Introduction
Burkitt’s lymphoma originates from follicular
germinal centers, it is the high-grade malignant B cell lymphoma.
Efforts are ongoing to develop innovative and effective therapies.
An important part of this process is to understand the mechanism of
cell death induced by potential chemotherapeutic agents.
Autophagy is a tightly regulated catabolic process
whereby cells degrade their own components by enveloping them in
double-membrane vesicles referred to as autophagosomes and
targeting them for lysosomal degradation. Furthermore, it plays an
important role in tumorigenesis and therapy (1). Studies of autophagy in different cell
types and under different conditions have provided conflicting
results regarding the influence of autophagy on cell death
(2,3). Autophagy is activated during
environmental stress, such as nutrient starvation or hypoxia,
thereby promoting cell survival; however, hyperactive autophagy
seriously disturbs the coordination of cell metabolism and finally
causes cell death, known as autophagic cell death or type II
programmed cell death (type II PCD) (4,5).
Autophagic cell death is a caspase-independent cell death pattern,
which is different from apoptosis (type I PCD). However, recent
reports suggest that autophagy and apoptosis often share similar
effectors and regulators and a complex crosstalk exists between the
two processes (6).
Arsenic trioxide (As2O3) has
been used successfully in the treatment of patients with newly
diagnosed acute promyelocytic leukemia (APL) (7,8). It
has also been reported that As2O3 can also be
used for treating other hematological malignancies and solid tumors
(9–11). Numerous studies have demonstrated
that the antitumor mechanism of As2O3 is very
complicated and it may result from causing cell cycle arrest and
inducing tumor cell apoptosis (12). However, the detailed mechanisms of
As2O3-mediated cell death are not fully
understood. In the present study, we evaluated the role of
autophagy and the relationship between autophagy and apoptosis
during Raji cell death induced by As2O3. Our
findings may provide a theoretical reference for further study on
the antitumor mechanism of As2O3.
Materials and methods
Cells and reagents
Burkitt’s lymphoma Raji cells were purchased from
Shanghai Institute of Biochemistry and Cell Biology (Shanghai,
China). As2O3, 3-methyladenine (3-MA), MTT,
RPMI-1640 medium and monodansylcadaverine (MDC) were purchased from
Sigma (St. Louis, MO, USA). Fetal bovine serum (FBS) was from
Sijiqing Biotechnology Co. (Hangzhou, China). The antibodies for
caspase-3, Beclin-1, Bcl-2, P62, LC3 and β-actin were obtained from
Cell Signaling Technology (Danvers, MA, USA). All primers for
beclin-1, bcl-2 and β-actin were synthesized by Takara Corporation
(Japan) and SYBR Premix Ex Taq and PrimeScript RT reagents were
also from Takara. Raji cells were cultured in RPMI-1640 medium with
10% (v/v) FBS, 100 U/ml penicillin and 100 μg/ml streptomycin at
37°C in the presence of 95% air, 5% CO2 with medium
changes every 2 days. Cells in the mid-log phase were used in the
experiments.
Cell proliferation analysis
Cells were seeded at a density of 5×104
cells/ml in 96-well plates. After treatment, cell viability was
evaluated by an MTT colorimetric assay. The spectrophotometric
absorbance of the sample was measured using a Powerwave X plate
reader (Bio-Tek, Winooski, VT, USA) at 570 nm. All samples were
carried out in sextuplicate.
Flow cytometric analysis of apoptosis and
cell cycle
For detection of As2O3-induced
apoptosis, 106 cells were collected and suspended in
binding buffer (400 μl) and incubated with Annexin V-FITC and
propidium iodide (PI) for 0.5 h and then suspended in binding
buffer. The samples were analyzed by flow cytometry (Beckman
Coulter, Miami, FL, USA). For cell cycle analysis,
~1×106 cells were collected and fixed overnight in 70%
ethanol at 4°C. Cells were then washed with PBS and stained with PI
in the presence of DNase-free RNase. After 30 min incubation at
room temperature in the dark, cells within the cell cycle
compartments were determined by flow cytometer.
Detection of autophagosome
Cells were fixed with 2% paraformaldehyde and 2%
glutaraldehyde in 0.1 mol/l phosphate buffer (pH 7.4), followed by
1% OsO4. After dehydration, thin sections were stained
with uranyl acetate and lead citrate for observation under JEM-1230
transmission electron microscopy (JEOL, Tokyo, Japan).
MDC fluorescent staining
MDC has been proposed as a special tracer for
autophagic vacuoles (13). The
autophagic vacuoles were labeled with 0.05 mmol/l MDC in PBS at
37°C for 1 h. After incubation, cells were washed with PBS and
immediately analyzed by fluorescence microscopy.
Western blot analysis
At the end of the designated treatments, Raji cells
were lysed in RIPA lysis buffer (Beyotime, P0013B) with 1 mM PMSF.
Equal amounts of protein were separated by SDS-PAGE and transferred
onto PVDF membranes. After blocking with 5% non-fat dried milk in
PBS with 0.5 ml/l Tween-20 for 2 h at room temperature, the
membrane was probed with primary antibodies against human Beclin-1,
LC3, P62, caspase-3, Bcl-2 or β-actin proteins. Then, the membranes
were incubated with the IRDye800CW or IRDye700DX conjugate
secondary antibodies (LI-COR, Lincoln, NE, USA). The protein bands
of immunoblot were visualized by an Odyssey double-color infrared
laser imaging system (LI-COR).
Real-time quantitative RT-PCR assay
Total RNA from the cells was extracted using the
TRIzol kit. From each sample, 2 μg of RNA was converted into cDNA
by oligo (dT) 18-primed reverse transcription using SuperScript II
RT First-Strand kit as described by the manufacturer. The levels of
the genes were analyzed on Rotor-Gene 3000 quantitative PCR
amplifier (Corbett, Australia).
Statistical analysis
All data are expressed as means ± SD. Statistical
analysis was performed using Student’s t-tests and SPSS 13.0.
P<0.05 and P<0.01 were considered to indicate a statistically
significant and highly statistically significant difference,
respectively.
Results
As2O3-mediated
inhibition of growth and induction of apoptosis in Raji cells
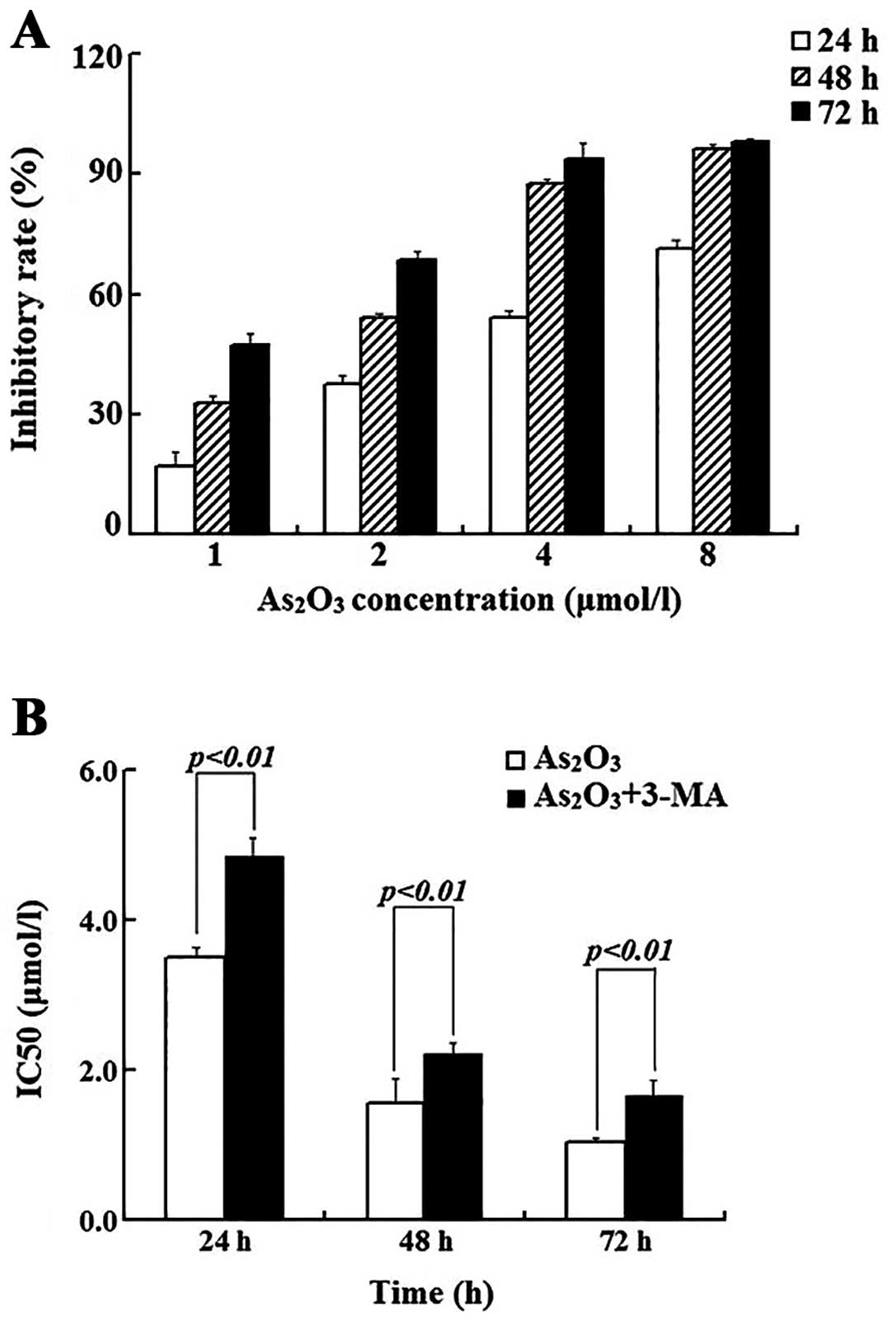
It has been shown that As2O3
induces growth arrest and apoptosis in many different cancer cell
lines. We treated Raji cells with various concentrations (0–8
μmol/l) of As2O3 for 24, 48 and 72 h; the
cell numbers were determined and the inhibitory rates are plotted
in Fig. 1A. It is clearly shown
that the cell growth was inhibited by As2O3
in a dose-and time-dependent manner. In addition, the
IC50 (μmol/l) of this agent at 24, 48 and 72 h was
calculated as 3.51±0.13, 1.57±0.32 and 1.03±0.08, respectively
(Fig. 1B). Since ~50% of the cell
growth was inhibited when the cells were treated with 3 μmol/l of
As2O3 for 24 h, we treated the cells with
As2O3 at this concentration and the results
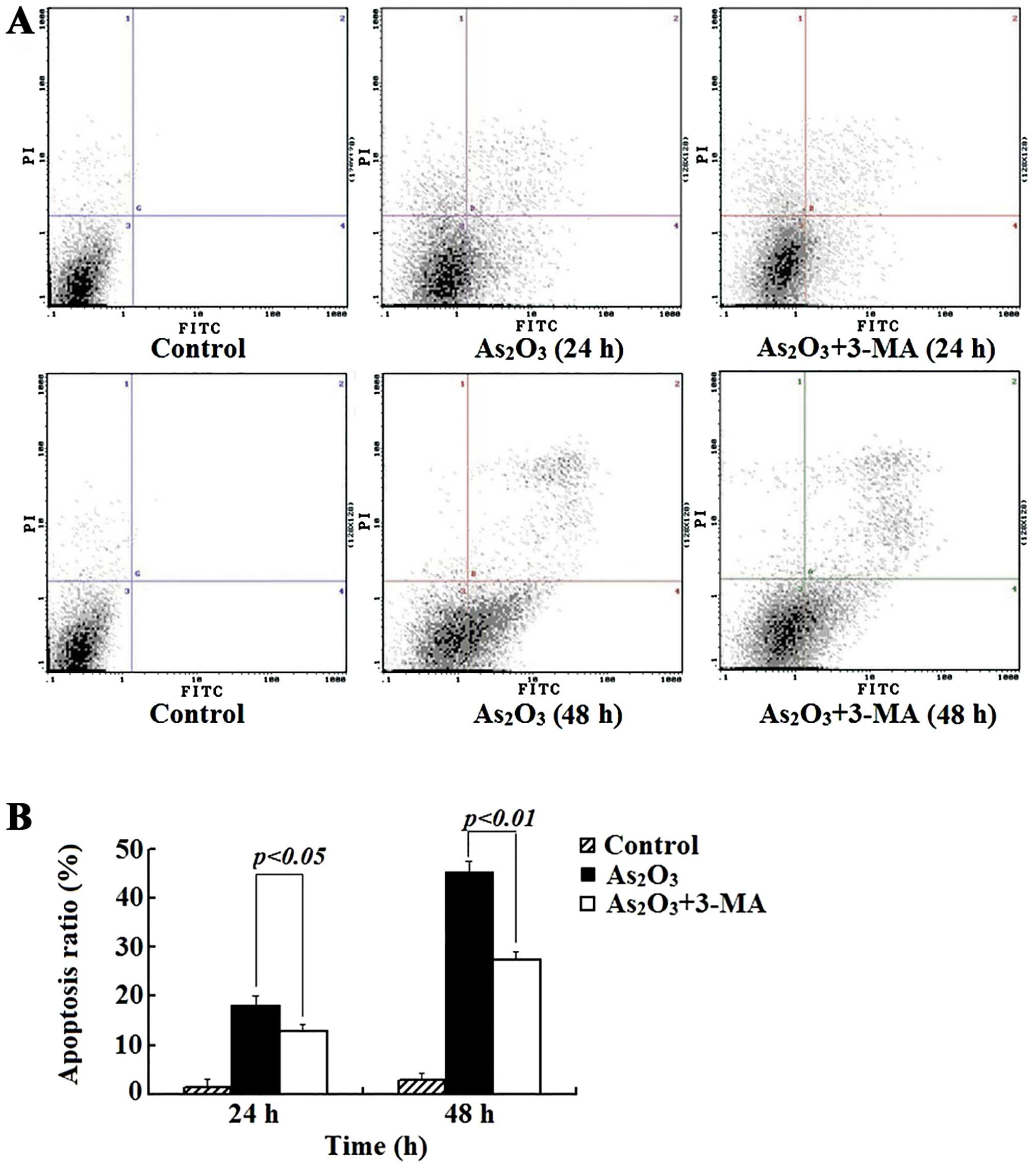
from the flow cytometry showed that when the cells were treated for
24 and 48 h, the apoptotic population increased from 2.72±1.09 to
23.5±1.32 and 40.8±2.48%, respectively (Fig. 2). In addition, the percentage of
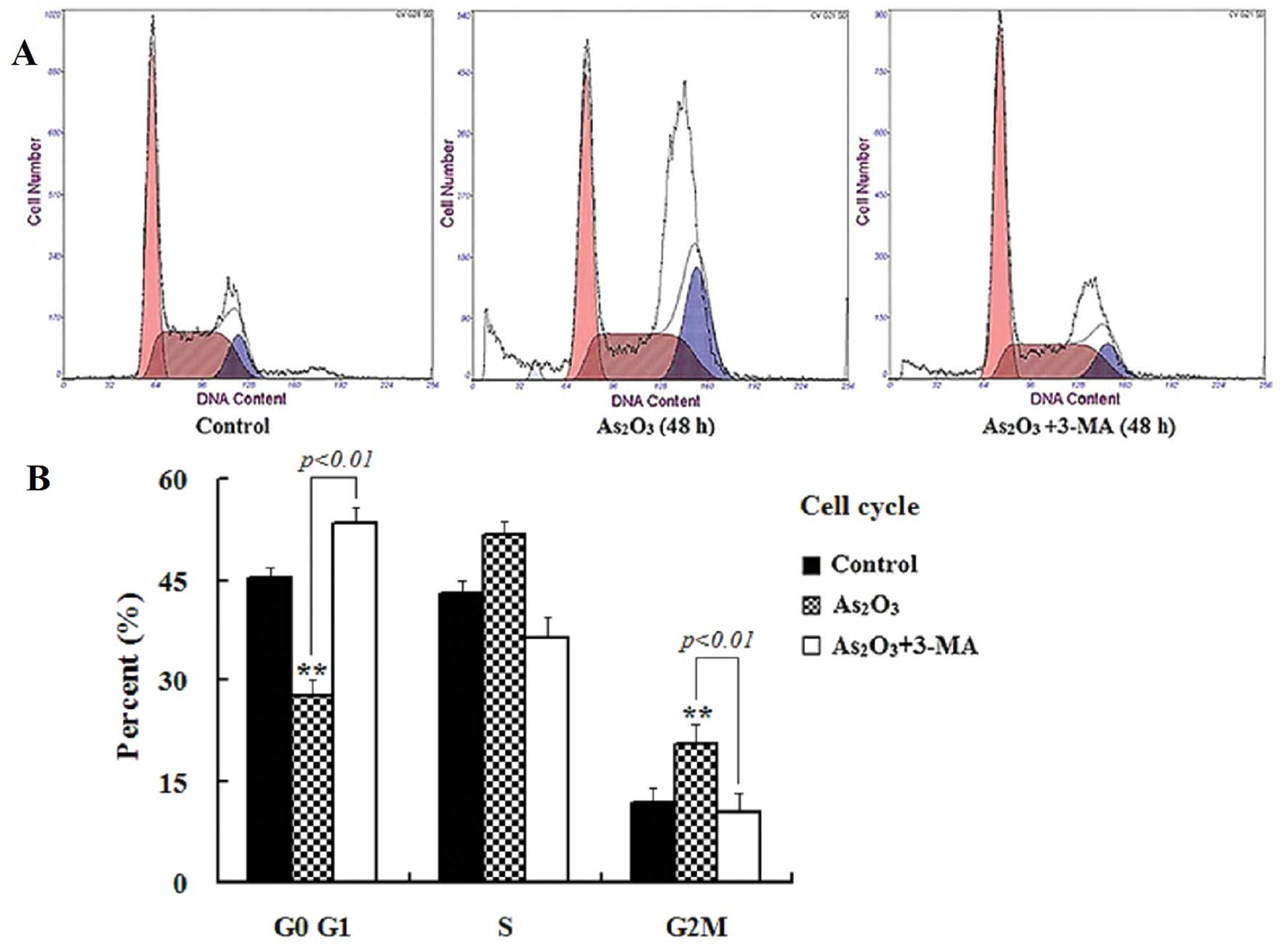
cells on different cell cycle stages was determined by flow
cytometry. As shown in Fig. 3, the
As2O3 treatment reduced the population in the
G0/G1 phase and increased that in both the S- and the G2/M-phase.
Compared to the control, the cells in the G2/M phase increased from
11.8 to 26.4% after they were treated with
As2O3 for 48 h. Consistent with the increased
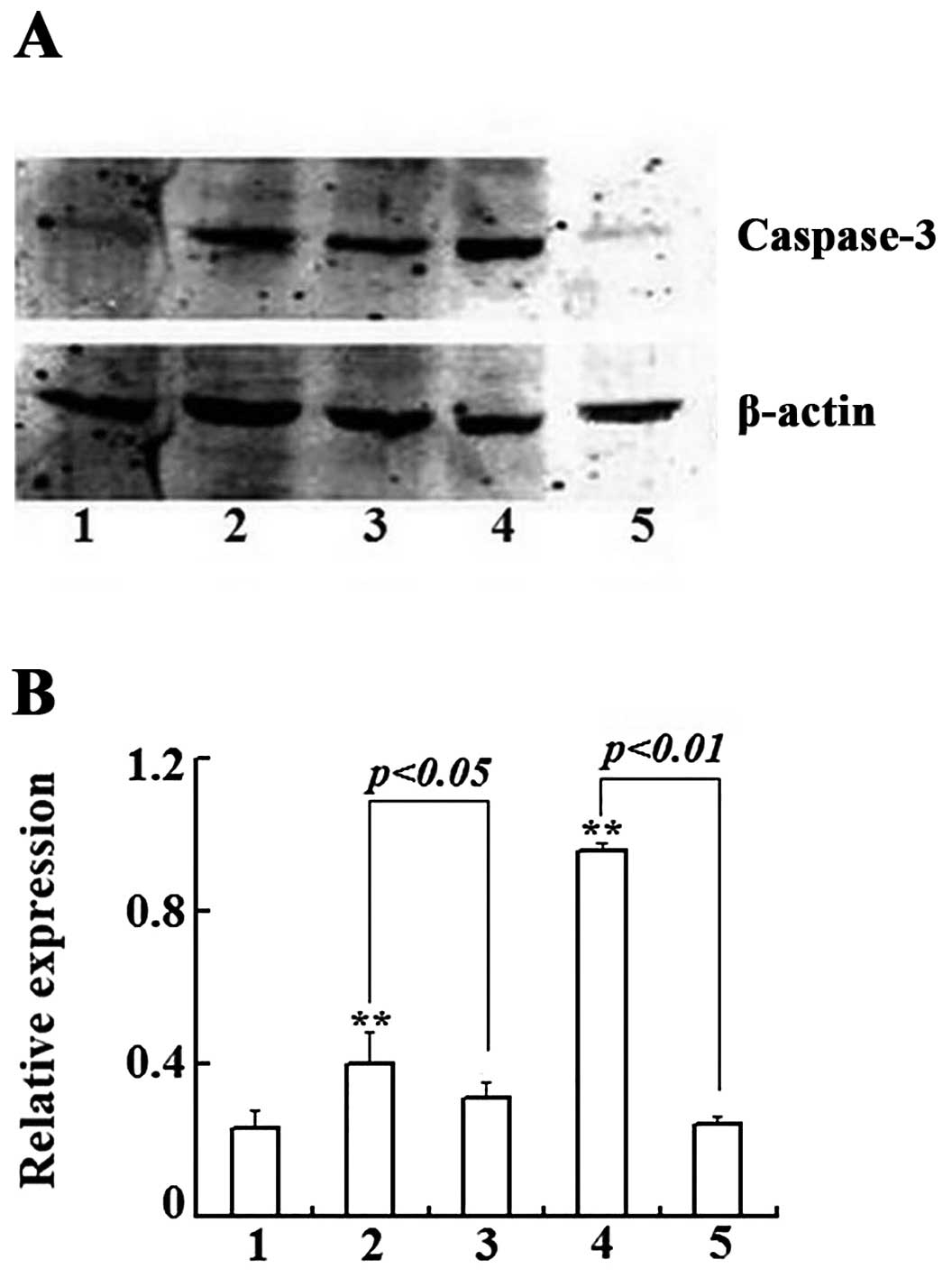
apoptotic population shown in the flow cytometry, western blot
assays showed the increased intensity of the cleaved and active
form of caspase-3 (Fig. 4A).
Quantification of the intensities of the bands and statistical
analyses showed that As2O3 treatment
increased the activated caspase-3 significantly (Fig. 4B). Collectively, these data
demonstrate that As2O3 inhibits Raji cell
growth by inducing G2/M arrest, eventually leading to
caspase-dependent apoptotic cell death.
3-MA reverses the inhibition of cell
proliferation and apoptosis induced by
As2O3
3-MA is a specific inhibitor of PI3K activity and
one of the most widely used inhibitors of the initial phase of
autophagy (5). We pre-treated Raji
cells with 3-MA (4 mmol/l) for 4 h, then different concentrations
of As2O3 were added into the pre-treated
cells. Compared with the As2O3 alone group,
the IC50 value (μmol/l) at 24, 48 and 72 h was increased
to 4.85±0.24, 2.22±0.15 and 1.65±0.22, increased by 38.2, 41.4 and
60.2%, respectively (Fig. 1B).
After treating Raji cells with 3-MA (4 mmol/l) and
As2O3 (3 μmol/l), the apoptotic ratio
decreased from 23.5 to 18.1% (24 h) and from 40.8 to 29.3% (48 h)
(P<0.05), compared with the As2O3 alone
group (Fig. 2). 3-MA also
alleviated the G2/M arrest caused by As2O3
and the percentage of cells in the G2/M phase decreased from 26.4
to 10.5%, accompanied by an increase of the cells in the G0/G1
phase from 36.3 to 53.2% (Fig. 3).
Meanwhile, the expression of caspase-3 protein (Fig. 4) markedly decreased in the combined
treatment group. These results showed that 3-MA reduced the
inhibition of cell proliferation and apoptosis induced by
As2O3.
As2O3-induced
autophagy in Raji cells is inhibited by 3-MA
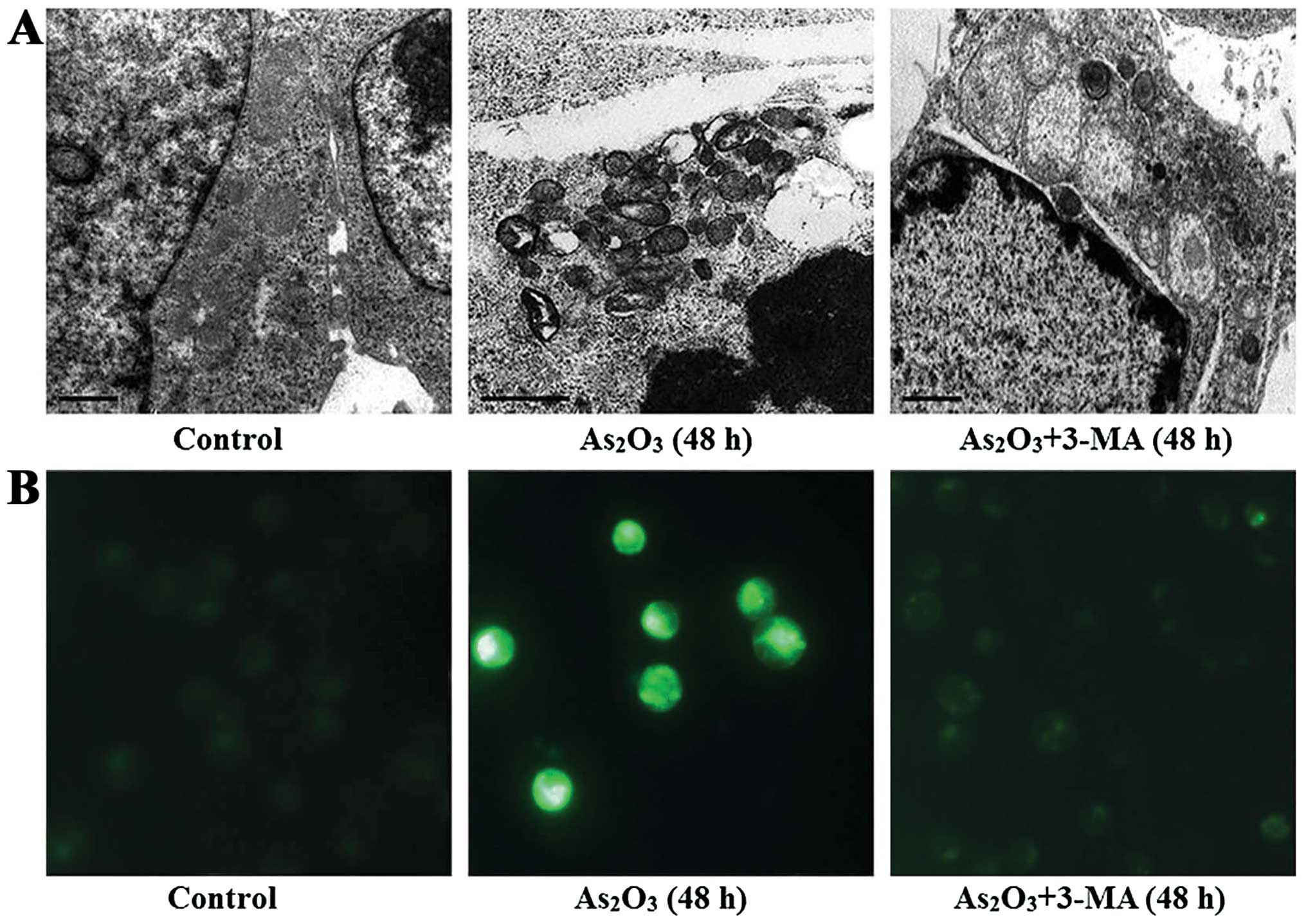
To assess the autophagic activity in the Raji cells
after being treated with 3 μmol/l As2O3, we
observed the formation of autophagosomes using the traditional
method transmission electron microscope and the fluorescence
intensity of MDC was detected by fluorescence microscope. The
number of autophagosomes and MDC-positive fluorescent points in the
As2O3-treated cells was much higher than in
the untreated cells and in some cells the autophagic vacuoles and
apoptotic changes co-existed. However, the addition of 3-MA (4
mmol/l) prior to As2O3 (3 μmol/l) treatment
decreased the number of autophagosomes and the fluorescence
intensity of MDC (Fig. 5).
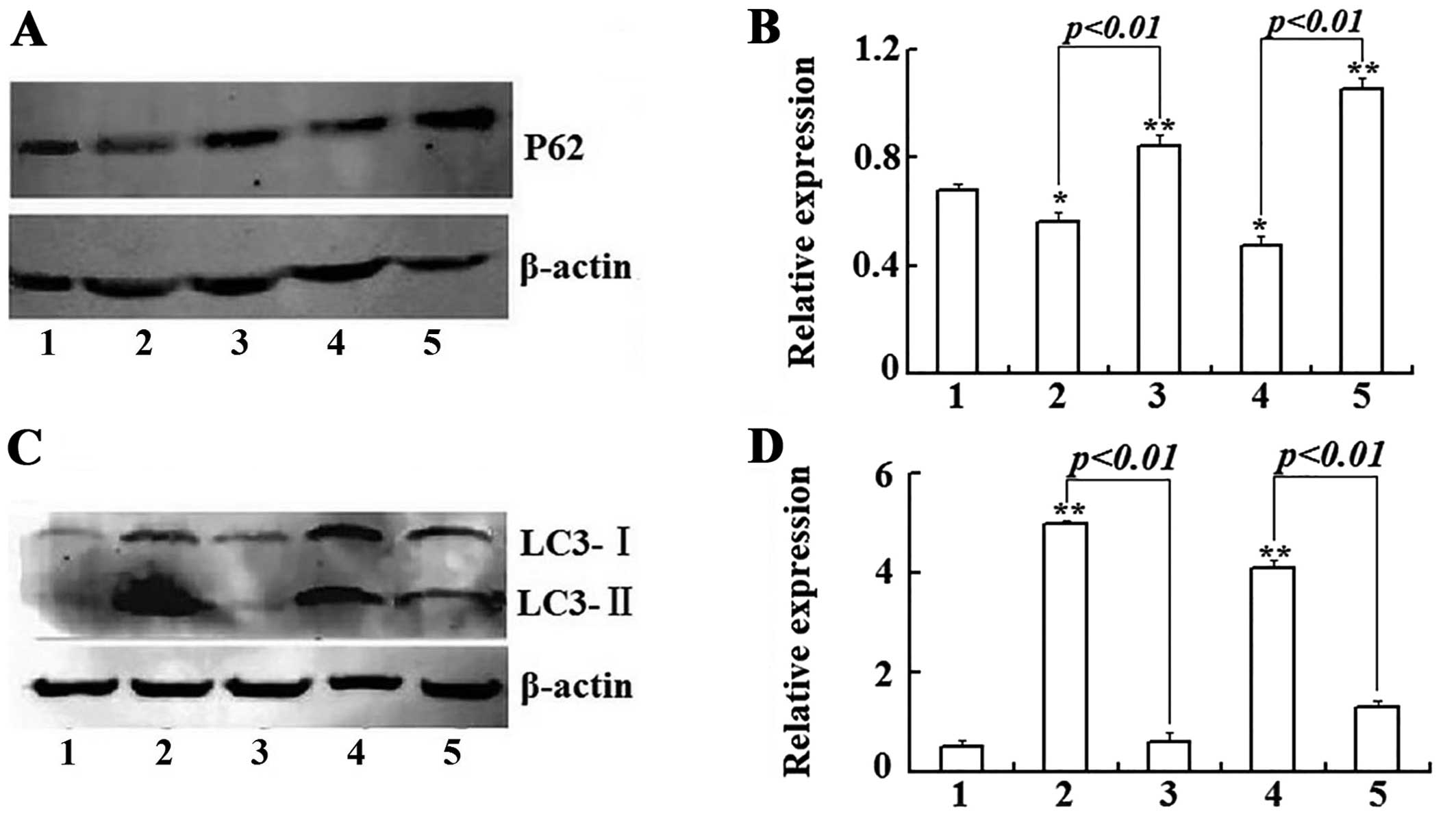
In order to distinguish whether autophagosome
accumulation was due to autophagy induction or a block in
downstream steps, we detected the expression level of P62, an
autophagy substrate that can be used to monitor autophagic flux,
and the expression levels of P62 inversely correlated with
autophagic activity (14). Our
research showed that after Raji cells were treated with 3 μmol/l
As2O3 for 24 and 48 h, the level of P62
protein was obviously decreased. On the contrary, 3-MA increased
the total level of P62 protein (Fig. 6A
and B). The results provide evidence that the autophagosome
accumulation was due to autophagy induction instead of a block in
downstream steps.
To further confirm the above result, we examined
expression of LC3-II (microtubule-associated protein 1, light chain
3, in its conjugated form) by western blotting, since LC3-II is
widely used as a specific marker of autophagic activity (15,16).
The results (Fig. 6C and D) showed
that As2O3 (3 μmol/l) treatment could result
in the upregulation of LC3-I and a considerable portion of LC3-I
was converted into LC3-II. However, after Raji cells were
co-treated with As2O3 and 3-MA, the
expression of LC3-I-II was significantly down-regulated and the
conversion of LC3-II from LC3-I was also inhibited.
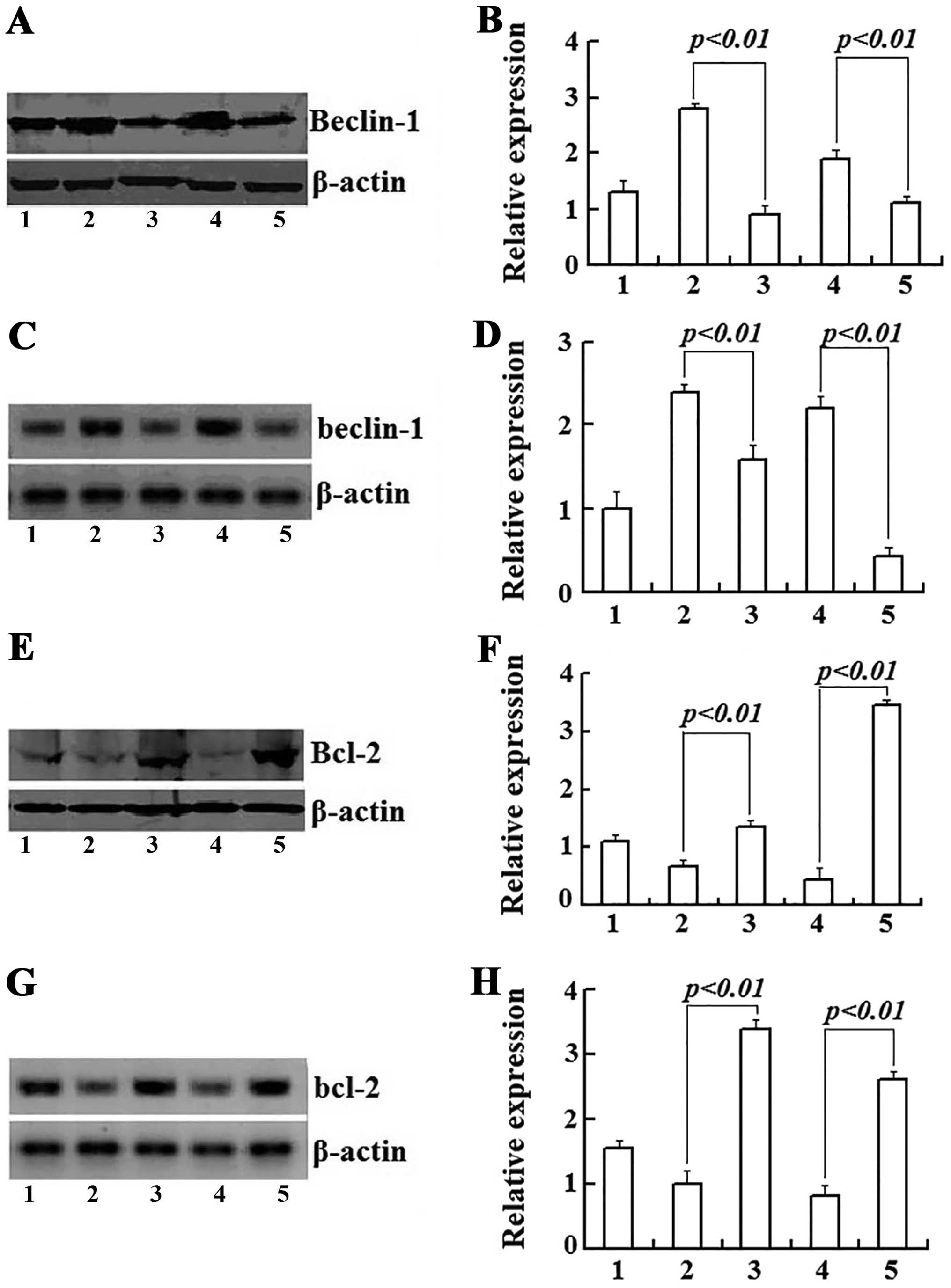
Interaction of Beclin-1 and Bcl-2
mediates the crosstalk between apoptosis and autophagy induced by
As2O3
To explore the necessary connection between
autophagy and apoptosis, we detected the expression levels of
Beclin-1 and Bcl-2 genes using western blotting and real-time
RT-PCR assay to prove the interaction of Beclin-1 and Bcl-2 in part
mediated the As2O3-induced apoptotic and
autophagic cell death in Raji cells. After treatment with 3 μmol/l
As2O3 for 24 and 48 h, the expression of
Beclin-1 protein was increased by 2.2- and 1.46-fold and the
Beclin-1 mRNA was increased by 2.38- and 2.19-fold respectively and
these actions were clearly inhibited by addition of 3-MA (Fig. 7A–D). On the contrary, Raji cells
treated with As2O3 exhibited a time-dependent
decrease in both Bcl-2 mRNA and protein expression, but 3-MA
upregulated their expression of both Bcl-2 mRNA and proteins
(Fig. 7E–H). The enhanced
expression of Beclin-1 was in parallel with suppression of Bcl-2
during As2O3-induced death in Raji cells.
Discussion
As2O3 is a clinically highly
relevant anticancer drug used for the treatment of various cancer,
especially leukemia. The mechanisms of
As2O3-induced cell death have been
extensively investigated. Apoptosis is considered to be one of the
important mechanisms (12,17). However, in recent years, the role of
autophagy in cancer therapy has also received increasing attention
from researchers, and many studies have shown that autophagy is
readily induced in response to certain stressful stimuli, such as
metabolic stress (18,19) and exposure to anticancer drugs
(20,21). Hence, it is believed that autophagy
may play an important role in tumorigenesis and cancer therapy.
However, the fundamental question, whether autophagy promotes
cancer cell death or protects them from unfavorable conditions,
remains controversial. It is certainly an intricate target for
cancer therapy (22).
In our study, we demonstrated that
As2O3 inhibited both proliferation and
viability of Raji cells in a dose- and time-dependent manner. Raji
cells treated with As2O3 underwent apoptosis
and cell cycle arrest. Moreover, As2O3
promoted the formation of autophagic vacuoles and the conversion of
soluble LC3-I to lipid bound LC3-II, as well as increased the
degradation of autophagy substrate P62 protein. The autophagic
vacuoles and apoptotic changes always co-existed in the same cells.
3-MA is one of the most widely used pharmacologic inhibitors of
autophagy; it can block class III phosphatidylinositol 3-kinase
(PI3K) activity, thereby reducing the number of autophagic vacuoles
and the conversion of LC3-I to LC3-II. Furthermore, 3-MA also
alleviated the proliferation inhibition, apoptosis and G2/M phase
arrest in Raji cells induced by As2O3. In
addition, 3-MA decreased the upregulation of caspase-3 protein
caused by As2O3. These results provide
evidence that the Raji cell death induced by
As2O3 shared characteristics of both
autophagic and apoptotic cell death.
Bcl-2 proto-oncogene is a well-known anti-apoptotic
mediator. However, its roles in inhibiting autophagy have attracted
increasing attention from researchers. Bcl-2 is well-documented to
inhibit autophagy via the interaction with Beclin-1 (23,24).
Akar et al (25) reported
that RNAi knockdown of Bcl-2 induced the autophagy and apoptosis in
MCF-7 breast cancer cells. Saeki et al (26) found that Bcl-2 silencing by
antisense RNA induced the autophagy-dependent cell death in human
leukemia HL-60 cells. The dual role of Bcl-2 in inhibiting both
apoptosis and autophagic-associated cell death makes this protein a
potential chemotherapeutic target. Beclin-1, a Bcl-2 homology 3
(BH3) domain only protein, is essential for the double-membrane
autophagosome formation, which is required during the initial steps
of autophagy (27,28). Beclin-1 recruits key autophagic
proteins to a pre-autophagosomal structure, thereby forming the
core complex consisting of Beclin-1, Vps15 and Vps34 (29–31).
In addition, Beclin-1 is a key determining factor with regard to
whether cells undergo autophagy or apoptosis. Nutrient deprivation
or other stress conditions result in the activation of the
stress-induced MAPK JNK, which phosphorylates three residues in the
regulatory loop of Bcl-2, disrupting its interaction with Beclin-1
to permit autophagy (32). In our
study, we found that As2O3 significantly
enhanced the expression of Beclin-1 protein and its mRNA; on the
contrary, the expression of Bcl-2 protein and the level of Bcl-2
mRNA were clearly downregulated and these effects were antagonized
by 3-MA. These findings indicate that both apoptotic and autophagic
pathways are involved in As2O3-induced death
in Burkitt’s lymphoma cells and the co-action or crosstalk of
Beclin-1 and Bcl-2 may play a key role in coordinating the
relationship between autophagic cell death and apoptosis.
Therefore, As2O3 may act as a joint activator
of apoptosis and autophagy and regulation of autophagic activity
may be a promising therapy for patients with Burkitt’s
lymphoma.
Acknowledgements
This study was supported by the Fundamental Research
Funds for the Central Universities from Northwest University for
Nationalities, China (no. zyz2011094) and Science and Technology
Planning Project from Chengguan District, Lanzhou, Gansu Province,
China (no. 2013-7-17).
References
|
1
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Debnath J, Baehrecke EH and Kroemer G:
Does autophagy contribute to cell death? Autophagy. 1:66–74. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kroemer G and Levine B: Autophagic cell
death: the story of a misnomer. Nat Rev Mol Cell Biol. 9:1004–1010.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen L, Liu Q, Huang Z, et al:
Tripchlorolide induces cell death in lung cancer cells by
autophagy. Int J Oncol. 40:1066–1070. 2012.PubMed/NCBI
|
|
5
|
Cho KH, Park JH, Kwon KB, et al: Autophagy
induction by low-dose cisplatin: The role of p53 in autophagy.
Oncol Rep. 31:248–254. 2014.PubMed/NCBI
|
|
6
|
Gordy C and He YW: The crosstalk between
autophagy and apoptosis: where does this lead? Protein Cell.
3:17–27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qian W, Liu J, Jin J, Ni W and Xu W:
Arsenic trioxide induces not only apoptosis but also autophagic
cell death in leukemia cell lines via up-regulation of Beclin-1.
Leuk Res. 31:329–339. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen GQ, Shi XG and Tang W: Use of arsenic
trioxide (As2O3) in the treatment of acute
promyelocytic leukemia (APL): I. As2O3 exerts
dose-dependent dual effects on APL cells. Blood. 89:3345–3353.
1997.PubMed/NCBI
|
|
9
|
Li YM and Broome JD: Arsenic targets
tubulins to induce apoptosis in myeloid leukemia cells. Cancer Res.
59:776–780. 1999.PubMed/NCBI
|
|
10
|
Hu XM, Hirano T and Oka K: Arsenic
trioxide induces apoptosis equally in T lymphoblastoid leukemia
MOLT-4 cells and P-gp-expressing daunorubicin-resistant MOLT-4
cells. Cancer Chemother Pharmacol. 51:119–126. 2003.PubMed/NCBI
|
|
11
|
Rousselot P, Labaume S, Marolleau JP, et
al: Arsenic trioxide and melarsoprol induce apoptosis in plasma
cell lines and in plasma cells from myeloma patients. Cancer Res.
59:1041–1048. 1999.PubMed/NCBI
|
|
12
|
Chen J, Wei H, Xie B, Wang B and Cheng J:
Endoplasmic reticulum stress contributes to arsenic
trioxide-induced apoptosis in drug-sensitive and -resistant
leukemia cells. Leuk Res. 36:1526–1535. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Biederbick A, Kern HF and Elsasser HP:
Monodansylcadaverine (MDC) is a specific in vivo marker for
autophagic vacuoles. Eur J Cell Biol. 66:3–14. 1995.PubMed/NCBI
|
|
14
|
Bjorkoy G, Lamark T, Brech A, Outzen H,
Perander M, et al: p62/SQSTM1 forms protein aggregates degraded by
autophagy and has a protective effect on huntingtin-induced cell
death. J Cell Biol. 171:603–614. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang Z, Ni L, Javidiparsijani S, et al:
Enhanced liver autophagic activity improves survival of septic mice
lacking surfactant proteins A and D. Tohoku J Exp Med. 231:127–138.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Galluzzi L, Aaronson SA, Abrams J, et al:
Guidelines for the use and interpretation of assays for monitoring
cell death in higher eukaryotes. Cell Death Differ. 16:1093–1107.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang W, Ohnishi K, Shigeno K, et al: The
induction of apoptosis and cell cycle arrest by arsenic trioxide in
lymphoid neoplasms. Leukemia. 12:1383–1391. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Degenhardt K, Mathew R, Beaudoin B and
Bray K: Autophagy promotes tumor cell survival and restricts
necrosis, inflammation, and tumorigenesis. Cancer Cell. 10:51–64.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Karantza-Wadsworth V, Patel S, Kravchuk O,
Chen G, et al: Autophagy mitigates metabolic stress and genome
damage in mammary tumorigenesis. Genes Dev. 21:1621–1635. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cheng J, Wei HL, Chen J and Xie B:
Antitumor effect of arsenic trioxide in human K562 and K562/ADM
cells by autophagy. Toxicol Mech Methods. 22:512–519. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pan J, Chen B, Su CH, Zhao R, Xu ZX, et
al: Autophagy induced by farnesyltransferase inhibitors in cancer
cells. Cancer Biol Ther. 7:1679–1684. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
de-Bruin EC and Medema JP: Apoptosis and
non-apoptotic death in cancer development and treatment response.
Cancer Treat Rev. 34:737–749. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shimizu S, Kanaseki T, Mizushima N, et al:
Role of Bcl-2 family proteins in a non-apoptotic programmed cell
death dependent on autophagy genes. Nat Cell Biol. 6:1221–1228.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pattingre S, Tassa A and Qu X: Bcl-2
antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell.
122:927–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Akar U, Chaves-Reyez A, Barria M, Tari A,
et al: Silencing of Bcl-2 expression by small interfering RNA
induces autophagic cell death in MCF-7 breast cancer cells.
Autophagy. 4:669–679. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Saeki K, Yuo A, Okuma E, et al: Bcl-2
down-regulation causes autophagy in a caspase-independent manner in
human leukemic HL60 cells. Cell Death Differ. 7:1263–1269. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Salminen A, Kaarniranta K and Kauppinen A:
Beclin 1 interactome controls the between apoptosis, autophagy and
inflammasome activation: impact on the aging process. Ageing Res
Rev. 12:520–534. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cao Y and Klionsky DJ: Physiological
functions of Atg6/Beclin 1: a unique autophagy-related protein.
Cell Res. 17:839–849. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Oberstein A, Jeffrey PD and Shi Y: Crystal
structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a
novel BH3-only protein. J Biol Chem. 282:13123–13132. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Takahashi Y, Coppola D, Matsushita N, et
al: Bif-1 interacts with Beclin 1 through UVRAG and regulates
autophagy and tumorigenesis. Nat Cell Biol. 9:1142–1151. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liang XH, Kleeman LK, Jiang HH, Gordon G,
et al: Protection against fatal Sindbis virus encephalitis by
beclin, a novel Bcl-2-interacting protein. J Virol. 72:8586–8596.
1998.PubMed/NCBI
|
|
32
|
Erlich S, Mizrachy L, Segev O, Lindenboim
L, et al: Differential interactions between Beclin 1 and Bcl-2
family members. Autophagy. 3:561–568. 2007. View Article : Google Scholar : PubMed/NCBI
|