity, another major determinant of breast cancer progression
(10). Using western blot analysis,
we verified that RO suppresses levels of progesterone receptor
(PR), the expression of which is directly controlled by ERα in
human breast cancer cells (11).
This confirms that the ERα-mediated signal transduction pathway is
inhibited when cells are exposed to RO.
Materials and methods
RO 48-8071 was purchased from Sigma, dissolved in
DMSO and stored in aliquots at −20°C prior to use.
Human tissue qPCR
qPCR-ready TissueScan™ cDNA Array (OriGene starter
kit, cat. no. TSRT101) was initially used to determine the
expression of OSC in human breast cancer tissues following
guidelines recommended by the manufacturer. Human breast cancer
tissue array (cat. no. BCRT101) was then used to measure OSC
expression in tissues that either expressed ER, PR and HER2
(hormone-dependent) or tissues lacking these receptors (triple
negative) at different stages of development (stage I–III). Human
OSC (Hs00158906_ml LSS) TaqMan Fam probes were obtained from
Applied Biosystems and normalized with human GAPDH (Hs03929097_g1
FG). Relative gene expression was determined using the following
formula: Fold-change in gene expression, 2-ΔΔCt = 2 - {ΔCt (cancer
tissue samples) - ΔCt (normal tissue)}, where ΔCt = Ct (OSC) - Ct
(GAPDH), where Ct represents threshold cycle number.
Luciferase activity
Receptor assay systems were obtained from Indigo
Biosciences (State College, PA, USA) and luciferase activity was
determined following the manufacturer’s recommended protocol. The
assays comprised non-human mammalian cells engineered to express
human ERα, ERβ, androgen receptor (AR) protein and luciferase
reporter gene functionally linked to the corresponding nuclear
receptor-responsive promoter. We initially used Indigo Biosciences
Receptor Assay (cat. no. IB00421-48P) to determine the effects of
RO on estradiol-induced ERα and ERβ activity. ICI 182,780 was used
as a reference antagonist to 17β-estradiol. Since our initial
results showed more pronounced effects of RO on ERα activity
compared with ERβ, we focused on ERα-promoter linked activity using
Indigo Biosciences Receptor Assay (cat. no. IB00401). We also
determined the effects of a commonly used cholesterol inhibitor,
atorvastatin (Ator) on ERα promoter-linked activity. Since there is
increasing evidence to support the role of AR in the development of
breast cancer (10), we conducted
studies to assess the effects of RO on AR-dependent luciferase
activity. A human AR reporter assay system (cat. no. IB03001;
Indigo Biosciences) was used with 6α-F1 testosterone as the
reference agonist for AR. For all reporter studies, we sequentially
used fluorescence-based LCM assays (cat. no. LCM-01; Indigo
Biosciences), following the manufacturer’s guidelines, to determine
the relative number of live cells at the assay endpoint. All
luciferase assays were read with GloMax®-Multi+
Microplate Multimode Reader system (Promega).
Western blotting
BT-474 breast cancer cells were grown in a
humidified atmosphere of 5% CO2 at 37°C in 100-mm cell
culture plates using phenol red-free Dulbecco’s modified Eagle’s
medium (DMEM/F12) supplemented with 10% fetal bovine serum. When
cells reached 50–60% confluence, they were washed with PBS and
switched into DMEM/F12 supplemented with 5% dextran-coated charcoal
(DCC) for 24 h. Cells were then washed, transferred into fresh
DMEM/F12 and treated with E2 (10 nM) in the absence and presence of
RO (5 μM) or with RO alone. Cells were treated with RO for 3
h prior to treatment with E2 for 16 h, after which cells were
harvested. Nuclear protein was extracted following the
manufacturer’s guidelines (cat. no. 40010; Active Motif, USA).
Protein aliquots (20 μg) were separated by SDS-PAGE,
transferred to polyvinylidene difluoride membrane and blotted with
the following human specific antibodies: ERα (SC-8005, 1:200
dilution), ERβ (SC-8974, 1:200 dilution), PR (SC-810, 1:200
dilution) (all from Santa Cruz Biotechnology), β-actin
(Sigma-Aldrich, St. Louis, MO, USA). Protein bands were detected
and quantified by blotting with anti-mouse secondary antibody
(SC-2005, 1:10,000 dilution), or anti-rabbit secondary antibody
(SC-2004, 1:10,000 dilution) (both from Santa Cruz Biotechnology),
and a chemiluminescent detection system according to the
manufacturer’s instructions (Amersham Pharmacia Biotech).
Real-time PCR
Total RNA was extracted from two distinct breast
cancer cell lines, BT-474 and T47D cells, which express both ERα
and ERβ. Cultured cells were treated with 20 μM RO for 1, 3,
6 and 24 h, respectively. Control samples were treated with
ethanol, the vehicle medium in which RO was dissolved. RNA was
extracted using an EZ-Bioresearch RNA Isolation kit (cat. no.
R1002-50) according to the manufacturer’s instructions. Two
micrograms of RNA were reverse transcribed into cDNA using a high
capacity DNA synthesis kit (cat. no. 4368814; Applied Biosystems).
cDNA was then amplified using an ABI 7300 Real-Time PCR Instrument
(Applied Biosystems), specific TaqMan primers and
TaqMan® Universal PCR Master Mix. Human ERα
(Hs00174860_m1), ER-β (Hs00230957_m1) and GAPDH (Hs03929097_g1 FG)
TaqMan Fam probes were used (Applied Biosystems). Relative gene
expression was determined using the following formula: Fold-change
in gene expression, 2-ΔΔCt = 2 - {ΔCt (treated samples) - ΔCt
(untreated control samples)}, where ΔCt = Ct (ERα or ERβ) - Ct
(GAPDH), where Ct represents threshold cycle number. All reactions
were carried out in duplicate for at least three independent
experiments.
Statistical analysis
Comparisons were made between multiple groups by
analysis of variance (ANOVA) with Neuman-Keuls post-hoc testing.
Where normality was not achieved, non-parametric ANOVA
(Kruskal-Wallis) and post-hoc Dunn’s test was performed.
Significance was defined as p<0.05. Unless indicated, values are
shown as mean ± SEM.
Results
OSC mRNA expression in human cancer
tissues
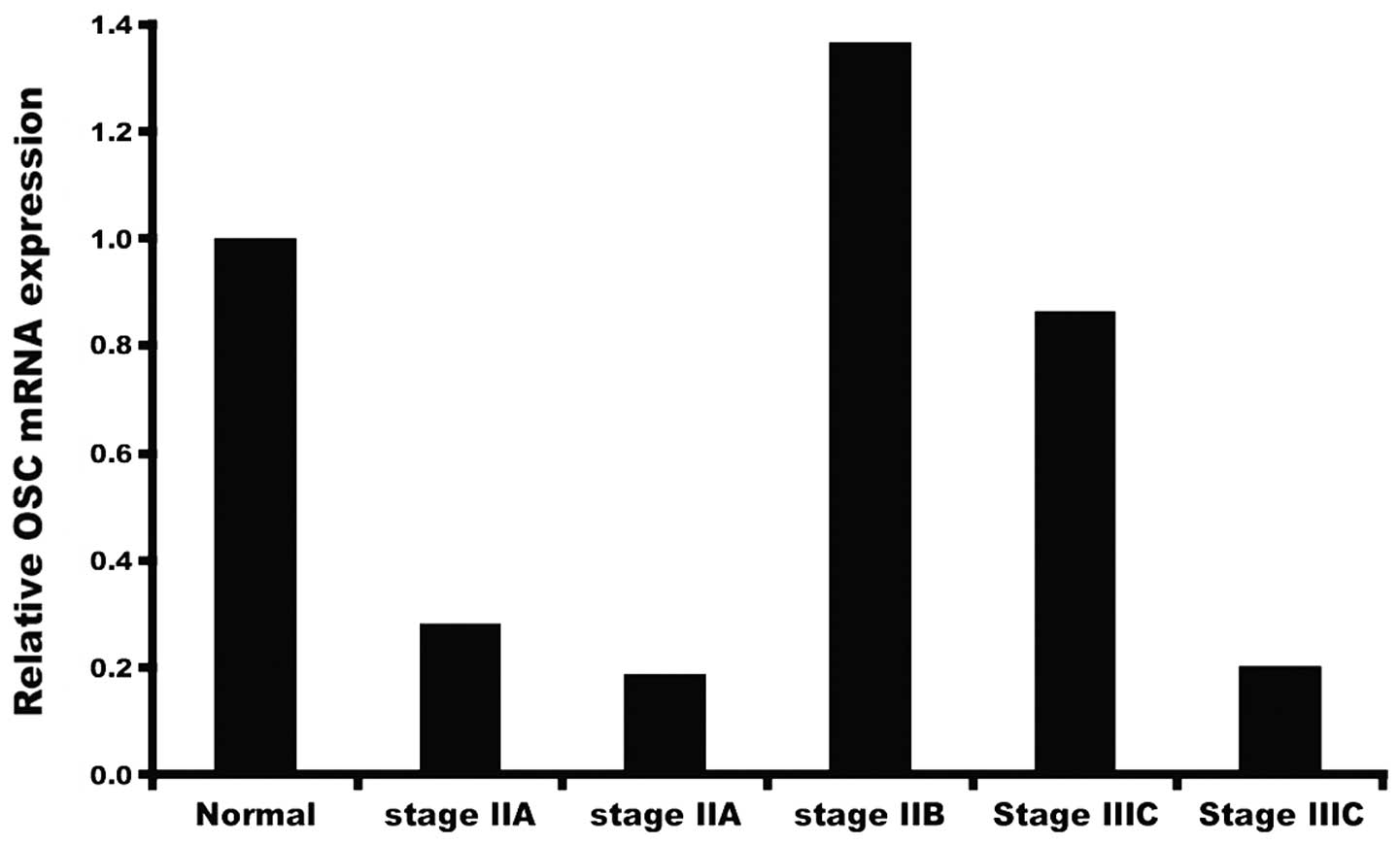
In order to measure OSC expression we employed ready
to use qRT-PCR human tissue cDNA arrays to obtain preliminary data
on levels of OSC mRNA in a limited number of tumors collected at
various stages of development (Fig.
1). Our initial study showed that varying levels of OSC message
are present in samples of tumor obtained at different stages, and
that, depending on the sample, mRNA levels may be higher or lower
than levels present in normal breast tissue. This prompted us to
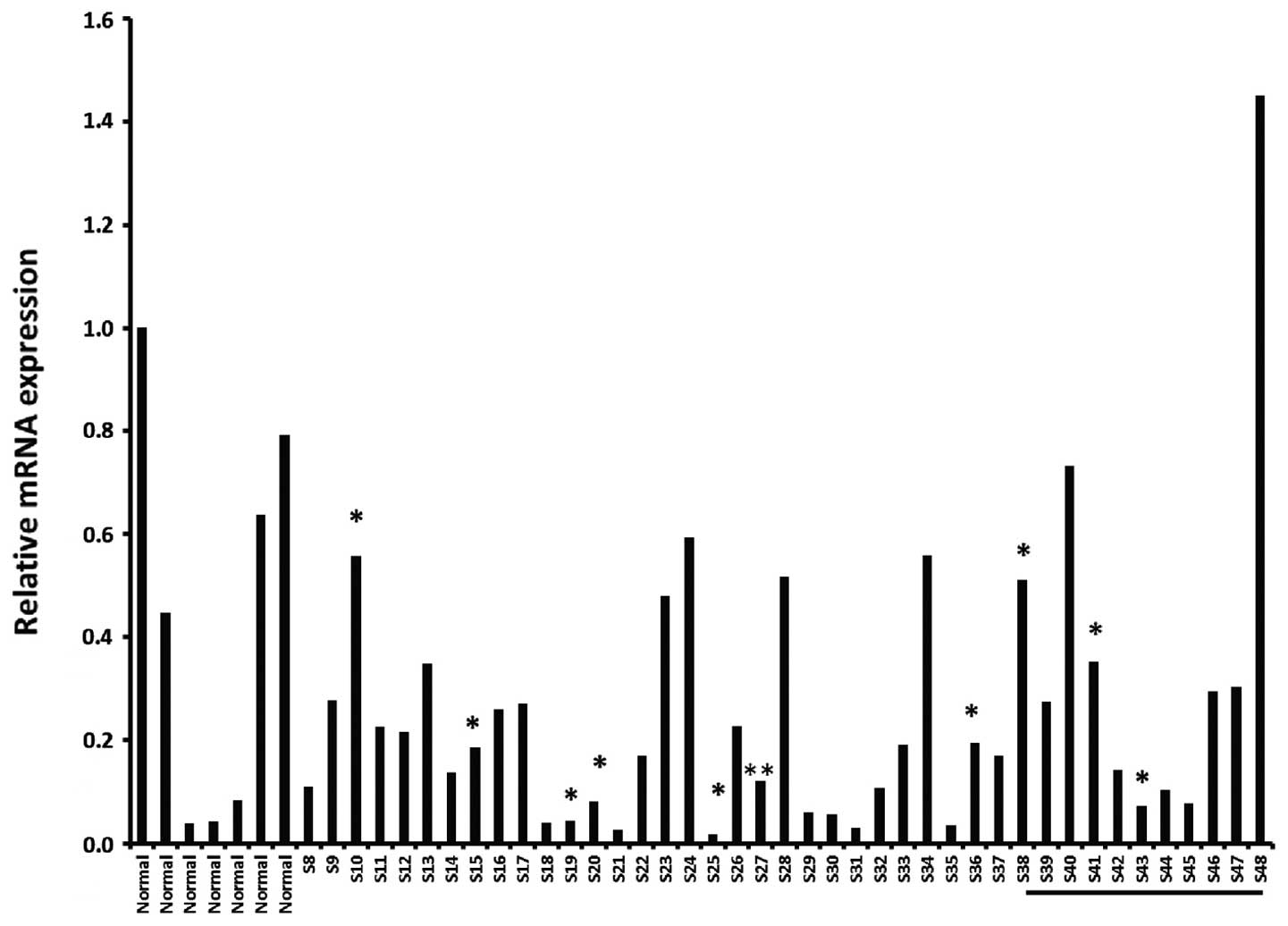
analyze a larger cohort of samples and to focus on different breast
cancer tissues at different stages (stage I–III) available from
OriGene as described in Materials and methods. As shown in Fig. 2, there was no consistency in levels
of OSC expression, either within tissue from normal breast, or
within breast tumor samples collected at various stages, including
triple negative cancers.
Effects of RO on ERα, ERβ and AR-promoter
linked hormone responsive luciferase activity
In a previous study, it was indicated that RO
reduces breast cancer cell viability in a dose-responsive manner
(8). However, since we found levels
of OSC mRNA in breast tumors to be inconsistent (Figs. 1 and 2), we concluded that it was unlikely that
RO exerts its effects on breast cancer cells primarily by targeting
OSC. Previously, we showed that pharmacological levels of RO
degrade ERα in breast cancer cells (Liang et al unpublished
data). In the present study, we examined whether lower levels of
the drug also influenced the transcriptional activity of ER,
without degrading the receptor. For these studies, we used steroid
receptor-driven luciferase reporter assays (Indigo Biosciences) and
concentrations of RO that did not affect cell viability, as
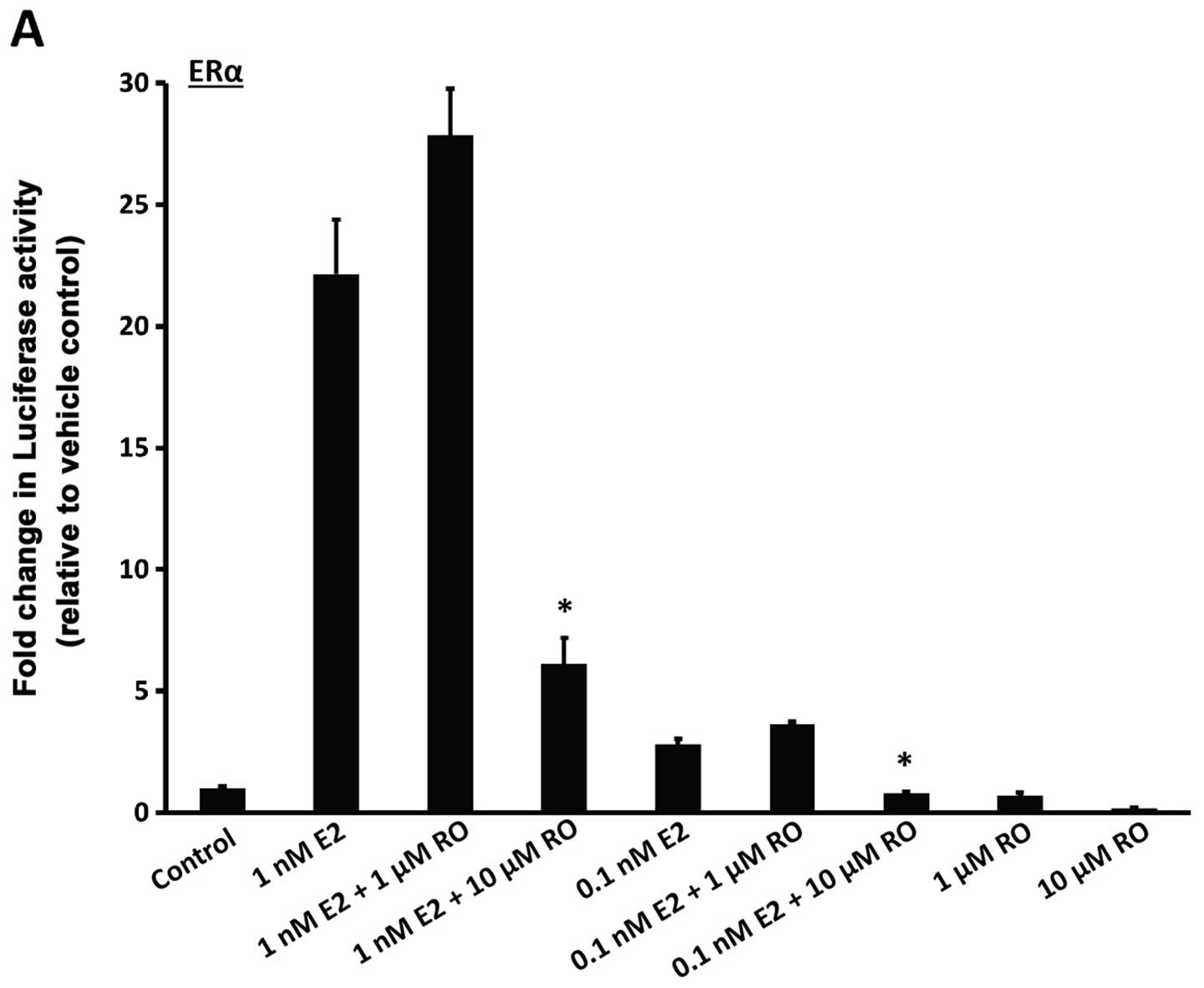
determined from a previous study (8), or ERα protein levels. We found that
treatment with 1 nM 17β-estradiol for 22 h increased luciferase
activity in both ERα and ERβ-linked reporter cells (Fig. 3A and B; bars on left). Induction was
much stronger with ERα than ERβ, an observation in accordance with
a previous study (12). RO caused a
decrease in E2-mediated transcriptional response in a manner that
was dose-dependent. Both ERα and ERβ luciferase activities were
reduced in response to RO (Fig. 3A and
B), although RO-mediated transcriptional suppression was much
greater with 10 μM RO when ERα activity was assessed
(>50%) than when ERβ induced transcription was measured
(<50%).
Having examined the effects of RO on transcriptional
activity mediated by both ERα and ERβ, we used ICI 182,780, a
well-characterized ERα antagonist, to specifically suppress
ERα-mediated transcription (Fig.
3C). ICI 182,780 inhibited transcriptional activity
considerably more potently than RO (~1,000-fold), although,
notably, atorvastatin, another cholesterol biosynthesis inhibitor
that acts on HMG-CoA reductase and is commonly used in humans to
lower cholesterol levels, inhibited ERα-mediated transcriptional
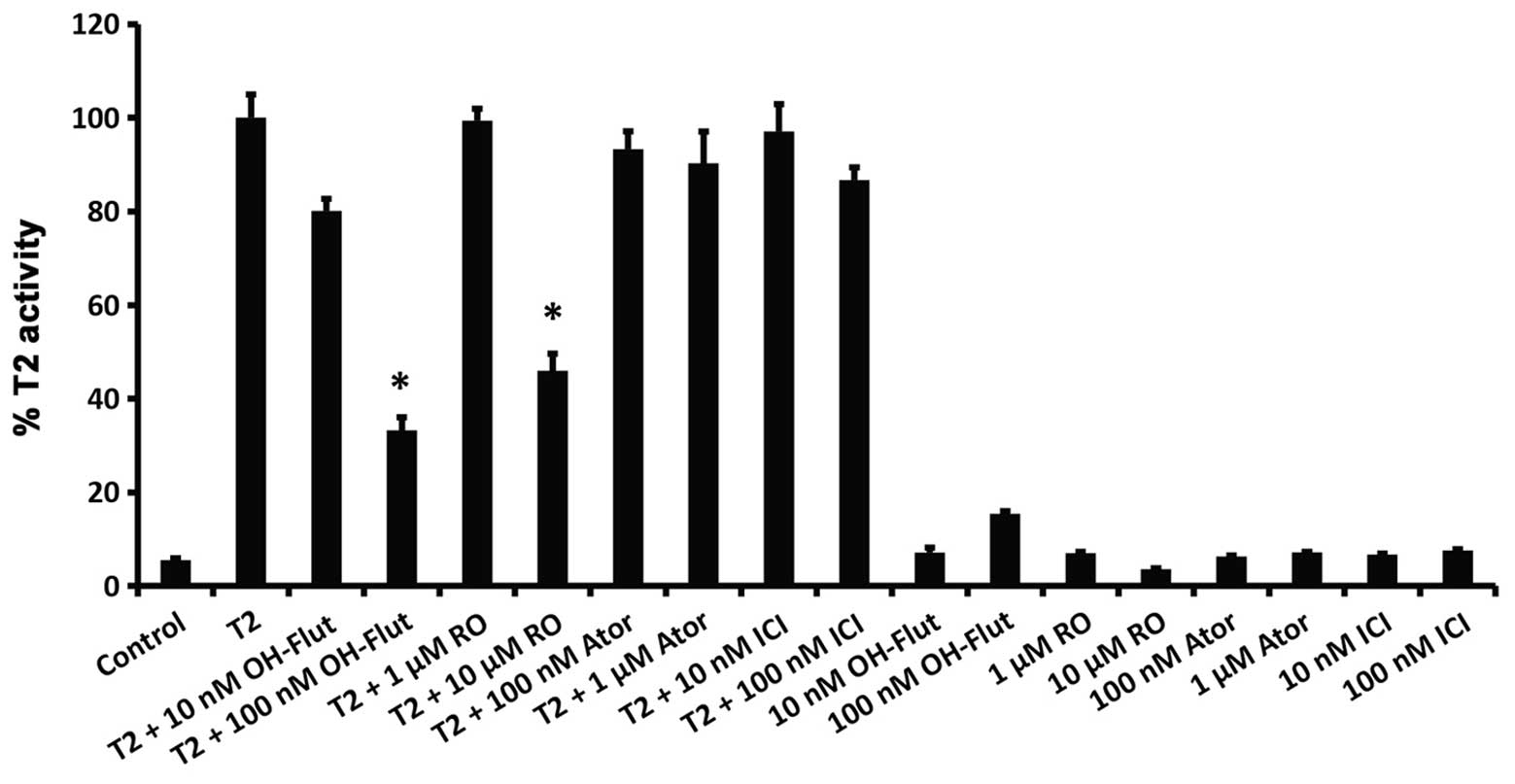
activity at a level comparable to RO (Fig. 3D). Markedly, RO also inhibited a
6α-testosterone-mediated increase in AR transcriptional activity,
which was also blocked by the AR antagonist hydroflutamide
(OH-flut; Fig. 4). However, in
contrast to their suppression of ERα-mediated transcriptional
activity (Fig. 3), neither
atorvastatin nor ICI 182,780 had any inhibitory effect on
AR-mediated transcription (Fig.
4).
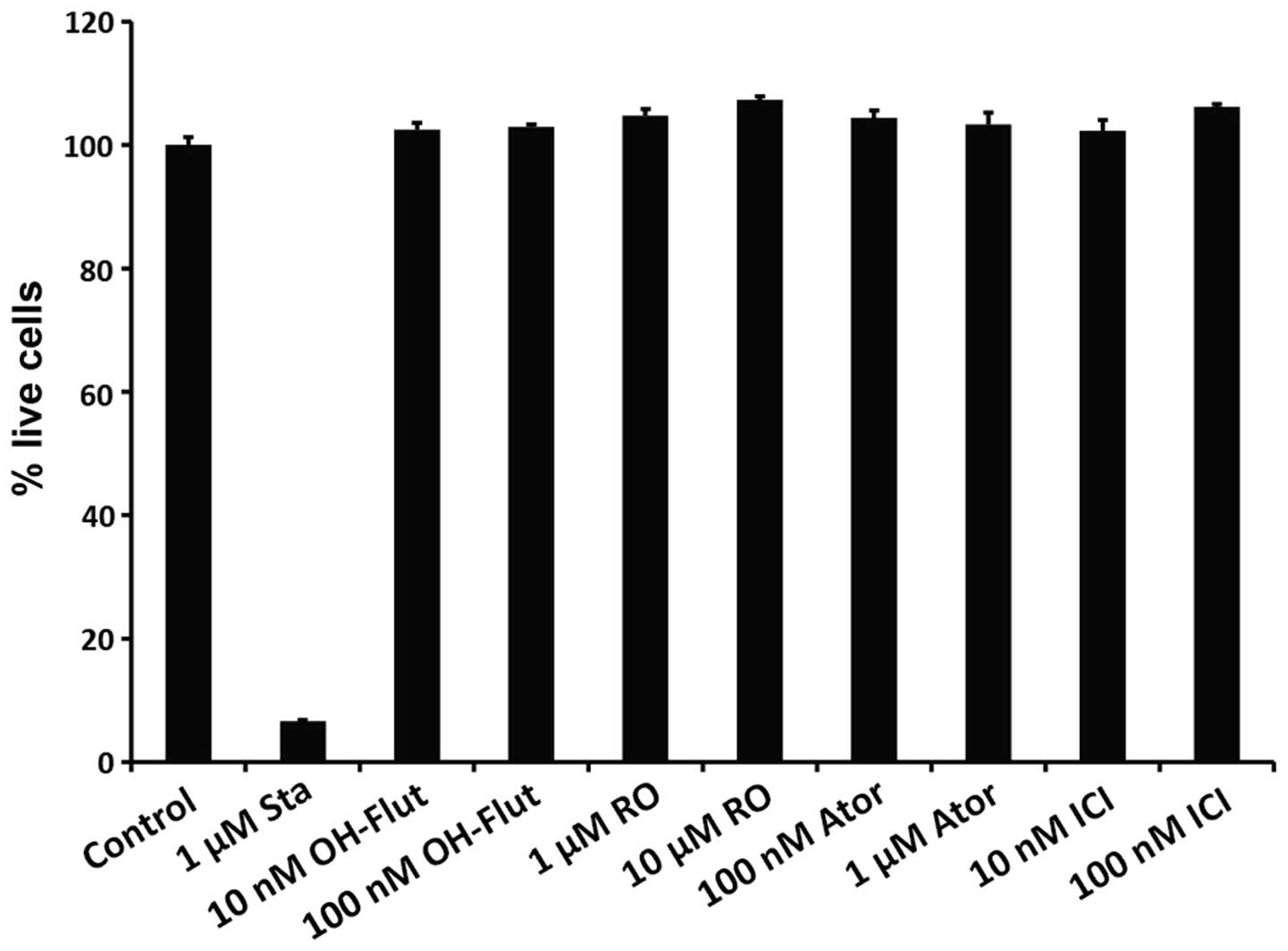
In order to ensure that the observed reduction in
luciferase reporter activity was not due to induced cell apoptosis,
a fluorescence- based LCM assay kit (LCM-01; Indigo Biosciences)
was used concurrently to measure the proportion of live cells in
wells where luciferase activity was measured. As shown in Fig. 5, while the proportion of live cells
was significantly reduced by the apoptosis-inducing compound
staurosporine, all the other compounds tested in the present study
were non-toxic at the concentrations used.
Effect of RO on estradiol-induced PR
expression in BT-474 human breast cancer cells
Confirmation that RO was able to modify the
biological activity of ERα was obtained in BT-474 cells in which
ERα regulates PR levels by increasing PR gene transcription
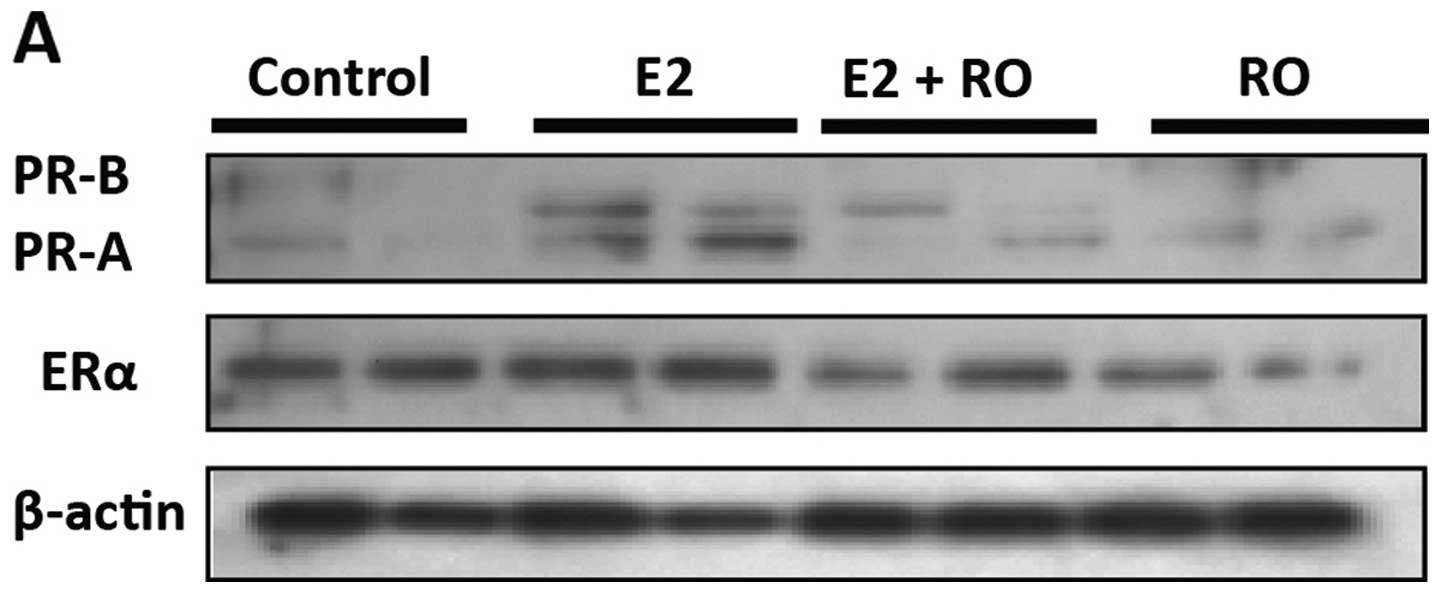
(11). As shown in Fig. 6A, treatment of BT-474 cells with 10
nM E2 for 16 h resulted in increased levels of both PRA and PRB.
E2-mediated increases in PR levels were blocked by RO (5
μM), without any significant loss of ERα. Real-time PCR
confirmed that 5 μM RO had no effect on ERα mRNA expression
in human BT-474 or T47-D breast cancer cells (Fig. 6B).
Discussion
There is growing evidence to suggest that
cholesterol and its metabolites play an important role in the
development of breast cancer (1–3).
Previous studies showed that inhibition of cholesterol synthesis at
different enzymatic steps of the biosynthetic pathway leads to a
suppression of cell growth (13,14),
although the significance of OSC in tumor progression remains
unclear. OSC is a critical enzyme which is involved in the
cyclization of 2,3-oxidosqualene to lanosterol and an increase in
its intra-tumor expression or activity has the potential to raise
the levels of cholesterol metabolites with estrogenic activities.
Such an increase could result in subsequent proliferative effects
within breast cancer cells. In earlier studies involving in
silico analysis, we showed that RO, an inhibitor of cholesterol
synthesis that targets OSC, is a potent ligand with
chemotherapeutic properties, which reduces breast cancer cell
viability (8). This led us to
investigate whether RO may have potential as a chemotherapeutic
agent against a broader range of breast cancer cells.
Our initial goal in the present study was to
determine whether hormone-responsive and hormone-independent tumors
express OSC differently. We hypothesized that differences in OSC
expression may partially account for both increased cholesterol
biosynthesis and higher levels of intracellular estrogen, which
could promote tumor growth. After extensive analysis of a variety
of tumor tissues collected at different stages of development and
from varying types of tumor, we concluded that there were no
significant differences in levels of OSC expression between normal,
hormone responsive, hormone-independent and triple-negative breast
cancers. It remains to be established, however, whether OSC protein
levels are different between the various types of tumor. Based on
these observations, we concluded that expression of OSC is unlikely
to be a prognostic marker for breast cancer.
In our previous study, we showed that RO
significantly reduced the viability of ERα-positive breast cancer
cells (8, Liang et al submitted). However, since levels of
OSC expression did not vary between different tumor types, we
concluded that it is unlikely that the ability of RO to disrupt
tumor cell proliferation is due entirely to its inhibition of OSC.
Since ERα is a major determinant for human breast cancer cell
proliferation, we conducted studies to determine whether RO targets
ERα, initially using non-human mammalian cells engineered to
express human ERα protein, and luciferase reporter gene
functionally linked to an ERα-responsive promoter to assess
17β-estradiol induced luciferase activity. Changes in luciferase
expression in cells treated with RO and other test compounds
provided a sensitive surrogate measure of changes in ERα
transcriptional activity without cellular toxicity. As expected,
ICI 182,780, a well characterized antagonist which acts partially
through downregulation of the receptor (15), blocked the effects of 17β-estradiol
on ERα. RO also inhibited 17β-induced ERα-linked luciferase
expression in a dose-dependent manner, although higher doses of RO
were required to achieve levels of inhibition comparable to ICI
182,780 (Figs. 3 and 4). RO also blocked ERβ activity, although
less potently than its inhibition of ERα. Notably, atorvastatin, an
alternative inhibitor of cholesterol biosynthesis which inhibits
HMG-CoA reductase, also blocked ERα-mediated transcriptional
activity. To determine whether RO, atorvastatin and ICI 182,780
exert differential effects on other steroid receptors, we examined
their capacity to inhibit transcriptional activity of AR, our
rationale being that AR is also involved in breast cancer
progression (10). Among the three
ligands tested (RO, atorvastatin and ICI 182,780) only RO inhibited
AR-mediated transcription, suggesting that the OSC inhibitor
possesses even broader effects than we first anticipated. The
ability to block both ER and AR effects possibly indicates that,
compared with other chemotherapeutic drugs, RO may have additional
advantages with respect to its properties as an anti breast cancer
agent.
In our studies using Indigo kits to measure
transcriptional activity of nuclear receptors, we utilized
non-mammalian cells transfected with human receptor. Following
these analyses, we confirmed that RO also affects human breast
cancer cells by determining its ability to modify the
transcriptional activity of ERα in human BT-474 cells. This was
achieved by assessing the effects of 17β-estradiol on ERα-mediated
induction of PR protein, measuring levels of the latter by western
blotting. PR is directly regulated by ERα in BT-474 breast cancer
cells in a ligand-dependent manner (11). Using western blot analysis, we found
that RO suppressed induction of PR by 17β-estradiol in human BT-474
breast cancer cells (Fig. 6A).
Concomitant analysis of ERα mRNA levels showed that under these
conditions, RO did not affect expression of ERα. This confirms, in
human breast cancer cells, that RO has the ability to suppress the
biological functions of ERα without reducing its levels (Fig. 6A and B).
Given that ERα-induced signal transduction controls
the growth of the majority of breast cancers (16), the results reported in the present
study suggest that RO has the potential to be an effective
chemotherapeutic agent against hormone-responsive breast cancer.
Based on our observations of its ability to inhibit the biological
activities of both ER and AR, further in-depth analysis of the
effects of RO on human breast cancer cells is warranted.
Acknowledgements
The present study was supported by a Department of
Defense Breast Cancer Program grant no. W81XWH-12-1-0191, and by a
Faculty Research Grant from the University of Missouri, Columbia.
S.M.H. is the Zalk Missouri Professor of Tumor Angiogenesis. Funds
to purchase the nanodrop instrument were provided through the
generosity of numerous donors to the Ellis Fischel Cancer Center.
The authors thank Mr. Jason Lee for his assistance with the
figures.
References
|
1
|
Sivente-Poirot S and Poirot M: Cholesterol
metabolism and cancer: the good, the bad and the ugly. Curr Opin
Pharmacol. 12:673–676. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Danilo C and Frank PG: Cholesterol and
breast cancer development. Curr Opin Pharmacol. 12:677–682. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wu Q, Ishikawa T, Sirianni R, Tang H, et
al: 27-Hydroxycholesterol promotes cell-autonomous, ER-positive
breast cancer. Cell Rep. 5:637–645. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Campagnoli C, Pasanisi P, Castellano I,
Abbà C, Brucato T and Berrino F: Postmenopausal breast cancer,
androgens, and aromatase inhibitors. Breast Cancer Res Treat.
139:1–11. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Umetani M, Domoto H, Gormley AK, Yuhanna
IS, et al: 27-Hydroxycholesterol is an endogenous SERM that
inhibits the cardiovascular effects of estrogen. Nat Med.
13:1185–1192. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
DuSell CD, Umetani M, Shaul PW,
Mangelsdorf DJ and McDonnell DP: 27-Hydroxycholesterol is an
endogenous selective estrogen receptor modulator. Mol Endocrinol.
22:65–77. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Michikawa M and Yanagisawa K: Inhibition
of cholesterol production but not of nonsterol isoprenoid products
induces neuronal cell death. J Neurochem. 72:2278–2285. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Grinter SZ, Liang Y, Huang SY, Hyder SM
and Zou X: An inverse docking approach for identifying new
potential anticancer targets. J Mol Graph Model. 29:795–799. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Geisler J, Suzuki T, Helle H, Miki Y, et
al: Breast cancer aromatase expression evaluated by the novel
antibody 677: correlations to intra-tumor estrogen levels and
hormone receptor status. J Steroid Biochem Mol Biol. 118:237–241.
2010. View Article : Google Scholar
|
|
10
|
Shah PD, Gucalp A and Traina TA: The role
of the androgen receptor in triple-negative breast cancer. Womens
Health. 9:351–360. 2013.PubMed/NCBI
|
|
11
|
Saceda M, Grunt TW, Colomer R, Lippman ME,
Lupu R and Martin MB: Regulation of estrogen receptor concentration
and activity by an erbB/HER ligand in breast carcinoma cell lines.
Endocrinology. 137:4322–4330. 1996.PubMed/NCBI
|
|
12
|
Klinge CM: Estrogen receptor interaction
with estrogen response elements. Nucleic Acids Res. 29:2905–2919.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen L, Monti S, Juszczynski P, Ouyang J,
et al: SYK inhibition modulates distinct PI3K/AKT-dependent
survival pathways and cholesterol biosynthesis in diffuse large B
cell lymphomas. Cancer Cell. 23:826–838. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fernández C, Martín M, Gómez-Coronado D
and Lasunción MA: Effects of distal cholesterol biosynthesis
inhibitors on cell proliferation and cell cycle progression. J
Lipid Res. 46:920–929. 2005.PubMed/NCBI
|
|
15
|
Bachelot T, McCool R, Duffy S, Glanville
J, et al: Comparative efficacy of everolimus plus exemestane versus
fulvestrant for hormone-receptor-positive advanced breast cancer
following progression/recurrence after endocrine therapy: a network
meta-analysis. Breast Cancer Res Treat. 143:125–133. 2014.
View Article : Google Scholar
|
|
16
|
Jensen EV and Jordan VC: The estrogen
receptor: a model for molecular medicine. Clin Cancer Res.
9:1980–1989. 2003.
|