Introduction
Lung cancer is one of the most common causes of
cancer-related mortality in the developed world (1,2).
Recurrence has been reported in a large proportion of lung cancer
patients in spite of successful resection of the primary tumor
(1,2). Although some lung tumors are known to
be sensitive to conventional chemotherapeutic agents or certain
molecular targeting agents, many are not (3,4). Thus,
further understanding of the molecular mechanisms involved in lung
carcinogenesis is essential for developing novel therapeutic
strategies.
Our previous studies, which involved a comprehensive
search for the downstream targets of oncogenic KRAS,
identified important molecules involved in lung carcinogenesis
(3,5). Mutations in driver oncogenes, such as
KRAS, EGFR, BRAF, and ALK, were found
to mutually and exclusively occur, and each of these mutations has
been shown to transmit the common essential oncogenic signal that
promotes carcinogenesis (6–8). Therefore, the downstream targets of
oncogenic KRAS participate not only in KRAS-mediated
carcinogenesis, but also in other oncogene-mediated carcinogeneses
(3,5). Thus, an investigation into the
downstream targets of oncogenic KRAS is considered to be an
available strategy that can identify common important molecular
mechanisms involved in lung cancer.
Small non-protein coding RNAs that regulate
messenger RNA levels, namely microRNAs (miRNAs), have been
identified and subsequently implicated in the pathogenesis of
various diseases. The present study investigated the expression
profiles of miRNAs modulated by oncogenic KRAS in airway epithelial
cells. We focused on miR-31 from the miRNAs that were
differentially expressed, and evaluated its potential role in the
development of lung cancer.
Materials and methods
Cell lines and culture
An immortalized human airway epithelial cell line
[16HBE14o, simian virus 40 (SV40)-transformed human bronchial
epithelial cells] described by Cozens et al (9) was kindly provided by D.C. Grunert
(California Pacific Medical Center Research Institute). A sub-clone
of 16HBE14o cells, described as NHBE-T in this study, was used.
Human lung cancer cell lines (A549, H358, H2087, H23, EKVX, H226,
H827, H1819, H441, H4006, HOP62, H1299 and H460) and a human
embryonic kidney cell line (HEK293T) were purchased from the
American Type Culture Collection (ATCC; Manassas, VA, USA). The
human lung cancer cell line LC2/ad was purchased from the Riken
Cell Bank (Tsukuba, Japan). The human lung cancer cell lines, PC1,
PC9 and HARA were from Immuno-Biological Laboratories Co. (Gunma,
Japan). The human lung cancer cell lines, TKB1, TKB2, TKB4, TKB5,
TKB6, TKB7, TKB8, TKB14 and TKB20 were obtained from Dr Hiroshi
Kamma via Dr Takuya Yazawa (Kyorin University School of Medicine)
(10). Primary small airway
epithelial cells (SAEC) were purchased from Sanko Kagaku (Tokyo,
Japan).
Plasmid construction
The pro-retrovirus vector bearing KRAS [wild-type
(WLD), V12, H61, V12/S35, V12/G37 and V12/C40 mutants] (11) and PIK3CA (WLD, E545K mutant, and
H1047R mutant) were previously described (12). Complementary DNA (cDNA) coding EGFR
(NM_001964) and BRAF (NM_004333) was PCR-amplified using cDNA from
NHBE-T cells as a template and inserted into the pro-retrovirus
vector pQCXIP (BD Clontech, Palo Alto, CA, USA). Mutant EGFR
(deletion: E745-A750) was PCR-amplified using cDNA from the PC9
lung cancer cell line. EGFR (L858R mutant) and BRAF (V600E mutant)
were generated by the method of site-directed mutagenesis. DNA
fragments including the miR-31 coding region (MIMAT0000089)
flanking approximately a 100 base-pair margin in both directions
was PCR-amplified and inserted into the pro-retrovirus vector pLHCX
(BD Clontech). Vectors bearing a sense and antisense strand of cDNA
were obtained. A pNsi-based miR-31 blocking vector was purchased
from Takara Bio Inc. (Kyoto, Japan).
Retroviral-mediated gene transfer
The pro-retrovirus vectors bearing the desired
constructs and the pLC10A1 retrovirus-packaging vector (Imgenex,
San Diego, CA, USA) were cotransfected into HEK293T cells with
Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA).
Forty-eight hours after the transfection, conditioned medium was
recovered as a viral solution. The desired genes were introduced by
incubating cells with the viral solution containing 10 μg/ml of
polybren (Sigma, St. Louis, MO, USA). Cells stably expressing the
desired genes were selected with 250 μg/ml of hygromycin B or 1,000
μg/ml of neomycin (both from Invitrogen). The pooled clones were
used for analyses as follows.
miRNA expression profiling
miRNA expression profiles in empty vector (MOCK)-,
KRAS/WLD-, KRAS/V12-, and KRAS/H61-vector transduced NHBE-T cells
were comprehensively evaluated with human miRNA microarray (Human
miRNA Microarray kit release 16.0, 8×60K, cat. no. G4870A; Agilent
Technologies, Santa Clara, CA, USA). Total RNA was extracted from
the cells immediately after completing the selection (5 days
post-transduction) using the RNA easy kit (Qiagen, Hilden,
Germany). Labeling and hybridization was performed according to the
manufacturer’s recommendations (Agilent Technologies).
Colony formation assays
Cells (appropriate count of 1.0×104 to
5.0×104) were seeded onto a 10 cm culture dish (Iwaki,
Tokyo, Japan), and grown for 10 days. Cells were fixed with
methanol and Giemsa-stained, and colonies visible in scanned images
were counted.
Growth curve assays
Cells (2.5×105) were seeded onto a 10 cm
culture dish, and were grown in DMEM (Sigma) with 10 FBS (Sigma) to
a semi-confluent state for 5–7 days. Cells were counted, and
2.5×105 cells were seeded again onto a 10 cm dish.
Several passages were repeated in the same manner. The sum of
population doublings (PDLs) at each point was calculated by the
formula SPDLn = log2
(countn/2.5×105) + SPDLn−1.
Soft agar colony formation assays
Cells (1.25×104) were cultured and grown
in 1 ml of DMEM-based 0.3% agar (agar noble; Becton-Dickinson,
Sparks, MD, USA) containing 10% FBS in 3.5 cm culture dishes
(Iwaki) for 4 weeks. The agars were fixed with a buffered 4%
paraformaldehyde solution, and colonies visible in scanned images
were counted.
Treatment with an inhibitor for MEK and
PI3K
Following infection of the retrovirus vector, cells
stably expressing KRAS were selected for 3 days, and were then
harvested. Cells were treated with an inhibitor for MEK (PD98059,
50 μM) or PI3K (LY294002, 50 μM) [both from Cell Signaling
Technology (CST), Danvers, MA, USA] and their combination for the
last 24 h before the harvest.
Comprehensive search for the downstream
target of miR-31
Gene expression in the empty vector (MOCK)-, miR-31
sense strand (SS)-, and miR-31 antisense (AS) strand-transduced
NHBE-T cells was comprehensively evaluated with a human gene chip
microarray (SurePrint G3 Human Gene Expression 8×60K v2 Microarray
kit, cat. no. G4851B; Agilent Technologies). Total RNA was
extracted from the cells immediately after completing the selection
(5 days post-transduction) using the RNA easy kit (Qiagen).
Labeling and hybridization were performed according to the
manufacturer’s recommendations (Agilent Technologies). Transcripts
whose signal values in the miR-31 SS-transduced cells were 5-fold
higher or lower than that in both the empty vector-transduced cells
and the miR-31 antisense strand-transduced cells were extracted
according to the same flow chart as described in a previous study
(13).
Results
miRNA expression profiling modulated by
oncogenic KRAS
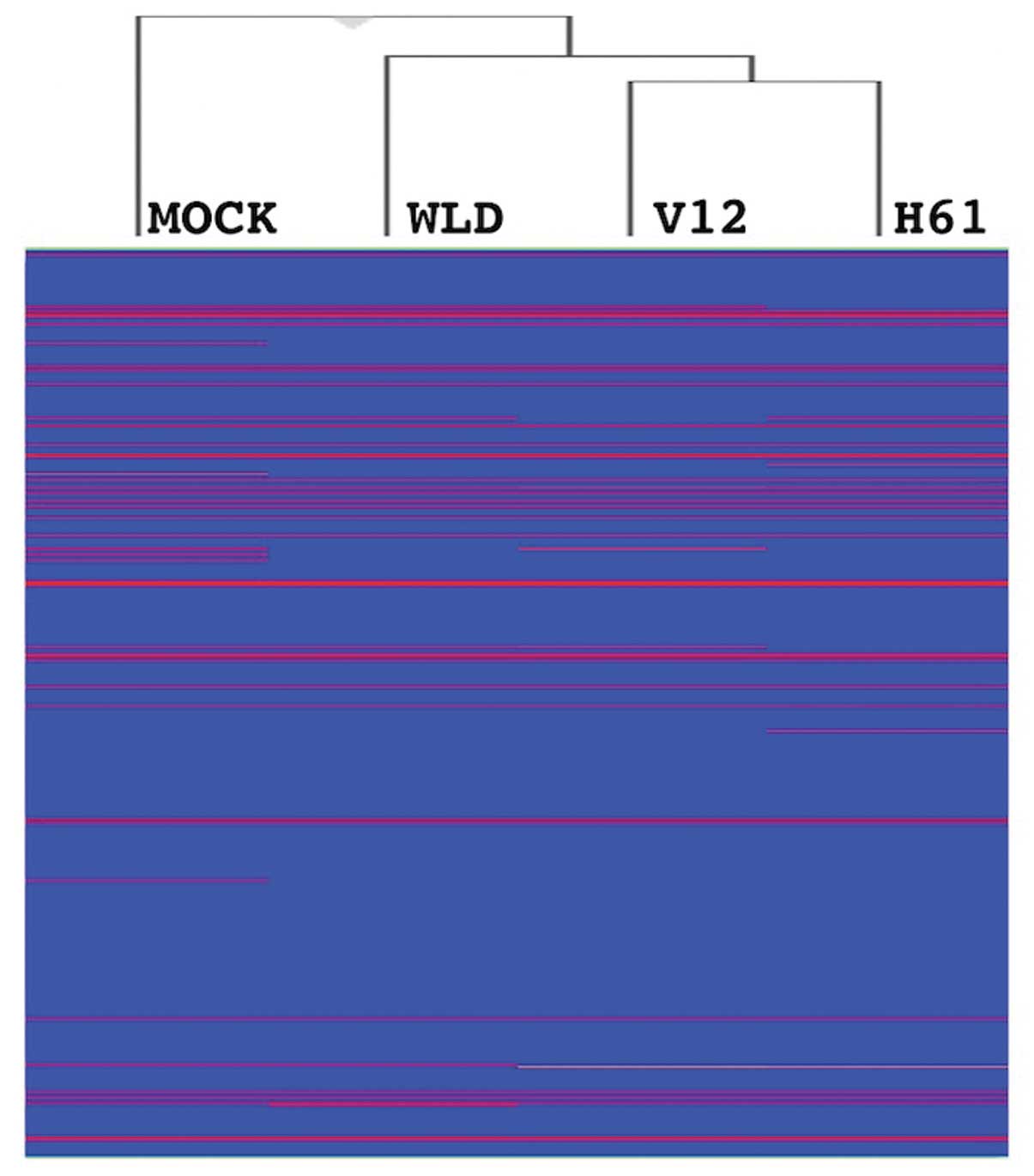
A comprehensive evaluation of the expression of
miRNA revealed that oncogenic KRAS-transduced cells had different
expression profiles from empty vector- and WLD KRAS-transduced
cells, as KRAS/V12- and KRAS/H61-transduced cells were classified
into a distant branch from the others on a dendrogram described by
hierarchical clustering analysis (Fig.
1). The miRNAs that had higher or lower levels in oncogenic
KRAS (KRAS/V12 and KRAS/H61)-transduced cells than in both mock
(MOCK)- and WLD KRAS (KRAS/WLD)-transduced cells are listed in
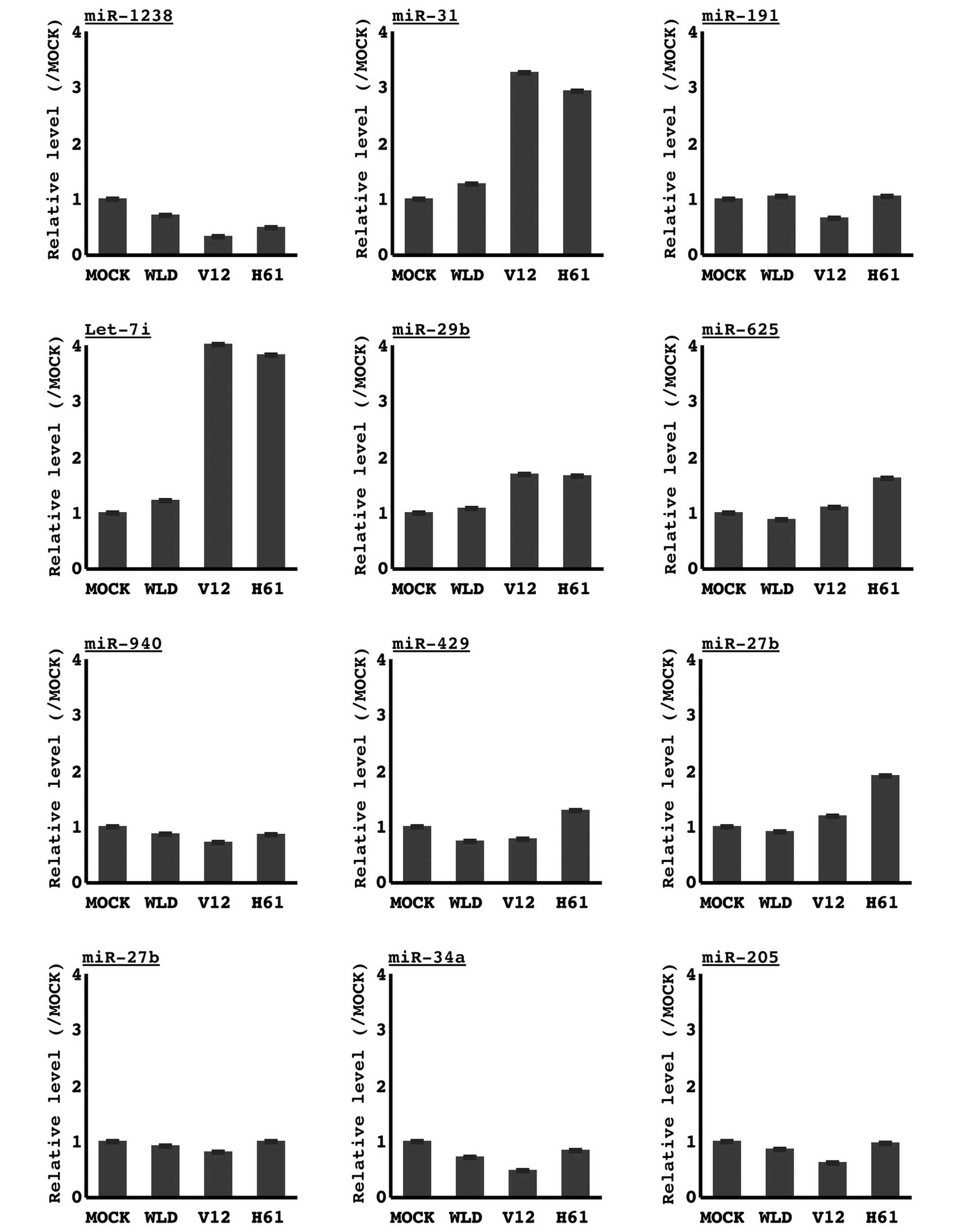
Table I. Quantitative RT-PCR
analysis confirmed that the transduction of oncogenic KRAS induced
the largest change in let-7i and miR-31 (Fig. 2). Therefore, we focused on these two
miRNAs.
 | Table IMicroRNAs differentially expressed by
the oncogenic KRAS. |
Table I
MicroRNAs differentially expressed by
the oncogenic KRAS.
| Name | Accession | MOCK | KRAS/WLD | KRAS/V12 | KRAS/H61 |
|---|
| Upregulated |
| hsa-miR-1238 | MIMAT0005593 | 4.53 | 8.45 | 11.49 | 9.58 |
| hsa-miR-31 | MIMAT0000089 | 6.40 | 5.96 | 16.53 | 12.68 |
|
hsa-miR-191* | MIMAT0001618 | 5.27 | 9.33 | 11.24 | 9.82 |
| hsa-let-7i | MIMAT0000415 | 109.44 | 98.74 | 224.70 | 164.02 |
|
hsa-miR-31* | MIMAT0004504 | 15.01 | 13.44 | 22.80 | 19.14 |
|
hsa-miR-29b* | MIMAT0004514 | 2.70 | 2.66 | 7.31 | 3.25 |
| hsa-miR-29b | MIMAT0000100 | 95.87 | 66.36 | 141.79 | 100.44 |
| hsa-miR-625 | MIMAT0003294 | 4.70 | 4.03 | 5.85 | 4.74 |
| hsa-miR-940 | MIMAT0004983 | 12.07 | 11.92 | 15.11 | 12.11 |
| Downregulated |
| hsa-miR-30c | MIMAT0000244 | 16.95 | 12.52 | 12.13 | 9.82 |
| hsa-miR-15b | MIMAT0000417 | 208.10 | 154.26 | 148.56 | 102.32 |
| hsa-miR-28-5p | MIMAT0000085 | 5.66 | 4.09 | 3.99 | 3.60 |
|
hsa-miR-30a* | MIMAT0000088 | 8.71 | 6.70 | 5.73 | 4.91 |
| hsa-miR-23b | MIMAT0000418 | 8.93 | 6.36 | 5.87 | 4.78 |
| hsa-miR-130a | MIMAT0000425 | 55.99 | 39.19 | 36.31 | 26.76 |
| hsa-miR-210 | MIMAT0000267 | 41.26 | 28.93 | 26.16 | 22.94 |
| hsa-miR-16 | MIMAT0000069 | 157.08 | 110.57 | 96.19 | 73.35 |
| hsa-miR-30e | MIMAT0000692 | 6.70 | 4.47 | 4.03 | 3.68 |
| hsa-miR-196a | MIMAT0000226 | 27.48 | 19.75 | 15.85 | 11.31 |
| hsa-miR-4286 | MIMAT0016916 | 264.56 | 205.35 | 150.73 | 138.68 |
| hsa-miR-15a | MIMAT0000068 | 57.49 | 40.25 | 31.70 | 29.54 |
| hsa-let-7b | MIMAT0000063 | 104.48 | 76.06 | 54.29 | 39.47 |
| hsa-miR-205 | MIMAT0000266 | 116.34 | 74.88 | 59.41 | 50.10 |
| hsa-miR-34a | MIMAT0000255 | 36.32 | 24.83 | 16.71 | 19.56 |
| hsa-miR-1246 | MIMAT0005898 | 52.63 | 36.00 | 23.66 | 21.83 |
| hsa-miR-27b | MIMAT0000419 | 12.85 | 8.09 | 5.49 | 4.76 |
| hsa-miR-429 | MIMAT0001536 | 11.46 | 7.44 | 4.71 | 3.73 |
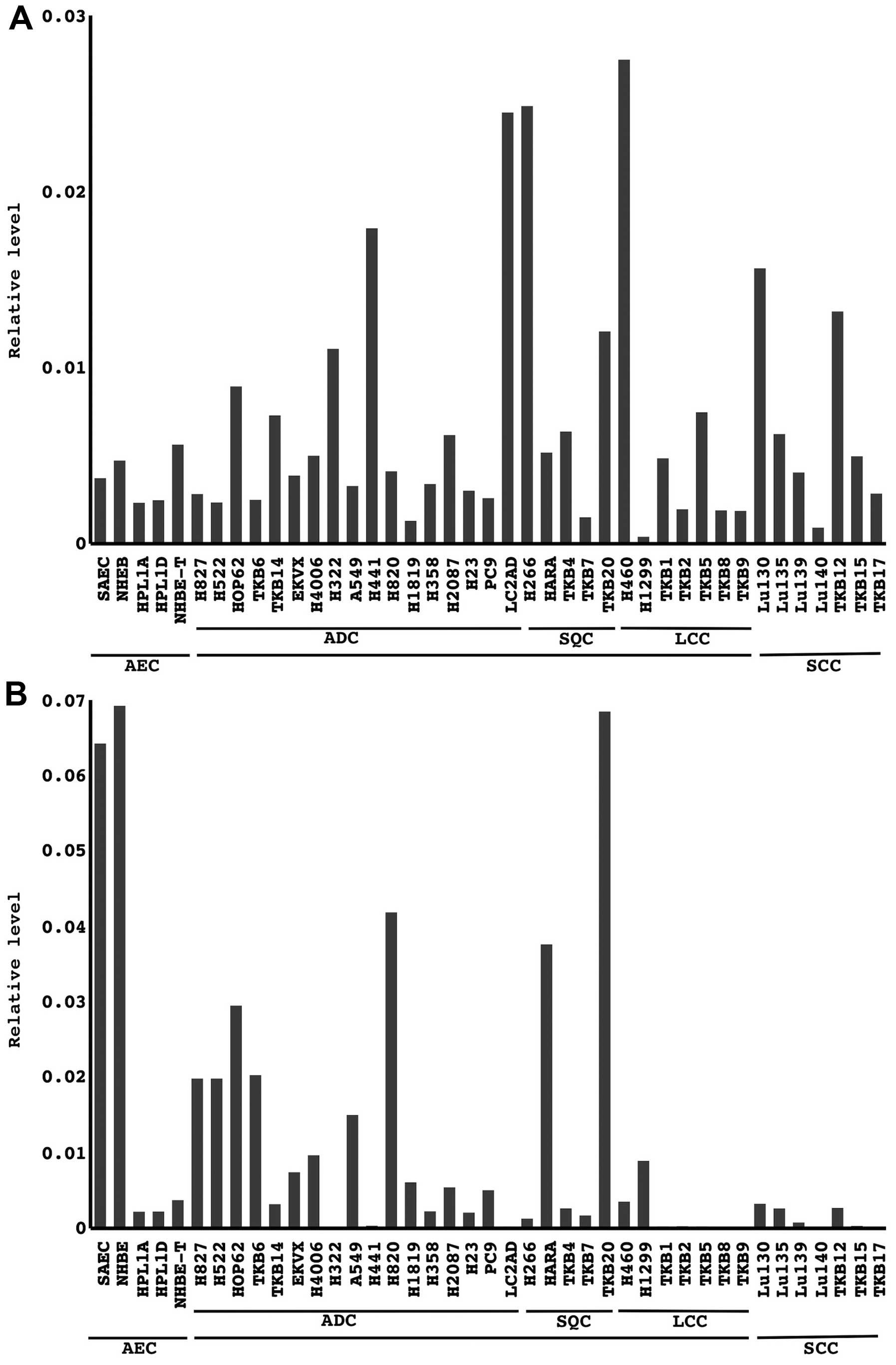
Let-7i and miR-31 expression in lung
cancer cell lines
Let-7i and miR-31 expression levels were analyzed by
quantitative RT-PCR analysis. Let-7i was expressed at various
levels in all the cells examined. Let-7i levels were higher in
cancer cell lines than in non-cancerous immortalized cell lines and
primary airway epithelial cells (Fig.
3). In contrast, miR-31 expression levels were lower in cancer
cell lines, and some cell lines [27.5%, 11/40, the loss of miR-31
expression was frequently detected in LCC cell lines (80.0%, 4/5)]
completely lost its expression (Fig.
2). These results prompted us to further investigate the
potential significance of the loss of miR-31 expression in lung
carcinogenesis.
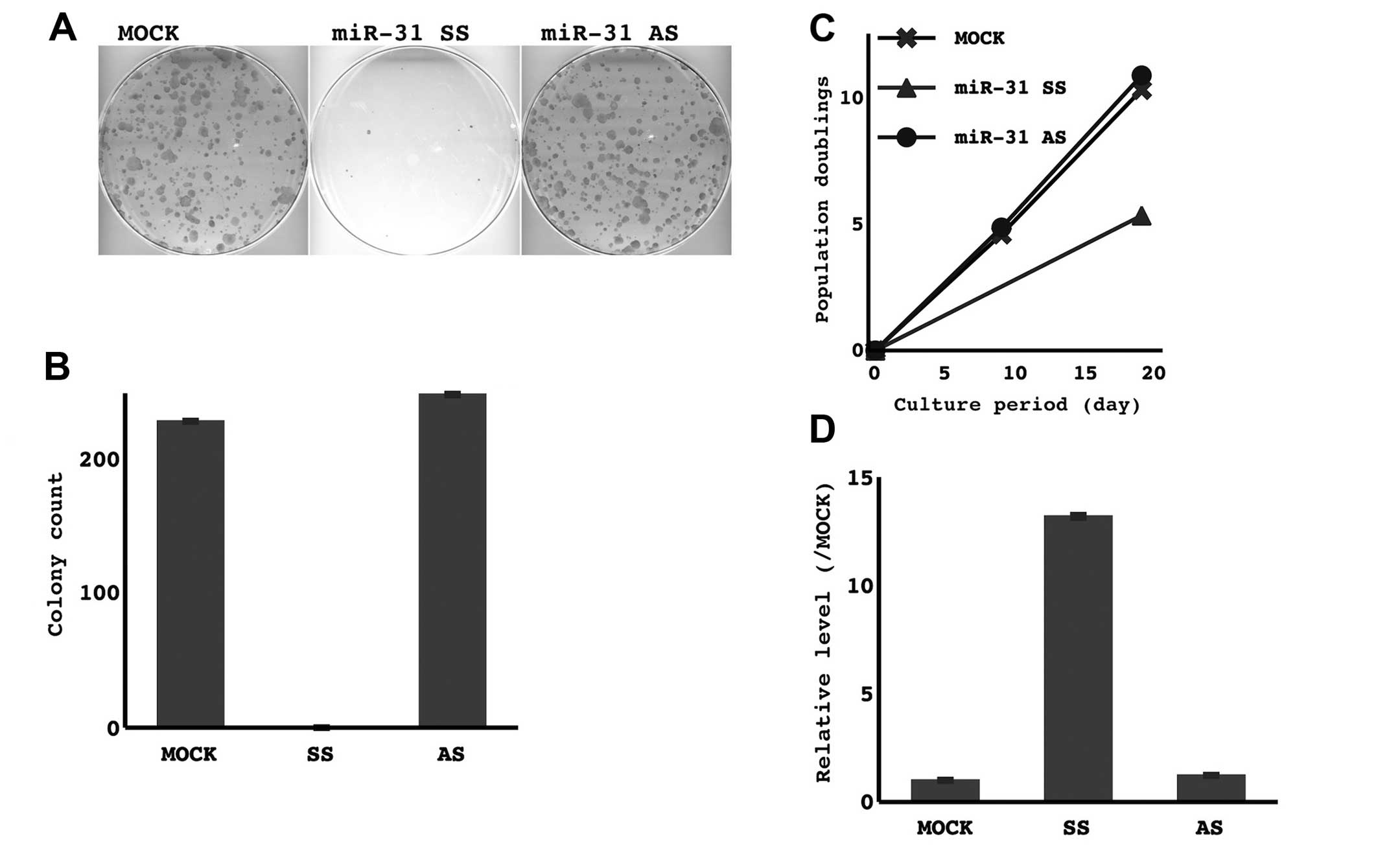
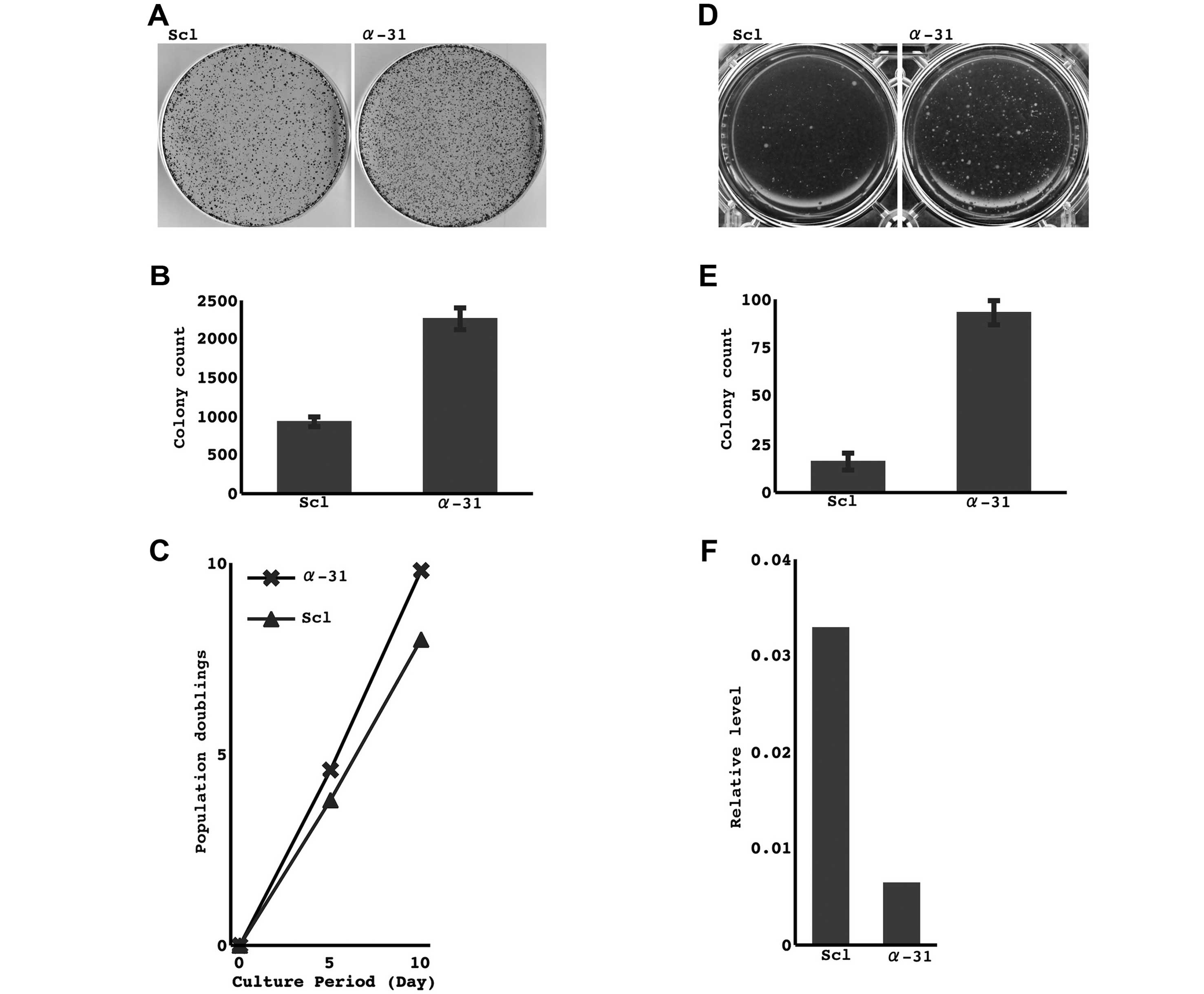
Effect of the restoration and knockdown
of miR-31 on cell growth
The restoration of the miR-31 SS, but not the
antisense strand, markedly suppressed the growth of cancer cell
lines that lost miR-31 expression, as it decreased the formation of
colonies and prolonged the doubling time (Fig. 4, TKB1 and H441 cells were examined.
A representative result in TKB1 cells is shown). On the other hand,
the knockdown of miR-31 expression in cell lines that retained its
expression resulted in an enhancement in their growth activities in
both ordinary media and soft agar (Fig.
5, HARA and A549 cells were examined. A representative result
in HARA cells is shown).
Comprehensive search for potential genes
modulated by miR-31
A comprehensive evaluation of gene expression
profiles revealed that miR-31 SS-transduced cells had a different
expression profile from empty vector (mock)- and miR-31 antisense
strand (AS)-transduced cells, as miR-31SS-transduced cells were
classified into the most distant branch on a dendrogram described
from a hierarchical clustering analysis (Fig. 6). Transcripts with expression levels
that were >5-fold higher in miR-31SS-transduced cells than in
mock- and miR-31AS-transduced cells were extracted (Tables II and III).
| Table IIUpregulated transcripts. |
Table II
Upregulated transcripts.
| | MOCK | miR-31SS | miR-31AS | | |
|---|
| |
|
|
| | |
|---|
| Symbol | Accession | Signal | Flag | Signal | Flag | Signal | Flag | miR-31SS/MOCK | miR-31SS/AS |
|---|
| NUDT10 | NM_153183 | 0.0119 | A | 1.1978 | P | 0.0124 | A | 100.8360 | 96.2198 |
| RIPK3 | NM_006871 | 0.0114 | A | 1.0102 | P | 0.0121 | A | 88.4318 | 83.3022 |
| C2CD4B | NM_001007595 | 0.0245 | A | 0.5805 | P | 0.0115 | A | 23.6809 | 50.5356 |
| SERPINI1 | NM_005025 | 0.0112 | A | 0.4145 | P | 0.0116 | A | 37.0894 | 35.6275 |
| KRTAP10-11 | NM_198692 | 0.0123 | A | 0.4065 | P | 0.0116 | A | 33.1742 | 35.1714 |
| SLC24A4 | NM_153647 | 0.0357 | A | 1.0444 | P | 0.0314 | A | 29.2833 | 33.2618 |
| CCDC140 | NM_153038 | 0.0110 | A | 0.3725 | P | 0.0115 | A | 33.9644 | 32.4076 |
| PLA2R1 | NM_007366 | 0.0103 | A | 0.2964 | P | 0.0106 | A | 28.7619 | 27.8916 |
| HTR3D | NM_182537 | 0.0106 | A | 0.3012 | P | 0.0111 | A | 28.3353 | 27.0245 |
| CXCR2P1 | NR_002712 | 0.0128 | A | 0.2771 | P | 0.0103 | A | 21.7125 | 27.0160 |
| STEAP2 | NM_001244945 | 0.0099 | A | 0.2577 | P | 0.0102 | A | 25.9085 | 25.1678 |
| GSG1L | NM_001109763 | 0.0148 | A | 0.2657 | P | 0.0108 | A | 17.9319 | 24.5156 |
| PLAC9 | NM_001012973 | 0.0111 | A | 0.2532 | P | 0.0109 | A | 22.7315 | 23.2710 |
| MPPED2 | NM_001584 | 0.0119 | A | 0.2784 | P | 0.0124 | A | 23.4476 | 22.5310 |
| NKX2-8 | NM_014360 | 0.0322 | A | 0.3877 | P | 0.0179 | A | 12.0409 | 21.6246 |
| ZMAT1 | NM_001011657 | 0.0112 | A | 0.2303 | P | 0.0116 | A | 20.6436 | 19.8318 |
| CYYR1 | NM_052954.2 | 0.0255 | A | 0.2456 | P | 0.0128 | A | 9.6355 | 19.2358 |
| MYL1 | NM_079420 | 0.0095 | A | 0.1850 | P | 0.0098 | A | 19.3932 | 18.7914 |
| ENTPD1 | NM_001776 | 0.0106 | A | 0.2072 | P | 0.0111 | A | 19.5553 | 18.6842 |
| OTOL1 | NM_001080440 | 0.0111 | A | 0.2118 | P | 0.0115 | A | 19.1511 | 18.4960 |
| BPIFB4 | NM_182519 | 0.0106 | A | 0.2031 | P | 0.0111 | A | 19.1675 | 18.3275 |
| STARD6 | NM_139171 | 0.0104 | A | 0.1980 | P | 0.0109 | A | 18.9708 | 18.2463 |
| FAM70A | NM_017938 | 0.0100 | A | 0.1856 | P | 0.0103 | A | 18.6179 | 18.0836 |
| SLC34A2 | NM_006424 | 0.0117 | A | 0.2084 | P | 0.0121 | A | 17.8522 | 17.1757 |
| TCL6 | NR_028288 | 0.0119 | A | 0.2186 | P | 0.0128 | A | 18.3689 | 17.0699 |
| TMEM64 | NM_001008495 | 0.0401 | A | 0.4789 | P | 0.0290 | A | 11.9343 | 16.5116 |
| SLC25A21 | NM_030631 | 0.0109 | A | 0.1860 | P | 0.0113 | A | 17.0261 | 16.4283 |
| FABP7 | NM_001446 | 0.0139 | A | 0.1728 | P | 0.0107 | A | 12.4500 | 16.2039 |
| ADAMTSL2 | NM_014694 | 0.0249 | A | 0.1737 | P | 0.0110 | A | 6.9690 | 15.7766 |
| OR2W5 | NM_001004698 | 0.0112 | A | 0.1846 | P | 0.0117 | A | 16.4571 | 15.7667 |
| MLANA | NM_005511 | 0.0094 | A | 0.1372 | P | 0.0098 | A | 14.5261 | 13.9842 |
| LDLRAD2 | NM_001013693 | 0.0119 | A | 0.1733 | P | 0.0124 | A | 14.6248 | 13.9382 |
| OR5K1 | NM_001004736 | 0.0095 | A | 0.1334 | P | 0.0098 | A | 14.0985 | 13.5742 |
| TMPRSS4 | NM_019894 | 0.0283 | A | 0.1621 | P | 0.0122 | A | 5.7287 | 13.2791 |
| GLT25D2 | NM_015101 | 0.0105 | A | 0.1410 | P | 0.0109 | A | 13.4839 | 12.8923 |
| FMN2 | NM_020066 | 0.0088 | A | 0.1484 | P | 0.0120 | A | 16.7833 | 12.4044 |
| SLC3A1 | NM_000341 | 0.0154 | A | 0.2346 | P | 0.0207 | A | 15.2038 | 11.3567 |
| AMICA1 | NM_153206 | 0.0185 | A | 0.2714 | P | 0.0247 | A | 14.6324 | 10.9989 |
| FAM186B | NM_032130 | 0.0919 | P | 1.1598 | P | 0.1056 | P | 12.6267 | 10.9792 |
| SNORD115-2 | NR_003294 | 0.0116 | A | 0.1296 | P | 0.0123 | A | 11.1625 | 10.5393 |
| PAX5 | NM_016734 | 0.0110 | A | 0.1198 | P | 0.0114 | A | 10.9324 | 10.5101 |
| MAGEC2 | NM_016249 | 0.0137 | A | 0.1294 | P | 0.0125 | A | 9.4181 | 10.3264 |
| IPW | NR_023915 | 0.0221 | A | 0.3631 | P | 0.0353 | A | 16.3945 | 10.2860 |
| TNFSF11 | NM_033012 | 0.0107 | A | 0.1121 | P | 0.0111 | A | 10.5095 | 10.0805 |
| SIGLECP3 | NR_002804 | 0.0627 | P | 0.3595 | P | 0.0359 | A | 5.7295 | 10.0019 |
| WDR49 | AK097556 | 0.0096 | A | 0.0992 | P | 0.0099 | A | 10.3910 | 9.9812 |
| SATL1 | NM_001012980 | 0.0120 | A | 0.1243 | P | 0.0127 | A | 10.3606 | 9.7853 |
| SLC19A3 | NM_025243 | 0.0121 | A | 0.1239 | P | 0.0127 | A | 10.2372 | 9.7655 |
| DPP6 | BC035912 | 0.0249 | A | 0.1543 | P | 0.0160 | A | 6.2027 | 9.6431 |
| OR5H2 | NM_001005482 | 0.0156 | A | 0.1053 | P | 0.0109 | A | 6.7301 | 9.6315 |
| CIB4 | NM_001029881 | 0.0117 | A | 0.1166 | P | 0.0122 | A | 9.9576 | 9.5572 |
| WDFY4 | NM_020945 | 0.0122 | A | 0.1211 | P | 0.0128 | A | 9.9557 | 9.4762 |
| SAMD13 | NM_001010971 | 0.0105 | A | 0.1029 | P | 0.0109 | A | 9.8322 | 9.4019 |
| NBPF6 | NM_001143988 | 0.0113 | A | 0.1090 | P | 0.0117 | A | 9.6855 | 9.2810 |
| FAM135B | NM_015912 | 0.0097 | A | 0.0922 | P | 0.0100 | A | 9.4969 | 9.2206 |
| OR4A47 | NM_001005512 | 0.0105 | A | 0.1659 | P | 0.0181 | A | 15.8434 | 9.1669 |
| CCL22 | NM_002990 | 0.0117 | A | 0.1145 | P | 0.0126 | A | 9.7614 | 9.0799 |
| SLC5A4 | NM_014227 | 0.0105 | A | 0.0987 | P | 0.0109 | A | 9.4252 | 9.0167 |
| SH3TC1 | NM_018986 | 0.0838 | P | 0.6744 | P | 0.0752 | P | 8.0521 | 8.9695 |
| MYH7 | NM_000257 | 0.0119 | A | 0.1112 | P | 0.0124 | A | 9.3740 | 8.9612 |
| TXLNG2P | NR_045129 | 0.0111 | A | 0.1045 | P | 0.0117 | A | 9.4000 | 8.9166 |
| NUBPL | NM_025152 | 0.0104 | A | 0.0950 | P | 0.0109 | A | 9.0971 | 8.7016 |
| CNGA3 | NM_001298 | 0.0094 | A | 0.1537 | P | 0.0177 | A | 16.2720 | 8.6960 |
| EGR4 | NM_001965 | 0.0105 | A | 0.0938 | P | 0.0109 | A | 8.9198 | 8.6280 |
| IL24 | NM_001185156 | 1.4258 | P | 9.1516 | P | 1.1788 | P | 6.4187 | 7.7634 |
| RXFP1 | NM_021634 | 0.0111 | A | 0.0888 | P | 0.0115 | A | 8.0062 | 7.7382 |
| CCDC85C | NM_001144995 | 0.0116 | A | 0.1157 | P | 0.0154 | A | 9.9729 | 7.4922 |
| TRIM36 | NM_001017397 | 0.0108 | A | 0.1024 | P | 0.0137 | A | 9.4777 | 7.4839 |
| TACC1 | BC041391 | 0.0128 | A | 0.1323 | P | 0.0177 | A | 10.3064 | 7.4633 |
| FAM24B | NM_152644 | 0.0140 | A | 0.0911 | P | 0.0126 | A | 6.5027 | 7.2256 |
| ITGAL | NM_002209 | 0.0668 | P | 0.4498 | P | 0.0641 | P | 6.7361 | 7.0141 |
| GMCL1P1 | NR_003281 | 0.0104 | A | 0.3532 | P | 0.0506 | A | 33.9062 | 6.9811 |
| IL24 | NM_001185156 | 10.1283 | P | 66.0420 | P | 9.7012 | P | 6.5205 | 6.8076 |
| PTPN20A | NM_001042387 | 0.0097 | A | 0.0671 | P | 0.0101 | A | 6.9039 | 6.6542 |
| SRPX | NM_006307 | 0.0090 | A | 0.0602 | P | 0.0093 | A | 6.6622 | 6.4412 |
| RORC | NM_005060 | 0.0096 | A | 0.0797 | P | 0.0124 | A | 8.3190 | 6.4372 |
| HDAC11 | NM_001136041 | 0.0688 | P | 0.5473 | P | 0.0856 | P | 7.9591 | 6.3925 |
| CRHBP | NM_001882 | 0.0094 | A | 0.0623 | P | 0.0098 | A | 6.6587 | 6.3675 |
| SPTSSB | NM_001040100 | 0.0111 | A | 0.0738 | P | 0.0117 | A | 6.6527 | 6.3147 |
| SERPINB11 | NM_080475 | 0.0110 | A | 0.1245 | P | 0.0201 | A | 11.2800 | 6.2069 |
| CC2D2B | XM_003403528 | 0.0102 | A | 0.0656 | P | 0.0106 | A | 6.4287 | 6.2016 |
| DNAH17 | NM_173628 | 0.0097 | A | 0.1610 | P | 0.0263 | A | 16.6227 | 6.1282 |
| DUOXA2 | BX537581 | 0.0087 | A | 0.0552 | P | 0.0090 | A | 6.3524 | 6.1045 |
| SLC35G3 | NM_152462 | 0.0088 | A | 0.0557 | P | 0.0092 | A | 6.3135 | 6.0811 |
| SLC17A4 | NM_005495 | 0.0105 | A | 0.0667 | P | 0.0110 | A | 6.3493 | 6.0686 |
| MEOX2 | NM_005924 | 0.0118 | A | 0.0726 | P | 0.0123 | A | 6.1790 | 5.8922 |
| CLDN8 | NM_199328 | 0.0106 | A | 0.0630 | P | 0.0109 | A | 5.9634 | 5.7753 |
| TMEM108 | NM_023943 | 0.0179 | A | 0.2513 | P | 0.0440 | A | 13.9997 | 5.7122 |
| DPP6 | NM_001039350 | 0.0124 | A | 0.2982 | P | 0.0533 | A | 23.9789 | 5.5933 |
| FAM164A | NM_016010 | 0.0103 | A | 0.0598 | P | 0.0107 | A | 5.8098 | 5.5618 |
| GSTTP1 | NR_003081 | 0.0118 | A | 0.0823 | P | 0.0148 | A | 6.9710 | 5.5445 |
| OR4F6 | NM_001005326 | 0.0118 | A | 0.0611 | P | 0.0113 | A | 5.1841 | 5.4305 |
| HEATR7B1 | XM_001721240 | 0.0153 | A | 0.1492 | P | 0.0277 | A | 9.7300 | 5.3768 |
| DPYS | NM_001385 | 0.0100 | A | 0.0554 | P | 0.0104 | A | 5.5306 | 5.3504 |
| OR10Z1 | NM_001004478 | 0.0121 | A | 0.0661 | P | 0.0127 | A | 5.4614 | 5.2210 |
| PARP10 | NM_032789 | 0.1746 | P | 1.1198 | P | 0.2158 | P | 6.4149 | 5.1885 |
| PIEZO2 | AK092226 | 0.0088 | A | 0.0466 | P | 0.0091 | A | 5.2934 | 5.0979 |
| ACCN5 | NM_017419 | 0.0090 | A | 0.0478 | P | 0.0094 | A | 5.2910 | 5.0893 |
| CAPS2 | AF251056 | 0.0105 | A | 0.0636 | P | 0.0126 | A | 6.0511 | 5.0303 |
| RBPMS | AK057533 | 0.0124 | A | 0.0900 | P | 0.0180 | A | 7.2453 | 5.0094 |
| Table IIIDownregulated transcripts. |
Table III
Downregulated transcripts.
| | MOCK | miR-31SS | miR-31AS | | |
|---|
| |
|
|
| | |
|---|
| Symbol | Accession | Signal | Flag | Signal | Flag | Signal | Flag | miR-31SS/MOCK | miR-31SS/AS |
|---|
| DYTN | NM_001093730 | 58.1445 | P | 7.8986 | P | 169.4681 | P | 0.1358 | 0.0466 |
| MGP | NM_000900 | 0.2369 | P | 0.0116 | A | 0.2249 | P | 0.0490 | 0.0516 |
| SAMD9L | NM_152703 | 0.1643 | P | 0.0149 | A | 0.2193 | P | 0.0908 | 0.0680 |
| SAA2 | NM_001127380 | 0.3662 | P | 0.0299 | A | 0.3758 | P | 0.0817 | 0.0796 |
| SAA4 | NM_006512 | 0.6052 | P | 0.0521 | P | 0.6040 | P | 0.0861 | 0.0863 |
| SAA1 | NM_000331 | 10.6803 | P | 1.0635 | P | 10.1069 | P | 0.0996 | 0.1052 |
| CCR1 | NM_001295 | 0.0807 | P | 0.0092 | A | 0.0858 | P | 0.1144 | 0.1077 |
| SAA2 | NM_030754 | 5.8777 | P | 0.6367 | P | 5.8671 | P | 0.1083 | 0.1085 |
| CIITA | NM_000246 | 0.0657 | P | 0.0104 | A | 0.0925 | P | 0.1590 | 0.1129 |
| SPTBN1 | NM_003128 | 0.0835 | P | 0.0101 | A | 0.0893 | P | 0.1210 | 0.1132 |
| MGP | NM_001190839 | 0.5979 | P | 0.0643 | P | 0.5636 | P | 0.1075 | 0.1141 |
| XAF1 | NM_017523 | 0.6196 | P | 0.0819 | P | 0.7122 | P | 0.1322 | 0.1150 |
| BTBD8 | NM_183242 | 0.0712 | P | 0.0083 | A | 0.0659 | P | 0.1162 | 0.1255 |
| PLAC8 | NM_001130715 | 0.0686 | P | 0.0086 | A | 0.0683 | P | 0.1259 | 0.1265 |
| OLFM4 | NM_006418 | 0.0751 | P | 0.0085 | A | 0.0667 | P | 0.1128 | 0.1269 |
| CLCA2 | NM_006536 | 0.1033 | P | 0.0105 | A | 0.0787 | P | 0.1017 | 0.1335 |
| CCL2 | NM_002982 | 0.5769 | P | 0.0771 | P | 0.5550 | P | 0.1337 | 0.1390 |
| SAMD9L | NM_152703 | 0.7186 | P | 0.1299 | P | 0.9136 | P | 0.1807 | 0.1421 |
| TRERF1 | NM_033502 | 0.0967 | P | 0.0131 | A | 0.0889 | P | 0.1354 | 0.1472 |
| PLEKHG4 | NM_015432 | 0.0871 | P | 0.0157 | A | 0.1017 | P | 0.1797 | 0.1539 |
| ITPK1-AS1 | NR_002808 | 0.0947 | P | 0.0120 | A | 0.0760 | P | 0.1262 | 0.1572 |
| DEFB127 | NM_139074 | 0.1531 | P | 0.0088 | A | 0.0538 | P | 0.0572 | 0.1627 |
| TFEC | NM_012252 | 0.2166 | P | 0.0095 | A | 0.0576 | P | 0.0438 | 0.1648 |
| MEOX1 | NM_004527 | 0.0839 | P | 0.0131 | A | 0.0792 | P | 0.1559 | 0.1652 |
| GGT5 | NM_001099781 | 1.6842 | P | 0.2891 | P | 1.7144 | P | 0.1717 | 0.1687 |
| RPGR | NM_001034853 | 0.0956 | P | 0.0179 | A | 0.1027 | P | 0.1869 | 0.1741 |
| CD99L2 | BC025729 | 0.0623 | P | 0.0109 | A | 0.0623 | P | 0.1749 | 0.1748 |
| CCNG2 | NM_004354 | 0.0846 | P | 0.0156 | A | 0.0878 | P | 0.1848 | 0.1781 |
| TNFSF15 |
NM_005118 | 3.6060 | P | 0.6136 | P | 3.4121 | P | 0.1702 | 0.1798 |
| IL7R | NM_002185 | 0.7080 | P | 0.1181 | P | 0.6427 | P | 0.1668 | 0.1837 |
| HSH2D | NM_032855 | 0.4028 | P | 0.0771 | P | 0.4128 | P | 0.1915 | 0.1869 |
| BDKRB1 | NM_000710 | 0.1140 | P | 0.0108 | A | 0.0578 | P | 0.0948 | 0.1869 |
| SPARC | NM_003118 | 12.4315 | P | 2.4446 | P | 12.9549 | P | 0.1966 | 0.1887 |
| HSD17B11 | NM_016245 | 0.1320 | P | 0.0245 | A | 0.1294 | P | 0.1856 | 0.1894 |
| EBI3 | NM_005755 | 0.0981 | P | 0.0150 | A | 0.0789 | P | 0.1531 | 0.1904 |
| ADRA1B | NM_000679 | 1.2211 | P | 0.2366 | P | 1.2080 | P | 0.1938 | 0.1959 |
Involvement of different oncogenic
pathways in the induction of miR-31
The expression of miR-31 was also induced by the
transduction of oncogenic EGFR [deletion mutation (deletion:
E746-A750), point mutation L858R], but not by oncogenic BRAF
(V600E) or oncogenic PIK3CA (E545K, H1047R) (Fig. 7). The expression of miR-31 was
consistently not induced by specific mutants in KRAS that activated
MEK, RAL-GDS or PI3K (Fig. 7).
Inhibitors for MEK (the BRAF-MEK-ERK pathway) or PI3Kinase (the
PIK3CA-mediated pathway) interfered with the induction of miR-31 by
oncogenic KRAS.
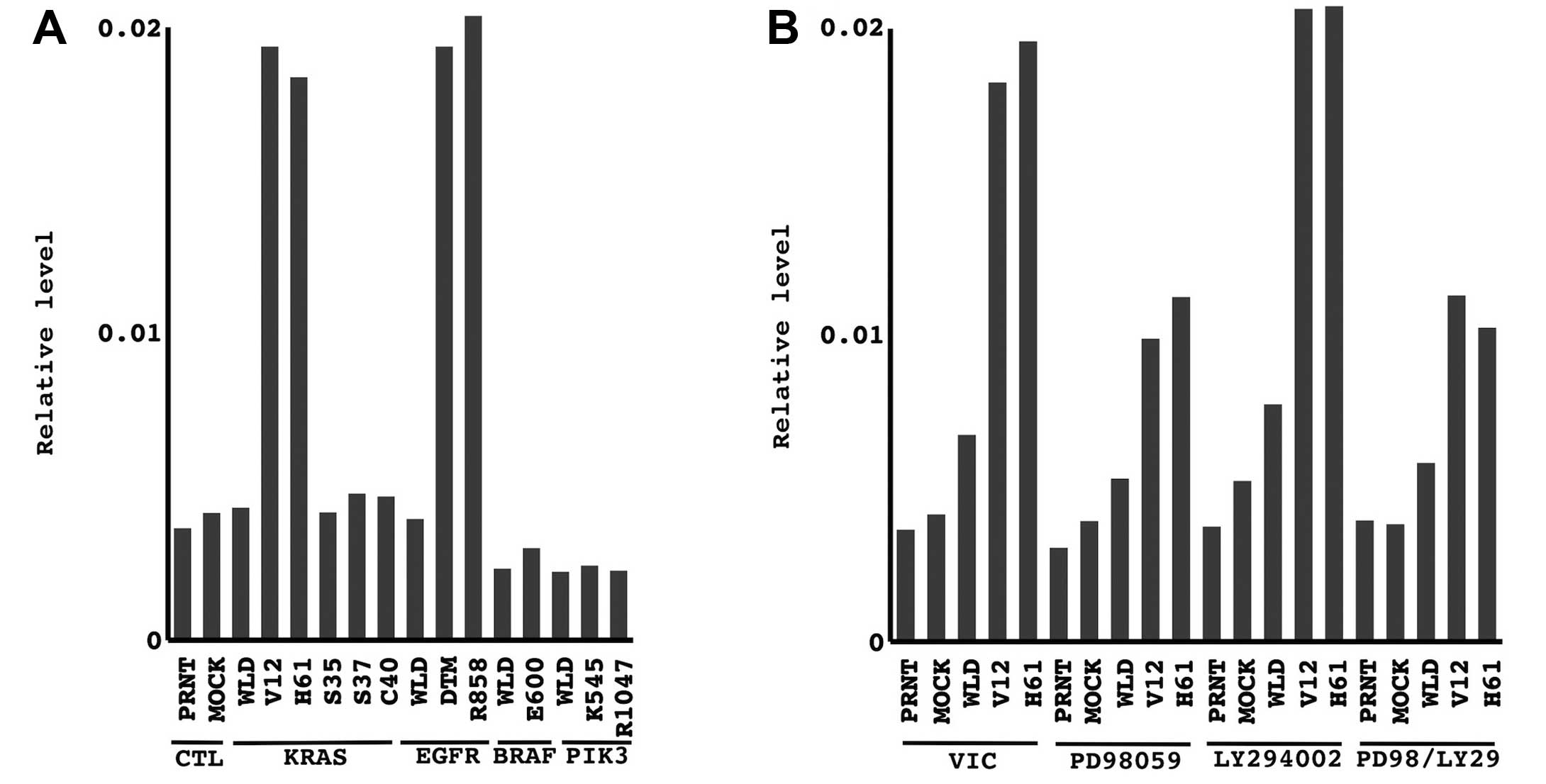 | Figure 7The modulation of miR-31 expression
through the essential oncogenic pathway. (A) The level of miR-31 in
NHBE-T cells transduced with different mutants of KRAS, EGFR, BRAF
and PIK3CA was evaluated. The copy number of miR-31 and U6 snRNA
was measured by quantitative RT-PCR. The level of miR-31 normalized
to that of U6 snRNA is presented. PRNT, parental NHBE-T; MOCK,
empty vector-transduced NHBE-T; WLD, wild-type KRAS-transduced
NHBE-T; V12, oncogenic mutant of KRAS/V12-transduced NHBE-T; H61,
oncogenic mutant of KRAS/H61-transduced NHBE-T; S35, a mutant of
KRAS activating only the MEK pathway-transduced NHBE-T; G37, a
mutant of KRAS activating only the Ral-GDS pathway-transduced
NHBE-T; C40, a mutant of KRAS activating only the PI3K
pathway-transduced NHBE-T. (B) The level of miR-31 in NHBE-T cells
treated with the inhibitors for MEK (PD98059, 50 μM), PI3K
(LY294002, 50 μM), and their combination (PD98/LY29, 50 μM each)
for 24 h was evaluated. CTL, control; VIC, vehicle. |
Discussion
The findings of an initial study on breast cancer
suggested that miR-31 could be a tumor suppressor that especially
inhibited the invasive and metastatic spread of neoplastic cells
(14). Previous studies have
supported this initial observation and identified the potential
molecular mechanisms responsible for the inhibition of invasion and
metastasis by miR-31 (15). miR-31
was also shown to be downregulated in gastric cancer and was
suggested to function as a tumor suppressor (16). In contrast, previous studies
reported that miR-31 was upregulated in colorectal cancer and may
also promote the invasive and metastatic spread of neoplastic cells
(17,18). Thus, the potential role of miR-31 in
carcinogenesis may differ depending on the type of cancer. The
expression of miR-31 in lung cancer has generally been reported to
be higher in tumor tissue than in the corresponding non-tumorous
tissue (19–25). However, miR-31 levels have been
shown to vary in primary lung tumors. Some tumors expressed miR-31
at lower levels than the corresponding non-tumorous tissue or did
not express it at all. miR-31 expression was markedly reduced or
completely lost in some lung cancer cell lines (Fig. 3). This result suggested that miR-31
may be a suppressor in lung carcinogenesis. The restoration of
miR-31 in lung cancer cell lines that had lost its expression
markedly attenuated their growth activities (Fig. 4). The knockdown of miR-31 expression
in cell lines that retained its expression enhanced oncogenic
phenotypes such as anchorage-independent growth activity (Fig. 5). These results supported miR-31
being a suppressor. However, a recent study demonstrated the
oncogenic role of miR-31 in lung carcinogenesis using experiments
with different cell lines from the ones used in the present study
(26). miR-31 may play a
pleiotropic role in the development of individual tumors. Further
investigations are required to resolve this issue.
In the present study, miR-31 was upregulated not
only by oncogenic KRAS, but also by oncogenic EGFR in vitro
(Fig. 7). Neither oncogenic BRAF
nor oncogenic PIK3CA induced its expression (Fig. 7). Similarly, the specific mutant of
KRAS that activated the MEK-mediated pathway only and another
specific mutant that activated the PI3K-mediated pathway only did
not induce the expression of miR-31 (Fig. 7). On the other hand, the MEK
inhibitor, but not PI3K inhibitor, attenuated the oncogenic
KRAS-induced expression of miR-31 (Fig.
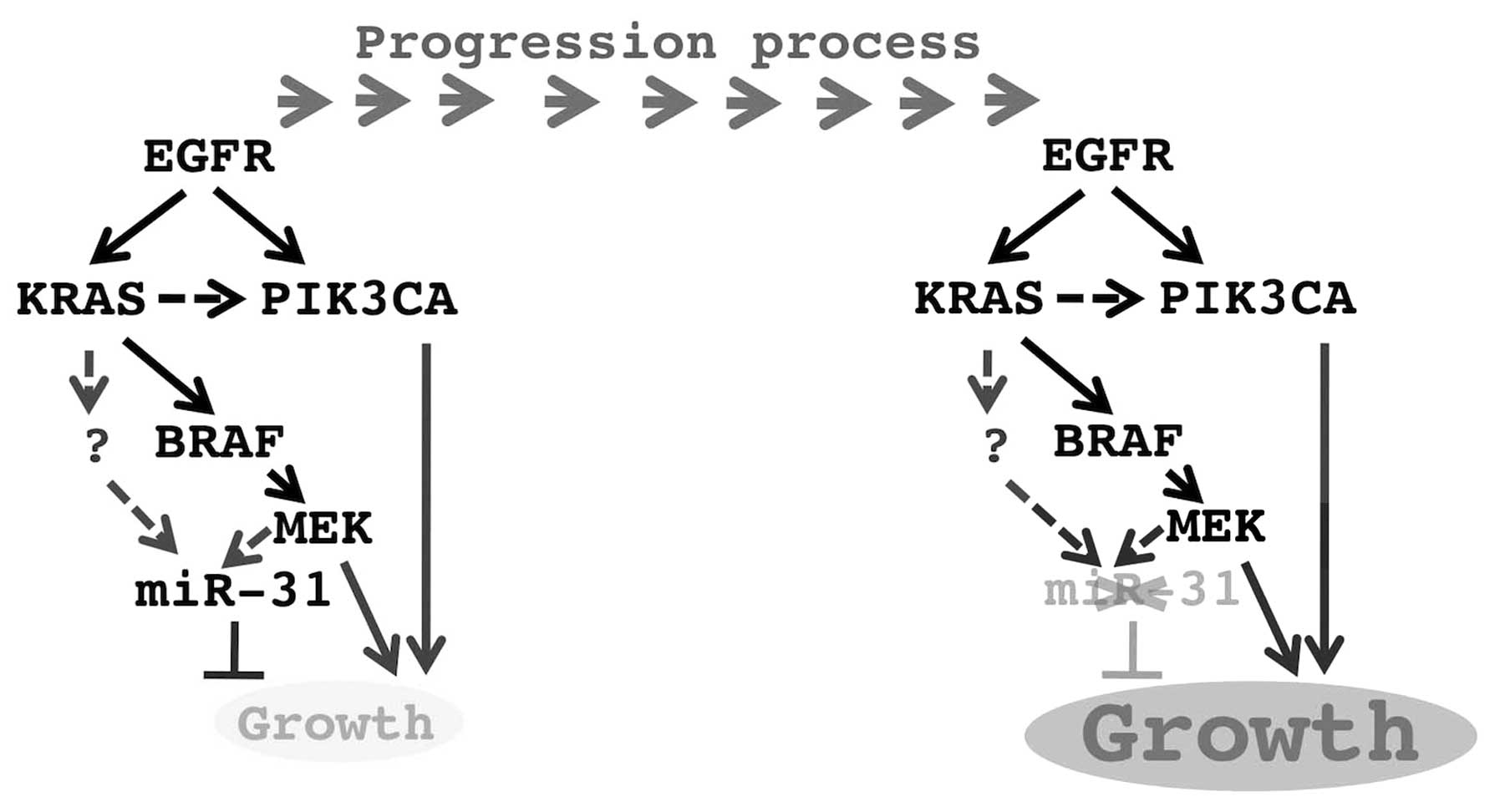
7). These results suggest that miR-31 could be a common player
that regulates the KRAS/EGFR-mediated essential oncogenic pathway
(Fig. 8). The MEK-mediated pathway
may be necessary, but not sufficient for the induction of miR-31
expression (Fig. 8). The
PI3K-mediated pathway may not be involved in its regulation
(Fig. 8). A novel pathway may be
inducing the expression of miR-31 in cooperation with the
MEK-mediated pathway.
This comprehensive search for the potential target
of miR-31 revealed that the downregulated transcripts included many
molecules mediating the cytokine/chemokine signaling pathway
(Tables II and III). TNFSF15, in particular, has been
published on an online site as the predicted downstream target for
miR-31 (http://mirdb.org/miRDB/index.html). TNFSF15, a member
of the tumor necrosis factor (TNF) and TNF receptor superfamilies,
regulates cell growth and apoptosis in an autocrine manner, and has
been reported to suppress cancer cell growth by inhibiting
angiogenesis (27). Thus, TNFSF15
could be a factor that participates in miR-31-induced growth
modulations. Future studies that focus on the potential downstream
targets of miR-31 may provide insight into the molecular mechanisms
involved in lung cancer.
The present study comprehensively searched miRNAs,
the expressions of which were regulated by oncogenic KRAS, and
focused on miR-31 in order to investigate its potential involvement
in lung carcinogenesis. The expression of miR-31 was markedly
attenuated in lung cancer cell lines. The restoration of miR-31 in
lung cancer cell lines that lost its expression attenuated their
growth activities. The knockdown of miR-31 expression in cell lines
that retained its expression enhanced oncogenic phenotypes. These
results suggest that miR-31 may be a suppressor, the loss of which
may promote lung carcinogenesis.
Acknowledgements
This study was supported by the Japanese Ministry of
Education, Culture, Sports and Science (Tokyo Japan), and by a
grant from the Yokohama Medical Facility (Yokohama, Japan). We
especially thank Emi Honda and Misa Otara (Kanagawa Prefectural
Cardiovascular and Respiratory Center Hospital, Yokohama, Japan)
for their assistance.
References
|
1
|
Hoffman PC, Cohen EE, Masters GA, et al:
Carboplatin plus vinorelbine with concomitant radiation therapy in
advanced non-small cell lung cancer: a phase I study. Lung Cancer.
38:65–71. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Spira A and Ettinger DS: Multidisciplinary
management of lung cancer. N Engl J Med. 350:379–392. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Okudela K, Woo T and Kitamura H: KRAS gene
mutations in lung cancer: particulars established and issues
unresolved. Pathol Int. 60:651–660. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Woo T, Okudela K, Yazawa T, et al:
Prognostic value of KRAS mutations and Ki-67 expression in stage I
lung adenocarcinomas. Lung Cancer. 65:355–362. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Okudela K, Yazawa T, Ishii J, et al:
Down-regulation of FXYD3 expression in human lung cancers: its
mechanism and potential role in carcinogenesis. Am J Pathol.
175:2646–2656. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pao W and Girard N: New driver mutations
in non-small-cell lung cancer. Lancet Oncol. 12:175–180. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
van Eijk R, Licht J, Schrumpf M, et al:
Rapid KRAS, EGFR, BRAF and PIK3CA
mutation analysis of fine needle aspirates from non-small-cell lung
cancer using allele-specific qPCR. PLoS One. 6:e177912011.
|
|
8
|
Xu J, He J, Yang H, et al: Somatic
mutation analysis of EGFR, KRAS, BRAF and PIK3CA in 861 patients
with non-small cell lung cancer. Cancer Biomark. 10:63–69.
2012.PubMed/NCBI
|
|
9
|
Cozens AL, Yezzi MJ, Kunzelmann K, et al:
CFTR expression and chloride secretion in polarized immortal human
bronchial epithelial cells. Am J Respir Cell Mol Biol. 10:38–47.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yazawa T, Kamma H, Fujiwara M, et al: Lack
of class II transactivator causes severe deficiency of HLA-DR
expression in small cell lung cancer. J Pathol. 187:191–199. 1999.
View Article : Google Scholar
|
|
11
|
Okudela K, Yazawa T, Suzuki T, Sugimura H
and Kitamura H: Role of 3′-phosphoinositides in oncogenic
KRAS-induced modulation of shape and motility of airway epithelial
cells. Pathol Int. 59:28–37. 2009.
|
|
12
|
Okudela K, Suzuki M, Kageyama S, et al:
PIK3CA mutation and amplification in human lung cancer.
Pathol Int. 57:664–671. 2007. View Article : Google Scholar
|
|
13
|
Okudela K, Yazawa T, Woo T, et al:
Down-regulation of DUSP6 expression in lung cancer: its mechanism
and potential role in carcinogenesis. Am J Pathol. 175:867–881.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Valastyan S, Reinhardt F, Benaich N, et
al: A pleiotropically acting microRNA, miR-31, inhibits breast
cancer metastasis. Cell. 137:1032–1046. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Augoff K, Das M, Bialkowska K, McCue B,
Plow EF and Sossey-Alaoui K: miR-31 is a broad regulator of
β1-integrin expression and function in cancer cells. Mol Cancer
Res. 9:1500–1508. 2011.
|
|
16
|
Zhang Y, Guo J, Li D, et al:
Down-regulation of miR-31 expression in gastric cancer tissues and
its clinical significance. Med Oncol. 27:685–689. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cottonham CL, Kaneko S and Xu L: miR-21
and miR-31 converge on TIAM1 to regulate migration and invasion of
colon carcinoma cells. J Biol Chem. 285:35293–35302. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Slaby O, Svoboda M, Fabian P, et al:
Altered expression of miR-21, miR-31, miR-143 and miR-145 is
related to clinicopathologic features of colorectal cancer.
Oncology. 72:397–402. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guan P, Yin Z, Li X, Wu W and Zhou B:
Meta-analysis of human lung cancer microRNA expression profiling
studies comparing cancer tissues with normal tissues. J Exp Clin
Cancer Res. 31:542012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jang JS, Jeon HS, Sun Z, et al: Increased
miR-708 expression in NSCLC and its association with poor survival
in lung adenocarcinoma from never smokers. Clin Cancer Res.
18:3658–3667. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gao W, Yu Y, Cao H, et al: Deregulated
expression of miR-21, miR-143 and miR-181a in non small cell lung
cancer is related to clinicopathologic characteristics or patient
prognosis. Biomed Pharmacother. 64:399–408. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Võsa U, Vooder T, Kolde R, et al:
Identification of miR-374a as a prognostic marker for survival in
patients with early-stage nonsmall cell lung cancer. Genes
Chromosomes Cancer. 50:812–822. 2011.PubMed/NCBI
|
|
23
|
Xing L, Todd NW, Yu L, Fang H and Jiang F:
Early detection of squamous cell lung cancer in sputum by a panel
of microRNA markers. Mod Pathol. 23:1157–1164. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu L, Todd NW, Xing L, et al: Early
detection of lung adenocarcinoma in sputum by a panel of microRNA
markers. Int J Cancer. 127:2870–2878. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tan X, Qin W, Zhang L, et al: A 5-microRNA
signature for lung squamous cell carcinoma diagnosis and hsa-miR-31
for prognosis. Clin Cancer Res. 17:6802–6811. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Meng W, Ye Z, Cui R, et al: MicroRNA-31
predicts the presence of lymph node metastases and survival in lung
adenocarcinoma patients. Clin Cancer Res. 19:5423–5433. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Duan L, Yang G, Zhang R, Feng L and Xu C:
Advancement in the research on vascular endothelial growth
inhibitor (VEGI). Target Oncol. 7:87–90. 2012. View Article : Google Scholar : PubMed/NCBI
|