Introduction
Neurofibromatosis type 1 (NF1; OMIM #162200) is a
commonly inherited autosomal dominant disorder, occurring with an
incidence of 1 in 3,000–3,500 individuals worldwide (1,2). NF1
is caused by loss of function mutations in the NF1 gene
(GenBank gene ID: NG_009018.1), which encodes a GTPase-activating
protein, neurofibromin (3). The
major clinical features of NF1 include cafe-au-lait spots,
freckling of the axillary or inguinal region, Lisch nodules, optic
nerve glioma, bone dysplasias and nerve sheath tumors in the
peripheral nervous system, including benign cutaneous
neurofibromas, benign plexiform neurofibromas (PNs), and malignant
peripheral nerve sheath tumors (MPNSTs) (4).
Since neurofibromin is a major RAS inactivator and
plays a role as a tumor suppressor, the lack of neurofibromin
resulting from NF1 mutation causes disruptions in the
RAS-mitogen-activated protein kinase (MAPK) and PI3K-AKT-mTOR
signaling pathways, which is implicated in the tumorigenesis and
tumor progression of PN to MPNST (5). MPNST, also called malignant schwannoma
or neurofibrosarcoma, develops in 8–13% of NF1 patients (6), and it represents a major cause of
mortality in NF1 patients (7).
Thus, the pathogenesis of the malignant transformation of PN to
MPNST in NF1 patients has attracted considerable attention
(8,9). However, the exact molecular mechanisms
of MPNST pathogenesis remain unclear.
Bi-allelic loss of NF1 gene function
(NF1−/−) due to the somatic loss of
heterozygosity (LOH) of the NF1 gene has been reported to be
essential for MPNST development (10). However, LOH at the NF1 locus
was also found in PNs (11),
suggesting that other genetic and/or epigenetic alterations may be
involved in tumor progression in NF1. Emerging evidence has
suggested that additional driver gene mutations and/or expression
alterations contribute to the tumor progression of PNs to MPNSTs
(5). Mutations in tumor suppressor
genes, including TP53, CDKN2A and RB are
commonly identified in MPNSTs (12–14).
It was also reported that several cell-cycle and signaling
regulation genes, including CDKN2A, CD44,
CXCR4, EGFR, HGF, MET, PDGFR,
PDGFRA, RB1, SOX9 and TP53, are
deregulated in MPNSTs (15,16). In addition, many differentially
expressed genes between benign neurofibromas and MPNSTs, such as
KIT, ERBB2, MET, TGFB1, HGF,
TNXB, TNC, MTOR, TSC2, PTEN,
BCL2L1, CDK4, FOXM1, BUB1B, PBK
and NEK2, have been identified in comparison studies with
immunohistochemical, immunoblotting, microarray-based, quantitative
reverse transcription PCR-based or comparative genomic
hybridization array-based analyses (17–24).
DNA methylation alterations in cancer-related genes
and microRNA (miRNA) dysregulation have been critically implicated
in the development and progression of many cancers (25,26).
Genes encoding miRNA can be epigenetically regulated by DNA
methylation or specific histone modifications (26). Recently, the emerging role of miRNAs
in the pathogenesis of NF1 tumorigenesis and MPNST development were
reported (27). Genome-wide miRNA
profiling analyses found a large number of dysregulated miRNAs in
MPNSTs, such as miR-34a against TP53 (28), miR-301a, miR-19a and miR-106b
against PTEN (29) and
miR-21 against PDCD4 (30).
However, only a few DNA methylation studies on NF1 have been
reported. In studies of miRNAs, genome-wide DNA methylation
analysis may be a new strategy for understanding the etiology of
MPNST development in NF1.
In the present study, we reported a TAGLN
gene encoding an actin-binding transgelin protein as a novel
candidate that plays a critical role in NF1-associated MPNST
pathogenesis. TAGLN was upregulated in MPNST tissues and
cells derived from NF1 patients. Notably, we found that this
upregulation was caused by an alteration of the DNA methylation in
the TAGLN genes in MPNSTs. Manipulation of TAGLN
expression in the NF1-deficient cells demonstrated the key
role of transgelin in MPNST pathogenesis. This finding may provide
insight into the underlying mechanisms of somatic tumor progression
in NF1 on the epigenetic level.
Materials and methods
Tumor tissue samples
We used the tumor tissues of eight patients
diagnosed with NF1 at the Ajou University Hospital according to NF1
diagnostic criteria (31) (Table I). The PN and MPNST tumor samples
were obtained through surgical resection at the Ajou University
Hospital. In the case of patients P7 and P8, two tumor specimens
obtained from surgeries at two different times were used in the
present study. The present study was approved by the Institutional
Review Board Committee of the Ajou University School of
Medicine.
 | Table IClinical and histological
characteristics of the patients with NF1. |
Table I
Clinical and histological
characteristics of the patients with NF1.
| Patient | Histological
findings | Clinical
features | |
|---|
|
|
| |
|---|
| ID | Gender | Age at
diagnosis | H&E | S100 | Transgelin | Cafe-au-lait
spots | Neurofibromas | Freckling | Optic glioma | Lisch nodule | Skeletal
dysplasia | Family history | Genotype NF1
gene mutation |
|---|
| P1 | Male | 59 | Benign | + | + | Y | Y | Y | N | N | N | Y | N/A |
| P2 | Male | 42 | Benign | + | + | Y | Y | Y | N | N | N | Y | N/A |
| P3 | Female | 5 | Benign | + | + | Y | Y | Y | N | N | N | Y | N/A |
| P4 | Male | 39 | Malignant | + | ++ | Y | Y | N | N | N | N | N | N/A |
| P5 | Male | 32 | Malignant | + | ++ | Y | Y | Y | N | N | N | N | c.4861_4862
GT>AG |
| P6 | Female | 41 | Malignant | + | ++ | Y | Y | N | N | N | N | N | N/A |
| P7 | Male | 12 | Benign | + | + | Y | Y | Y | N | Y | Y | N | c.4537C>T |
| | 17 | Benign | + | ++ | Y | Y | Y | N | Y | Y | N | c.4537C>T |
| P8 | Male | 21 | Benign | + | + | Y | Y | Y | N | Y | N | N | c.6792C>A |
| | 23 | Malignant | + | ++ | Y | Y | Y | N | Y | N | N | c.6792C>A |
Primary tissue cultured cells and cell
lines
We used three types of previously established
primary tissue-cultured NF1-deficient cells: normal
phenotype tissues (PC-N), benign PN tissues (PC-B) and the MPNST
tissues (PC-M) of NF1 patient P8 (32). The cellular characteristics,
including GTP-RAS activity and its downstream effectors in the
three cell types, were previously demonstrated (32). Cells were grown in Dulbecco’s
modified Eagle’s medium (DMEM) supplemented with 15% fetal bovine
serum (FBS) (both from HyClone Laboratories, Logan, UT, USA),
penicillin (100 U/ml) and streptomycin (100 μg/ml). Cells were used
from passages 5 through 10. The NF1-deficient MPNST cell
lines, sNF02.2 and sNF96.2, were purchased from the American Type
Culture Collection (Manassas, VA, USA) and grown in DMEM media
supplemented with 10% FBS. All cultured cells were incubated at
37°C in a humidified atmosphere containing 5% CO2.
Hematoxylin and eosin (H&E) staining
and immunohistochemistry (IHC)
The tumor tissue specimens from eight patients with
NF1 were formalin-fixed and embedded in paraffin wax for
pathological evaluation by H&E staining and IHC. Serial 3 μm
sections were prepared on glass using a cryostat and the slides
were stained with H&E. For IHC, the blocks were cut at 10-μm
thickness and adhered to glass slides. They were deparaffinized in
xylene and rehydrated with graded ethanol, which was followed by
antigen retrieval in boiling 10 mM citrate buffer (pH 6.0) for 4
min. Immunostaining was carried out using the UltraVision LP
Detection System and the HRP Polymer and DAB Plus Chromogen (Thermo
Fisher Scientific, Fremont, CA, USA) according to the
manufacturer’s instructions. Briefly, the sections were incubated
with Ultra V Block for 5 min at room temperature to reduce the
nonspecific background, and they were then treated with hydrogen
peroxide to block endogenous peroxidase activity. The sections were
incubated with the primary antibody for 1 h at room temperature and
then incubated with HRP polymer for 20 min at room temperature. The
reaction product was visualized with the DAB chromogen.
Pathological evaluation was performed under light microscopy.
Anti-transgelin and anti-S100 antibodies were purchased from Thermo
Science (Rockford, IL, USA) and Abcam (Cambridge, UK),
respectively.
Plasmid constructs and small interfering
RNAs (siRNAs)
Full-length human TAGLN cDNA was amplified by
reverse transcription polymerase chain reaction (RT-PCR) using the
primers: 5′-AGTGCAGTCCAAAATCGAGAAG-3′ and
5′-CTTGCTCAGAATCACGCCAT-3′, which were from the total RNAs of the
human skin tissue-cultured fibroblast cells. The cDNAs were
subcloned into the pcDNA3.1(−) vector (Clontech, Palo Alto, CA,
USA) using the XhoI and BamHI restriction enzyme
sites. The siRNAs were synthesized by Genolution Pharmaceuticals,
Inc. (Seoul, South Korea). The target sequences for the siRNAs
were: 5′-CCAAAATCGA GAAGAAGTATT-3′ for the TAGLN gene, and
5′-CCTACGC CACCAATTTCGT-3′ for the non-specific scramble siRNA
control. Cell transfection of the siRNAs and plasmid constructs was
conducted using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA,
USA) and Lipofectamine 2000 (Invitrogen), respectively, according
to the manufacturer’s instructions.
Reverse transcription-PCR (RT-PCR)
Total RNAs were isolated from the cultured cells
using TRIzol reagent and they were treated with RNase-free DNase I
(both from Invitrogen) to avoid amplification of the genomic DNA,
and were subsequently reverse transcribed using the RevertAid™ H
Minus First Strand cDNA Synthesis kit (Fermentas, Burlington, ON,
Canada) with the oligo(dT)15–18 primer. PCR
amplification was carried out using the Ex-Taq DNA polymerase
(Takara, Shiga, Japan) at an annealing temperature of 60°C for 25
cycles. The gene specific primers used were: 5′-AGTGCAGTCCAA
AATCGAGAAG-3′ and 5′-CTTGCTCAGAATCACGCCAT-3′ for the TAGLN
gene; and 5′-TGTTGCCATCAATGACCC CTT-3′, and
5′-CTCCACGACGTACTCAGCG-3′ for the GAPDH gene (a
control).
Western blot analysis
Cultured cells were lysed in RIPA buffer [150 mM
NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium
dodecyl sulfate (SDS) and 50 mM Tris buffer, pH 8.0]. The proteins
were heated at 100°C for 10 min and analyzed by SDS-polyacrylamide
gel electrophoresis on 8–12% polyacrylamide gels. The proteins were
electroblotted onto PVDF membranes (Millipore, Milford, MA, USA).
The membrane blots were blocked with 5% (w/v) non-fat dried milk,
incubated with primary and secondary antibodies, and then
visualized with the enhanced chemiluminescence western blotting
detection system (WEST-ZOL plus; iNtRON Biotechnology, Daejeon,
Korea). Anti-transgelin was purchased from Abcam, while the
anti-extracellular regulated kinase (ERK)1/2 and
anti-phosphorylated ERK1/2 antibodies were purchased from Cell
Signaling Technology (Danvers, MA, USA). Anti-α-tubulin,
HRP-conjugated goat anti-rabbit IgG and HRP-conjugated goat
anti-mouse IgG antibodies were purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA).
Ras activation assay
Ras activation was assayed using the RAS activation
assay kit (Upstate Biotechnology, Lake Placid, NY, USA). Briefly,
cells were lysed in lysis buffer (25 mM HEPES pH 7.5, 150 mM NaCl,
1% Igepal CA-630, 10 mM MgCl2, 1 mM EDTA, 2% glycerol,
25 mM NaF and 1 mM Na3VO4) on ice and
centrifuged at 14,000 × g for 5 min at 4°C. A total of 300 μg
cellular lysates were incubated with Raf-RBD agarose beads at 4°C
for 1 h. The beads were washed 3 times with 1 ml of ice-cold lysis
buffer, resuspended in 2X Laemmli sample buffer, boiled and
separated on SDS-PAGE gels, which was followed by western blot
analysis using an anti-ras antibody.
DNA methylation analysis
DNA methylation analysis was performed using the
Infinium HumanMethylation27 BeadChip (Illumina Inc., San Diego, CA,
USA; catalog no: WG-311–1201), which contains 27,578 CpG loci
located in the promoter and subpromoter regions (5′-UTR, exon 1 and
intron 1) of 14,495 human RefSeq genes, as previously described
(33). Genomic DNA was isolated
from cells using the QIAamp DNA Mini kit (Qiagen, UK). The
bisulfite conversion of genomic DNA (1 μg) was carried out using
the EZ DNA Methylation-Gold kit (Zymo Research, Orange, CA, USA).
Bisulfite-converted DNA was amplified and hybridized to the
HumanMethylation27 BeadChip according to the manufacturer’s
standard protocols. Each interrogated locus is represented by
specific oligomers linked to two bead types: one representing the
sequence for methylated DNA (M) and the other for unmethylated DNA
(U). For each specific CpG island region, the methylation status is
calculated from the intensity of the M and U alleles, as the ratio
of the fluorescent signals β = Max(M,0)/[Max(M,0)+Max(U,0)+100].
The DNA methylation β-value is a way to represent the scores of DNA
methylation. Quantitative scores of DNA methylation levels range
from 0 (methylation absent) to 1 (completely methylated).
Results
Upregulation of transgelin in MPNST
tissues and primary MPNST cells from NF1 patients
Based on the analysis of the RT-PCR-based
differential display data of the normal phenotype, benign PN and
MPNST tissues of an NF1 patient, we previously found transgelin
(SM22) to be significantly upregulated in MPNST tissues (34). Here, we sought to confirm this
finding in more tumor tissues of NF1 patients using IHC analysis.
The clinical features and genotypes of the eight NF1 patients
analyzed in the present study are summarized in Table I. First, the PN and MPNST tumor
specimens of eight NF1 patients were evaluated using
histopathological analysis with H&E staining (Table I). Since Schwann cells are the
primary pathogenic cell source in PN and MPNST tumors, we further
evaluated tumor specimens through IHC analysis using the Schwann
cell lineage marker S100 (Table
I).
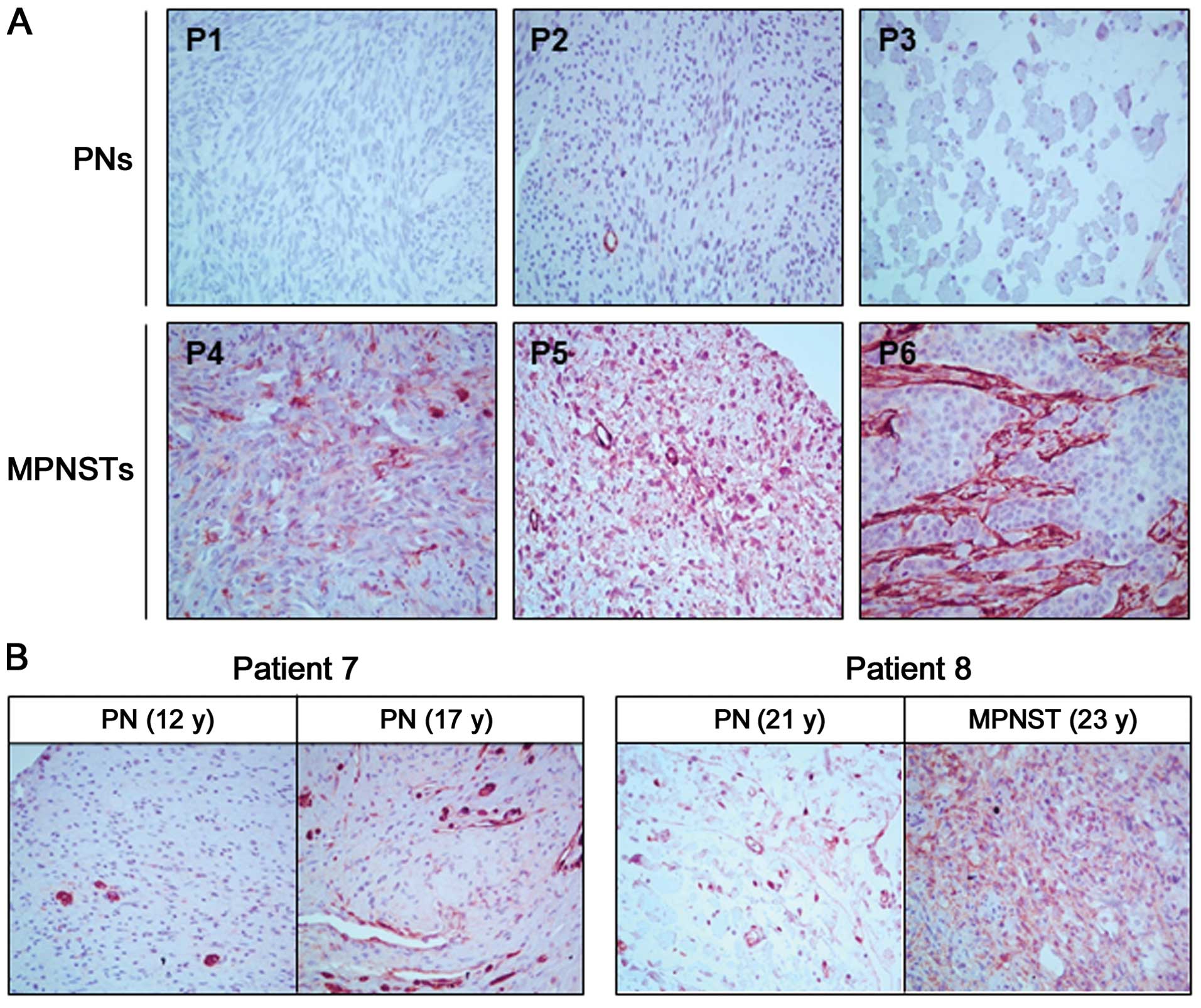
We compared the transgelin protein levels between
the PN and MPNST tumor tissues using IHC analysis. The expression
levels of transgelin were higher in the MPNST tissues (P4–P6) than
in PN tissues (P1–P3) (Fig. 1A).
Next, to investigate whether the transgelin expression level was
altered in accordance with tumor progression, we compared the
transgelin levels in the two tumor tissues resected at different
times from the same patient. In patient P7, different transgelin
levels were observed in two PN tumors with a five-year interval;
the latter showed higher transgelin levels than the former
(Fig. 1B). In patient P8, we
compared the PN tumor from the first surgery and the MPNST tumor
from the second surgery (a 2-year interval) and found significantly
higher transgelin expression in the MPNST tumor than in the PN
tumor (Fig. 1B).
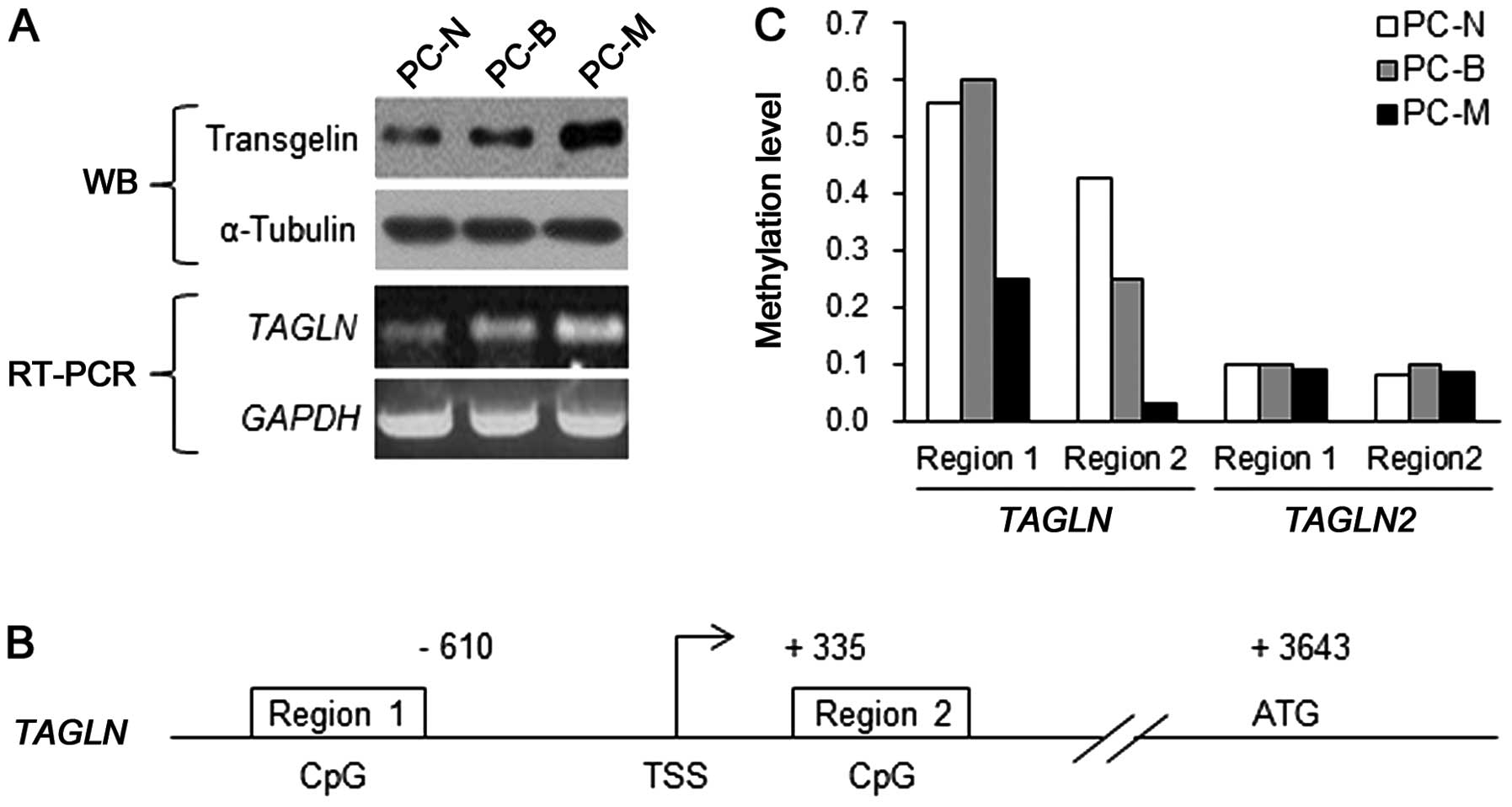
Subsequently, we confirmed this result in the
primary NF1-deficient cells from NF1 patient P8 (32): normal-phenotypic cells (PC-N),
benign PN cells (PC-B) and MPNST cells (PC-M). Western blot
analysis revealed that transgelin expression gradually increased
from PC-N to PC-M (Fig. 2A). To
determine whether the differential expression of transgelin among
the cells was responsible for the changes in the transcriptional
expression of the TAGLN gene, we examined the TAGLN
mRNA levels by RT-PCR. Consistent with the results for the protein
level, the mRNA expression level of TAGLN also gradually
increased from PC-N to PC-M (Fig.
2A).
Hypomethylation in the promoter and
subpromoter regions of the TAGLN gene in primary MPNST cells from
NF1 patients
Since DNA methylation alteration of the TAGLN
gene was reported in cancers (35,36),
we attempted to investigate the TAGLN gene methylation level
in the primary NF1-deficient cells. Using the
HumanMethylation27 BeadChip microarray, we performed genome-wide
DNA methylation analysis. Following analysis of the methylation
data of 27,578 CpG loci located in the promoter and subpromoter
regions of 14,495 human genes (data not shown), we intensively
examined the methylation status of two CpG island regions in the
TAGLN gene (Fig. 2B). We
also examined the DNA methylation status of the TAGLN2 gene,
which is closely related to TAGLN (37), as a comparison control. The results
revealed that both CpG island regions, regions 1 and 2, in the
TAGLN gene were less methylated in the primary PC-M cells
than in the PC-N and PC-B cells (Fig.
2C). The DNA methylation level in region 2 of the TAGLN
gene gradually decreased from PC-N to PC-M. In particular, region 2
of the PC-M cells was extremely hypomethylated. In contrast, the
DNA methylation status of the TAGLN2 gene was not different
in the three types of cells (Fig.
2C). These results indicate that the upregulation of transgelin
in the MPNSTs was caused by the hypomethylation of the TAGLN
gene.
Involvement of transgelin in RAS and
ERK1/2 activation in primary MPNST cells and MPNST cell lines
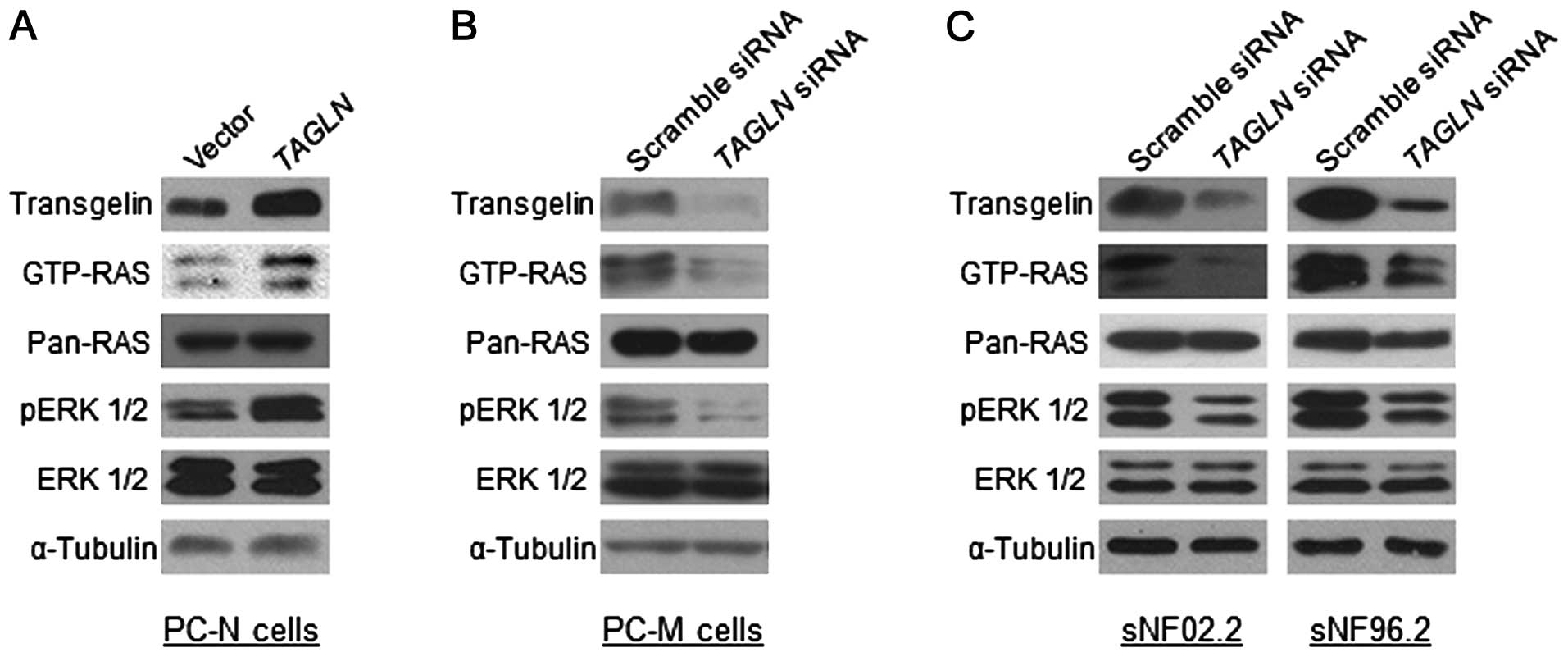
To examine the functional role of transgelin in
MPNST pathogenesis, we manipulated the TAGLN gene expression
and investigated if the RAS signaling was altered according to the
transgelin expression level in the normal-phenotypic cells and
malignant tumor cells. Overexpression of TAGLN in the
primary PC-N cells caused an increase in Ras activation (GTP-RAS)
and the downstream ERK1/2 activation (phosphorylated ERK1/2,
pERK1/2) (Fig. 3A). In contrast,
the downregulation of TAGLN expression in the primary PC-M
cells through the treatment of the short interfering siRNAs
(siRNAs) resulted in a decrease in RAS and ERK1/2 activation
(Fig. 3B). Next, we carried out a
further downregulation experiment in two Schwann-like MPNST cell
lines, sNF02.2 (NF1+/−) and sNF96.2
(NF1−/−) (38),
that express a high level of transgelin (Fig. 3C). Both cell lines treated with
TAGLN siRNAs showed decreased GTP-RAS and pERK1/2, as in the
primary PC-M cells (Fig. 3C). These
results indicate that the transgelin level is closely involved with
the Ras/Raf/Mek/Erk signaling pathway.
Discussion
Aberrations of DNA methylation in cancer-related
genes play a key role in cancer development (39). Hypermethylation of the promoter or
first exon of tumor suppressor genes causes transcriptional
silencing. In contrast, hypomethylation in proto-oncogenes induces
transcriptional activation. Hypermethylation could explain the
somatic loss of the tumor suppressor gene function without gene
mutations in cancer. However, how hypomethylation contributes to
carcinogenesis is less clear (40).
In addition, DNA methylation is crucially involved in the
dysregulation of miRNAs, which are small non-coding RNAs that act
as post-transcriptional regulators of target gene expression, in
cancer (41).
Only a few methylation studies have been conducted
on NF1. A methylation study on the monozygotic twin pairs with NF1
that presented with several discordant features indicated that
differences in the methylation patterns of the normal NF1
allele in twins may result in NF1 expression difference,
thereby causing a modification of the NF1 phenotype (42). Comparing NF1 patients with a low
number of cutaneous neurofibromas to those with a high number of
cutaneous neurofibromas, using methylation-specific PCR and
pyrosequencing, indicated that the promoter methylation of the
mismatch repair MSH2 gene in the blood cells of patients was
significantly different (43).
Another methylation study on normal Schwann cells and
NF1-associated PN tumor samples reported that a low level of
methylation in NF1 gene promoters was found in PN tumors
(44). In a methylation analysis of
MPNSTs, although no significant methylation changes in the
NF1 gene promoter were found in the MPNST tissues (45), frequent PTEN promoter
methylation was detected in NF1-associated MPNST tissues and
NF1-deficient MPNST cell lines, although not in benign
tumors (46).
In the present study, we found hypomethylation of
the TAGLN gene in the primary MPNST cells and a high
expression of transgelin in the MPNST tissues, primary MPNST cells
and MPNST cell lines (Figs. 1 and
2). Since the methylation levels in
CpG island region 2 of the TAGLN gene were in inverse
proportion to the mRNA expression levels of the TAGLN gene
in three types of primary cells (Fig.
2), we concluded that upregulated transgelin in the MPNSTs was
a result of the hypomethylation of the TAGLN gene. Although
the human promoter region of the TAGLN gene contains two
CArG boxes, a binding site for the serum response factor and other
transcription factor binding sites for AP-1 and SP1 (47), the epigenetic modification of
TAGLN through DNA methylation was also reported to be
important in the regulation of transgelin transcription (48). Contrary to our results, however,
hypermethylation of TAGLN was found in hepatocellular and
colorectal carcinoma (35,36).
Transgelin (SM22) is a 22-kDa actin-binding protein
of the calponin family that is found abundantly in the smooth
muscle tissues of adult vertebrates (49). Although the precise function of the
protein remains unclear, transgelin is suggested to be involved in
cell differentiation, cell migration, podosome formation, tissue
invasion and matrix remodeling (50,51).
Most previous studies reported that transgelin acted as a tumor
suppressor (49). However, recent
data have indicated that it has a pro-tumorigenic role (50). The upregulation of transgelin was
observed in gastric and pancreatic cancers (52,53).
Based on these controversial results, the pathological role of
transgelin appears to be different between cancer types and could
change during tumor progression (50).
Neurofibromas and MPNSTs consist mostly of Schwann
cells and fibroblasts and they also contain other cell types,
including perineural and mast cells, pericytes, endothelial and
smooth muscle cells (54).
Fibroblasts are known to express transgelin (53) and Schwann cells also express
transgelin based on our data (Fig.
3C), indicating that two of the major cells forming MPNST
tumors, Schwann cells and fibroblasts, express transgelin. Thus,
the hyperexpression of transgelin in the MPNSTS in the present
study may reflect the hyperexpression in these two types of cells,
as well as in smooth muscle cells. Schwann cells are well known to
contribute fundamentally to MPNST development (55). Fibroblasts are associated with
cancer cells in all stages of cancer progression through the
production of growth factors, chemokines and the extracellular
matrix, and therefore fibroblasts are a key determinant in the
malignant progression of cancer (56). It was reported that the upregulation
of transgelin in stromal fibroblasts promoted gastric cancer cell
migration and invasion by inducing the expression of matrix
metalloproteinase-2 (MMP-2) (53).
Further studies are required to elucidate how transgelin
upregulation in these cells is implicated in tumor progression in
NF1.
The molecular mechanisms of transgelin in cancer
development remain poorly understood (49,50).
The involvement of transgelin in transforming growth factor β
(TGF-β) and MMP-9 has been reported (49). In addition, RAS-dependent and
RAS-independent downregulation of transgelin in human breast and
colon carcinomas has been reported (57). Although this study showed results
opposite to our results, in that transgelin is downregulated in
tumors, it is consistent with our results on how activation of the
RAS-RAF-MEK-ERK signaling pathway antagonizes transgelin
expression. Our data demonstrated that the upregulation of
transgelin in normal-phenotypic primary cells by TAGLN
overexpression caused an increase in RAS and ERK1/2 activation,
while the downregulation of transgelin in MPNST primary cells and
cell lines by siRNA treatment resulted in decreased RAS and ERK1/2
activation (Fig. 3). These results
indicate that transgelin is closely involved in the RAS-MAPK
signaling pathway.
Our data indicated that transgelin acts as a
proto-oncogene rather than as a tumor suppressor gene in
NF1-associated MPNST development, which suggests that transgelin
may be a suitable target molecule to be inhibited for therapeutic
treatment and/or prevention of MPNSTs. Pancreatic cancer patients
with high transgelin expression showed a shorter 5-year overall
survival rate than those with low transgelin expression (52). Since reduced transgelin expression
was accompanied by significantly decreased RAS-MAPK signaling in
MPNST cells, the inhibition of transgelin expression may attenuate
MPNST development. The upregulation of the miR-145 miRNA enhanced
transgelin expression (58);
therefore, inhibitors of miR-145 could also be therapeutic drugs in
MPNSTs. Gene-specific and genome-wide DNA methylation profiling may
be useful biomarkers for diagnosis, risk assessment, prognosis,
therapy and therapy-response prediction in cancer (59,60).
Recent advances in the field of DNA methylation have begun to
elucidate the underlying mechanisms of DNA methylation modification
in cancers and to identify novel biomarker targets (60). The methylation profile of the
TAGLN gene may be another biomarker that is useful for
decision making with regard to the tumor progression of
NF1-associated tumors.
Acknowledgements
This study was supported by a grant (03-2013-0190)
from the SNUH Research Fund, and a National Research Foundation of
Korea (NRF) grant funded by the Korean government
(2014-001483).
References
|
1
|
Boyd KP, Korf BR and Theos A:
Neurofibromatosis type 1. J Am Acad Dermatol. 61:1–14. 2009.
View Article : Google Scholar
|
|
2
|
McClatchey AI: Neurofibromatosis. Annu Rev
Pathol. 2:191–216. 2007. View Article : Google Scholar
|
|
3
|
Cawthon RM, Weiss R, Xu GF, et al: A major
segment of the neurofibromatosis type 1 gene: cDNA sequence,
genomic structure, and point mutations. Cell. 62:193–201. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jett K and Friedman JM: Clinical and
genetic aspects of neurofibromatosis 1. Genet Med. 12:1–11. 2010.
View Article : Google Scholar
|
|
5
|
Katz D, Lazar A and Lev D: Malignant
peripheral nerve sheath tumour (MPNST): the clinical implications
of cellular signalling pathways. Expert Rev Mol Med. 11:e302009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Evans DG, Baser ME, McGaughran J, Sharif
S, Howard E and Moran A: Malignant peripheral nerve sheath tumours
in neurofibromatosis 1. J Med Genet. 39:311–314. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Grobmyer SR, Reith JD, Shahlaee A, Bush CH
and Hochwald SN: Malignant peripheral nerve sheath tumor: molecular
pathogenesis and current management considerations. J Surg Oncol.
97:340–349. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Brems H, Beert E, de Ravel T and Legius E:
Mechanisms in the pathogenesis of malignant tumours in
neurofibromatosis type 1. Lancet Oncol. 10:508–515. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thway K and Fisher C: Malignant peripheral
nerve sheath tumor: pathology and genetics. Ann Diagn Pathol.
18:109–116. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
U padhyaya M, Kluwe L, Spurlock G, et al:
Germline and somatic NF1 gene mutation spectrum in
NF1-associated malignant peripheral nerve sheath tumors (MPNSTs).
Hum Mutat. 29:74–82. 2008.
|
|
11
|
Kluwe L, Friedrich RE and Mautner VF:
Allelic loss of the NF1 gene in NF1-associated plexiform
neurofibromas. Cancer Genet Cytogenet. 113:65–69. 1999.
|
|
12
|
Legius E, Dierick H, Wu R, et al: TP53
mutations are frequent in malignant NF1 tumors. Genes Chromosomes
Cancer. 10:250–255. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nielsen GP, Stemmer-Rachamimov AO, Ino Y,
Moller MB, Rosenberg AE and Louis DN: Malignant transformation of
neurofibromas in neurofibromatosis 1 is associated with
CDKN2A/p16 inactivation. Am J Pathol. 155:1879–1884. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mawrin C, Kirches E, Boltze C, Dietzmann
K, Roessner A and Schneider-Stock R: Immunohistochemical and
molecular analysis of p53, RB, and PTEN in malignant peripheral
nerve sheath tumors. Virchows Arch. 440:610–615. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Upadhyaya M: Genetic basis of
tumorigenesis in NF1 malignant peripheral nerve sheath tumors.
Front Biosci. 16:937–951. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mo W, Chen J, Patel A, et al: CXCR4/CXCL12
mediate autocrine cell-cycle progression in NF1-associated
malignant peripheral nerve sheath tumors. Cell. 152:1077–1090.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lévy P, Ripoche H, Laurendeau I, et al:
Microarray-based identification of tenascin C and
tenascin XB, genes possibly involved in tumorigenesis
associated with neurofibromatosis type 1. Clin Cancer Res.
13:398–407. 2007.
|
|
18
|
Watanabe T, Oda Y, Tamiya S, Masuda K and
Tsuneyoshi M: Malignant peripheral nerve sheath tumour arising
within neurofibroma. An immunohistochemical analysis in the
comparison between benign and malignant components. J Clin Pathol.
54:631–636. 2001. View Article : Google Scholar
|
|
19
|
Johannessen CM, Reczek EE, James MF, Brems
H, Legius E and Cichowski K: The NF1 tumor suppressor critically
regulates TSC2 and mTOR. Proc Natl Acad Sci USA. 102:8573–8578.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Park HJ, Lee SJ, Sohn YB, et al:
NF1 deficiency causes Bcl-xL upregulation in Schwann cells
derived from neurofibromatosis type 1-associated malignant
peripheral nerve sheath tumors. Int J Oncol. 42:657–666. 2013.
|
|
21
|
Carroll SL and Ratner N: How does the
Schwann cell lineage form tumors in NF1? Glia. 56:1590–1605. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gregorian C, Nakashima J, Dry SM, et al:
PTEN dosage is essential for neurofibroma development and malignant
transformation. Proc Natl Acad Sci USA. 106:19479–19484. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu J, Deshmukh H, Payton JE, et al:
Array-based comparative genomic hybridization identifies
CDK4 and FOXM1 alterations as independent predictors
of survival in malignant peripheral nerve sheath tumor. Clin Cancer
Res. 17:1924–1934. 2011.PubMed/NCBI
|
|
24
|
Stricker TP, Henriksen KJ, Tonsgard JH,
Montag AG, Krausz TN and Pytel P: Expression profiling of 519
kinase genes in matched malignant peripheral nerve sheath
tumor/plexiform neurofibroma samples is discriminatory and
identifies mitotic regulators BUB1B, PBK and NEK2 as
overexpressed with transformation. Mod Pathol. 26:930–943. 2013.
View Article : Google Scholar
|
|
25
|
Kulis M and Esteller M: DNA methylation
and cancer. Adv Genet. 70:27–56. 2010. View Article : Google Scholar
|
|
26
|
Malumbres M: miRNAs and cancer: an
epigenetics view. Mol Aspects Med. 34:863–874. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sedani A, Cooper DN and Upadhyaya M: An
emerging role for microRNAs in NF1 tumorigenesis. Hum Genomics.
6:232012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Subramanian S, Thayanithy V, West RB, et
al: Genome-wide transcriptome analyses reveal p53 inactivation
mediated loss of miR-34a expression in malignant peripheral nerve
sheath tumours. J Pathol. 220:58–70. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Masliah-Planchon J, Pasmant E, Luscan A,
et al: MicroRNAome profiling in benign and malignant
neurofibromatosis type 1-associated nerve sheath tumors: evidences
of PTEN pathway alterations in early NF1 tumorigenesis. BMC
Genomics. 14:4732013. View Article : Google Scholar
|
|
30
|
Itani S, Kunisada T, Morimoto Y, et al:
MicroRNA-21 correlates with tumorigenesis in malignant peripheral
nerve sheath tumor (MPNST) via programmed cell death protein 4
(PDCD4). J Cancer Res Clin Oncol. 138:1501–1509. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ferner RE, Huson SM, Thomas N, et al:
Guidelines for the diagnosis and management of individuals with
neurofibromatosis 1. J Med Genet. 44:81–88. 2007. View Article : Google Scholar
|
|
32
|
Lee SJ, Park HJ, Kim YH, et al: Inhibition
of Bcl-xL by ABT-737 enhances chemotherapy sensitivity in
neurofibromatosis type 1-associated malignant peripheral nerve
sheath tumor cells. Int J Mol Med. 30:443–450. 2012.PubMed/NCBI
|
|
33
|
Bibikova M and Fan JB: Genome-wide DNA
methylation profiling. Wiley Interdiscip Rev Syst Biol Med.
2:210–223. 2010. View Article : Google Scholar
|
|
34
|
Jeong SY, Han JH, Park YY and Kim HJ:
Identification of differentially expressed genes related to
NF1-associated malignant transformation from a patient with
neurofibromatosis type 1. Genes Genomics. 30:407–418. 2008.
|
|
35
|
Hirasawa Y, Arai M, Imazeki F, et al:
Methylation status of genes upregulated by demethylating agent
5-aza-2′-deoxycytidine in hepatocellular carcinoma. Oncology.
71:77–85. 2006.
|
|
36
|
Zhao L, Wang H, Deng YJ, et al: Transgelin
as a suppressor is associated with poor prognosis in colorectal
carcinoma patients. Mod Pathol. 22:786–796. 2009.PubMed/NCBI
|
|
37
|
Stanier P, Abu-Hayyeh S, Murdoch JN,
Eddleston J and Copp AJ: Paralogous sm22α (Tagln)
genes map to mouse chromosomes 1 and 9: further evidence for a
paralogous relationship. Genomics. 51:144–147. 1998.
|
|
38
|
Li Y, Rao PK, Wen R, et al: Notch and
Schwann cell transformation. Oncogene. 23:1146–1152. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Grønbaek K, Hother C and Jones PA:
Epigenetic changes in cancer. APMIS. 115:1039–1059. 2007.
|
|
40
|
Ehrlich M and Lacey M: DNA hypomethylation
and hemimethylation in cancer. Adv Exp Med Biol. 754:31–56. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Suzuki H, Maruyama R, Yamamoto E and Kai
M: DNA methylation and microRNA dysregulation in cancer. Mol Oncol.
6:567–578. 2012. View Article : Google Scholar
|
|
42
|
Harder A, Titze S, Herbst L, et al:
Monozygotic twins with neurofibromatosis type 1 (NF1) display
differences in methylation of NF1 gene promoter elements, 5′
untranslated region, exon and intron 1. Twin Res Hum Genet.
13:582–594. 2010.PubMed/NCBI
|
|
43
|
Titze S, Peters H, Währisch S, et al:
Differential MSH2 promoter methylation in blood cells of
Neurofibromatosis type 1 (NF1) patients. Eur J Hum Genet. 18:81–87.
2010.
|
|
44
|
Fishbein L, Eady B, Sanek N, Muir D and
Wallace MR: Analysis of somatic NF1 promoter methylation in
plexiform neurofibromas and Schwann cells. Cancer Genet Cytogenet.
157:181–186. 2005.PubMed/NCBI
|
|
45
|
Harder A, Rosche M, Reuss DE, et al:
Methylation analysis of the neurofibromatosis type 1 (NF1)
promoter in peripheral nerve sheath tumours. Eur J Cancer.
40:2820–2828. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bradtmöller M, Hartmann C, Zietsch J, et
al: Impaired Pten expression in human malignant peripheral nerve
sheath tumours. PLoS One. 7:e475952012.PubMed/NCBI
|
|
47
|
Camoretti-Mercado B, Forsythe SM, LeBeau
MM, et al: Expression and cytogenetic localization of the human
SM22 gene (TAGLN). Genomics. 49:452–457. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yamamura H, Masuda H, Ikeda W, et al:
Structure and expression of the human SM22α gene, assignment of the
gene to chromosome 11, and repression of the promoter activity by
cytosine DNA methylation. J Biochem. 122:157–167. 1997.
|
|
49
|
Assinder SJ, Stanton JA and Prasad PD:
Transgelin: an actin-binding protein and tumour suppressor. Int J
Biochem Cell Biol. 41:482–486. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dvorakova M, Nenutil R and Bouchal P:
Transgelins, cytoskeletal proteins implicated in different aspects
of cancer development. Expert Rev Proteomics. 11:149–165. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Dos Santos Hidalgo G, Meola J, Rosa E,
Silva JC, Paro de Paz CC and Ferriani RA: TAGLN expression is
deregulated in endometriosis and may be involved in cell invasion,
migration, and differentiation. Fertil Steril. 96:700–703.
2011.PubMed/NCBI
|
|
52
|
Zhou L, Zhang R, Zhang L, et al:
Upregulation of transgelin is an independent factor predictive of
poor prognosis in patients with advanced pancreatic cancer. Cancer
Sci. 104:423–430. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yu B, Chen X, Li J, et al: Stromal
fibroblasts in the microenvironment of gastric carcinomas promote
tumor metastasis via upregulating TAGLN expression. BMC Cell Biol.
14:172013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gottfried ON, Viskochil DH, Fults DW and
Couldwell WT: Molecular, genetic, and cellular pathogenesis of
neurofibromas and surgical implications. Neurosurgery. 58:1–16.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Carroll SL: Molecular mechanisms promoting
the pathogenesis of Schwann cell neoplasms. Acta Neuropathol.
123:321–348. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kalluri R and Zeisberg M: Fibroblasts in
cancer. Nat Rev Cancer. 6:392–401. 2006. View Article : Google Scholar
|
|
57
|
Shields JM, Rogers-Graham K and Der CJ:
Loss of transgelin in breast and colon tumors and in RIE-1 cells by
Ras deregulation of gene expression through Raf-independent
pathways. J Biol Chem. 277:9790–9799. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Adammek M, Greve B, Kässens N, et al:
MicroRNA miR-145 inhibits proliferation, invasiveness, and stem
cell phenotype of an in vitro endometriosis model by targeting
multiple cytoskeletal elements and pluripotency factors. Fertil
Steril. 99:1346–1355.e5. 2013. View Article : Google Scholar
|
|
59
|
Vairaktaris E, Kalokerinos G, Goutzanis L,
et al: Diabetes enhances cell proliferation but not
Bax/Bcl-2-mediated apoptosis during oral oncogenesis. Int J Oral
Maxillofac Surg. 37:60–65. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Sinčić N and Herceg Z: DNA methylation and
cancer: ghosts and angels above the genes. Curr Opin Oncol.
23:69–76. 2011.PubMed/NCBI
|