Introduction
Bee venom (BV) therapy is the part of apitherapy
that utilizes BV in the treatment of inflammatory conditions
(1). BV has been used as a
traditional medicine to treat back pain, rheumatism and skin
diseases by its antibacterial, antiviral and anti-inflammatory
effects (2–4). BV is a rich source of enzymes and
peptides, including melittin, phospholipase A2 (PLA2), apamin,
adolapin and mast cell-degranulating peptide (MCDP) (5–7). There
are at least 18 active components in the venom which have some
pharmaceutical properties (8).
Among these compounds, melittin, a small linear peptide consisting
of 26 amino acids, is the major potent toxin of BV (9), which comprises ~50% of BV (10).
BV can induce apoptosis in synovial fibroblasts via
caspase-3 activation and inhibition of cyclooxygenase (Cox)-2
expression in human lung cancer cells (11). Moreover, several studies have
demonstrated that BV and/or melittin have anticancer effects in
prostate (12), liver (13,14),
breast (15), cervical (16) and renal cancer cells (17). Recently, it was shown that melittin
can poke holes in the protective envelope that surrounds human
immunodeficiency virus (HIV), and other viruses as well as tumor
cells by melittin-loaded nanoparticles (18). Many viruses, including hepatitis B
and C, rely on the same type of protective envelope and would be
vulnerable to melittin-guided BV therapy. Secreted phospholipases
A2 from BV have potent anti-HIV activities (19). A nanoscale delivery vehicle for
cytolytic peptides was demonstrated by incorporating the
non-specific amphipathic cytolytic peptide melittin into the outer
lipid monolayer of a perfluorocarbon nanoparticle (20). The nanovehicles were delivered
significant payloads of melittin i.v. and targeted and killed
precancerous lesions in K14-HPV16 mice with squamous dysplasia and
carcinoma harboring human papilloma virus (HPV) transgenic elements
(E6 and E7 oncogenes). However, experiments demonstrating the
molecular mechanisms of the antiviral effects of BV in cervical
cancer cells have not been reported. It is becoming increasingly
uncertain whether there are wide variations in tumorigenic
inhibitory effects among different cell types.
In the present study, we investigated the anticancer
effects of BV on cervical cancer cells well-known for having two
viral oncogenic proteins, E6 and E7, that play a critical role in
inducing cervical cancer. We observed that there is a significant
difference in sensitivity to BV, and HPV16/18 E6/E7 are
downregulated by BV in the cervical cancer cells. Moreover, the
downregulation is likely to be dependent upon the cervical cancer
cell line used. Thus, BV plays a differential role in suppressing
HPV16-infected cells (CaSki cells) and HPV18-infected cells (HeLa
cells) by inducing cervical cancer cell growth arrests by the
downregulation of E6/E7 protein of HPV16/18.
Materials and methods
Ethics statement
All procedures of animal research were conducted in
accordance with the Laboratory Animals Welfare Act (protocol no.
8852), the Guide for the Care and Use of Laboratory Animals
(protocol nos. 9025 and 21370), and the Guidelines and Policies for
Rodent Experiment provided by the Institutional Animal Care and Use
Committee (IACUC) of the School of Medicine, The Catholic
University of Korea. The present study was reviewed and approved by
the Catholic University of Korea’s IACUC (CUMC-2012-0054-07:
Effects of BV on the inhibition of HPV E6 and E7 expression in
cervical cancer cells). All rodents used for surgeries were
initially anesthetized using isoflurane in desiccators then
followed by isoflurane as required.
Cell culture conditions
C33A (HPV-uninfected cervical cancer cell line),
CaSki and HeLa (HPV-infected cervical cancer cell lines) were
purchased from the Korean Cell Line Bank (KCLB; Seoul, Korea). TC-1
cells were prepared by transformation of C57BL/6 primary mouse lung
cells with HPV16 E6/E7 oncogene and activated HRAS (21,22).
All cell lines were incubated in RPMI-1640 cell culture medium
supplemented with 10% (v/v) heat-inactivated fetal bovine serum
(FBS) and 1% penicillin/streptomycin (all from Gibco) at 37°C in a
humidified 5% CO2 atmosphere.
Cell viability assay
The cytotoxicity and sensitivity of BV was first
determined by measuring the conversion of the tetrazolium salt
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
(MTT; Sigma) to formazan. Briefly, CaSki, HeLa and C33A were seeded
1×104 cells/well in 96-well culture plates and cultured
overnight. The cells were then treated with 1–15 μg/ml of BV. The
control group was treated with the same volume of
phosphate-buffered saline (PBS). After 12 and 24 h of incubation,
20 μl of MTT stock solution (2 mg/ml in PBS) was added to each
well. After 4 h incubation at 37°C, the supernatant was discarded
and the precipitate was dissolved with 200 μl of dimethyl sulfoxide
(DMSO). The absorbance of the wells was measured at 570 nm using
Soft Max ELISA (Molecular Devices, Sunnyvale, CA, USA). The optical
density (OD) was calculated as the difference between the reference
wavelength and the test wavelength. Percent of cell viability =
[A570 nm absorbance of drug-treated cells/A570 nm absorbance of
control cells] × 100.
Reverse transcription (RT)-PCR and
real-time PCR (qRT-PCR) analysis
Total RNA was extracted from CaSki, HeLa and C33A
cells or TC-1 tumors treated by BV using TRIzol (Invitrogen,
Carlsbad, CA, USA) and purified using RNeasy columns (Qiagen,
Valencia, CA, USA) according to the manufacturer’s protocol. After
processing with DNase digestion, clean-up procedures, RNA samples
were quantified and stored in 10 μl aliquot at −80°C until use. For
quality control, RNA purity and integrity were evaluated by
denaturing gel electrophoresis, OD 260/280 ratio, and analyzed on
Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA,
USA). The cDNA was synthesized using PrimeScript reagent kit
(Takara, Japan) according to the manufacturer’s instructions.
Briefly, 1 μg of RNA was reverse-transcribed to cDNA using 200 U
moloney murine leukemia virus (M-MLV) reverse transcriptase and
oligo(dT) primer in a total reaction volume of 20 μl for 1 h at
37°C. The cDNA was amplified using HPV16 E6, HPV16 E7, HPV18 E6 and
HPV18 E7 primers. The E6 primers were 5′-GAGAACTGCAATGTTTCAG GAC-3′
and 5′-CCACCGACCCCTTATATTATGG-3′ for HPV-16, and
5′-AATACTATGGCGCGCTTTGA-3′ and 5′-CTGGATTCAACGGTTTCTGG-3′ for
HPV-18. The E7 primers were 5′-GCAACCAGAGACAACTGATCTCTAC-3′ and
5′-GGTCTTCCAAAGTACGAATGTCTACG-3′ for HPV-16, and
5′-TGCATGGACCTAAGGCAA-3′ and 5′-GCT GGGATGCACACCA-3′ for HPV-18.
Amplified products were analyzed using an image documentation
system (GelDoc 2000) with image analysis software (Quantity One)
(both from Bio-Rad, Hercules, CA, USA). DNA size markers
(Fermentas, Pittsburgh, PA, USA) were run in parallel to validate
the predicted sizes of the amplified bands. For qRT-PCR analysis,
cDNA was amplified using these specific primers and SYBR Premix Ex
Taq (2X) kit (Takara, Japan) according to the manufacturer’s
instructions. HPV E6/E7 expression analysis was carried out using
LightCycler 480 II (Roche, Palo Alto, CA, USA) and gene expression
raw data were extracted using the software provided by the
manufacturer (BeadStudio v.3.0; Partek® Genomics). HPV
E6/E7 gene expression was determined compared to GAPDH gene
expression.
Western blotting
For immunoblots, confluent monolayers of CaSki, HeLa
and C33A or TC-1 tumors were lysed with cell extraction buffer (10
mM Tris, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM NaF, 20 mM
Na4P2O7, 2 mM
Na3VO4, 1% Triton X-100, 10% glycerol, 0.1%
SDS, 0.5% deoxycholate) (BioSource International, Camarillo, CA,
USA) in the presence of protease inhibitors. Protein concentration
was determined using the BCA protein assay (Bio-Rad).
Electrophoration was performed on 10–12% SDS-polyacrylamide gels
for 2 h, and the gels were then transferred onto PVDF membranes
(Millipore, Temecula, CA, USA). Membranes were blocked in
Tris-buffered saline with 0.1% Tween-20 (TBST) containing 5% skim
milk for 1 h at room temperature. After blocking, membranes were
incubated for 1 h at room temperature or overnight at 4°C with
polyclonal rabbit anti-HPV16/18 E6, anti-HPV16 E7 and anti-HPV18
E7, and monoclonal mouse anti-β-actin antibodies (both from Santa
Cruz Biotechnology, Santa Cruz, CA, USA) in a 1:200–500 dilution
buffer (5% skim milk in TBST). Membranes were washed three times
with TBST and then incubated with goat anti-mouse IgG-HRP and goat
anti-rabbit IgG (H+L) HRP-conjugated antibodies (Zymed, San
Francisco, CA, USA) for 1 h at room temperature. Immunoblots were
developed with enhanced chemiluminescence agents according to the
manufacturer’s instructions (SuperSignal West Pico Chemiluminescent
Substrate; Pierce, Rockford, IL, USA) and exposed to imaging
film.
Antitumor effects of TC-1 tumor
models
TC-1 tumors were implanted in the abdomens of 4- to
5-week old female C57BL/6 mice by subcutaneous injection of
5×105 TC-1 cells in 100 μl of serum and antibiotic-free
DMEM media (Gibco-BRL). When tumors reached a volume of 150–200
mm3, mice were randomly assigned to one of three groups
to receive PBS, 1 mg/kg BV and 2 mg/kg BV (5 mice/group). The first
day of treatment was designated as day 1. BV or PBS was
administered three times via intra-tumoral injection (for TC-1
tumors, BV diluted in 100 μl of PBS) on days 1, 2 and 3. Tumor
growth delay was assessed by taking measurements every day or every
2 days. Tumor volume was calculated by the following formula:
Volume = 0.523 LW2, where L is
length and W is width. Tumor responses to each treatment
were compared by use of the Mann-Whitney test.
Statistical analysis
The data were obtained by three or more independent
experiments and are expressed as means ± standard error of the mean
(SEM). Statistical comparisons were analyzed by Mann-Whitney test
(non-parametric method) using StatView software (Abacus Concepts,
Inc., Berkeley, CA, USA). Survival was assessed with the
Kaplan-Meier method, and results were compared with a log-rank test
SPSS software version 13.0 (SPSS Inc., Chicago, IL, USA).
Statistical significance was defined as P<0.05.
Results
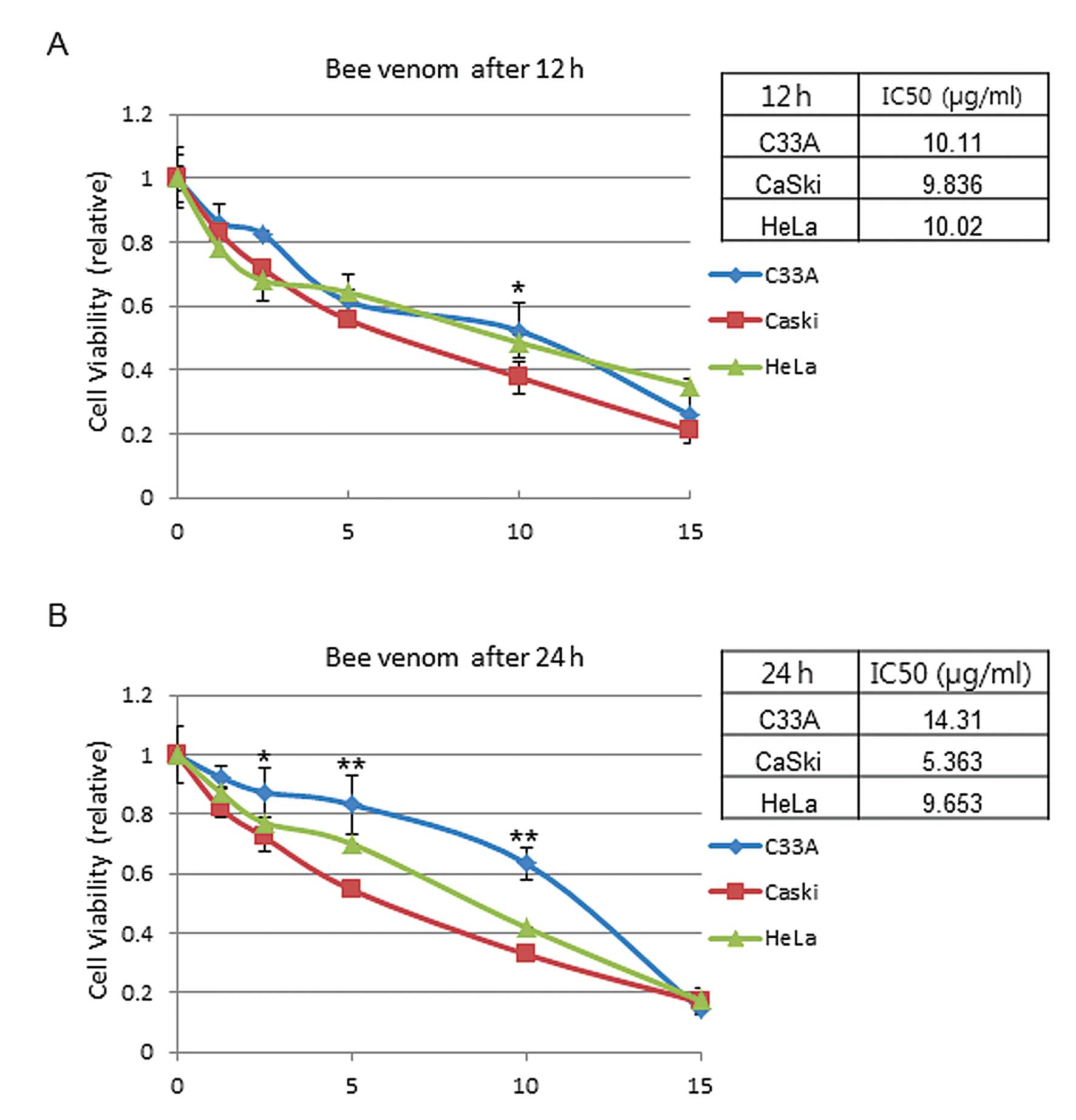
Cell viability by BV on HPV-infected
cervical cancer cells
CaSki (HVP16-infected), HeLa (HPV18-infected) and
C33A (HPV-negative) cells were treated by various concentrations of
BV for 12 or 24 h. To find out the half maximal inhibitory
concentration (IC50) of BV, cell viability was
determined by MTT assay using various BV concentrations (1, 2.5, 5,
10 and 15 μg/ml). The results showed that cell viability was
affected in a dose-dependent manner (1–15 μg/ml). The
IC50 value of CaSki, HeLa and C33A was 9.8, 10.0 and
10.1 μg/ml after 12 h of BV treatment, respectively (Fig. 1A). After 24 h of BV treatment, the
IC50 value was 5.4, 9.7 and 14.3 μg/ml in CaSki, HeLa
and C33A, respectively (Fig. 1B).
The represented data are means ± SEM from three independent
experiments and are expressed in terms of the control value. These
data support the hypothesis that there is a significant difference
in sensitivity to BV between HeLa and CaSki cells. In contrast, in
the case of C33A, relatively less suppression of cell growth was
observed, suggesting that inhibition of cell growth is mediated by
antiviral effects of BV.
Inhibition of HPV E6/E7 mRNA expression
by BV
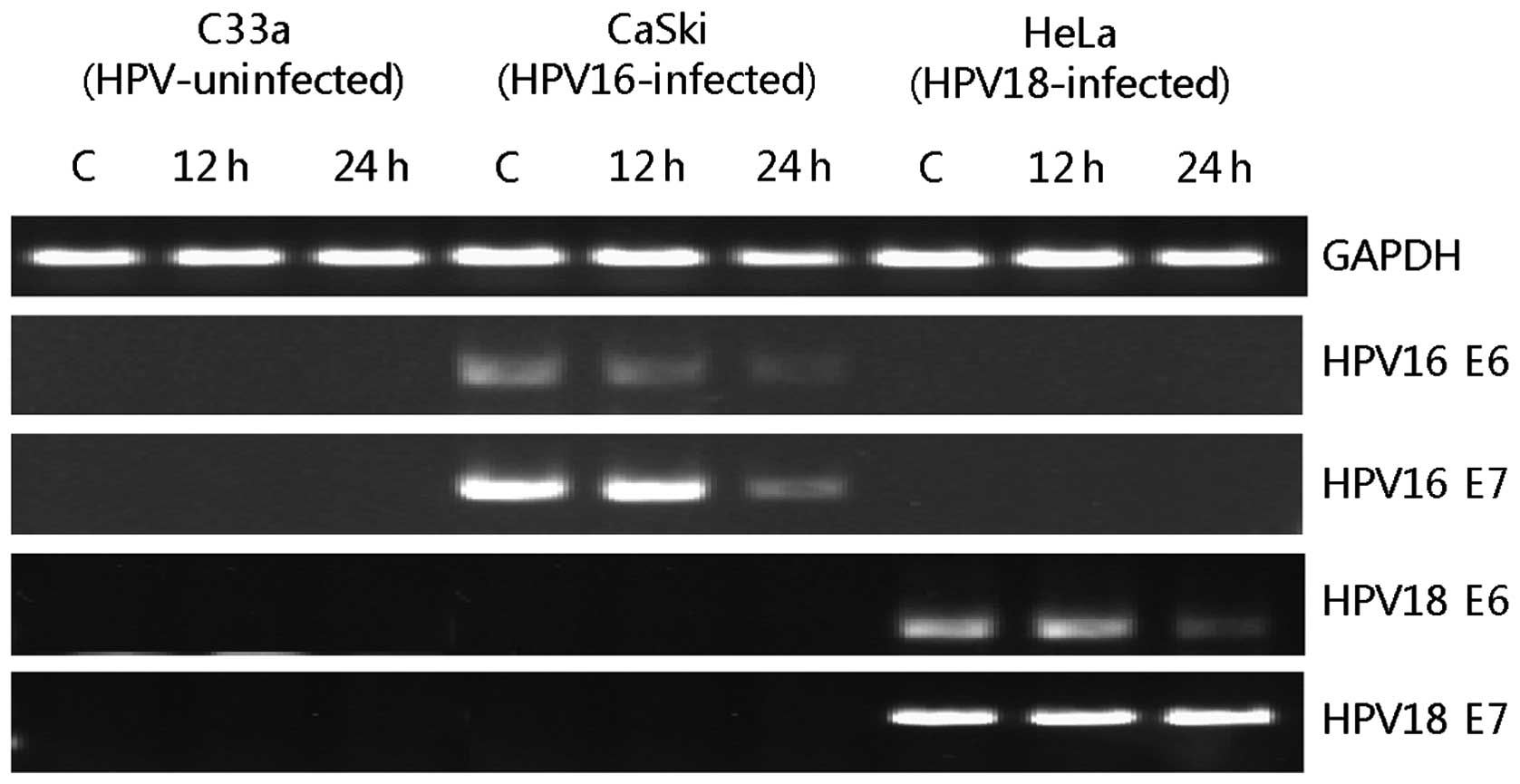
To examine whether BV treatment downregulates HPV
E6/E7 mRNA expression in CaSki and HeLa cells, the levels of mRNA
were analyzed by RT-PCR (Fig. 2).
The mRNA expression of HPV16 E6 was gradually decreased until 24 h
and that of HPV16 E7 was decreased at 24 h after 10 μg/ml BV was
treated to CaSki cells. In the case of HeLa, the mRNA expression of
HPV18 E6 was decreased at 24 h after 10 μg/ml BV treatments but
that of HPV18 E7 was not altered by BV. We also evaluated the mRNA
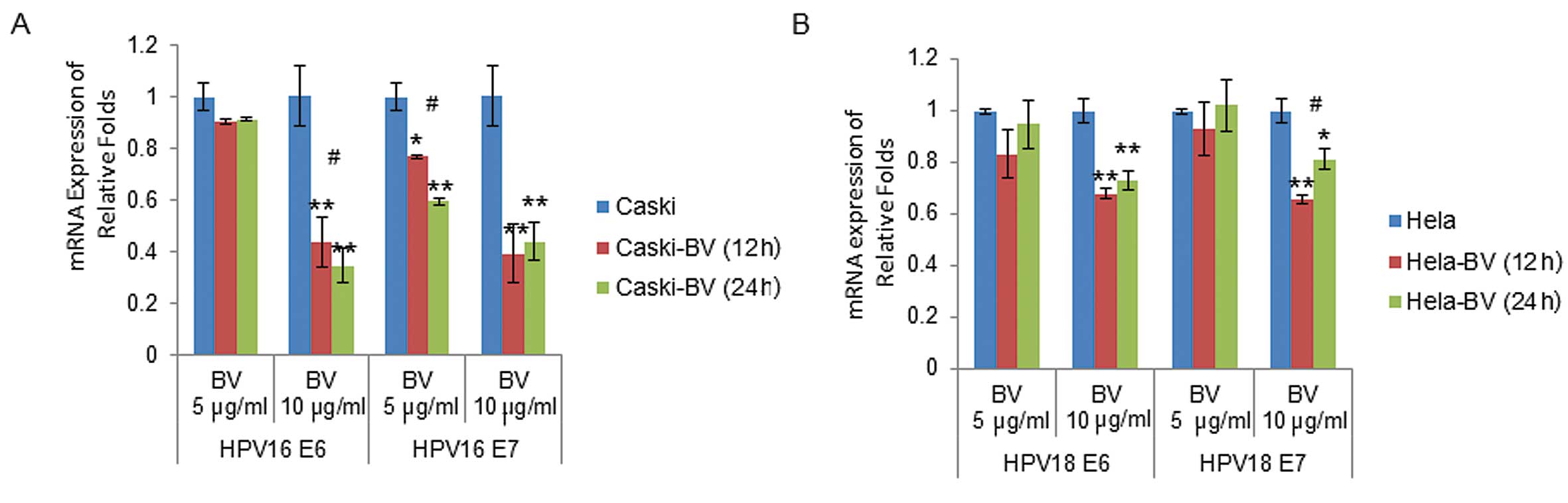
expression by qRT-PCR that analyzed HPV16/18 E6/E7 mRNA expression
levels as compared with GAPDH in BV-treated CaSki and HeLa cells.
The mRNA expression levels were obtained for the threshold cycle
(Ct) for each gene and normalized using the average of the GAPDH
gene and determined quantified relative folds. HPV16 E6 and E7 mRNA
expressions were 0.90±0.01 and 0.77±0.01-fold after 12 h, and
0.91±0.01 and 0.59±0.01-fold after 24 h in 5 μg/ml BV-treated CaSki
cells, respectively. In 10 μg/ml BV-treated CaSki cells, the mRNA
expression levels of HPV16 E6 and E7 were significantly decreased
by BV: 0.44±0.10 and 0.39±0.11-fold (after 12 h), and 0.35±0.06 and
0.44±0.07-fold (after 24 h) in BV-treated CaSki cells (Fig. 3A). HPV18 E6 and E7 mRNA expression
levels were not significantly altered by 5 μg/ml BV-treated HeLa
cells. In 10 μg/ml BV-treated HeLa cells, HPV18 E6 and E7 mRNA
expressions were 0.68±0.02 and 0.66±0.02-fold after 12 h, and
0.73±0.03 and 0.81±0.04-fold after 24 h, respectively (Fig. 3B). When the HPV16/18 E6/ E7 mRNA
expression levels at 5 μg/ml BV-treatment were compared between
CaSki and HeLa cells, more HPV16 E7 was observed in CaSki cells, as
compared to HeLa cells. Moreover, when the cervical cancer cells
were treated with 10 μg/ml BV, the inhibition of HPV16/18 E6/E7
mRNA expression levels was significantly observed. Collectively,
these data further suggest that BV treatment can induce these
different cell lines in a different manner.
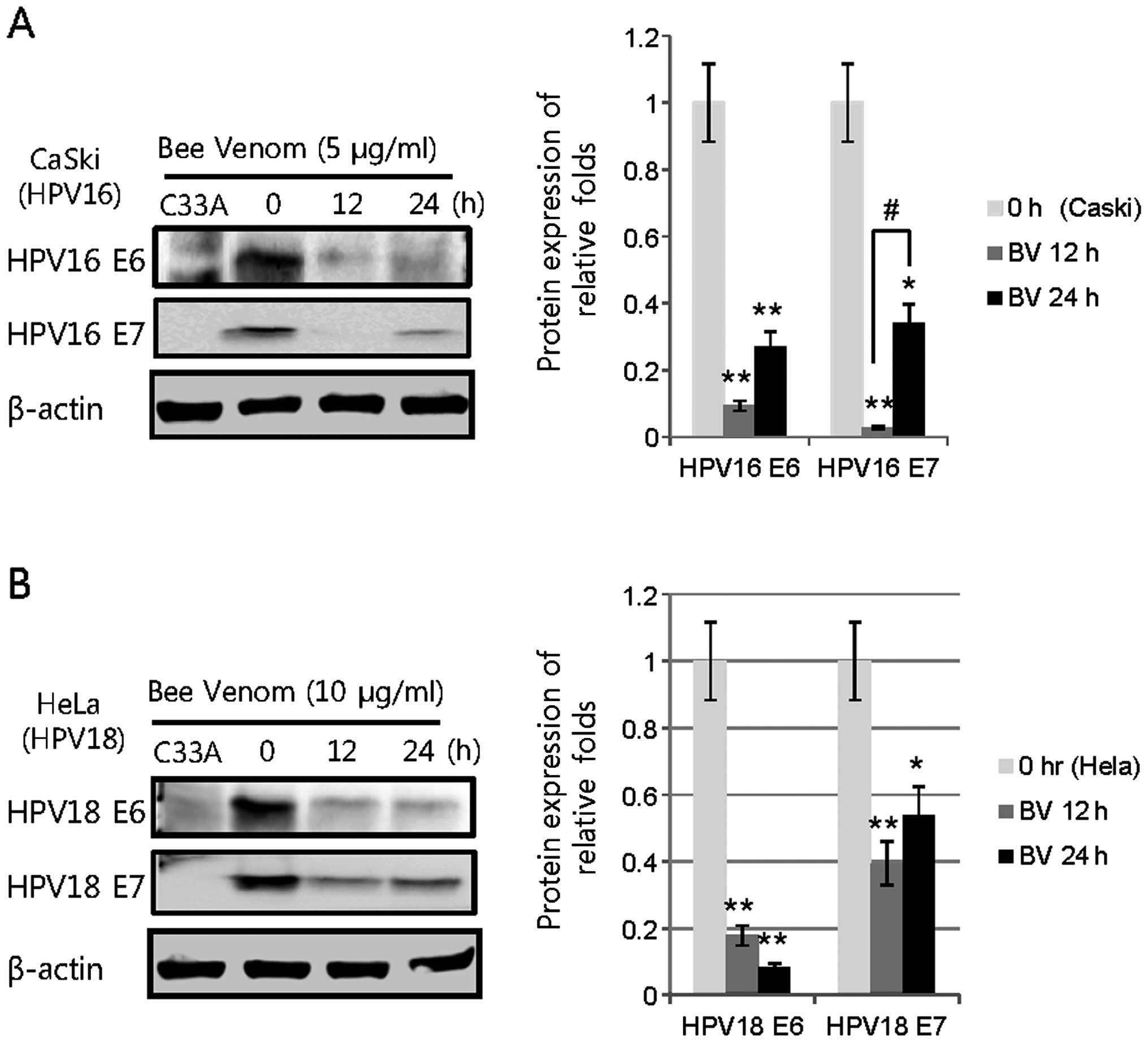
Protein expression of HPV E6/E7 in CaSki
and HeLa with BV treatments
To investigate whether HPV E6/E7 protein expression
by BV treatment displayed a different level, we performed western
blot analyses that estimated HPV16/18 E6/E7 protein expression as
compared with β-actin. The protein expression levels were also
quantified by densities of blotted bands and were measured by
relative folds compared to those of HPV16/18 E6/E7 of BV-untreated
CaSki and HeLa cells. In BV-treated CaSki and HeLa cells, the
levels of these protein expressions were significantly lower than
those of BV-untreated cells. Protein expressions of HPV16 E6 and E7
were significantly decreased at 5 μg/ml BV-treated CaSki cells for
24 h, compared to the BV-treated CaSki cells for 12 h (Fig. 4A). In 10 μg/ml BV-treated HeLa
cells, HPV18 E6 and E7 were less expressed at 12 and 24 h as
compared to the expression of HPV16 E6 and E7 by BV-treated CaSki
cells (Fig. 4B). These data support
the theory that HPV16/18 E6 and E7 are downregulated by BV in the
cervical cancer cells.
Antitumor effect of TC-1 tumor model by
BV treatments
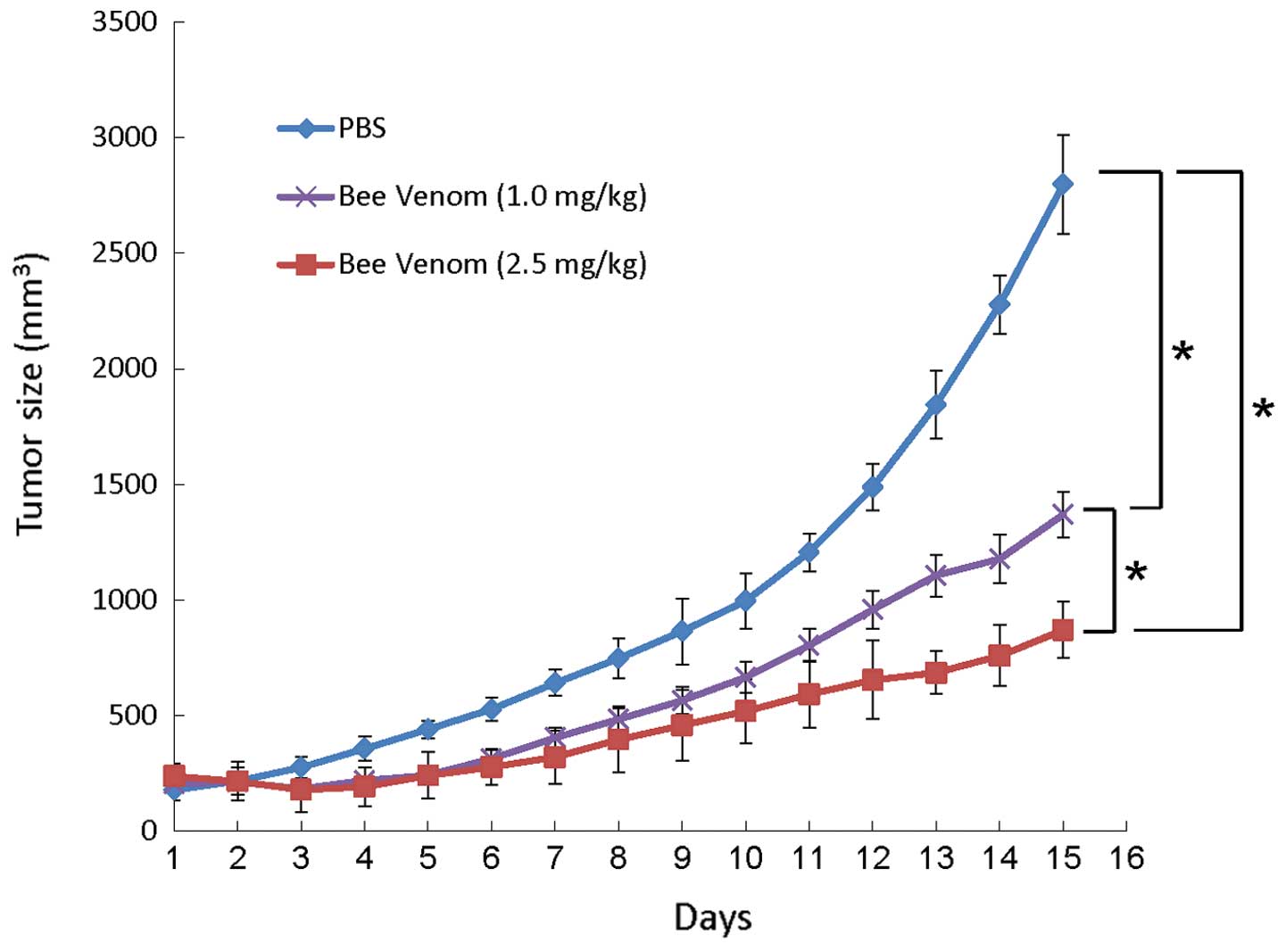
To determine the antitumor effects of BV in
vivo, C57BL/6 mice were challenged s.c. with TC-1 cells and
then treated with BV. As shown in Fig.
5, BV treatment was initiated on day 1 when tumors were
relatively small in size. The growth of TC-1 tumors was
significantly decreased compared with that of tumors treated with
PBS. Specifically, by day 15 after treatment, PBS-treated tumors
reached an average tumor volume of 2,797±215 mm3, while
those treated with 1 mg/kg of BV reached 1,369±98 mm3,
and those treated with 2.5 mg/kg of BV reached 839±122
mm3. In addition, at day 15 after treatment, the 2.5
mg/kg BV-treated TC-1 tumor group showed that the tumor growth was
significantly suppressed as compared with the 1.0 mg/kg BV-treated
group (P<0.0001) and with the PBS group (P<0.0001). The
antitumor effects of BV in vivo were consistent with the MTT
assays and protein expression analyses in vitro.
HPV16 E6/E7 expression of TC-1 tumor
model by BV treatments
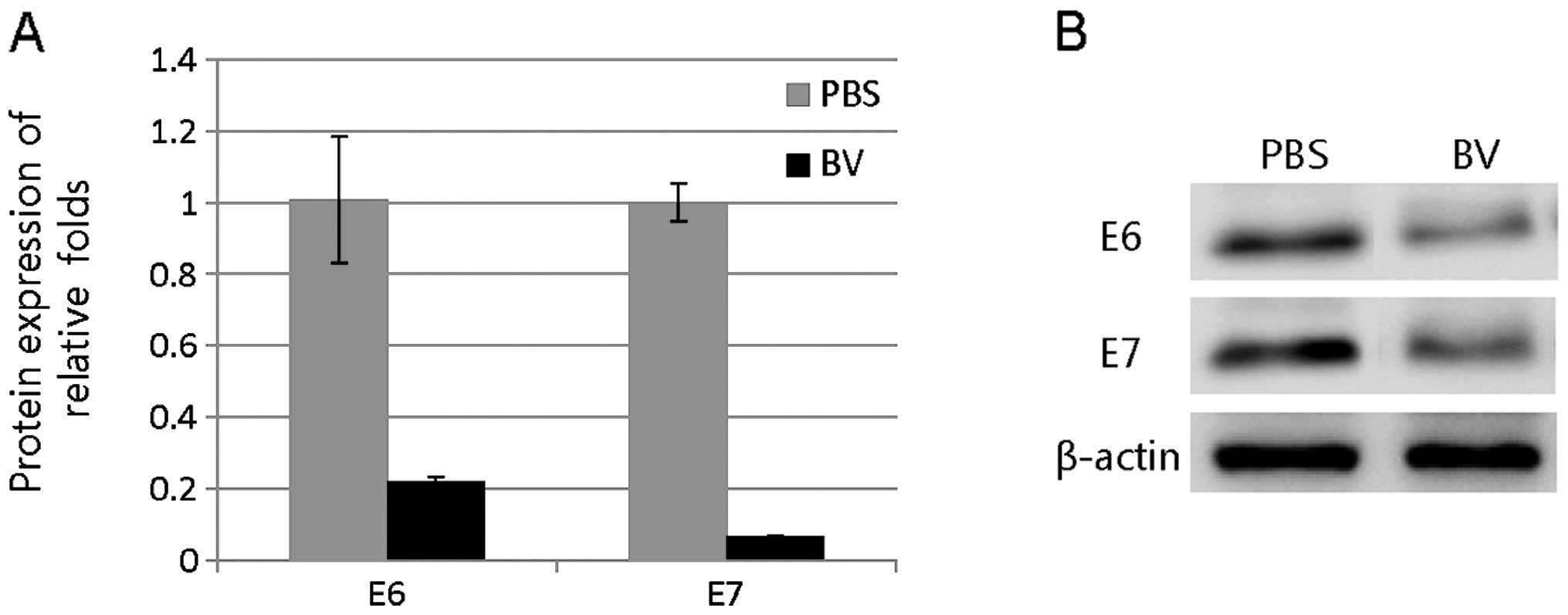
To analyze HPV16 E6/E7 mRNA and protein levels in
vivo by BV treatment in TC-1 tumors, we evaluated mRNA and
protein expression by qRT-PCR and western blotting, respectively.
The mRNA expression levels of HPV16 E6 and E7 were decreased by BV.
HPV16 E6 gene in the TC-1 tumors was 0.22±0.02-fold and E7 gene was
0.07±0.004-fold compared with PBS-treated tumor (Fig. 6A). Protein expression levels of
HPV16 E6 and E7 in the TC-1 tumors were significantly decreased by
BV (Fig. 6B). Overall, these data
suggest that BV treatments induce downregulation of HPV16 E6 and E7
depending on the cervical cancer cell lines.
Discussion
Cervical carcinoma is caused mostly by infection
with a high-risk group of human papillomaviruses (HPVs). After
high-risk HPV infection, E6 and E7 oncoproteins, which are
essential for immortalization and transformation of human squamous
epithelial cells, are consistently expressed. In HPV type 16,
mutations on the open reading frame of E6 or E7, or on their
upstream sequences, alter the oncogenicity of the virus, suggesting
an important role of these viral proteins in oncogenesis. In most
cervical cancers, the function of p53 is downregulated by the E6
protein of HPV16; E6 binds to p53 and leads to degradation of
E6-p53 complexes due to E6-activated ubiquitin-dependent protease
digestion. In the present study, we evaluated the effect of BV on
cervical cancer cells to get an insight into the molecular basis of
tumor-specific growth inhibition in vitro and in
vivo. BV induced an increase in the levels of p53 and decreased
the survival of cancer cells (16).
BV downregulated the levels of anti-apoptotic genes such as Bcl-2
and Bcl-XS/L; however, the levels of Bax, a pro-apoptotic gene,
were upregulated (23). Moreover,
the efficacy of BV therapy has been confirmed clinically (14). We also observed a significant growth
suppression of cervical cancer cells when BV was treated into the
cells. In particular, HPV16-infected cell types were more
susceptible to BV-mediated cell growth inhibition than
HPV18-infected cell types. In one study, HPV16-infected cells
(SiHa, 1–2 copies of HPV/cell) were more susceptible to growth
inhibition exerted by adeno-associated virus, as compared to
HPV18-infected cells (HeLa, 50 copies of HPV/cells) (24). In agreement with this, our study
showed that CaSki cells (600 copies of HPV/ cell) resulted in more
susceptibility to growth inhibition by BV treatments compared to
HeLa cells. Furthermore, similar suppression of cancer cell growth
was observed between C33A and HeLa cells after BV treatments,
supporting the theory that the effects of BV were significantly
mediated by the downregulation of E6/E7 protein of HPV 16. On the
other hand, it was notable that there was local recurrence in mice
from day 7. The loss of inhibitory properties of BV began with the
increased expression of E6. A possible explanation for this may be
that the continual release of E6 oncogene overcomes the inhibitory
effects of BV, suggesting BV treatments could be transient as the
host immune system plays a major role in preventing sustained
effects of BV. Thus, an advanced strategy in the development of
long-term persistent effects of BV is required.
Although the mechanisms behind the antiviral
activity of BV remain unclear, BV showed very similar inhibition of
pro-inflammatory cytokines (25).
It was reported that a BV-derived peptide could provide a scaffold
for p53 inhibitors to treat cancer. Further studies are required to
define a possible mechanism of the adenoviral p53-like
anti-inflammatory activity of BV. Although BV treatment induced
tumor suppressor p53 as well as cyclin-dependent kinase inhibitor
p21WAF1/CIP1 expression in a dose-dependent manner, BV is not a
well-defined inflammatory mediator. Melittin is known as one of the
principal active components of BV and is a powerful stimulator of
phospholipase A2 (26). Treatment
of melittin also potently induced pro-apoptotic protein p53, Bax
and caspase-3 expression but decreased anti-apoptotic protein Bcl-2
expression (27). We also observed
that p53 overexpression induced apoptosis in CaSki and HeLa cells,
as determined by Annexin V staining (28). This is consistent with our
observation that both CaSki and HeLa cells displayed cell death
upon BV treatment with the downregulation of E6/E7 protein of
HPV16/18, and BV treatment behaves differently in HeLa and CaSki
cells. It could be suggested that, in these cells, endogenous p53
is not exhausted with the downregulation of viral E6 protein that
binds to p53, resulting in degradation of p53-E6 complexes via the
ubiquitin pathway. It was reported that treatment with
adeno-associated virus induced different levels of cell growth
inhibition among cervical cancer cell types (24), suggesting a different nature of each
cervical cancer cell’s HPV infection status.
BV plays an important role in maintaining regulation
of pro-inflammatory cytokines and was suggested to be involved in
distinct signaling pathways (29).
For example, BV inhibited the expression of specific inflammatory
genes that were upregulated by nuclear factor-κB in the presence of
LPS, including mitogen-activated protein kinase 8 (MAP3K8), TNF,
TNF-α-induced protein 3 (TNFAIP3), suppressor of cytokine signaling
3 (SOCS3), TNF receptor-associated factor 1 (TRAF1), JUN and CREB
binding protein (CBP). However, it should be noted that BV could
inhibit both pro- and anti-inflammatory cytokine expression
(4,30). It may be noteworthy to identify
certain biological compounds in BV that are responsible for
altering transcription expression of both inflammatory cytokines
and the mechanism whereby BV induces growth arrest in different
phases in cervical cells.
Taken together, we observed that BV treatments exert
a differential effect in suppressing cervical cancer cell growth
through the downregulation of E6/E7 protein of HPV16/18. In
particular, HPV16-infected cells (CaSki cells) were more
susceptible to growth inhibition exerted by BV, as compared to
HPV18-infected cells (HeLa cells), suggesting that the growth
inhibition phase is dependent on the cervical cancer cell line.
Thus, these data support the hypothesis that BV plays an important
role in suppressing cancer cell growth by the downregulation of
E6/E7 protein of HPV16/18, depending on the cancer cell line.
Acknowledgements
We deeply thank PhD student Gantumur Battogtokh for
his active advice and support. This study was supported by the
National Research Foundation of Korea (NRF) grant funded by the
Korean government (MEST) (NRF-2012R1A2A1A 03670430 and
NRF-2013R1A1A2063115), the MSIP (Ministry of Science, ICT and
Future Planning) under the ‘IT Consilience Creative Program’
(NIPA-2014-H0201-14-1002) supervised by the NIPA (National IT
Industry Promotion Agency), and a grant (Industry-Academic
Cooperation Foundation program) from Dong Sung Biopharm Co.
References
|
1
|
Billingham ME, Morley J, Hanson JM,
Shipolini RA and Vernon CA: Letter: An anti-inflammatory peptide
from bee venom. Nature. 245:163–164. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Orlov BN, Omarov ShM, Gelashvili DB,
Korneva NV and Asafova NN: Chemistry and pharmacology of bee venom
(a review of the literature). Farmakol Toksikol. 41:358–369.
1978.(In Russian). PubMed/NCBI
|
|
3
|
Hockenhull J, Elremeli M, Cherry MG, et
al: A systematic review of the clinical effectiveness and
cost-effectiveness of Pharmalgen® for the treatment of
bee and wasp venom allergy. Health Technol Assess. 16:III–IV.
1–110. 2012.
|
|
4
|
Park HJ, Lee HJ, Choi MS, et al: JNK
pathway is involved in the inhibition of inflammatory target gene
expression and NF-kappaB activation by melittin. J Inflamm.
5:72008. View Article : Google Scholar
|
|
5
|
Habermann E: Bee and wasp venoms. Science.
177:314–322. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schmidt JO: Biochemistry of insect venoms.
Annu Rev Entomol. 27:339–368. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Son DJ, Lee JW, Lee YH, Song HS, Lee CK
and Hong JT: Therapeutic application of anti-arthritis,
pain-releasing, and anti-cancer effects of bee venom and its
constituent compounds. Pharmacol Ther. 115:246–270. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Texier C, Pouvelle S, Busson M, et al:
HLA-DR restricted peptide candidates for bee venom immunotherapy. J
Immunol. 164:3177–3184. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kleinschmidt JH, Mahaney JE, Thomas DD and
Marsh D: Interaction of bee venom melittin with zwitterionic and
negatively charged phospholipid bilayers: a spin-label electron
spin resonance study. Biophys J. 72:767–778. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Przybilla B: Insect venom allergy.
Removing the sting of killer bees! MMW Fortschr Med. 143:41–44.
2001.(In German). PubMed/NCBI
|
|
11
|
Hong SJ, Rim GS, Yang HI, et al: Bee venom
induces apoptosis through caspase-3 activation in synovial
fibroblasts of patients with rheumatoid arthritis. Toxicon.
46:39–45. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park MH, Choi MS, Kwak DH, et al:
Anti-cancer effect of bee venom in prostate cancer cells through
activation of caspase pathway via inactivation of NF-κB. Prostate.
71:801–812. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Park JH, Kim KH, Kim SJ, Lee WR, Lee KG
and Park KK: Bee venom protects hepatocytes from tumor necrosis
factor-α and actinomycin D. Arch Pharm Res. 33:215–223. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu XD, Zhang JL, Zheng HG, Liu FY and
Chen Y: Clinical randomized study of bee-sting therapy for
rheumatoid arthritis. Zhen Ci Yan Jiu. 33:197–200. 2008.(In
Chinese). PubMed/NCBI
|
|
15
|
Ip SW, Liao SS, Lin SY, et al: The role of
mitochondria in bee venom-induced apoptosis in human breast cancer
MCF7 cells. In Vivo. 22:237–245. 2008.PubMed/NCBI
|
|
16
|
Ip SW, Wei HC, Lin JP, et al: Bee venom
induced cell cycle arrest and apoptosis in human cervical
epidermoid carcinoma Ca Ski cells. Anticancer Res. 28:833–842.
2008.PubMed/NCBI
|
|
17
|
Han HJ, Lee JH, Park SH, et al: Effect of
bee venom and its melittin on apical transporters of renal proximal
tubule cells. Kidney Blood Press Res. 23:393–399. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hood JL, Jallouk AP, Campbell N, Ratner L
and Wickline SA: Cytolytic nanoparticles attenuate HIV-1
infectivity. Antivir Ther. 18:95–103. 2013. View Article : Google Scholar
|
|
19
|
Fenard D, Lambeau G, Maurin T, Lefebvre JC
and Doglio A: A peptide derived from bee venom-secreted
phospholipase A2 inhibits replication of T-cell tropic
HIV-1 strains via interaction with the CXCR4 chemokine receptor.
Mol Pharmacol. 60:341–347. 2001.PubMed/NCBI
|
|
20
|
Soman NR, Baldwin SL, Hu G, et al:
Molecularly targeted nanocarriers deliver the cytolytic peptide
melittin specifically to tumor cells in mice, reducing tumor
growth. J Clin Invest. 119:2830–2842. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jemon K, Young V, Wilson M, et al: An
enhanced heterologous virus-like particle for human papillomavirus
type 16 tumour immunotherapy. PLoS One. 8:e668662013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim YW, Bae SM, Battogtokh G, Bang HJ and
Ahn WS: Synergistic anti-tumor effects of combination of
photodynamic therapy and arsenic compound in cervical cancer cells:
in vivo and in vitro studies. PLoS One. 7:e385832012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim SK, Park KY, Yoon WC, et al: Melittin
enhances apoptosis through suppression of IL-6/sIL-6R
complex-induced NF-κB and STAT3 activation and Bcl-2 expression for
human fibroblast-like synoviocytes in rheumatoid arthritis. Joint
Bone Spine. 78:471–477. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Su PF and Wu FY: Differential suppression
of the tumorigenicity of HeLa and SiHa cells by adeno-associated
virus. Br J Cancer. 73:1533–1537. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kwon YB, Kim HW, Ham TW, et al: The
anti-inflammatory effect of bee venom stimulation in a mouse air
pouch model is mediated by adrenal medullary activity. J
Neuroendocrinol. 15:93–96. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ownby CL, Powell JR, Jiang MS and Fletcher
JE: Melittin and phospholipase A2 from bee (Apis
mellifera) venom cause necrosis of murine skeletal muscle in vivo.
Toxicon. 35:67–80. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Son DJ, Ha SJ, Song HS, et al: Melittin
inhibits vascular smooth muscle cell proliferation through
induction of apoptosis via suppression of nuclear factor-κB and Akt
activation and enhancement of apoptotic protein expression. J
Pharmacol Exp Ther. 317:627–634. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ahn WS, Han YJ, Bae SM, et al:
Differential suppression of human cervical cancer cell growth by
adenovirus delivery of p53 in vitro: arrest phase of cell cycle is
dependent on cell line. Jpn J Cancer Res. 93:1012–1019. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jang HS, Chung HS, Ko E, et al: Microarray
analysis of gene expression profiles in response to treatment with
bee venom in lipopolysaccharide activated RAW 264.7 cells. J
Ethnopharmacol. 121:213–220. 2009. View Article : Google Scholar
|
|
30
|
Han S, Lee K, Yeo J, et al: Effect of
honey bee venom on microglial cells nitric oxide and tumor necrosis
factor-α production stimulated by LPS. J Ethnopharmacol.
111:176–181. 2007. View Article : Google Scholar
|