Introduction
Alcohol is one of the most widely used psychoactive
substances worldwide. Alcohol consumption carries a risk of adverse
health effects since alcohol cannot be excreted and must be
metabolized, primarily by the liver. If alcohol is consistently
consumed at high levels, the metabolic byproducts can promote the
development of steatosis, which can progress to steatohepatitis,
fibrosis, cirrhosis, liver failure and/or hepatocellular carcinoma
(1). Although diverse mechanisms
are involved in alcohol-induced hepatotoxicity, accumulating
evidence has supported that oxidative stress is crucial in the
pathogenesis and progression of alcoholic liver disease (2). Oxidative stress refers to the enhanced
generation of reactive oxygen species (ROS) and/or depletion of the
antioxidant defense system, causing an imbalance between
pro-oxidants and antioxidants (3).
Thus, antioxidant therapy, as a promising strategy for alcoholic
liver disease treatment, has been gaining attention.
Chitosan is derived from the deacetylation of
chitin, which is the structural element in the exoskeleton of
crustaceans (such as crabs and shrimp) and cell walls of fungi
(4). Chitooligosaccharides (COS)
can be generated by the chemical or enzymatic hydrolysis of
chitosan (5). COS has attracted
interest in pharmaceutical and medicinal applications due to high
solubility and non-toxicity. In a previous study, COS significantly
attenuated alcohol-induced cytotoxicity in human HepG2 cells
(6). However, the molecular
mechanisms for the cytoprotection of COS against alcohol are poorly
understood (6).
Nuclear factor erythroid-2-related factor-2 (Nrf2)
is a basic leucine zipper transcription factor that binds to
antioxidant response element (ARE) sequences in the promoter
regions of specific genes (7). The
levels of intracellular antioxidants and antioxidant enzymes are
regulated by Nrf2. Under normal conditions, Nrf-2 is combined with
Kelch-like ECH-associated protein 1 (Keap-1) in the cytosol. Under
oxidative stress, Nrf-2 dissociates from Keap-1 and the unbound
Nrf-2 translocates into the nucleus where it binds to the ARE of
antioxidant genes (8). These genes
include the rate-limiting enzyme in the glutathione (GSH) synthesis
pathway, superoxide dismutase (SOD), heme oxygenase-1 (HO-1) and
NAD(P)H:quinone oxidoreductase 1 (NQO1) (9). Findings of recent studies identified
that Nrf-2 activation protected against alcohol-induced liver
damage (10,11). However, whether the beneficial
effects of COS are associated with the activation of Nrf-2 remains
to be clarified.
Mitogen-activated protein kinase (MAPK) cascades are
another major signaling pathway that is activated in response to
various cellular stimuli. Evidence has shown that the MAPK family
is essential in the initiation of cell processes such as
proliferation, differentiation, development, apoptosis, oxidative
stress and inflammatory responses (12). The major MAPK subfamilies identified
are p38 MAPK, c-Jun N-terminal kinase (JNK) and extracellular
signal-regulated kinase (ERK) (13). One type of stress that induces the
potential activation of MAPK pathways is oxidative stress caused by
ethanol (14). Therefore, drugs,
which exert effects against the ethanol-induced activation by
attenuating MAPK, may be candidates for therapeutic use in liver
diseases.
In the present study, we investigated whether COS
could confer protection against ethanol-induced oxidative damage in
human L02 normal liver cells. To determine the underlying
mechanisms of COS, we assessed the effects of COS on Nrf2
activation and MAPK phosphorylation following ethanol exposure.
Materials and methods
Cell culture and drug treatment
COS (degree of deacetylation ≥95%; average molecular
weight, <1,000 Da) was obtained from the Dalian Institute of
Chemical Physics (Dalian, China). Human L02 normal liver cells were
obtained from the Chinese Academy of Science (Shanghai, China), and
maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco,
Grand Island, NY, USA) containing 10% fetal bovine serum (Sijiqing,
Hangzhou, China) at 37°C in a 5% CO2 incubator. Cultures
were allowed to reach 80–90% confluence prior to experiments being
initiated. The cells were pretreated with different concentrations
of COS (0.25, 0.5 and 1.0 mg/ml) and incubated in a humidified
incubator at 37°C for 1 h. Ethanol (80 mM) was then added as a
final concentration and incubated for 24 h. Untreated cells served
as the control.
MTT assay
L02 cells were seeded in a 96-well plate at a
density of 5.0×103 cells/ml. Following treatment with
the abovementioned methods, the medium was removed and 50 μl of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
(5 mg/ml; Sigma, St. Louis, CA, USA) was added to each well. The
cells were then incubated in the dark at 37°C for an additional 4
h. The reaction was stopped by the addition of 150 μl DMSO (Sigma)
and the absorbance of samples at 570 nm was measured with a
microplate reader (Molecular Devices, Sunnyvale, CA, USA). Relative
cell viability was determined by the amount of MTT converted to the
insoluble formazan salt. The optical density of the formazan formed
in the control cells was taken as 100%. The viability of L02 cells
in other groups was presented as a percentage of the control
cells.
Detection of cell apoptosis by flow
cytometry
Cells were collected, followed by the addition of 5
μl FITC-labeled Annexin V and 2 μl propidium iodine (PI) (both from
Beyotime Biotechnology, Nantong, China). The cells were mixed well
and bathed in warm water for 15 min at 37°C in the dark, followed
by the addition of 400 μl buffer solution to detect cell apoptosis.
Stained cells were filtered through a nylon-mesh sieve to remove
cell clumps and analyzed by a FACScan flow cytometer and CellQuest
analysis software (Becton-Dickinson, Franklin Lakes, CA, USA).
Measurement of ROS production
2′7′-Dichlorodihydrofluorescein diacetate (DCFH-DA)
is able to diffuse through the cell membrane and become
enzymatically hydrolysed by intracellular esterases to produce
non-fluorescent DCFH. The oxidation of DCFH by intracellular ROS
results in fluorescent DCF which stains the cells. Thus, the
intracellular ROS generation of cells can be detected and
quantified by DCFH-DA. Cell samples were incubated in the presence
of 10 μM DCFH-DA (Sigma) in phosphate-buffered saline (PBS) at 37°C
for 30 min, washed two times with PBS and centrifuged at 1,200 rpm
to remove the extracellular DCFH-DA. Stained cells were examined
under a fluorescence spectrophotometer (Hidex Oy, Turku, Finland)
at 498/530 nm. The percentage of DCF fluorescence was compared with
the control, which was arbitrarily assigned as 100%.
Determination of lipid peroxidation and
reduced GSH
Accumulated intracellular lipid peroxidation was
measured as MDA equivalent generated, as an indicator of lipid
peroxidation in cultured cell lysates (10). The levels of MDA and GSH were
detected using commercial kits (Jiancheng, Nanjing, China). The MDA
levels were measured at 535 nm based on the reaction of
thiobarbituric acid (TBA) with MDA, while the GSH levels were
measured at 412 nm following the reaction of GSH with
5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB).
Transfection of small interfering
RNA
The small interfering RNA (siRNA) directed against
Nrf2 (Nrf2-siRNA) or non-targeting negative control siRNA
(NC-siRNA) was purchased from GenePharma (Shanghai, China). L02
cells were transfected with the Nrf2- or NC-siRNA with
Lipofectamine according to the manufacturer’s instructions
(Invitrogen, Carlsbad, CA, USA). Briefly, the cells were seeded in
6-well plates at a density of 2×105 cells/well in 2 ml
of complete DMEM. The cells were allowed to grow to 60–80%
confluence prior to transfection with siRNA. For each well, 100
pmol of siRNA were mixed with 2 μl of Lipofectamine. Serum- and
antibiotic-free DMEM medium (500 μl) was subsequently added. The
cells were exposed to the transfection mixture for 6 h. At the end
of the incubation period, 1.5 ml of antibiotic-free complete medium
was added and the cells were cultured for an additional 18 h.
Following treatment with COS and/or ethanol, the cells were
harvested and Nrf2 expression was determined by quantitative PCR
(qPCR) and western blotting.
qPCR analysis
Total RNA of cells was extracted using TRIzol
reagent (Invitrogen). Total RNA (2 μg) was reverse-transcribed in a
total volume of 20 μl containing 200 units of SuperScript II RNase
H Reverse Transcriptase (Invitrogen) at 42°C for 50 min. This was
followed by inactivation at 70°C for 15 min. The products were
amplified for 40 cycles for 10 sec at 95°C for denaturation and 30
sec at 56°C for annealing. GAPDH expression was used as endogenous
control for the normalization of gene expression. The primer
sequences for the genes are shown in Table I and were synthesized by Sangon
(Shanghai, China).
 | Table IOligonucleotide sequences for qPCR of
Nrf2 and antioxidative enzymes. |
Table I
Oligonucleotide sequences for qPCR of
Nrf2 and antioxidative enzymes.
| Gene | Sequences
(5′-3′) |
|---|
| HO-1 | F:
CTGACCCATGACACCAAGGAC
R: AAAGCCCTACAGCAACTGTCG |
| SOD | F:
CGGATGAAGAGAGGCATGTT
R: CACCTTTGCCCAAGTCATCT |
| NQO1 | F:
TTCTCTGGCCGATTCAGAGT
R: GGCTGCTTGGAGCAAAATAG |
| Nrf2 | F:
ACACGGTCCACAGCTCATC
R: TGTCAATCAAATCCATGTCCTG |
| GAPDH | F:
CCAACCGCGAGAAGATGA
R: CCAGAGGCGTACAGGGATAG |
Western blot analysis
The treated cells were lysed and centrifuged at
12,000 × g for 5 min at 4°C. The supernatant was collected and 5X
protein loading buffer was added followed by boiling for 10 min.
After SDS-PAGE, the cells were transferred to a PVDF membrane
(Millipore, Bedford, MA, USA). The membranes were blocked with 5%
non-fat milk solution for 1 h at room temperature, and then
incubated with the specific primary antibodies overnight at 4°C.
After washing three times, the membranes were incubated with
horseradish peroxidase-conjugated secondary antibodies (Beyotime
Biotechnology) for 1 h at room temperature. The ECL (Beyotime
Biotechnology) chemiluminescence method was used to visualize the
protein bands. Antibodies directed against phospho-p38 MAPK
(Thr180/Tyr182), phospho-JNK (Thr183/Tyr185), phospho-ERK, p38
MAPK, JNK and ERK were obtained from Cell Signaling Technology
(Beverly, MA, USA). SOD, NQO1, HO-1, Nrf2, histone H3 and GAPDH
antibodies were obtained from Santa Cruz Biotechnology, Inc. (Santa
Cruz, CA, USA).
Statistical analysis
Statistical analysis was performed using SPSS 13.0
software. Each assay was performed at least 3 times. Data are
presented as means ± SD. The Student’s t-test was used to evaluate
the significance of data. P<0.05 was considered to indicate a
statistically significant result.
Results
COS effectively suppresses
ethanol-induced cytotoxicity in L02 cells
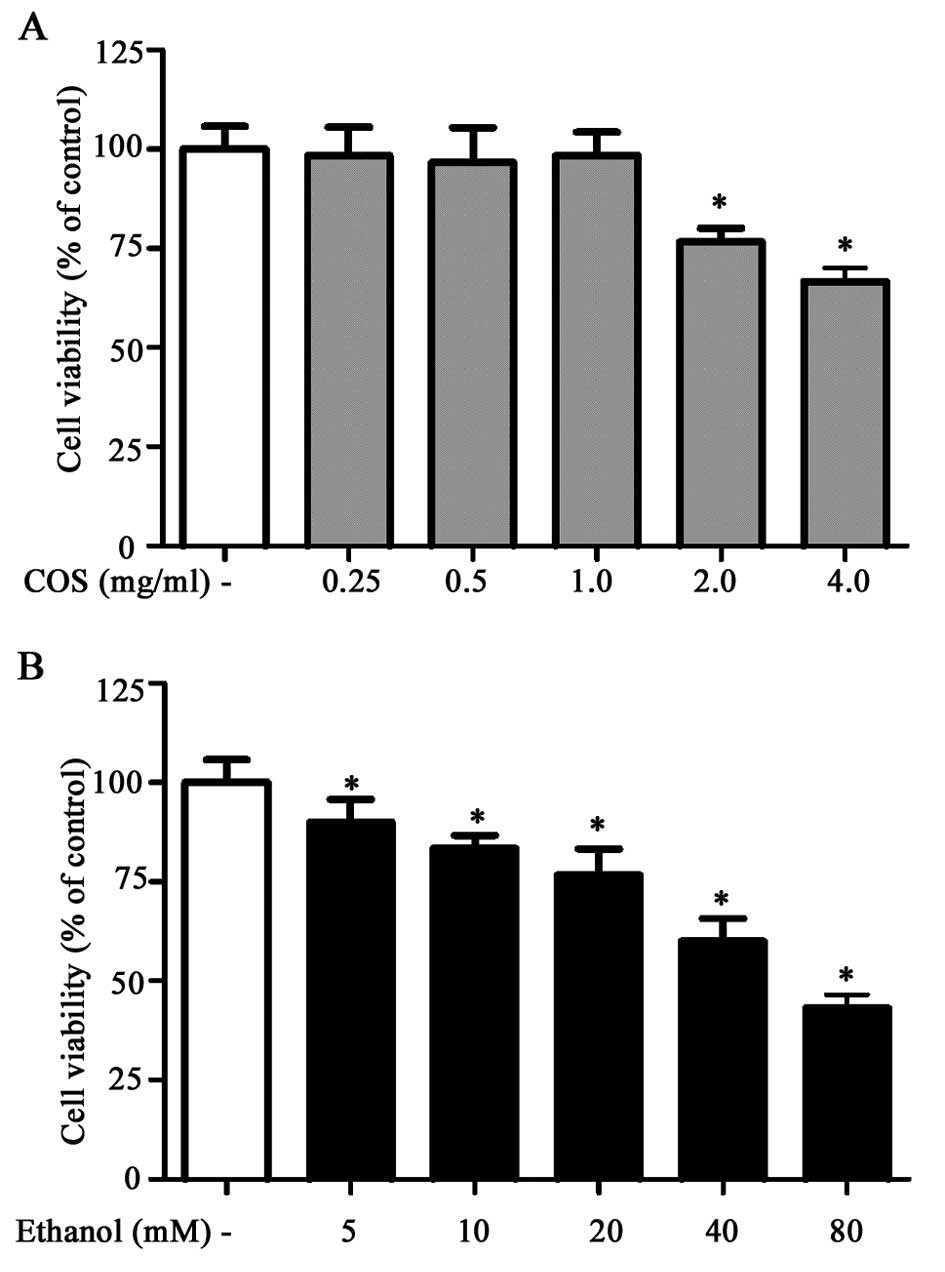
Prior to in vitro hepatoprotective studies,
the cytotoxic effect of COS was examined in human L02 normal liver
cells. Results of the MTT assay showed that L02 cells treated with
increasing concentrations of COS (0.25, 0.5, 1.0, 2.0 and 4.0
mg/ml) for 24 h did not show any cytotoxic effect up to the
concentration of 1.0 mg/ml. The concentration of ethanol required
to induce oxidative stress and cell death in L02 cells was also
examined. Among the various concentrations tested (5, 10, 20, 40
and 80 mM), ethanol caused loss of cell viability at a
concentration of 80 mM over a period of 24 h incubation (Fig. 1B). Therefore, the non-cytotoxic
concentrations of COS (0.25, 0.5 and 1.0 mg/ml) and cytotoxic
concentration of ethanol (80 mM) were selected as the standard
concentrations for the subsequent assessments.
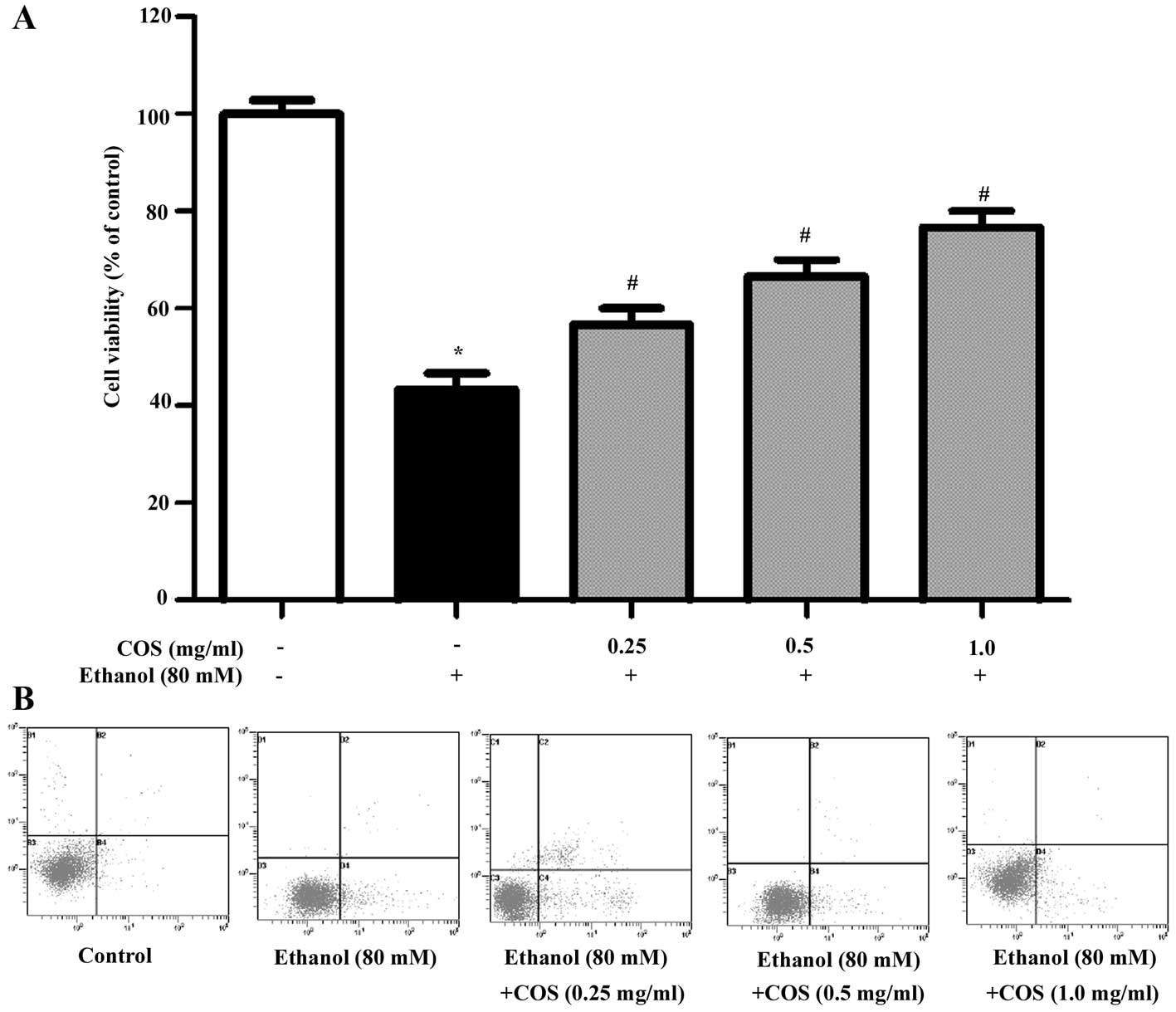
Different concentrations of COS were evaluated for
its protective effect against ethanol-induced toxicity employed in
L02 cells. As shown in Fig. 2A,
ethanol treatment markedly decreased L02 cell viability to
46.3±3.2% compared with the control group (P<0.05). However,
pretreatment with COS significantly attenuated the decrease of cell
viability, which was 66.1± 2.1, 70.5±4.3 and 78.6±2.5% for COS
(0.25, 0.5 and 1.0 mg/ml) (P<0.05). Furthermore, the apoptosis
triggered by ethanol with or without pre-incubation of COS was
detected by flow cytometry (Fig.
2B). The results showed that the apoptosis ratio of L02 in the
control group was 6.22%, and it increased to 18.47% (P<0.05)
following induction by ethanol for 24 h. After the cells were
pretreated with COS (0.25, 0.5 and 1.0 mg/ml) for 1 h and induced
by ethanol for 24 h, the cell apoptotic ratios were 16.15, 12.91
and 9.73%, respectively (P<0.05). These results suggested that
COS was able to efficiently protect L02 cells against oxidative
stress induced cell damage in a dose-dependent manner.
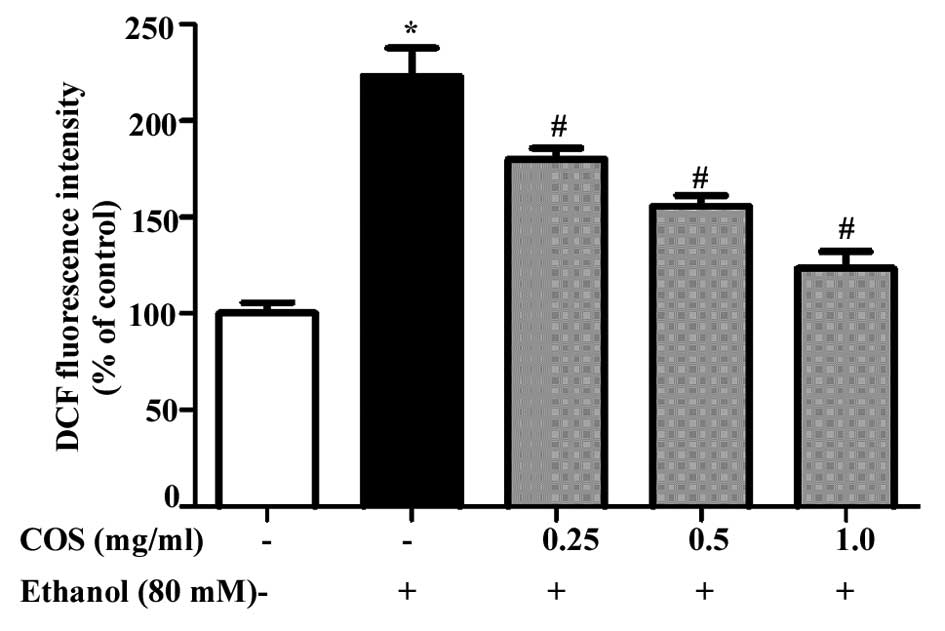
COS pretreatment prevents ethanol-induced
ROS generation in L02 cells
Sustained intracellular ROS production has been
recognized as a crucial step for ethanol-induced oxidative stress
(15). Fig. 3 shows the mean values of DCF
fluorescence, an indicator of intracellular ROS generation. L02
cells exposed to ethanol at 80 mM exhibited a significant increase
in the intracellular level of ROS as compared with that in the
control cells (P<0.05). However, the increase in intracellular
ROS (235%) caused by ethanol was significantly reduced to 181, 157
and 129% by 0.25, 0.5 and 1.0 mg/ml of COS, respectively
(P<0.05).
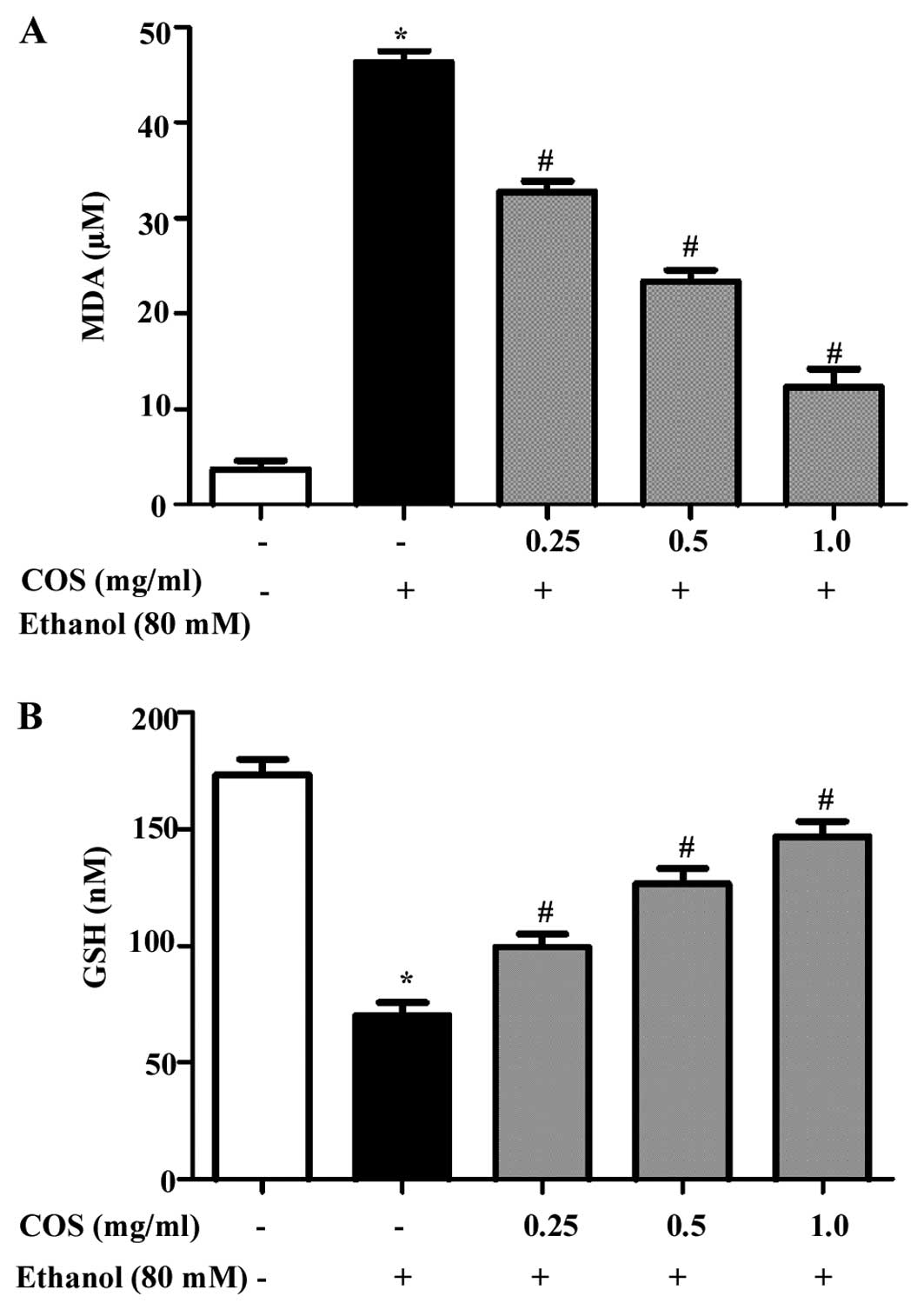
COS inhibits ethanol-induced lipid
peroxidation and GSH depletion in L02 cells
Inhibitory effect of COS on oxidative damage in L02
cells induced by ethanol was evaluated by lipid peroxidation assay.
As shown in Fig. 4A,
ethanol-exposure significantly (P<0.05) increased the
intracellular MDA level (45±1.2 μM) compared to the control group
(4±0.8 μM). However, the elevated MDA levels were significantly
(P<0.05) decreasesd to 33±0.5, 23±1.7 and 14±0.6 μM at 0.25, 0.5
and 1.0 mg/ml of COS, respectively. By contrast, the effect of COS
on ethanol-induced GSH depletion was investigated. Fig. 4B shows that ethanol depleted the GSH
levels (71±2.1 nM) significantly (P<0.05) compared to that of
the control cells (169±3.6 nM). However, the levels were
effectively restored by pretreatment with COS in a dose-dependent
manner (99±2.3, 127±1.9 and 145±2.8 nM at concentrations of 0.25,
0.5 and 1.0 mg/ml, respectively).
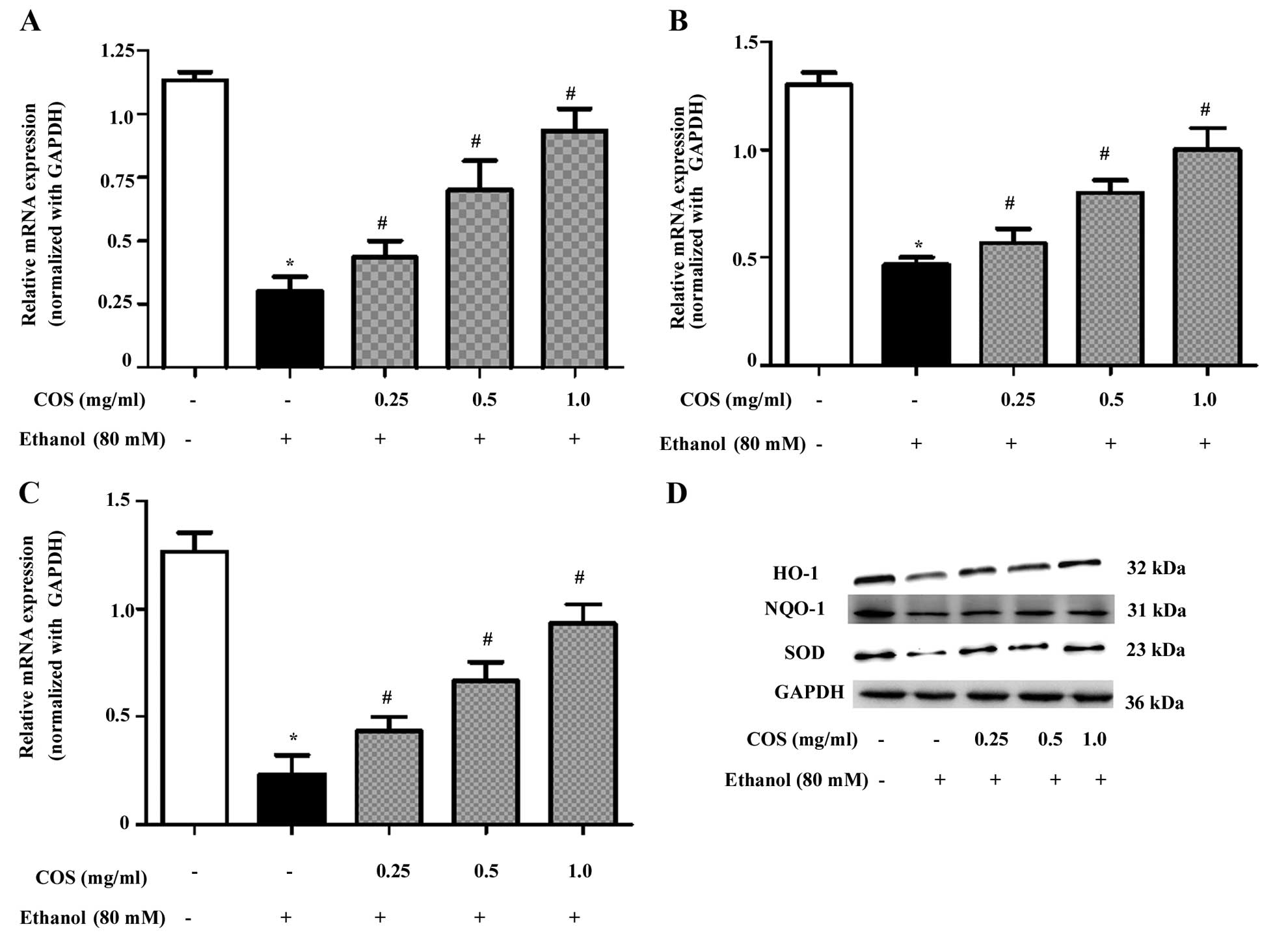
COS upregulates antioxidant gene
expression in ethanol-induced L02 cells
To determine whether the inhibitory effect of COS on
ethanol-induced oxidative stress was associated with the induction
of antioxidant genes, we examined the mRNA and protein expression
of HO-1, NQO-1 and SOD. The qPCR analysis showed that pretreatment
with COS significantly (P<0.05) increased the mRNA expression
levels of HO-1 (Fig. 5A), NQO-1
(Fig. 5B) and SOD (Fig. 5C) in a dose-dependent manner. In
addition, the western blot analysis confirmed that pretreatment
with COS significantly (P<0.05) increased HO-1, NQO-1 and SOD
protein expression in ethanol-induced L02 cells (Fig. 5D).
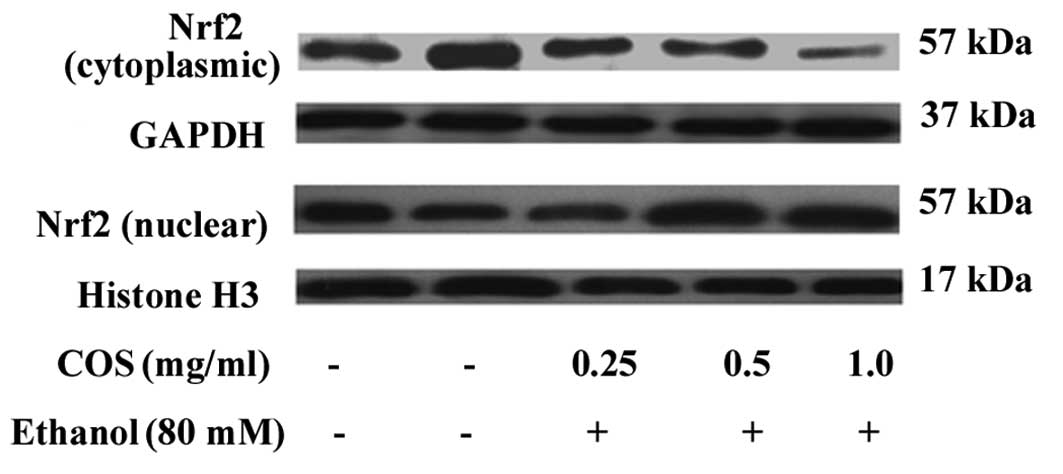
COS stimulates the nuclear translocation
of Nrf-2 in L02 cells
It is well known that antioxidant genes including
HO-1, NQO-1 and SOD are transcribed by Nrf2, a major transcription
factor regulating ARE-driven phase II gene expression (9). To investigate the effect of COS on the
nuclear translocation of Nrf-2, cytosolic and nuclear fractions
were prepared, and the protein levels of Nrf-2 were measured by
western blot analysis. The results indicated that the nuclear
protein levels of Nrf-2 were dose-dependently increased in L02
cells treated with COS, while the cytosolic protein levels of Nrf-2
were decreased (Fig. 6).
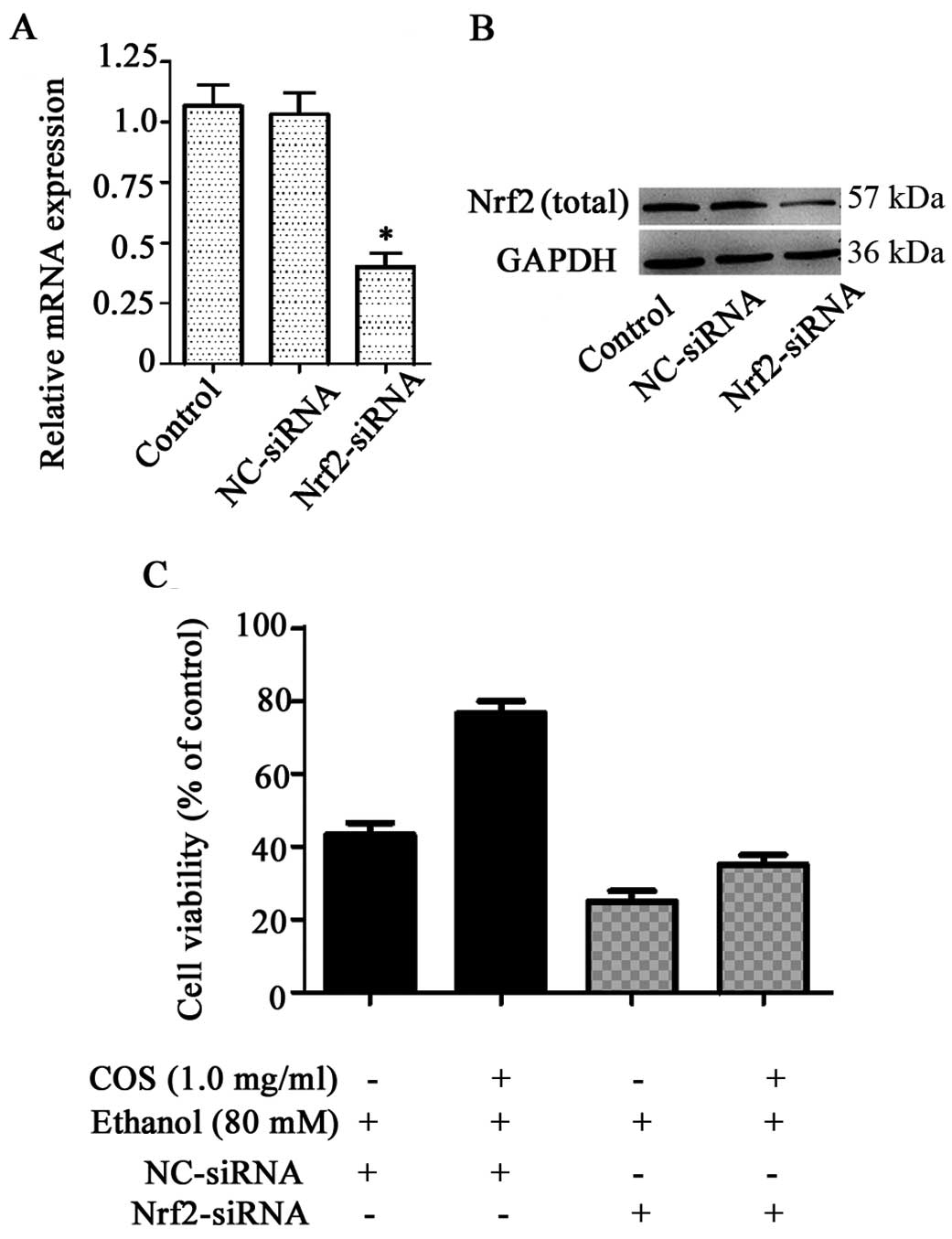
Nrf2 knockdown decreases the protective
effects of COS in L02 cells
To investigate whether the protection of COS against
ethanol-induced toxicity was attributed to the activation of Nrf2,
we developed an Nrf2 gene knockdown model in L02 cells using siRNA
transfection. As shown in Fig. 7A and
B, L02 cells transfected with Nrf2 siRNA exhibited a direct
reduction at the levels of Nrf2 mRNA and protein (P<0.05),
whereas no significant inhibitory effect was observed in the
control group and cells treated with NC-siRNA (P>0.05). Nrf2
knockdown cells were treated with COS (1.0 mg/ml) for 1 h prior to
the challenge with 80 mM ethanol. Cell viability was measured by
MTT assay. Compared to ethanol-treated cells (45.1±2.2%), COS
pretreatment significantly increased the amount of viable cells to
74.9±1.8% in the NC-siRNA system, whereas the percentage of viable
cells in the Nrf2 knockdown system was further decreased to
24.1±2.3% by ethanol. COS, therefore, failed to protect liver cells
from ethanol-induced cell death in the Nrf2 knockdown system, as
shown by the 36.4±2.4% cell viability observed (Fig. 7C). We found Nrf2 siRNA treatment,
not only aggravated the adverse effects of ethanol, but also
abrogated the protective effects of COS.
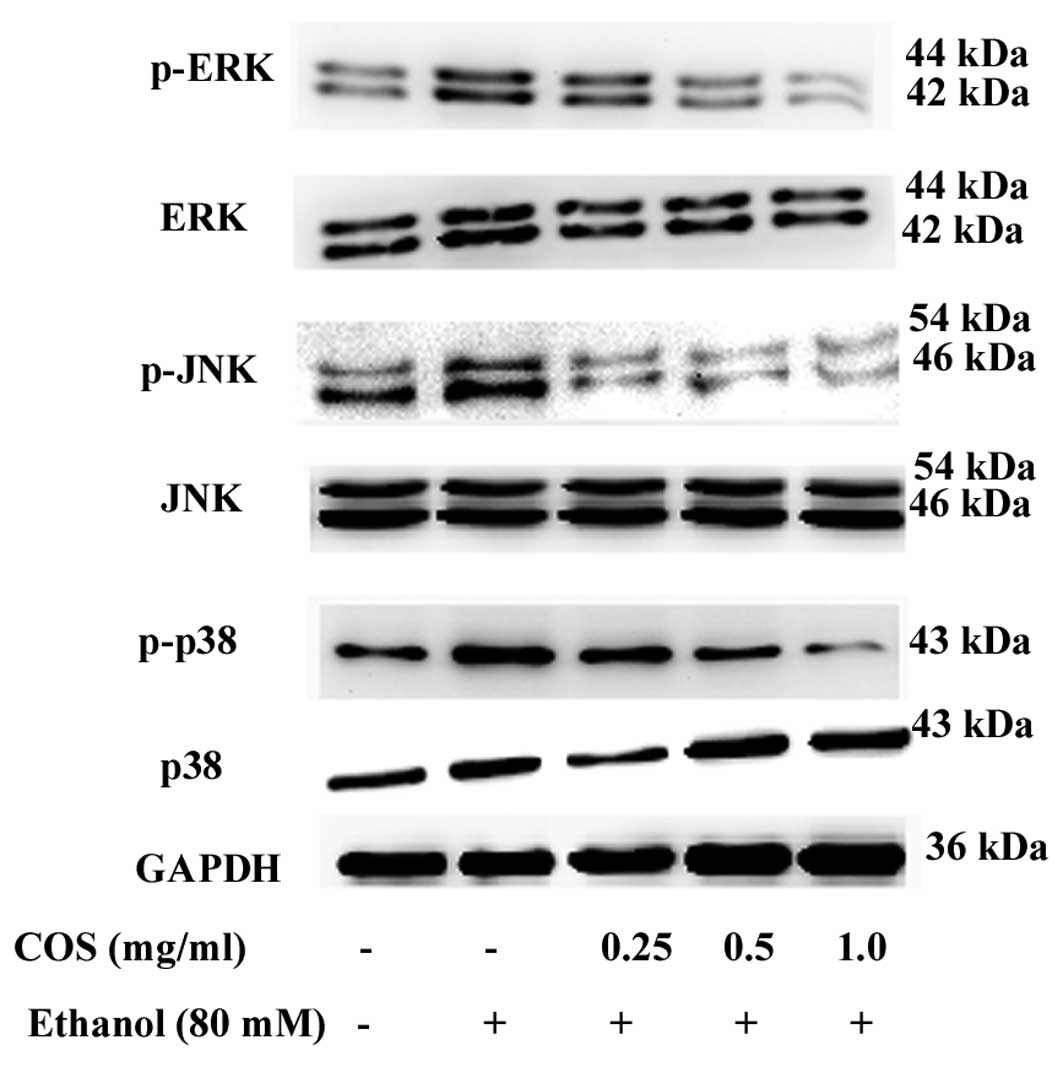
COS treatment attenuates ethanol-induced
MAPK phosphorylation in L02 cells
MAPK cascades are key signaling pathways involved in
the regulation of normal cell proliferation and survival (12). Therefore, we measured the level of
phosphorylated MAPKs in the presence and/or absence of COS. As
shown in Fig. 8, p38 MAPK, JNK and
ERK were highly phosphorylated in L02 cells when treated with
ethanol alone. By contrast, the increase of p38 MAPK, JNK and ERK
phosphorylation were suppressed by COS pretreatment. COS reduced
MAPK activation in a dose-dependent manner, suggesting that MAPK
inhibition by COS can result in increased survival in L02
cells.
Discussion
Ethanol-induced liver disease remains a worldwide
health concern without effective therapies. Since oxidative stress
is a well-established factor in the progression of liver damage due
to alcohol intake, using antioxidants to ameliorate oxidative
stress and alleviate ethanol hepatotoxicity is a logical approach.
This approach has shown a high rate of success in in vitro
and in vivo models. For example, EGCG is the major and most
active polyphenolic antioxidant present in green tea. It has been
confirmed that EGCG exerted a protective action against
ethanol-induced toxicity in Chang liver cells (16). Diosmin, a naturally occurring
flavone glycoside readily obtained by dehydrogenation of
hesperidin, alleviated alcoholic liver injury via inhibition of
oxidative stress markers in female wistar rats (17). Lucidone, isolated from the fruits of
Lindera lucida, also protected HepG2 cells against
ethanol-induced cytotoxicity (10).
In the present study, we have demonstrated that COS effectively
inhibited ethanol-induced oxidative damage in human L02 normal
liver cells. A similar effect was observed in an in vitro
system in which ethanol was used to induce oxidative stress in
cultured human hepatic (HepG2) cells (6). These results suggest that oxidative
stress critically contributes to ethanol hepatotoxicity, and that
antioxidant protection is an effective strategy to prevent alcohol
liver diseases.
Oxidative stress-induced cell death is associated
with an increase in ROS, such as hydrogen peroxide, nitric oxide,
superoxide anion, hydroxyl radicals and singlet oxygen (18). The accumulation of ROS in the liver
was found to cause dysfunction of cellular membrane systems,
protein and DNA oxidation and eventually hepatocyte injury. This
damage if unrepaired irreversibly caused cells to undergo apoptosis
(19). Ethanol promoted oxidative
stress, by the increased formation of ROS and by the depletion of
oxidative defenses in the cell (20). Thus, removal of excess ROS or
suppression of their generation by antioxidants may be effective in
preventing oxidative cell death induced by ethanol. Studies have
been conducted to investigate the antioxidant effects of natural
products and their findings showed significant results in ROS
production (21,22). In the present study, we have shown
from the results obtained that COS is a potential source that can
be used in medical- or pharmaceutical-related fields. Ethanol
exposure was found to induce the overproduction of ROS in L02
cells, leading to oxidative stress. However, treatment with COS
significantly suppressed ethanol-induced ROS generation and
increased the survival rate of L02 cells probably through potent
antioxidant activity.
Lipid peroxidation is a prominent manifestation of
ROS and oxidative stress in biological systems. Exposure to
chemical or physical agents triggers membrane-free radical
reactions in living cells, which accelerates lipid peroxidation
(23). A large number of toxic
byproducts are formed during lipid peroxidation such as MDA,
4-hydroxynonenal, conjugated-dienes, lipid hydroperoxides and
isoprostanes (24). The
concentration of MDA in cells or tissue lysates was considered to
be a major lipid peroxidation marker (10). We observed that ethanol exposure
sustained MDA levels in L02 cells, whereas the increase of MDA
levels was significantly inhibited by COS pretreatment. In
addition, GSH can act as a non-enzymatic antioxidant by direct
interaction of its sulfhydryl group with ROS or it can be involved
in the enzymatic detoxification of ROS, as a cofactor or coenzyme
(25). Therefore, reduced GSH
content was also used to evaluate oxidative stress in biological
systems. In the present study, we found that ethanol exposure
decreased GSH levels in L02 cells, which is in agreement with a
previous report (26). Notably,
results of this study also show that COS pretreatment prevented
ethanol-induced GSH depletion in liver cells.
Besides exogenous antioxidant defense, the body
depends on several endogenous defense mechanisms to protect against
oxidative stress. Among these antioxidant molecules, the phase II
enzymes including HO-1, NQO-1 and SOD play important roles in the
exclusion of ROS (27). HO-1 is a
stress inducible protein. Various stimuli, such as thiol
scavengers, ultraviolet radiation and oxidative stress act as
inducers of HO-1 (28). Increased
HO-1 expression has been reported to reduce cell injury such as
oxidative stress, pro-inflammatory cytokine production and the
activation of pro-apoptotic inducers (29). NQO-1 is an antioxidant that is
upregulated in response to hyperoxia and oxidative stress (30). SOD is the major enzyme for
scavenging ROS, which can catalyze the conversion of the superoxide
anion into oxygen and hydrogen peroxide (31). In the present study, we found that
COS treatment significantly enhanced HO-1, NQO-1 and SOD expression
at the mRNA and protein levels. As HO-1, NQO-1 and SOD can be
activated by Nrf-2, a major transcription factor regulating
ARE-driven phase-II gene expression, the cytosolic and nuclear
protein levels of Nrf-2 were then detected by western blotting. The
data showed that the nuclear protein levels of Nrf-2 were
dose-dependently increased in L02 cells treated with COS, while the
cytosolic protein levels of Nrf-2 were decreased, which indicated
that COS was able to enhance the nuclear translocation of Nrf-2.
Previous studies have shown that increasing Nrf-2 activity in
hepatic tissues was highly hepatoprotective during ethanol-induced
oxidative stress (32). We also
found Nrf2 knockdown decreased the protective effects of COS in L02
cells. These results strongly suggested that COS protects liver
cells from oxidative stress and cell death via the Nrf2-dependent
pathway. In a previous study, COS was found to increase cellular
antioxidant defense through the Nrf2 pathway in rat
pheochromocytoma (PC12) cells (12). Thus, it can be concluded that Nrf2
activation by COS is not cell type-dependent and it can be elicited
in cell lines of human and rat origin.
MAPKs are serine-threonine protein kinases that play
a significant role in signal transduction from the cell surface to
the nucleus (12). Diverse cellular
functions, ranging from cell survival to cell death, are regulated
by MAPK signaling. A number of extracellular and intracellular
stimuli that induce ROS production concomitantly can activate MAPK
pathways in multiple cell types (33). For example, direct exposure of
vascular endothelial cells to lipopolysaccharide (LPS), to mimic
oxidative stress, led to the activation of MAPK pathways (34). PC12 cells treated with hydrogen
peroxide, a ROS-generating agent, increased the phosphorylation of
MAPKs. Ethanol itself and ROS produced by ethanol also modulated
MAPKs in hepatocyte-like VL-17A cells (35). Therefore, we detected the
phosphorylation of MAPK family proteins including ERK, JNK and p38
MAPK. Results of the western blot analysis revealed that ethanol
significantly activated the phosphorylation of these MAPKs.
However, COS pretreatment markedly inhibited the phosphorylated
levels of MAPK in ethanol-induced L02 cells. It has been reported
that COS suppressed p38 MAPK and ERK phosphorylation in LPS-induced
N9 microglial cells (36). Based on
these results, we conclude that the protective effects of COS on
L02 cells may be attributed to the inhibition of phosphorylated
levels of MAPKs.
In conclusion, our findings have demonstrated that
COS inhibited ethanol-induced oxidative stress in L02 cells through
inhibition of ROS generation and upregulation of antioxidant genes
including HO-1, NQO1 and SOD at the transcription and translation
levels. The fact that COS regulated Nrf2 activation and MAPK
phophorylation, along with its established antioxidant effect,
makes this compound a candidate to be studied towards the
development of a therapeutic strategy for alcoholic liver
disease.
Acknowledgements
This study was supported by grants from the Health
Department of Hubei Province (JX3A20), the Young Talent Project of
Hubei Provincial Education Department (Q200724004), the Outstanding
Youth Scientific Innovation Team of Hubei University of Medicine
(2011CXG03), and the Scientific Research Foundation of Hubei
University of Medicine (nos. 2010QDJ20 and 2010QDJ21).
References
|
1
|
Lee HI, McGregor RA, Choi MS, et al: Low
doses of curcumin protect alcohol-induced liver damage by
modulation of the alcohol metabolic pathway, CYP2E1 and AMPK. Life
Sci. 93:693–699. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhu H, Jia Z, Misra H and Li YR: Oxidative
stress and redox signaling mechanisms of alcoholic liver disease:
updated experimental and clinical evidence. J Dig Dis. 13:133–142.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zeng T, Zhang CL, Song FY, et al: The
activation of HO-1/Nrf-2 contributes to the protective effects of
diallyl disulfide (DADS) against ethanol-induced oxidative stress.
Biochim Biophys Acta. 1830:4848–4859. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hajji S, Younes I, Ghorbel-Bellaaj O, et
al: Structural differences between chitin and chitosan extracted
from three different marine sources. Int J Biol Macromol.
65:298–306. 2014.PubMed/NCBI
|
|
5
|
Lu X, Guo H, Sun L, Zhang L and Zhang Y:
Protective effects of sulfated chitooligosaccharides with different
degrees of substitution in MIN6 cells. Int J Biol Macromol.
52:92–98. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cho SY, Yun JW, Park PJ, et al: Effects of
chitooligosaccharide lactate salt on activity of acetaldehyde
dehydrogenase. J Med Food. 13:1061–1068. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qiang W, Cahill JM, Liu J, et al:
Activation of transcription factor Nrf-2 and its downstream targets
in response to moloney murine leukemia virus ts1-induced
thiol depletion and oxidative stress in astrocytes. J Virol.
78:11926–11938. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ryu EY, Park AJ, Park SY, et al:
Inhibitory effects of Ginkgo biloba extract on inflammatory
mediator production by Porphyromonas gingivalis
lipopolysaccharide in murine macrophages via Nrf-2 mediated heme
oxygenase-1 signaling pathways. Inflammation. 35:1477–1486.
2012.
|
|
9
|
Gao D, Gao Z and Zhu G: Antioxidant
effects of Lactobacillus plantarum via activation of
transcription factor Nrf2. Food Funct. 4:982–989. 2013.
|
|
10
|
Senthil Kumar KJ, Liao JW, Xiao JH, Gokila
Vani M and Wang SY: Hepatoprotective effect of lucidone against
alcohol-induced oxidative stress in human hepatic HepG2 cells
through the up-regulation of HO-1/Nrf-2 antioxidant genes. Toxicol
In Vitro. 26:700–708. 2012.PubMed/NCBI
|
|
11
|
Kumar KJ, Chu FH, Hsieh HW, et al:
Antroquinonol from ethanolic extract of mycelium of Antrodia
cinnamomea protects hepatic cells from ethanol-induced
oxidative stress through Nrf-2 activation. J Ethnopharmacol.
136:168–177. 2011.PubMed/NCBI
|
|
12
|
Joodi G, Ansari N and Khodagholi F:
Chitooligosaccharide-mediated neuroprotection is associated with
modulation of Hsps expression and reduction of MAPK
phosphorylation. Int J Biol Macromol. 48:726–735. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Woo CC, Hsu A, Kumar AP, Sethi G and Tan
KH: Thymoquinone inhibits tumor growth and induces apoptosis in a
breast cancer xenograft mouse model: the role of p38 MAPK and ROS.
PLoS One. 8:e753562013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ku BM, Lee YK, Jeong JY, et al:
Ethanol-induced oxidative stress is mediated by p38 MAPK pathway in
mouse hippocampal cells. Neurosci Lett. 419:64–67. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barzegar A and Moosavi-Movahedi AA:
Intracellular ROS protection efficiency and free radical-scavenging
activity of curcumin. PLoS One. 6:e260122011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kaviarasan S, Ramamurthy N, Gunasekaran P,
Varalakshmi E and Anuradha CV:
Epigallocatechin-3-gallate(−)protects Chang liver cells against
ethanol-induced cytotoxicity and apoptosis. Basic Clin Pharmacol
Toxicol. 100:151–156. 2007.
|
|
17
|
Tahir M, Rehman MU, Lateef A, et al:
Diosmin protects against ethanol-induced hepatic injury via
alleviation of inflammation and regulation of TNF-α and NF-κB
activation. Alcohol. 47:131–139. 2013.PubMed/NCBI
|
|
18
|
Shen Y, Yang T, Guo S, et al: Increased
serum ox-LDL levels correlated with lung function, inflammation,
and oxidative stress in COPD. Mediators Inflamm.
2013:9723472013.PubMed/NCBI
|
|
19
|
Prietsch RF, Monte LG, da Silva FA, et al:
Genistein induces apoptosis and autophagy in human breast MCF-7
cells by modulating the expression of proapoptotic factors and
oxidative stress enzymes. Mol Cell Biochem. 390:235–242. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hoek JB and Pastorino JG: Ethanol,
oxidative stress, and cytokine-induced liver cell injury. Alcohol.
27:63–68. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim YS, Lee SJ, Hwang JW, et al: In vitro
protective effects of Thymus quinquecostatus Celak extracts
on t-BHP-induced cell damage through antioxidant activity.
Food Chem Toxicol. 50:4191–4198. 2012.PubMed/NCBI
|
|
22
|
Kim YS, Hwang JW, Han YK, et al:
Antioxidant activity and protective effects of Trapa
japonica pericarp extracts against
tert-butylhydroperoxide-induced oxidative damage in Chang
cells. Food Chem Toxicol. 64:49–56. 2014.PubMed/NCBI
|
|
23
|
Gokila Vani M, Kumar KJ, Liao JW, et al:
Antcin C from Antrodia cinnamomea protects liver cells
against free radical-induced oxidative stress and apoptosis in
vitro and in vivo through Nrf2-dependent mechanism. Evid Based
Complement Alternat Med. 2013:2960822013.
|
|
24
|
De K, Roy K and Sengupta C: Inhibition of
lipid peroxidation induced by hydroxyprogesterone caproate by some
conventional antioxidants in goat liver homogenates. Acta Pol
Pharm. 64:201–210. 2007.PubMed/NCBI
|
|
25
|
Townsend DM, Tew KD and Tapiero H: The
importance of glutathione in human disease. Biomed Pharmacother.
57:145–155. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Narasimhan M, Rathinam M, Patel D,
Henderson G and Mahimainathan L: Astrocytes prevent ethanol induced
apoptosis of Nrf2 depleted neurons by maintaining GSH homeostasis.
Open J Apoptosis. 1: View Article : Google Scholar : 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Matés JM: Effects of antioxidant enzymes
in the molecular control of reactive oxygen species toxicology.
Toxicology. 153:83–104. 2000.PubMed/NCBI
|
|
28
|
Birrane G, Li H, Yang S, Tachado SD and
Seng S: Cigarette smoke induces nuclear translocation of heme
oxygenase 1 (HO-1) in prostate cancer cells: Nuclear HO-1 promotes
vascular endothelial growth factor secretion. Int J Oncol.
42:1919–1928. 2013.
|
|
29
|
Bao W, Li K, Rong S, et al: Curcumin
alleviates ethanol-induced hepatocytes oxidative damage involving
heme oxygenase-1 induction. J Ethnopharmacol. 128:549–553. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim JY, Kim do Y, Son H, Kim YJ and Oh SH:
Protease-activated receptor-2 activates NQO-1 via Nrf2
stabilization in keratinocytes. J Dermatol Sci. 74:48–55. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
You H, Wei L, Sun WL, et al: The green tea
extract epigallocatechin-3-gallate inhibits irradiation-induced
pulmonary fibrosis in adult rats. Int J Mol Med. 34:92–102.
2014.PubMed/NCBI
|
|
32
|
Yao P, Nussler A, Liu L, et al: Quercetin
protects human hepatocytes from ethanol-derived oxidative stress by
inducing heme oxygenase-1 via the MAPK/Nrf2 pathways. J Hepatol.
47:253–261. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Son Y, Cheong YK, Kim NH, Chung HT, Kang
DG and Pae HO: Mitogen-activated protein kinases and reactive
oxygen species: how can ROS activate MAPK pathways? J Signal
Transduct. 2011:7926392011.PubMed/NCBI
|
|
34
|
Li J, He J and Yu C: Chitosan
oligosaccharide inhibits LPS-induced apoptosis of vascular
endothelial cells through the BKCa channel and the p38 signaling
pathway. Int J Mol Med. 30:157–164. 2012.PubMed/NCBI
|
|
35
|
Venugopal SK, Chen J, Zhang Y, Clemens D,
Follenzi A and Zern MA: Role of MAPK phosphatase-1 in sustained
activation of JNK during ethanol-induced apoptosis in
hepatocyte-like VL-17A cells. J Biol Chem. 282:31900–31908. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wei P, Ma P, Xu QS, et al: Chitosan
oligosaccharides suppress production of nitric oxide in
lipopolysaccharide-induced N9 murine microglial cells in vitro.
Glycoconj J. 29:285–295. 2012. View Article : Google Scholar : PubMed/NCBI
|