Introduction
Cervical cancer is the second leading cause of death
among women worldwide. High-risk human papillomavirus (HR-HPV)
infection is one of the major risk factors for cervical cancer; in
particular, type 16 and 18 HR-HPV are responsible for more than 68%
of cervical cancer cases (1). It
has been well demonstrated that HPV persistence and the chromosomal
rearrangements of two early genes, E6 and E7, are critical
occurrences in HPV-associated cancers. The oncogenes E6 and E7 can
disturb the cell cycle by inhibiting tumor-suppressor genes, namely
p53 and retinoblastoma tumor-suppressor protein (pRB), respectively
(2–5). E6 binds to the central region of p53
and induces its degradation via the ubiquitin pathway (6), while E7 protein binds and induces the
degradation of the growth suppressive form of pRB and promotes cell
cycle progression by destabilizing the pRb-E2F complex. This action
of E7 causes persistent activation of E2F transcription factors.
All of the mechanisms mentioned above help HPV evade host
immuno-surveillance, allow viral persistence, and alter cell cycle
and apoptosis control thereby facilitating the accumulation of DNA
damage or mutations (6). Although
cervical cancer is intimately related to HPV infection, not all
patients infected with HPV eventually develop cervical cancer. HPV
has been reported to be a necessary but not sufficient cause of
cervical cancer. Therefore, the question still remains as to what
is the critical step in carcinogenesis.
The epithelial-mesenchymal transition has been shown
to be involved in carcinogenesis, cancer progression and cancer
metastasis (7–11). Downregulation of E-cadherin has been
regarded as the vital marker for the loss of connection between
cells in epithelial-mesenchymal transition (EMT) events (10–12).
E-cadherin transcription and protein levels can be downregulated by
EMT-activating transcription factors, such as the Twist family of
bHLH factors (Twist1 and Twist2) which act as transcription
repressors to regulate metastasis. At the same time, Twist2
promotes EMT through downregulation of E-cadherin in subsets of
breast cancer (12–14). Moreover, highly expressed Twist2
protein is mainly found in cervical intraepithelial neoplasias and
cervical squamous cell carcinoma tissues (13–15).
Recently, studies have confirmed that HPV16 can
induce the EMT-like process. For example, HPV16 E6/E7-immortalized
human gingival keratinocytes cells demonstrated EMT-like properties
(16). Forced expression of HPV16
E7 can mediate EMT in the process of cervical carcinogenesis
(17,18). Caberg et al (19) discovered that silencing of the E7
gene could upregulate the expression of E-cadherin. Mühlen et
al (20) demonstrated that
HPV16 E2 activates targets such as ERK1/2, AP-1 and MMP-9,
suggesting that it plays a role in cancer carcinogenesis and
metastasis. It remains to be determined whether HPV16 E2/E6/E7
induce EMT in cervical cancer cells.
In the present study, we used tissue microarray and
immunohistochemical staining to evaluate the protein levels of
E-cadherin and Twist2 and found that Twist2 expression was
gradually increased during the progression from normal cervical
squamous epithelium to cervical intraepithelial neoplasia (CIN) and
cervical squamous cell carcinoma. At the same time, E-cadherin
expression was downregulated. We found significant differences in
the expression of E-cadherin and Twist2 between the 31 cases of
cervical tumor tissues and tumor adjacent tissues. In addition, we
explored the impact of HPV16 oncogenes on the EMT process of
cervical cancer cells. Thus, our results provide novel insight into
cervical carcinogenesis and a new evaluation for cervical cancer
progression.
Materials and methods
Cell culture and transfection
The human cervical carcinoma SiHa cell line
(HPV16-positive) purchased from ATCC, was cultured in Dulbecco’s
modified Eagle’s medium (DMEM) F12 1:1 medium containing 10% fetal
bovine serum (FBS) (both from Gibco, Gaithersburg, MD, USA) and a
100 μg/ml penicillin/streptomycin antibiotic mixture at 37°C with
5% CO2.
Before transfection, the SiHa cells were cultured on
6-ml dishes in 5 ml of growth medium. The p-CAG-MYC-HPV16 E2
plasmid, p-CAG-MYC-HPV16 E6 plasmid, p-CAG-MYC-HPV16 E7 plasmid and
the empty vector were donated by Dr Sufang Wu (Shanghai First
People’s Hospital, Shanghai Jiaotong University). After serum
starvation for 24 h, the SiHa cells were transfected with the
plasmids using Lipofectamine™ 2000 (Invitrogen) according to the
manufacturers’ protocol.
RT-PCR
Total RNA was extracted with TRIzol (Invitrogen).
For microarray hybridization, cDNA was produced from 2.5 μg of
total RNA according to the Takara manuscript. Thirty-four PCR
cycles were used for HPV16 E2, E6, E7, E-cadherin, vimentin, Twist2
and 28 PCR cycles for GAPDH. The objective gene was amplified using
appropriate primers (Table I).
 | Table IPrimers used in this study. |
Table I
Primers used in this study.
| Primer | Sequence (5′ to
3′) |
|---|
| E2 | F:
TCTGTGTTTAGCAGCAACGAA
R: TAATCCGTCCTTTGTGTGAGC |
| E6 | F:
CGACCCAGAAAGTTACCACAGT
R: AATCCCGAAAAGCAAAGTCATA |
| E7 | F:
GAGGAGGAAGATGAAATAGATGG
R: AACCGAAGCGTAGAGTCACAC |
| E-cadherin | F:
TTGCTACTGGAACAGGGACAC
R: CCCGTGTGTTAGTTCTGCTGT |
| Vimentin | F:
AGATGGCCCTTGACATTGAG
R: CCAGAGGGAGTGAATCCAGA |
| Twist2 | F:
ACAAGCTGAGCAAGATCCAGAC
R: GCTGGTCATCTTATTGTCCATCT |
| GAPDH | F:
AGAAGGCTGGGGCTCATTTG
R: AGGGGCCATCCACAGTCTTC |
Western blot analysis
After the harvested cells were lysed and the
supernatant was collected, then, 60 μg of protein was loaded onto
an SDS-PAGE and transferred to polyvinylidene fluoride (PVDF)
membranes. The membranes were blocked with 5% skimmed milk for 1.5
h and incubated overnight with one of the following primary
antibodies: anti-E-cadherin (diluted at 1:500), anti-vimentin
(diluted at 1:200), anti-Twist2 (diluted at 1:500) (all from Abcam)
and anti-GAPDH (diluted at 1:1,000; Cell Signaling Technology)
rabbit polyclonal antibody, anti-HPV16 E2 (diluted at 1:500),
anti-HPV16 E6 (diluted at 1:500) (both from Abcam) and anti-HPV16
E7 (diluted at 1:100; Beijing, Biosynthesis Biotechnology Co.)
rabbit polyclonal antibody. The reactions were then incubated with
the appropriate secondary antibody (1:5,000).
Immunofluorescence
A total of 2.5×104 cells were seeded on a
glass cover in a 24-well microtiter plate for each time period.
Thirty-six hours after introduction of the plasmids, the cells were
fixed with 4% paraformaldehyde in PBS, permeabilized by incubation
with PBS + 0.01% Triton X-100 (Sigma) for 5 min, and stained with
anti-Twist2 antibody (ab66031; Abcam) in PBS + 5% sheep serum for 1
h. The secondary antibody was Alexa Fluor 488-conjugated
anti-rabbit antibody (#A11001; Invitrogen) in PBS + 5% sheep serum.
The nuclei were counterstained with 4′,6-diamidino-2-phenylindole
(DAPI). Photographs were captured using a Zeiss Axiophot
fluorescence microscope equipped with Axiovision software version
4.6 (Carl Zeiss).
Immunohistochemistry of the tissue
microarray
Cervical intraepithelial neoplasia tissue microarray
(CIN481) and cervical squamous cancer tissue microarray (CXC962 and
Utr03-003) were purchased from Shanghai Zuocheng Biology Co.
containing normal cervical squamous epithelial tissue (6 cases),
CIN I–II (19 cases), CIN III (11 cases), and cervical squamous cell
carcinoma tissue (70 cases). There were 31 pairs of cervical tumor
tissues and tumor adjacent tissues. The tumor tissues were also
included in the 70 cases of cervical squamous cell carcinoma
tissues but the tumor adjacent tissues were not included in the 6
cases of normal cervical squamous epithelial tissue since they
encompassed not only normal cervical tissues but also CIN tissues
that we did not distinguish.
Immunohistochemistry was performed according to the
manufacturer’s instructions. Rabbit anti-E-cadherin Ab 1:100 and
anti-Twist2 Ab 1:100 were used for staining.
Invasion assay
To perform the invasion assays, the cells were
plated (105 cells/chamber) in BD BioCoat Matrigel
invasion chambers (BD Biosciences). In the upper chamber, the
medium was supplemented with 2% heat-inactivated FBS. In the lower
chamber, 20% FBS was used as a chemoattractant. After 24 h, the
medium was removed and the chambers were washed twice with PBS.
Non-invading cells were removed from the upper surface of the
membrane by scrubbing with a cotton-tipped swab, and invading cells
were fixed with 3.7% formaldehyde in PBS for 2 min, washed with PBS
twice, permeabilized with methanol for 20 min, washed twice with
PBS, stained with 0.05% crystal violet for 15 min, and washed twice
with PBS. For each chamber, five fields for each chamber were
photographed using a digital camera mounted on an inverted
microscope (magnification, ×40) and the invading cells were counted
in each field.
Evaluation of immunohistochemistry
results
The immunohistochemistry sections were scored
independently by three experienced pathologists who had no prior
knowledge of the patient data. To evaluate the staining results,
all areas of each sample were examined and only the section with
the greatest immunoreactivity was selected for quantification. For
Twist2, cytoplasmic and nuclear immunoreactivity were determined.
Positive cells had brown granules in the cytoplasm (15). Cytoplasmic staining was scored based
on the staining intensity and percentage of positive cell nuclei.
The cytoplasmic expression of Twist2 and membrane expression of
E-cadherin protein were assessed by a semi-quantitative method: the
sections were assessed for the intensity of the staining (0–3) and
the percentage of positively stained cells (0–3). Index of Twist2
and E-cadherin expression was calculated as percentage × intensity
of the staining. Therefore, score 0 presents negative (−), 1–3
serves as weak positive (+), 4–6 as positive (++), and 7–9 as
strong positive (+++) expression (15–19,21–23).
Statistical analysis
Statistical analyses were performed using SPSS 17.0
software. Clinical and histopathologic information and the results
from the immunohistochemical studies of the tissue microarray were
entered into a database. The Twist2 and E-cadherin expression among
different tissues was analyzed using the Kruskal-Wallis test and
the clinicopathological data were analyzed with the Mann-Whitney U
test. The differences between the 31 pairs of tumor and para-tumor
tissues were campared using the Wilcoxon’s sign rank test. For all
of the statistical analyses, a two-tailed p-value of ≤0.05 was
considered statistically significant.
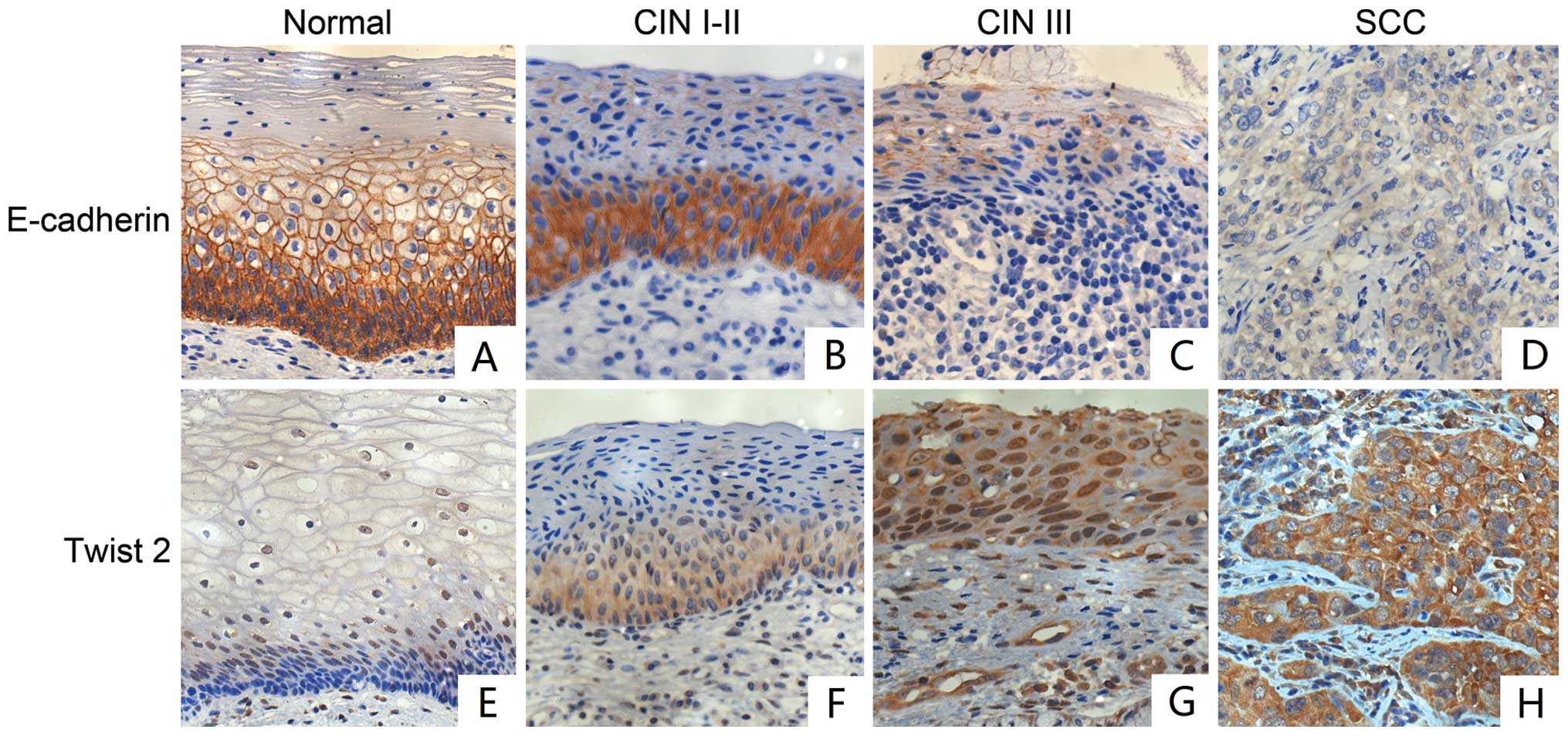
Results
Correlation between Twist2 and E-cadherin
expression and the clinicopathological parameters in all grades of
cervical disease
To investigate the relationship of Twist2 and
E-cadherin expression with the pathological characteristics, we
performed immunohistochemical staining for Twist2 and E-cadherin on
tissue microarrays, which included 6 cases of cervical squamous
epithelial tissue, 23 cases of CIN I–II tissue, 12 cases of CIN III
tissue, and 70 cases of cervical squamous cell carcinoma tissue.
Twist2 expression was mainly located in the cytoplasm. The rate of
Twist2 expression gradually increased from 0% in the normal
cervical squamous epithelial to 57.9% in CIN I–II, 72.7% in CIN III
and 95.5% in the cervical squamous cell carcinoma tissue. The
differences between the four groups were statistically significant,
with p-values <0.05. In addition, we found no relationship
between Twist2 expression and patient age, tumor grade, or lymph
node metastasis (Table II).
| Table IIAssociation between the
clinicopathological features and the expression of Twist2 in
cervical disease tissues. |
Table II
Association between the
clinicopathological features and the expression of Twist2 in
cervical disease tissues.
| Twist2 expression
in cytoplasm | | | |
|---|
|
| | | |
|---|
| − | + to +++ | Positive rates | χ2 | P-value |
|---|
| Tissue type | | | | 36.938 | 0.000 |
| Normal | 6 | 0 | 0.0% | | |
| CIN I–II | 8 | 11 | 57.9% | | |
| CIN III | 3 | 8 | 72.7% | | |
| SCC | 3 | 67 | 95.5% | | |
| Total (n=106) | 20 | 86 | | | |
| Age (years) | | | | 0.220 | 0.801 |
| ≤45 | 13 | 51 | 79.7% | | |
| >45 | 7 | 35 | 83.3% | | |
| Total (n=106) | 20 | 86 | | | |
| Differentiation
grade of tumor cells | | | | 2.178 | 0.588 |
| High | 0 | 2 | 100.0% | | |
| Moderate | 1 | 42 | 97.7% | | |
| Low | 2 | 23 | 92.0% | | |
| Total (n=70) | 3 | 67 | | | |
| Lymph node
metastasis | | | | 0.264 | 0.521 |
| Negative | 2 | 53 | 96.4% | | |
| Positive | 1 | 14 | 93.3% | | |
| Total (n=70) | 3 | 67 | | | |
The rate of E-cadherin expression in cervical
squamous cell carcinoma was 68.6%, which was lower than that in the
normal cervical squamous epithelial and CIN tissues (p<0.05). In
contrast to Twist2 expression, we found that the expression of
E-cadherin was related to lymph node metastasis (p<0.05);
however, there was no significant relationship with patient age or
tumor grade (Table III).
| Table IIIAssociation between the
clinicopathological features and the expression of E-cadherin in
cervical disease tissues. |
Table III
Association between the
clinicopathological features and the expression of E-cadherin in
cervical disease tissues.
| E-cadherin
expression in membrane | | | |
|---|
|
| | | |
|---|
| − | + to +++ | Positive rate | χ2 | P-value |
|---|
| Tissue type | | | | 14.156 | 0.001 |
| Normal | 0 | 6 | 100.0% | | |
| CIN I–II | 0 | 19 | 100.0% | | |
| CIN III | 0 | 11 | 100.0% | | |
| SCC | 22 | 48 | 68.6% | | |
| Total (n=106) | 22 | 84 | | | |
| Age (years) | | | | 1.25 | 0.264 |
| ≤45 | 11 | 53 | 82.8% | | |
| >45 | 11 | 31 | 73.8% | | |
| Total (n=106) | 22 | 84 | | | |
| Differentiation of
tumor cells | | | | 3.802 | 0.148 |
| High | 1 | 1 | 50.0% | | |
| Moderate | 10 | 33 | 76.7% | | |
| Low | 11 | 14 | 56.0% | | |
| Total (n=70) | 22 | 48 | | | |
| Lymph node
metastasis | | | | 7.231 | 0.007 |
| Negative | 13 | 42 | 76.4% | | |
| Positive | 9 | 6 | 40.0% | | |
| Total (n=70) | 22 | 48 | | | |
Expression of Twist2 and E-cadherin in
the progression of cervical cancer
To determine whether there was a statistically
significant difference in Twist2 expression between each grade of
cervical disease, we compared the following groups: normal cervical
squamous epithelial vs. CIN I–II; normal cervical squamous
epithelial vs. CIN III; normal cervical squamous epithelial vs.
squamous cell carcinoma (SCC); CIN I–II vs. CIN III; CIN I–II vs.
SCC; and CIN III vs. SCC. Our results demonstrated statistically
significant differences in the above groups with the exception of
the following two pairings: CIN I–II and CIN III, CIN III and SCC
(Tables IV and V, Fig.
1).
| Table IVProtein expression of Twist2 in
various lesions of cervical squamous epithelial. |
Table IV
Protein expression of Twist2 in
various lesions of cervical squamous epithelial.
| Twist2 expression
in cytoplasm | | | |
|---|
|
| | | |
|---|
| − | + | ++ | +++ | Total | H | P-value |
|---|
| Tissue group | | | | | | 31.738 | 0.000 |
| Normal | 6 | 0 | 0 | 0 | 6 | | |
| CIN I–II | 8 | 4 | 6 | 1 | 19 | | |
| CIN III | 3 | 7 | 0 | 1 | 11 | | |
| SCC | 3 | 18 | 33 | 16 | 70 | | |
| Total | 20 | 29 | 39 | 18 | 106 | | |
| Table VDifference in the expression of
Twist2 between the groups of cervical tissues. |
Table V
Difference in the expression of
Twist2 between the groups of cervical tissues.
| Tissue groups
compared | Z | P-value |
|---|
| Normal | CIN I–II | 2.766 | 0.011 |
| CIN III | 2.389 | 0.037 |
| SCC | 3.174 | 0.002 |
| CIN I–II | CIN III | −0.652 | 0.553 |
| SCC | 5.027 | 0.000 |
| CIN III | SCC | 1.674 | 0.094 |
We also compared E-cadherin expression in each grade
of cervical disease using the same comparison groups with Twist2.
We found statistically significant differences in all pairs, except
for the CIN I–II and CIN III pair (Tables VI and VII, Fig.
1).
| Table VIProtein expression of E-cadherin in
various lesions of cervical squamous epithelial. |
Table VI
Protein expression of E-cadherin in
various lesions of cervical squamous epithelial.
| E-cadherin
expression in membrane | | | |
|---|
|
| | | |
|---|
| − | + | ++ | +++ | Total | H | P-value |
|---|
| Tissue group | | | | | | 18.776 | 0.000 |
| Normal | 0 | 0 | 0 | 6 | 6 | | |
| CIN I–II | 0 | 2 | 11 | 6 | 19 | | |
| CIN III | 0 | 4 | 3 | 4 | 11 | | |
| SCC | 22 | 16 | 16 | 16 | 70 | | |
| Total | 22 | 22 | 30 | 32 | 106 | | |
| Table VIIDifference in the expression of
E-cadherin between the groups of cervical tissues. |
Table VII
Difference in the expression of
E-cadherin between the groups of cervical tissues.
| Tissue groups
compared | Z | P-value |
|---|
| Normal | CIN I–II | 2.336 | 0.036 |
| CIN III | 2.724 | 0.015 |
| SCC | 4.095 | 0.000 |
| CIN I–II | CIN III | 0.182 | 0.856 |
| SCC | 4.095 | 0.000 |
| CIN III | SCC | 3.420 | 0.001 |
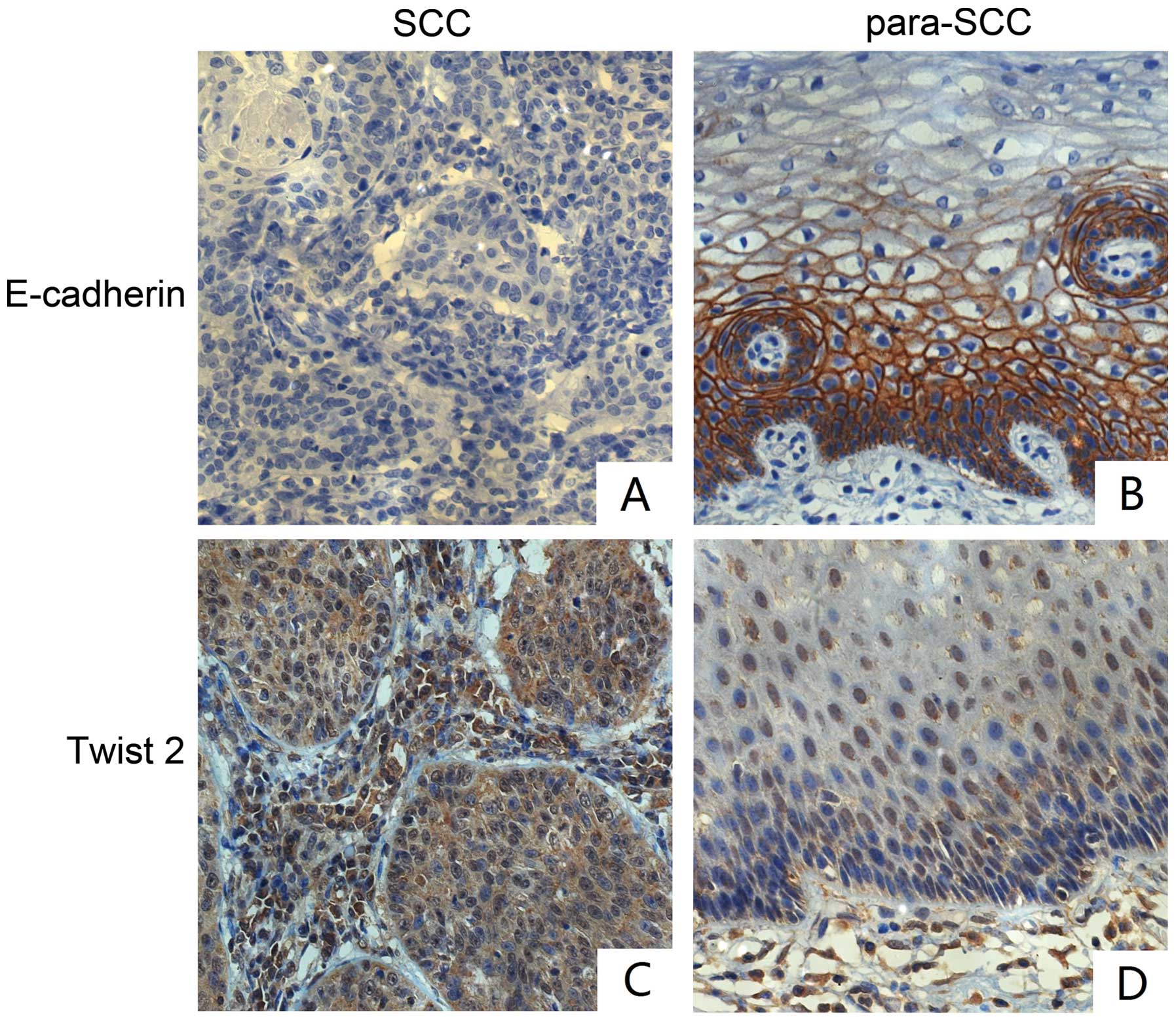
The difference in Twist2 and E-cadherin
expression between cervical tumor and tumor adjacent tissues
To evaluate the potential metastasis of cervical
cancer, we analyzed the expression of Twist2 and E-cadherin in 31
pairs of cervical tumor tissues and tumor adjacent tissues. In the
cervical tumor tissues, the rate of E-cadherin expression was 77.4%
(24/31), which was lower than the rate of 96.8% (for 30/31) in the
tumor adjacent tissues (p<0.05). In addition, the rate of Twist2
expression in tumor tissues (96.8% 30/31) was higher than that in
the tumor adjacent tissues (51.6% 16/31), (p<0.05) (Tables VIII and IX, Fig.
2). In addition, we found that the difference in E-cadherin
expression between the cervical tumor and tumor adjacent tissues
was statistically significant (Z=−4.602, p<0.05); the same
result was found when evaluating Twist2 expression (Z=−3.383,
p<0.05) (Table X and XI).
| Table VIIIExpression of E-cadherin in 31 pairs
of cervical tumor and tumor adjacent tissues. |
Table VIII
Expression of E-cadherin in 31 pairs
of cervical tumor and tumor adjacent tissues.
| E-cadherin
expression in membrane | | | |
|---|
|
| | | |
|---|
| − | + | ++ | +++ | Total | Z | P-value |
|---|
| Pairs of cervical
disease tissue | | | | | | −3.172 | 0.001 |
| SCC | 7 | 8 | 8 | 8 | 31 | | |
| Para-SCC | 1 | 0 | 15 | 15 | 31 | | |
| Total | 8 | 8 | 23 | 23 | 62 | | |
| Table IXExpression of Twist2 in 31 pairs of
cervical tumor and tumor adjacent tissues. |
Table IX
Expression of Twist2 in 31 pairs of
cervical tumor and tumor adjacent tissues.
| Twist2 expression
in cytoplasm | | | |
|---|
|
| | | |
|---|
| − | + | ++ | +++ | Total | Z | P-value |
|---|
| Pairs of cervical
disease tissue | | | | | | −5.662 | 0.000 |
| SCC | 1 | 4 | 8 | 18 | 31 | | |
| Para-SCC | 15 | 10 | 6 | 0 | 31 | | |
| Total | 16 | 14 | 14 | 18 | 62 | | |
| Table XDifference in the expression of
Twist2 between tumor and tumor adjacent tissue. |
Table X
Difference in the expression of
Twist2 between tumor and tumor adjacent tissue.
| Para
tumor-tumor |
|---|
| Z | −4.602a |
| Exact Sig.
(two-tailed) | 0.000 |
| Table XIDifference in the expression of
E-cadherin between tumor and tumor adjacent tissue. |
Table XI
Difference in the expression of
E-cadherin between tumor and tumor adjacent tissue.
| Para
tumor-tumor |
|---|
| Z | −3.383a |
| Exact Sig.
(two-tailed) | 0.000 |
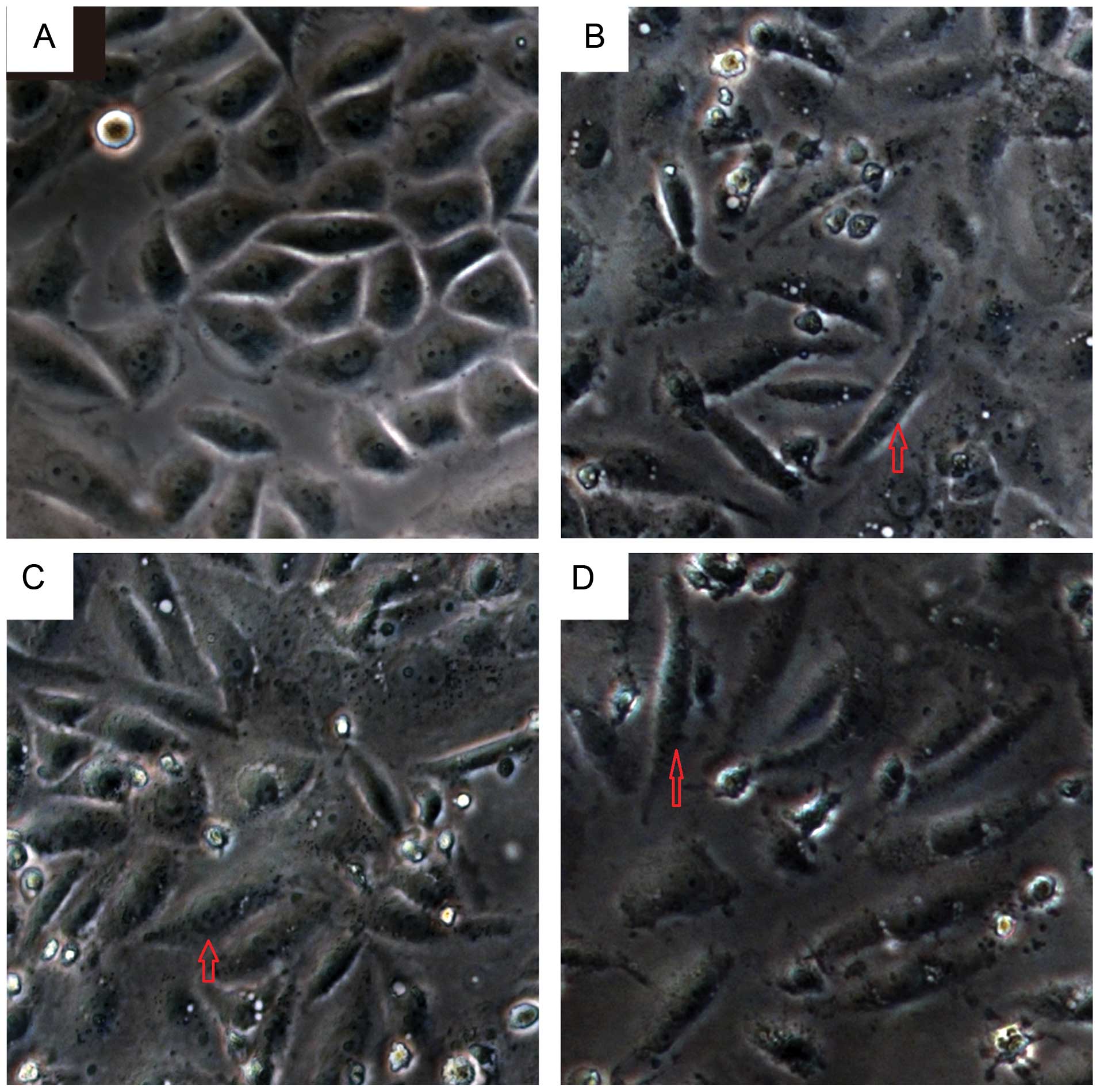
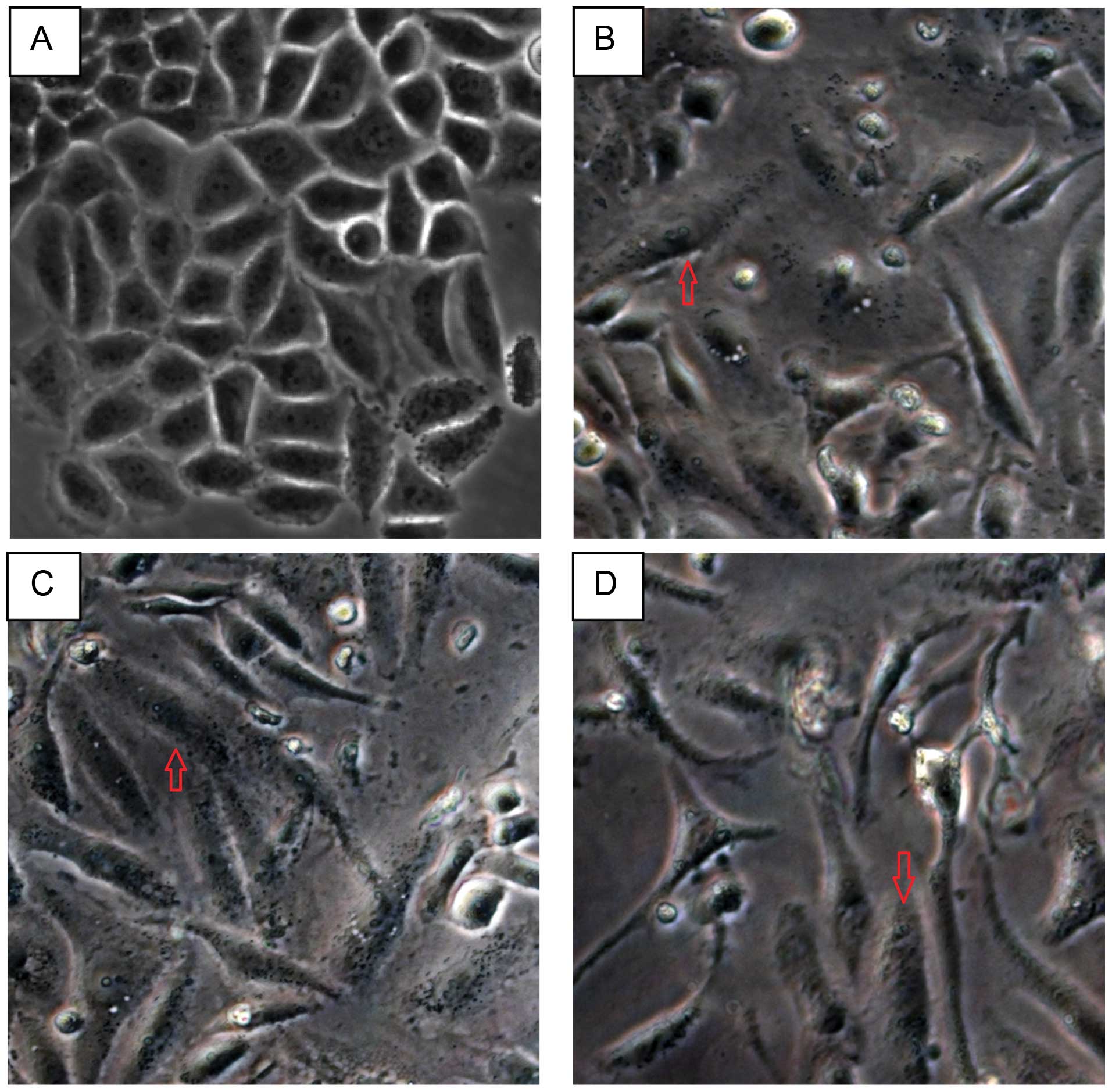
HPV16 oncogenes induce EMT cell
morphological alterations of SiHa cells in vitro
Above we described the clinical significance of
Twist2 and E-cadherin expression in the progression of cervical
cancer. We next aimed to identify if HPV16 oncogenes affect the EMT
process. Firstly, we transfected HPV16 E2/E6/E7 into SiHa cells
in vitro, for 24 and 48 h, respectively, to investigate
whether the cellular morphology is able to be altered due to the
overexpression of HPV16 oncogenes. Compared with the vector group,
cells transfected with the target genes became elongated,
spindle-shaped and scattered and adopted a fibroblast-like
morphology. Moreover, the intracellular gap was wider than the
vector control. The transition was even more obvious when cells
were transfected for 48 h (Figs. 3
and 4).
HPV16 oncogenes upregulate the expression
of Twist2 and vimentin and downregulate E-cadherin in vitro
To validate the effect of HPV16 oncogenes on Twist2
and vimentin, the most important genes controlling EMT in cervical
cancer, we applied RT-PCR and western blotting, to detect their
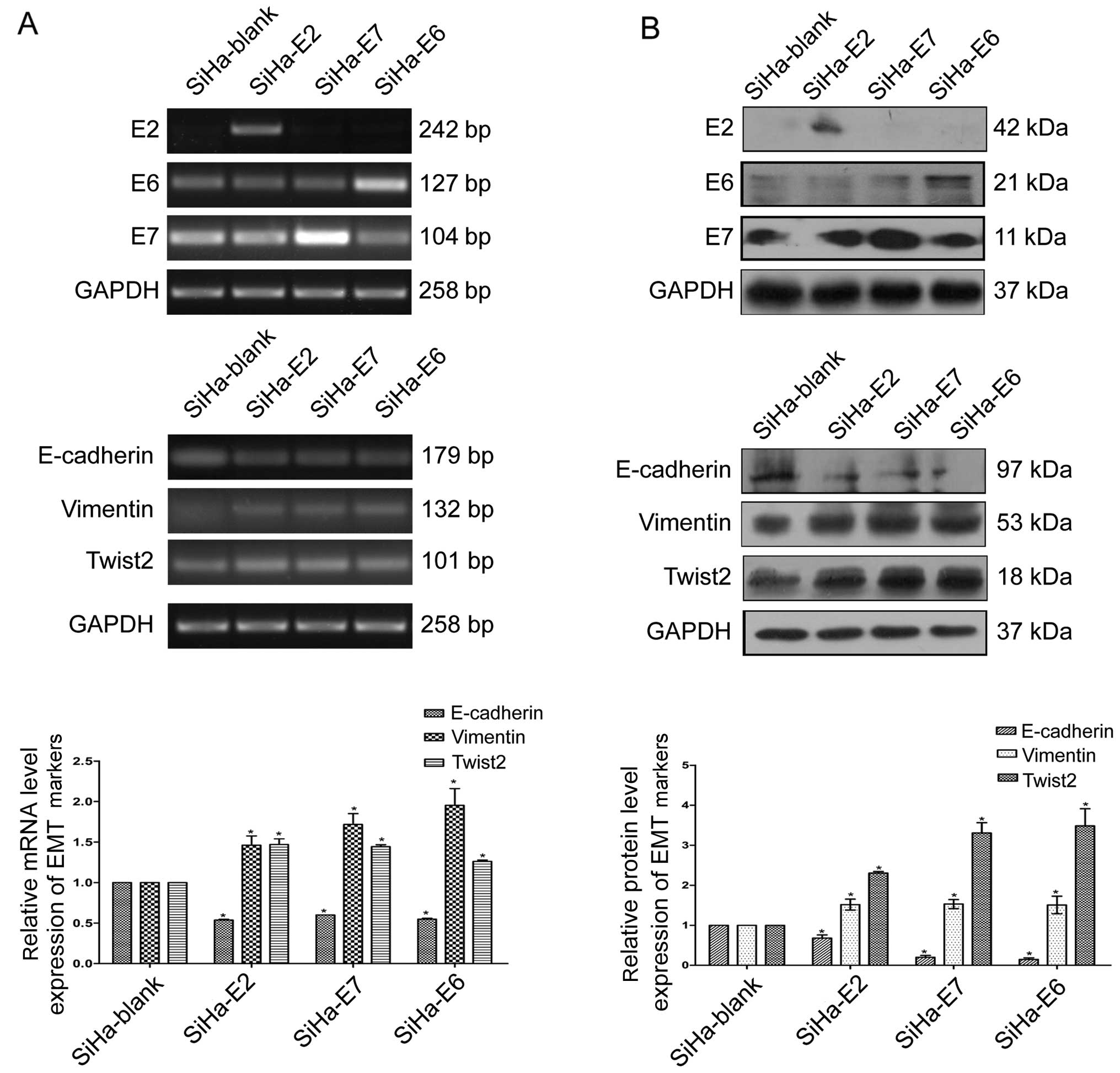
mRNA and protein levels, respectively. Fig. 5 demonstrates that both the mRNA and
protein levels of Twist2 and vimentin were significantly elevated
upon HPV16 E2/E6/E7 overexpression. In contrast, the HPV16 E2/E6/E7
genes downregulated the mRNA and protein levels of E-cadherin
(Fig. 5).
HPV16 E2 promotes cytonuclear
translocation of Twist2
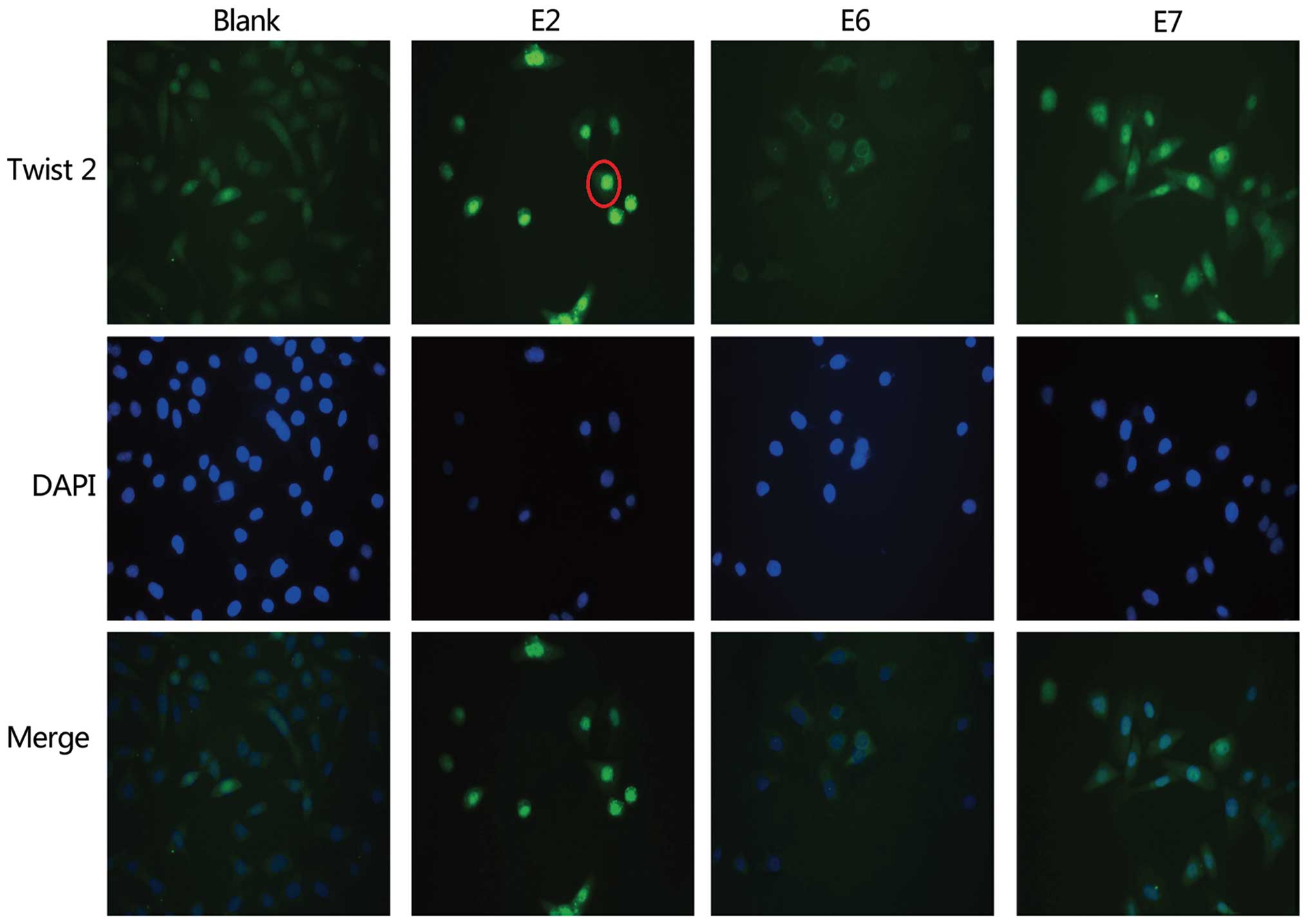
To explore whether HPV16 genes affect cytonuclear
translocation of the Twist2 protein, we performed an
immunofluorescence assay in SiHa cells. When the SiHa cells were
transfected with the HPV16E2 gene, the nuclear Twist2 protein level
was elevated, and the cytoplasmic Twist2 level was somewhat
decreased. However, when the E6 and E7 genes were transfected into
the SiHa cells, no significant change was observed in the
distribution of Twist2 in the cytoplasm and nucleus (Fig. 6). This intriguing result may
indicate that Twist2 may, in some degree, help Twist2 translocate
from the cytoplasm to the nucleus.
Promoting effect of HPV16 oncogenes on
the invasion of SiHa cells
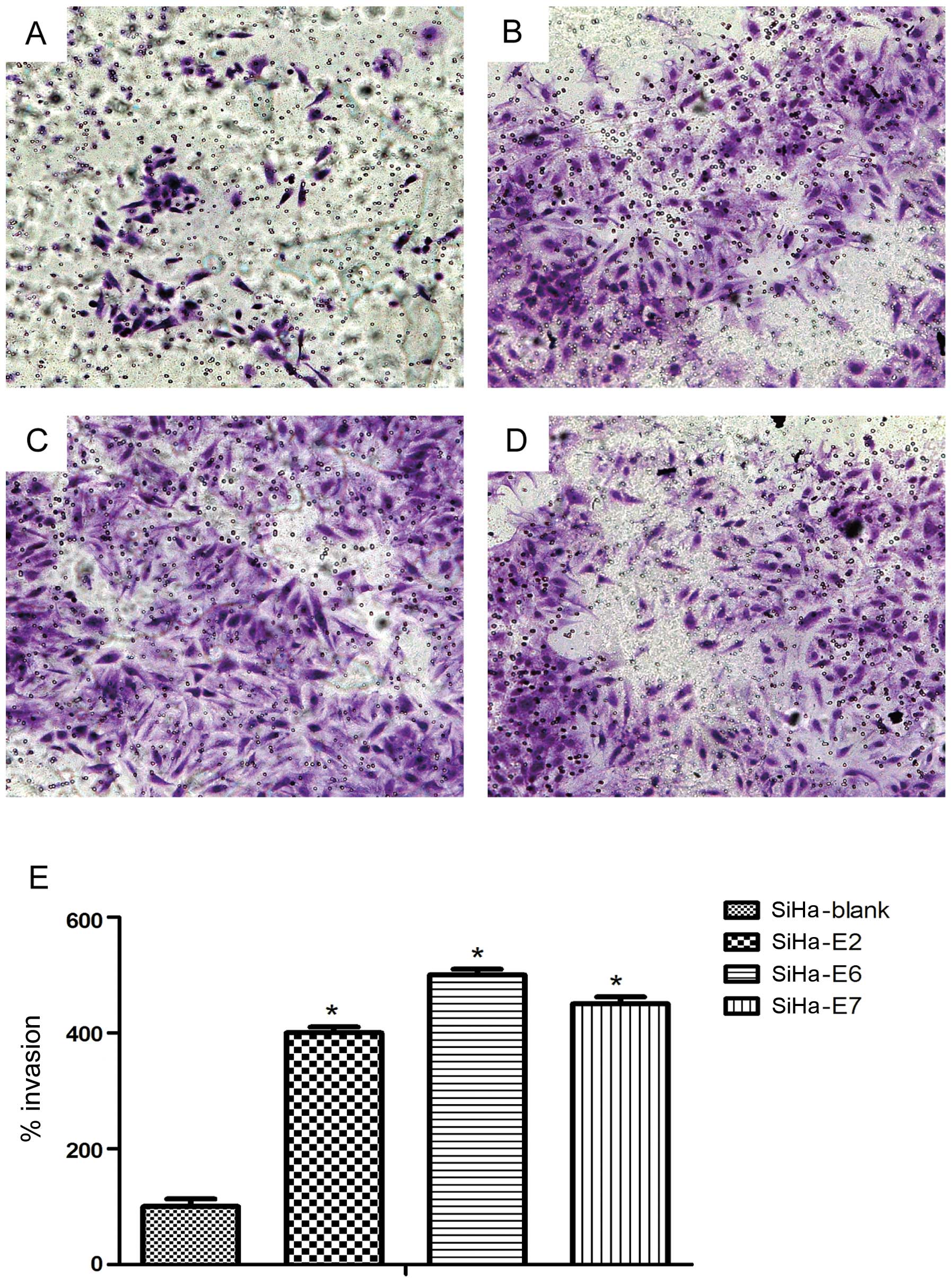
We then investigated whether treatment with HPV16
oncogenes promotes the invasion of SiHa cell lines using
Matrigel-coated Boyden chambers. First, SiHa cells were introduced
with P-CAG-MYC, P-CAG-MYC-HPV16 E2, P-CAG-MYC-HPV16 E6, and
P-CAG-MYC-HPV16 E7 plasmids. After 24 h, the HPV16 E2/E6/E7-treated
SiHa cells were markedly more invasive than their untreated
counterparts (p<0.05) (Fig.
7).
Discussion
It has been proposed that tumor cells lose homotypic
adhesion, change morphology, and acquire migratory capacity to
become ‘invasive’ through the activation of an evolutionarily
conserved developmental process known as epithelial-mesenchymal
transition (EMT) (24,25). It is well known that
epithelial-mesenchymal transition is closely involved in
carcinogenesis, invasion, metastasis, recurrence and
chemoresistance (7–14). Some molecular changes are reported
to be found in the EMT process, including the following: i)
decreasing expression of epithelial markers, such as E-cadherin;
ii) increasing expression of mesenchymal-related proteins, such as
vimentin; iii) cytoskeleton rearrangement mediated by Rho small
GTPases; and iv) upregulation and nuclear translocation of
transcription factors, such as Twist2, Snail and Zeb (14).
The transcription factors Twist2 and Twist1 are
vital members of the basic helix-loop-helix (bHLH) family. Both of
these molecules are able to bind to the E-boxes of E-cadherin to
repress the expression of the CDH1 (E-cadherin) gene and induce the
EMT process (13,14,23).
Recently, a great deal of effort has been made in investigating
whether Twist2 is an effective biomarker with which to predict and
evaluate cancer progression. Gasparotto et al (26) confirmed that overexpression of
Twist2 is intimately related to the poor prognosis of patients with
head and neck squamous cell carcinoma. Li et al (15) found that upregulation of Twist2, in
combination with aberrant E-cadherin expression in primary cervical
cancer tissues, may indicate malignant transformation and distant
metastasis.
It has been suggested that the Twist2 protein is
located in both the cytoplasm and nucleus (15,27).
In the present study, we did not evaluate the 7 cases in which
Twist2 was located mainly in the nucleus, which mostly was noted in
the normal cervical squamous epithelial and CIN I–II tissues. We
primarily focused on cytoplasmic staining. This analysis
demonstrated that Twist2 staining was gradually increased from 0%
in normal cervical squamous epithelial to 57.9% in CIN I–II (mild
to moderate atypical hyperplasia ), 72.7% in CIN III (severe
atypical hyperplasia or carcinoma in situ) and 95.5% in cervical
squamous cell carcinoma. This intriguing finding indicates that
Twist2 upregulation is a possible predictive indicator of the
malignant potential for cervical disease. Moreover, the expression
of Twist2 could also partly be valuable for evaluating the grades
of cervical lesions, although there was no statistical significance
between CIN I–II and CIN III (p>0.05), or CIN III and SCC (CIN
III only 11 cases) (p>0.05).
It is known that a reduction in the epithelial
adhesion protein E-cadherin and an increase in N-cadherin and the
mesenchymal intermediate filament protein vimentin mark the EMT
process (17). A similar pattern
was observed in a study performed by Myong et al (28). In this study it was proven that
E-cadherin and vimentin were effective indicators of EMT in
evaluating cervical cancer progression by immunohistochemical
staining. In the present study, although the expression of
E-cadherin could not be differentiated between CIN I–II and CIN III
(p>0.05), it is still quite obvious that the expression of
E-cadherin in cervical cancer tissues was lower than that in normal
cervical squamous epithelial and cervical squamous cell carcinoma
tissues. Cancer cells can convert to the epithelial state through
the process of mesenchymal to epithelial transition (MET), which is
highly indicative of the reversibility of EMT (29). This reversible process allows gene
expression to be rapidly switched according to developmental and
micro-environmental signals. A hybrid cell which contains both
epithelial and mesenchymal traits can be generated by incomplete
EMT into an epithelial cell (30).
This may account for the reason why there was no significant
difference in Twist2 expression between the CIN I–II and CIN III,
or CIN III and SCC tissues and why there was no significant
difference in E-cadherin expression between the CIN I–II and CIN
III tissues in the immunohistochemical staining. The limited number
of cases of immunohistochemistry samples may be another reason.
Lee et al (31) and Hsu et al (32) demonstrated that EMT is intimately
related to the invasion of cervical cancer and particularly the
progression of malignant tumors through clinical tissue
observations. It is believed that EMT plays a vital role in the
intravasation of cells into the vascellum as well as the lymph
vessels, thus shaping micrometastases as the most essential process
during EMT is the degradation of the basal membrane (29). Needless to say, it is widely
believed that EMT plays an indispensable role during the process of
cancer malignant metastasis (7–14,23,26–28).
In the present study, we found that E-cadherin expression was lower
in the tumor tissues than that in the tumor adjacent tissues
(p<0.05), and Twist2 expression was higher in tumor tissues than
that in the tumor adjacent tissues (p<0.05) (Tables VIII and IX, Fig.
2). In addition, the difference in the expression of Twist2
between the tumor tissues and tumor adjacent tissues was
statistically significant (Z=−4.602, p<0.05); the same was true
for the difference in E-cadherin expression (Z=−3.383, p<0.05).
Based on the multiple anti-proliferative functions, anti-invasion
and anti-metastasis properties of E-cadherin, the loss of
E-cadherin tends to enhance the metastatic diffusion in a large
number of cancer types (33). There
are several mechanisms involved in the loss of E-cadherin, such as
genetic mutation, epigenetic silencing and transcription repression
(14). By evaluating the
differences in the expression of Twist2 and E-cadherin at the same
time between 31 pairs of cervical tumor and tumor adjacent tissues,
we were able to demonstrate that joint detection of the expression
of Twist2 and E-cadherin could better predict cervical
carcinogenesis and progression.
Previous studies have suggested that high-risk HPV
oncoproteins may contribute to EMT. Recently, studies have reported
that HPV16 oncogenes are correlated with the EMT process during
cervical progression and metastasis. Jung et al (34) found that HPV16 induces EMT-like
processes via induction of EMT transcription factors which may
contribute to tumor progression and metastasis. HPV18 E6 expression
was found to be correlated with a fibroblastoid morphology in
SV40-immortalized human keratinocytes (35). A microarray analysis suggested that
the modulation of a great number of genes are involved in
keratinocyte differentiation and EMT mediated by HPV16 E6 (36). In addition, Hellner et al
(17) reported that the oncogenic
HPV16 E7 protein in normal human epithelial cells leads to
increased levels of vimentin as well as reduced levels of the
epithelial adhesion protein E-cadherin. In the present study, HPV16
E6/E7 induced the morphological conversion of SiHa cells from a
cobblestone-shaped epithelium-like phenotype to a spindle-shaped
mesenchyme-like phenotype. Corresponding to the morphological
alterations, HPV16 E6/E7 also influenced the expression of
EMT-relative indicators, such as upregulation of Twist2 and
vimentin and downregulation of E-cadherin. It is worth noting that
these results were also manifested in the SiHa cells transfected
with the HPV16 E2 plasmid. The HPV16 E2 protein is a
multifunctional DNA-binding transcription factor that plays a
pivotal role in transcriptional regulation and viral DNA
replication. In addition, this protein modulates host cells through
direct protein interactions (37–41)
and is therefore potentially responsible for HPV-induced
carcinogenesis (42). According to
our results, HPV16 E2 may induce Twist2 translocation from the
cytoplasm to the nucleus to fully exert its function as a
transcription factor (Fig. 6).
Since Twist2 serves as a transcription factor which binds to the
E-boxes of E-cadherin to repress its expression and induce the EMT
process (13,14,23),
our findings indicate that the HPV oncogene E2 can actually
upregulate the expression of Twist2 and downregulate E-cadherin. We
then speculated that Twist2 may pass through the nuclear envelope
to interact with HPV E2 and continue to extend the function of
E-cadherin suppression and EMT induction. While the mechanism of
E-cadherin downregulation in HPV16 E2/E6/E7-transfected SiHa cells
remains to be explored, our results are similar to a recent
investigation that demonstrated that E-cadherin expression was
preserved when HPV16 E7 was silenced by siRNA in HPV16-transformed
human keratinocytes, irrespective of the transcription factors slug
and snail (19). Moreover, in the
present study, we found that HPV16 oncogenes could promote the
invasive ability of SiHa cells in vitro. This once again
identified a new metastatic potential of HPV16 oncogenes in
cervical carcinogenesis. Thus, we believe that HPV16 oncogenes can
actually induce EMT-like processes which may contribute to cancer
carcinogenesis and progression. However, the specific mechanism
remains to be elucidated.
In conclusion, we demonstrated that joint detection
of Twist2 and E-cadherin expression can be used to predict and
evaluate carcinogenesis and progression of cervical cancer. HPV16
oncogenes can induce EMT-like processes in SiHa cells and
contribute to the EMT process via regulating the expression of
Twist2, E-cadherin and vimentin. These results provide new insight
into the mechanism of cervical carcinogenesis and progression. In
addition, joint detection of E-cadherin and Twist2 expression could
aid in distinguishing and diagnosing early stage cervical cancer to
improve the survival rate of cervical cancer patients.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (81201541), the Shanghai
Pujiang Program (12PJD002), and the Shanghai Science and Technology
Committee Foundation (09411962500).
References
|
1
|
zur Hausen H: Papillomaviruses and cancer:
from basic studies to clinical application. Nat Rev Cancer.
2:342–350. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Faridi R, Zahra A, Khan K and Idrees M:
Oncogenic potential of human papillomavirus (HPV) and its relation
with cervical cancer. Virol J. 8:2692011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Panjković M and Ivković-Kapicl T: Etiology
and pathogenesis of precancerous lesions and invasive cervical
carcinoma. Med Pregl. 61:364–368. 2008.(In Serbian). View Article : Google Scholar
|
|
4
|
Snijders PJ, Steenbergen RD, Heideman DA
and Meijer CJ: HPV-mediated cervical carcinogenesis: concepts and
clinical implications. J Pathol. 208:152–164. 2006. View Article : Google Scholar
|
|
5
|
Javier MG and Sastre-Garau X: Uterine
cervix carcinoma: recent biological data and update for improving
follow-up and treatment. Isr Med Assoc J. 14:700–704.
2012.PubMed/NCBI
|
|
6
|
Ghittoni R, Accardi R, Hasan U, Gheit T,
Sylla B and Tommasino M: The biological properties of E6 and E7
oncoproteins from human papillomaviruses. Virus Genes. 40:1–13.
2010. View Article : Google Scholar
|
|
7
|
De Craene B and Berx G: Regulatory
networks defining EMT during cancer initiation and progression. Nat
Rev Cancer. 13:97–110. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stewart CJ and McCluggage WG:
Epithelial-mesenchymal transition in carcinomas of the female
genital tract. Histopathology. 62:31–43. 2013. View Article : Google Scholar
|
|
9
|
Lim J and Thiery JP:
Epithelial-mesenchymal transitions: insights from development.
Development. 139:3471–3486. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Le Bras GF, Taubenslag KJ and Andl CD: The
regulation of cell-cell adhesion during epithelial-mesenchymal
transition, motility and tumor progression. Cell Adh Migr.
6:365–373. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rodriguez FJ, Lewis-Tuffin LJ and
Anastasiadis PZ: E-cadherin’s dark side: possible role in tumor
progression. Biochim Biophys Acta. 1826:23–31. 2012.PubMed/NCBI
|
|
12
|
Iwatsuki M, Mimori K, Yokobori T, et al:
Epithelial-mesenchymal transition in cancer development and its
clinical significance. Cancer Sci. 101:293–299. 2010. View Article : Google Scholar
|
|
13
|
Sanchez-Tillo E, Liu Y, de Barrios O, et
al: EMT-activating transcription factors in cancer: beyond EMT and
tumor invasiveness. Cell Mol Life Sci. 69:3429–3456. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee MY and Shen MR: Epithelial-mesenchymal
transition in cervical carcinoma. Am J Transl Res. 4:1–13.
2012.PubMed/NCBI
|
|
15
|
Li Y, Wang W, Wang W, et al: Correlation
of TWIST2 up-regulation and epithelial-mesenchymal transition
during tumorigenesis and progression of cervical carcinoma. Gynecol
Oncol. 124:112–118. 2012. View Article : Google Scholar
|
|
16
|
Chamulitrat W, Schmidt R, Chunglok W, Kohl
A and Tomakidi P: Epithelium and fibroblast-like phenotypes derived
from HPV16 E6/E7-immortalized human gingival keratinocytes
following chronic ethanol treatment. Eur J Cell Biol. 82:313–322.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hellner K, Mar J, Fang F, Quackenbush J
and Munger K: HPV16 E7 oncogene expression in normal human
epithelial cells causes molecular changes indicative of an
epithelial to mesenchymal transition. Virology. 391:57–63. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cheng YM, Chou CY, Hsu YC, Chen MJ and
Wing LY: The role of human papillomavirus type 16 E6/E7
oncoproteins in cervical epithelial-mesenchymal transition and
carcinogenesis. Oncol Lett. 3:667–671. 2012.PubMed/NCBI
|
|
19
|
Caberg JH, Hubert PM, Begon DY, et al:
Silencing of E7 oncogene restores functional E-cadherin expression
in human papillomavirus 16-transformed keratinocytes.
Carcinogenesis. 29:1441–1447. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mühlen S, Behren A, Iftner T and Simon C:
Influence of HPV16 E2 and its localisation on the expression of
matrix metalloproteinase-9. Int J Oncol. 37:337–345.
2010.PubMed/NCBI
|
|
21
|
Zhang J, Yang Y, Zhang Z, et al: Gankyrin
plays an essential role in estrogen-driven and GPR30-mediated
endometrial carcinoma cell proliferation via the PTEN/PI3K/AKT
signaling pathway. Cancer Lett. 339:279–287. 2013. View Article : Google Scholar
|
|
22
|
Wang YX, Zhang XY, Zhang BF, Yang CQ and
Gao HJ: Study on the clinical significance of Argonaute2 expression
in colonic carcinoma by tissue microarray. Int J Clin Exp Pathol.
6:476–484. 2013.PubMed/NCBI
|
|
23
|
Franco HL, Casasnovas J, Rodriguez-Medina
JR and Cadilla CL: Redundant or separate entities? - roles of
Twist1 and Twist2 as molecular switches during gene transcription.
Nucleic Acids Res. 39:1177–1186. 2011. View Article : Google Scholar :
|
|
24
|
Zeisberg M and Neilson EG: Biomarkers for
epithelial-mesenchymal transitions. J Clin Invest. 119:1429–1437.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gasparotto D, Polesel J, Marzotto A, et
al: Overexpression of TWIST2 correlates with poor prognosis in head
and neck squamous cell carcinomas. Oncotarget. 2:1165–1175.
2011.PubMed/NCBI
|
|
27
|
Mao Y, Zhang N, Xu J, Ding Z, Zong R and
Liu Z: Significance of heterogeneous Twist2 expression in human
breast cancers. PloS One. 7:e481782012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Myong NH: Loss of E-cadherin and
acquisition of vimentin in epithelial-mesenchymal transition are
noble indicators of uterine cervix cancer progression. Korean J
Pathol. 46:341–348. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Thiery JP and Sleeman JP: Complex networks
orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell
Biol. 7:131–142. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reik W: Stability and flexibility of
epigenetic gene regulation in mammalian development. Nature.
447:425–432. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee MY, Chou CY, Tang MJ and Shen MR:
Epithelial-mesenchymal transition in cervical cancer: correlation
with tumor progression, epidermal growth factor receptor
overexpression, and snail up-regulation. Clin Cancer Res.
14:4743–4750. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hsu YM, Chen YF, Chou CY, et al: KCl
cotransporter-3 down-regulates E-cadherin/beta-catenin complex to
promote epithelial-mesenchymal transition. Cancer Res.
67:11064–11073. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
van Roy F and Berx G: The cell-cell
adhesion molecule E-cadherin. Cell Mol Life Sci. 65:3756–3788.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jung YS, Kato I and Kim HR: A novel
function of HPV16-E6/E7 in epithelial-mesenchymal transition.
Biochem Biophys Res Commun. 435:339–344. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Watson RA, Thomas M, Banks L and Roberts
S: Activity of the human papillomavirus E6 PDZ-binding motif
correlates with an enhanced morphological transformation of
immortalized human keratinocytes. J Cell Sci. 116:4925–4934. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Duffy CL, Phillips SL and Klingelhutz AJ:
Microarray analysis identifies differentiation-associated genes
regulated by human papillomavirus type 16 E6. Virology.
314:196–205. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Thierry F: Transcriptional regulation of
the papillomavirus oncogenes by cellular and viral transcription
factors in cervical carcinoma. Virology. 384:375–379. 2009.
View Article : Google Scholar
|
|
38
|
Forma E, Wójcik-Krowiranda K, Bieńkiewicz
A and Bryś M: Molecular basis of gynecological oncology - TopBP1
protein and its participation in the transcription process. Ginekol
Pol. 83:363–367. 2012.(In Polish). PubMed/NCBI
|
|
39
|
Bodily JM, Hennigan C, Wrobel GA and
Rodriguez CM: Regulation of the human papillomavirus type 16 late
promoter by E7 and the cell cycle. Virology. 443:11–19. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xue Y, Lim D, Zhi L, He P, Abastado JP and
Thierry F: Loss of HPV16 E2 protein expression without disruption
of the E2 ORF correlates with carcinogenic progression. Open Virol
J. 6:163–172. 2012. View Article : Google Scholar
|
|
41
|
Ramirez-Salazar E, Centeno F, Nieto K,
Valencia-Hernandez A, Salcedo M and Garrido E: HPV16 E2 could act
as down-regulator in cellular genes implicated in apoptosis,
proliferation and cell differentiation. Virol J. 8:2472011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bellanger S, Tan CL, Xue YZ, Teissier S
and Thierry F: Tumor suppressor or oncogene? A critical role of the
human papillomavirus (HPV) E2 protein in cervical cancer
progression. Am J Cancer Res. 1:373–389. 2011.PubMed/NCBI
|