Introduction
Pancreatic cancer is a malignant tumor caused by the
mutation of normal pancreatic tissues, and 95% of pancreatic
cancers are adenocarcinomas (1). In
the US, pancreatic cancer is the fourth highest cause of
cancer-related mortality each year, and the eighth highest in the
world (2). In China, pancreatic
cancer death ranks sixth among cancer-related deaths each year
(3). Since the pathogenesis is not
clear, there are no effective drugs for the treatment of pancreatic
cancer. Currently, surgical resection is the most common treatment
for pancreatic cancer. However, there are no convincing results
concerning clinically relevant improvements in the quality of life
and survival of these patients. Therefore, it is imperative to
identify drugs that exhibit low toxicity and are relatively
inexpensive.
Epigenetic changes in cancer have gained great
attention. Epigenetic modifications mainly include DNA methylation
and hydroxymethylation, histone methylation and acetylation,
chromatin remodeling, genomic imprinting and RNA interference. DNA
methylation is catalyzed by a group of DNA methyltransferase (DNMT)
enzymes: mainly DNMT1, DNMT3a and DNMT3b (4). Studies have shown that DNA methylation
plays an important role in the occurrence and development of many
malignant tumors. In recent years, it has been discovered that
under the catalysis of the Tets family, 5mC is transformed into
5hmC and 5fmC. The content of 5hmC in many types of cancer is low,
and is closely related to the occurrence and development of
melanoma (6).
Studies have found that many tumor-suppressor genes
have abnormal methylation in pancreatic cancer, including CDKN1C
(7), SPARC (8), P16 (9), RASSF1A (10) and ppENK (11). Methylated tumor-suppressor genes
have a different degree of methylation and are not able to express
the corresponding mRNAs. Among these tumor-suppressor genes, P16,
RASSF1A and ppENK have been extensively studied. Ueki et al
(11) and Fukushima et al
(12) found an increase of more
than 90% of ppENK gene methylation level in pancreatic cancer.
Schutte et al (13) reported
that of 95% of P16 gene inactivation, 15% was related with
methylation in pancreatic cancer. Moore et al (14) reported that the P16 gene was
methylated in 27% of pancreatic cancer cells. Dammann et al
(10) reported that after culturing
various pancreatic cells with the demethylation drug 5-Aza-CdR, the
RASSF1A gene methylation level was increased in 64% of primary
pancreatic ductal cell carcinoma, 83% of pancreatic endocrine
tumors and 88% of pancreatic cancer cell lines. P16 and RASSF1A
were re-expressed at various degrees to play an antitumor
function.
Currently, the most commonly used demethylation
drugs are 5-azacytidine (5-AzaC) and decitabine
(5-aza-2-deoxycyt-idine; 5-Aza-CdR), which are two different types
of nucleoside analogs (15). In the
US, 5-AzaC and 5-aza-2′-deoxycytidine have been approved by the
Food and Drug Administration (FDA). These drugs are mainly used for
the treatment of hematological malignancies. Other approved
demethylation drugs include zebularine (16), DHAC (17), hydralazine (18), selenite (19), RG108 (20) and arsenic trioxide (21). However, clinical trials have
reported that the demethylation effects of certain drugs are not
specific. Genomic hypomethylation was found to occur when the drug
concentration was too high, and side effects, such as drug toxicity
and inhibition of bone marrow, limit its application in the
clinical treatment of tumors. The development of a demethylation
drug with strong specificity, high safety and low toxicity has
become one of the important tasks in tumor treatment.
In recent years, Chinese herbal medicines have been
recognized as having antitumor efficacy. Emodin
(1,3,8-trihy-droxy-6-methyl anthraquinone), one such Chinese herbal
medicine, has extensive pharmacological effects such as immune
regulation (22), and antibacterial
(23), ant-inflammatory and
antitumor activities (24). Liu
et al (25) reported that
emodin inhibits pancreatic cancer cell growth through different
modes of action, but the detailed mechanism remains unclear.
Studies have found that curcumin (26) and epidermal catechins (EGCG)
(27) exert similar antitumor
efficacy through demethylation. Therefore, we hypothesized that
emodin may exert its antitumor effect though participating in the
regulation of the DNA methylation level.
Materials and methods
Chemicals and reagents
Emodin (purity ≥98%) and 5-Aza-CdR were both
purchased from Sigma (St. Louis, MO, USA). Emodin was dissolved in
dimethyl sulfoxide (DMSO) to create a stock solution at a
concentration of 10 mmol/l and was stored at −70°C. The DMSO
concentration was maintained below 0.1% in all of the cell cultures
and did not exert any detectable effect on cell growth or cell
death. The Cell Counting Kit-8 (CCK-8) was purchased from Gibco. A
cell and tissue genomic DNA extraction kit was purchased from
Fastagen Biotech (Shanghai, China). The EpiTect®
Bisulfite and EpiTect® MSP kits were purchased from
Qiagen. The RNA extraction kit was purchased from Tiangen (Beijing,
China). The antibodies, anti-RASSF1a and anti-ppENK were purchased
from Abcam. The anti-P16/INK4a antibody and anti-β-actin were
purchased from Epitomics.
Cell line and culture
Human pancreatic cancer cell line PANC-1 was
obtained from the American Type Culture Collection (ATCC;
Manassasas, VA, USA). The cells were cultured in Dulbecco’s
modified Eagle’s medium (DMEM) supplemented with 10% FBS, 100 U/ml
penicillin and 100 μg/ml streptomycin. Cells were maintained
at 37°C in a humidified atmosphere of 5% CO2. The medium
was changed every 2–3 days, and the cells were subcultured when
confluency reached 70–80% in 0.25% trypsin at 37°C.
Cell proliferation assay
Cell survival was determined using CCK-8. Briefly,
logarithmic phase PANC-1 cells were plated in 96-well culture
plates (~5×103 cells/well). After 24 h of incubation,
the cells were treated with the vehicle alone (0.1% DMSO) and
various concentrations (10, 20, 40 and 80 μM) of emodin,
followed by a 24-, 48- and 72-h cell culture. Each group had 6
wells. A total of 10 μl CCK-8 was added to each well 1 h
before the end of the incubation period. The absorbance at 450 nm
was read using the Bio-Tek EL×800 absorbance microplate reader. The
experiment was repeated 3 times. The degree of cellular inhibition
by each drug was calculated by the following formula: Relative %
inhibition = 1 - (dosing absorbance - blank absorbance)/(control
absorbance - blank absorbance) × 100%.
Dot-blot assay
The PANC-1 cells were treated with emodin (0, 10, 20
and 40 μM) and 5-Aza-CdR (1 μM) for 72 h. The control
cells were treated with 0.1% DMSO only. The total DNA was isolated
from the cultured cells using the cell/tissue genomic DNA
extraction kit according to the manufacturer’s instructions. The
concentrations of DNA were determined by fluorometry using the
Qubit® dsDNA HS kit and fluorometer (both from
Invitrogen). The procedure for the dot-blot assay was performed
with reference to a previous study (28).
mRNA-sequence
The PANC-1 cells were treated with emodin (0, 10, 20
and 40 μM) and 5-Aza-CdR (1 μM) for 72 h. The control
cells were treated with 0.1% DMSO only. The cells were collected
and sent to Major Biotechnology Company (Shanghai, China), where
the mRNA-seq was analyzed.
Bisulfite sequencing PCR (BSP)
The PANC-1 cells were treated with emodin (0, 10, 20
and 40 μM) and 5-Aza-CdR (1 μM) for 72 h. The control
cells were treated with 0.1% DMSO only. Genomic DNA was extracted
using the cell/tissue genomic DNA extraction kit according to the
manufacturer’s instructions. When the DNA was concentrated to 1
μg, bisulfite modification of genomic DNA was performed
using the EpiTect® Bisulfite kit. The sequences of the
methylation-specific primers for P16, RASSF1A and ppENK are shown
in Table I. Bisulfite-modified DNA
(4 μl), methylation-specific primers (3 μl), 2X
Taq PCR Master Mix (12.5 μl) and DEPC-H2O
(5.5 μl) were added to achieve a final volume of 25
μl. PCR amplification conditions were as follows: 95°C for 5
min, 94°C for 30 sec, annealing for 45 sec, and extension at 72°C
for 45 sec; a total of 40 cycles; followed by a final extension at
72°C for 10 min. A total of 10 μl of the PCR product was
separated on a 2% agarose gel electrophoresis, and the results were
photographed. BSP products were extracted from agarose gel, then
purified and sequenced (ShangHai Maipu Biotechnology Co., Ltd.,
China). The methylation of the sample was analyzed using BiQ
Analyzer software.
 | Table IPrimer sequences for PCR and
bisulfite sequencing. |
Table I
Primer sequences for PCR and
bisulfite sequencing.
| Genes | Primer
pairs
(5′→3′) | Product size
(bp) |
|---|
| P16 | F:
GCCGATCCAGGTCATGATGAT
R: GCATCTATGCGGGCATGGTTA | 300 |
| RASSF1A | F:
TGGGGAGGTGAACTGGGAC
R: ACACGGCACGCACTTGG | 217 |
| ppENK | F:
GCGGTTCCTGACACTTTGC
R: GGGTGCTGGTGCCATCTT | 245 |
| DNMT1 | F:
GACCCATCTCTTGAAGGTGGTGTT
R: CCTCGTCATAACTCTCCACCTGCT | 164 |
| DNMT3a | F:
AGGTGGACCGCTACATTGCC
R: GAGATGTCCCTCTTGTCACTAACG | 143 |
| P16 | BS-F:
TTGTTGTTTAGGTTGGAGTGTAGTG
BS-R: TCAAAAACATATATTAATAACAACCATCAA | 256 |
| ppENK | BS-F:
AAAGAGTTTTTGGAAATAGGGGATA
BS-R: CATCAACAATTTCCCACTAAAAAAT | 241 |
| RASSF1A | BS-F:
GTATGTAAGGGTTGGATGTGTAGAGA
BS-R: CCCCAAATAAAATCTCCACAAAAATC | 298 |
Real-time PCR
The PANC-1 cells were treated with emodin (0, 10, 20
and 40 μM) and 5-Aza-CdR (1 μM) for 72 h. The control
cells were treated with 0.1% DMSO only. Total RNA was isolated from
the cells using the TRIzol reagent (Invitrogen) according to the
manufacturer’s instructions. For reverse transcriptase analysis, 1
μg of total RNA was reversely transcribed in a 20 μl
volume using RevertAid™ First Strand cDNA Synthesis kit
(Fermentas). Real-time PCR amplification with 1 μl of the
reverse transcriptase reaction mixture was performed with SYBR
Green Real-Time PCR Master Mix-Plus (Toyobo, Japan). The initial
denaturation step was 95°C for 60 sec followed by 40 cycles of
amplification at 95°C for 15 sec, 60°C for 15 sec, and 72°C for 45
sec. All samples were performed in triplicate, and the relative
amount of the target gene was normalized to GAPDH. The primers are
listed in Table I.
Western blot analysis
The PANC-1 cells were treated with emodin (0, 10, 20
and 40 μM) and 5-Aza-CdR (1 μM) for 72 h. The control
cells were treated with 0.1% DMSO only. Total proteins were
extracted from the cells using cell lysis buffer (20 mmol/l
Tris-HCl pH 7.5, 150 mmol/l NaCl, 1 mmol/l Na2EDTA, 1
mmol/l EGTA, 1% Triton, 2.5 mmol/l sodium pyrophosphate, 1 mmol/l
β-glycerophosphate, 1 mmol/l Na3VO4, 1
μg/ml leupeptin, and 1 mmol/l PMSF). After centrifugation at
14,000 × g for 10 min at 4°C, the supernatant was collected and the
protein concentration was determined using the BCA protein assay
kit according to the manufacturer’s instructions. The protein
lysates (20-μg-lane) were separated on 12% SDS
polyacrylamide gel and transferred onto a nitrocellulose membrane.
Each membrane was blocked with 5% skim milk and then incubated with
the indicated primary antibodies against P16, RASSF1A, ppENK and
β-actin over night at 4°C. Subsequently, the membrane was incubated
with the secondary antibodies, goat anti-rabbit and anti-mouse IgG
conjugated with HRP, for 1 h at room temperature, and the formed
immunocomplex was visualized by enhanced chemiluminescence reagent
and exposed to X-ray film. Quantitative data are expressed as a
percentage of the mean ± standard deviation (SD) of the relative
levels of the objective protein and control β-actin of each group
of cells from three independent experiments.
Statistical analysis
All results were repeated in at least three separate
experiments. The data are expressed as the mean ± SD. Statistical
comparisons were conducted using one-way analysis of variance,
which revealed significant differences between groups, and the
Student’s t-test which revealed significant differences between two
sample means. Statistical analyses were carried out using SPSS
version 17.0 software (SPSS Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Effect of emodin on PANC-1 cell
proliferation
To investigate the effect of emodin on cell growth,
PANC-1 cells were cultured with 0, 10, 20, 40 and 80 μM
emodin for 24, 48 and 72 h. Cell proliferation was determined by
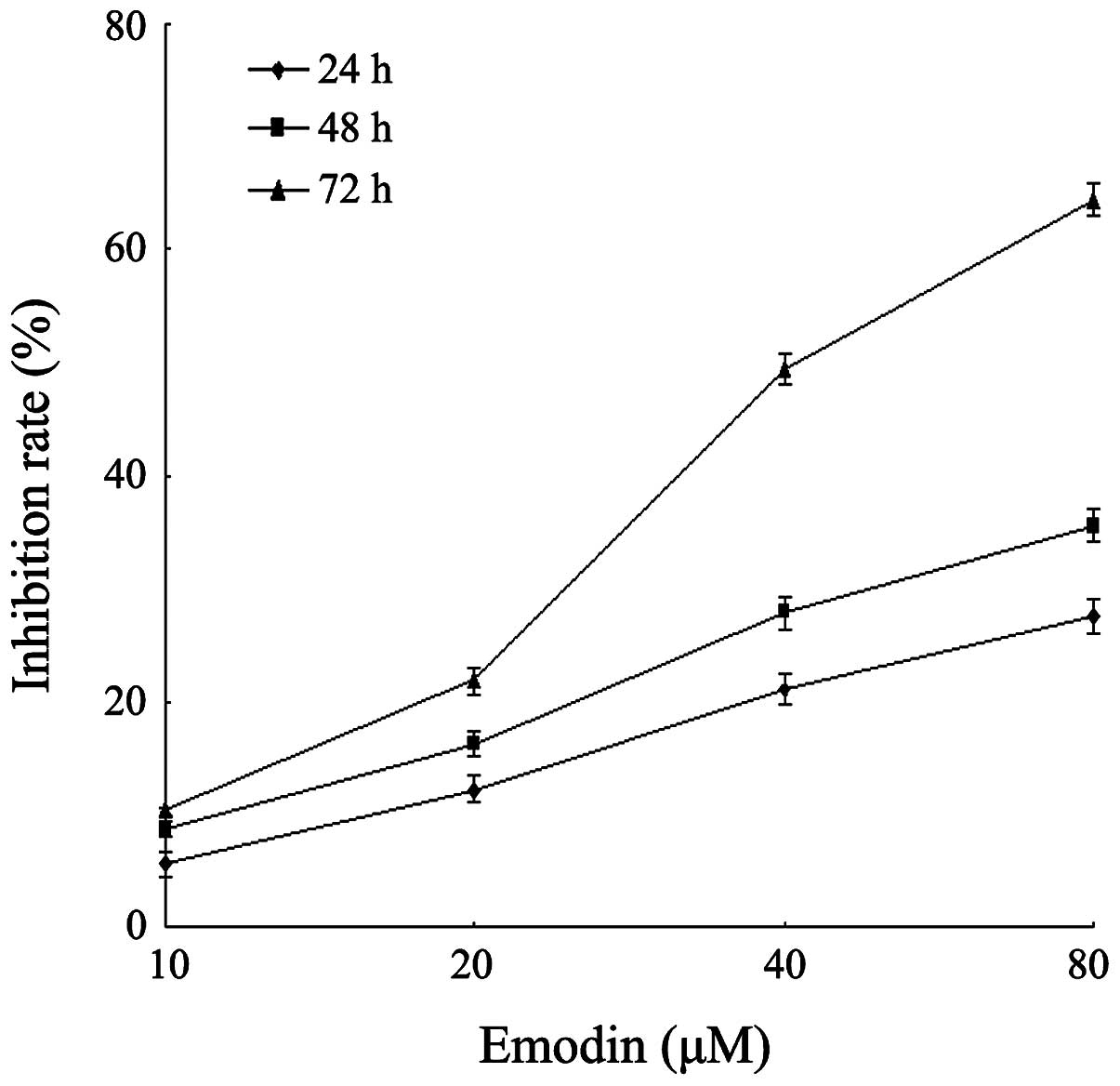
the CCK-8 assay. As demonstrated in Fig. 1, emodin was shown to inhibit the
growth of the cells in a dose- and time-dependent manner. This
result was similar with a previous study (29). The inhibition rate of emodin at a
concentration of 40 μM for 72 h was 49.4%, while the
inhibition rate of emodin at a concentration of 80 μM for 72
h reached 64.4%. Since the methylation efficiency of drugs is
usually exerted under the appropriate low drug concentration,
emodin at a concentration of 10, 20 and 40 μM was used for
the following experimental research.
Emodin decreases the level of genome 5mC,
but has no significant effect on 5hmC
To determine the effects of emodin on the genomic
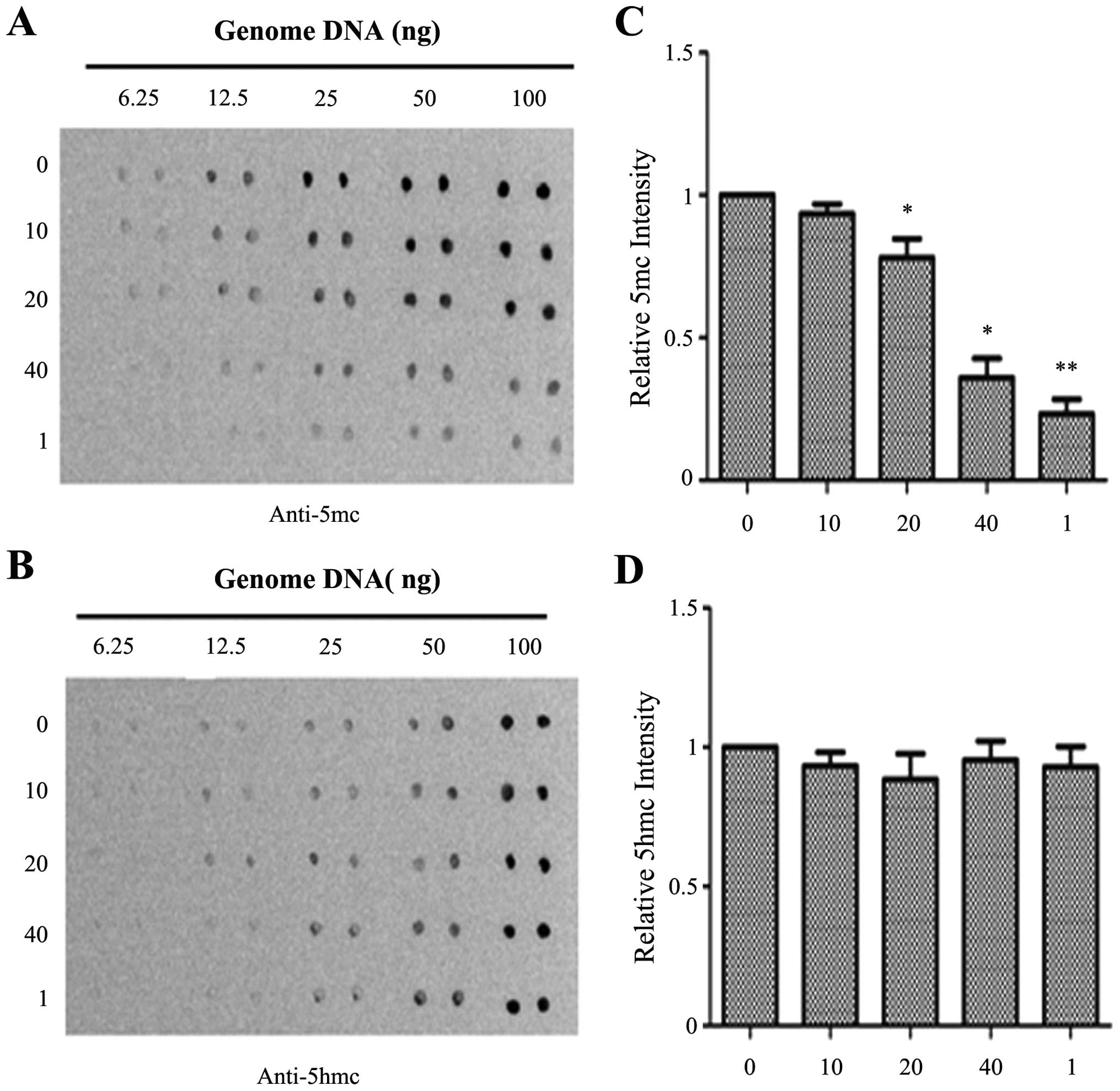
DNA methylation level in the PANC-1 cells, a dot-blot assay was
performed. As shown in Fig. 2,
emodin at a concentration of 40 μM and 1 μM 5Aza-CdR
significantly reduced the 5mC level when compared with the level in
the control group. However, there were no significant differences
in 5hmC levels. The levels of 5hmC were slightly decreased by
emodin at concentrations of 10 and 20 μM, but the effect was
not obvious. To further confirm this result, the effects of emodin
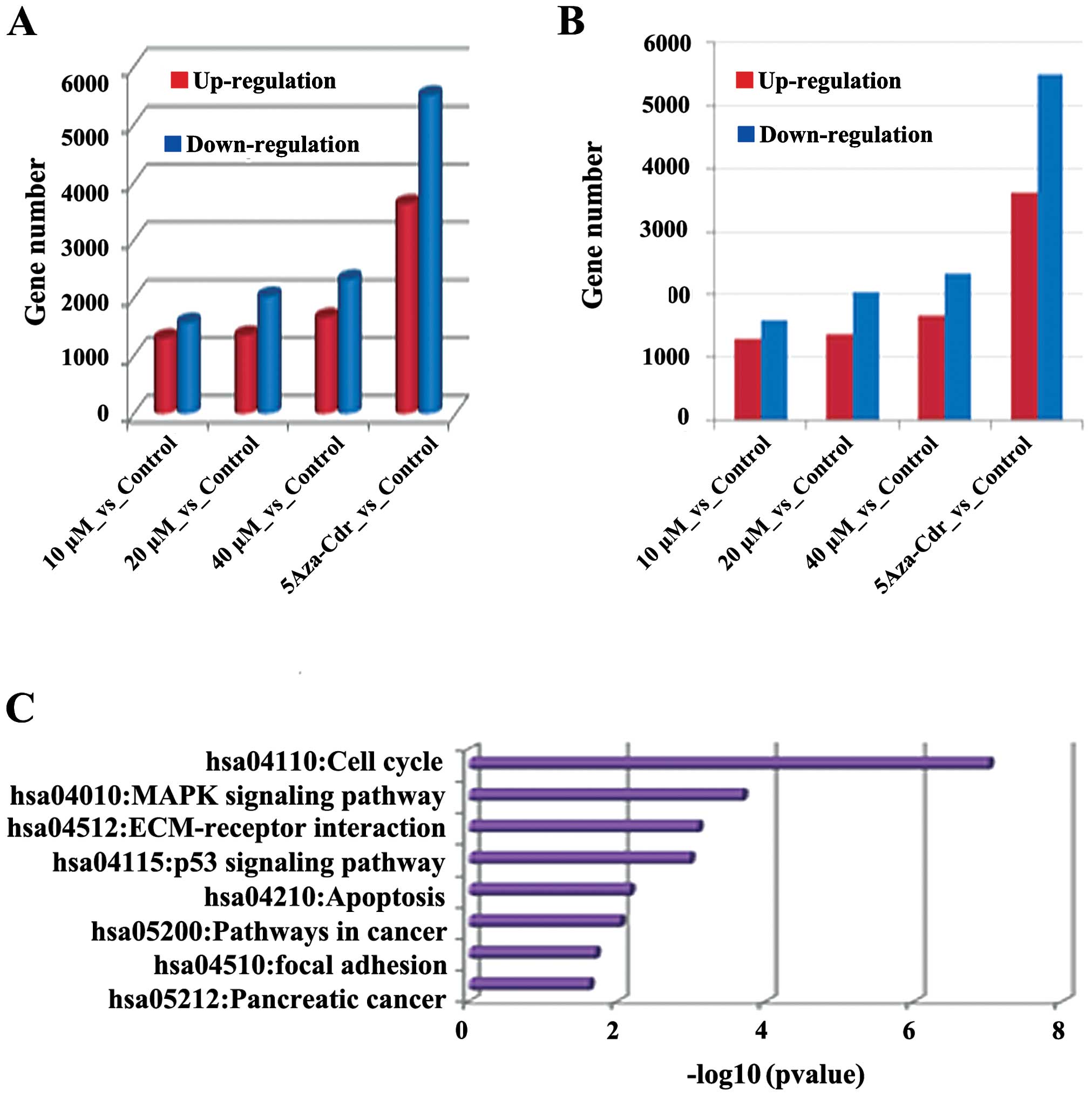
on gene expression profiles were detected by mRNA-Seq. As shown in
Fig. 3, different concentrations of
emodin promoted differences between changes in the gene expression
profiles. The number of differentially expressed genes was also
increased in a dose-dependent manner. In each group, the number of
downregulated genes was higher than the upregulated genes. For
example, in the emodin (40 μM) vs the control group, the
expressed genes were mainly concentrated in the KEGG pathway which
was closely related to tumor pathways such as cell cycle, p53
signaling, pathways in cancer and apoptosis, cell division, DNA
metabolic processes and regulation of cell death. The data suggest
that emodin treatment has significant effects on gene expression,
and plays an important role in regulating metabolic processes such
as cell differentiation and proliferation.
Pyrophosphate sequencing PCR (BSP)
In order to verify the differential expression of
gene methylation after emodin treatment, BSP was used to detect the
methylation levels of P16, RASSF1A and ppENK in the promoter
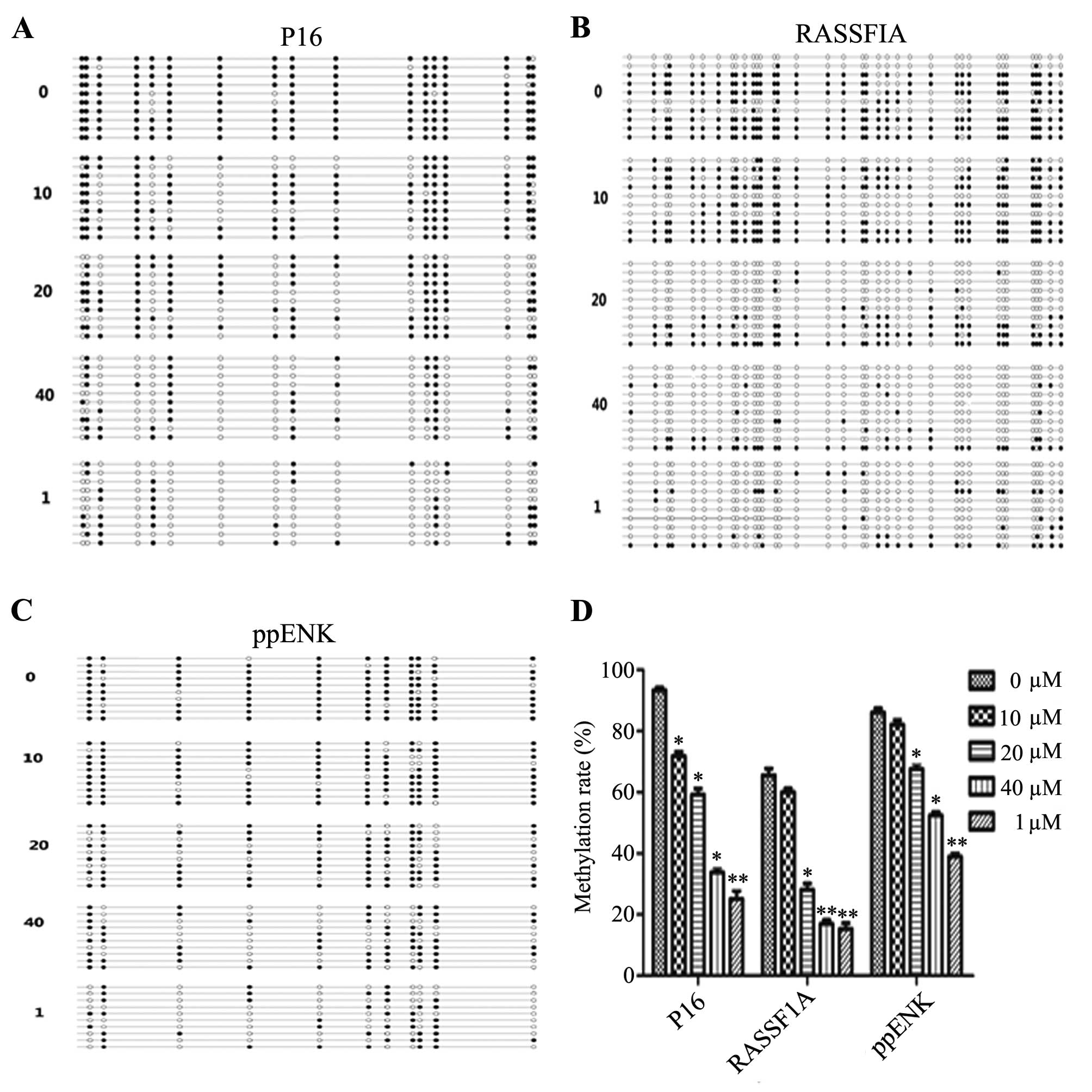
region. As shown in Fig. 4,
treatment with emodin led to different degree of demethylation
levels of P16, ppENK and RASSF1A in the promoter regions. Scopes of
P16, ppENK, RASSF1A gene sequencing were respectively composed of
17, 11 and 36 CpG islands, 10 of which were randomly selected to
clone and sequence. After treatment of PANC-1 cells with various
concentrations of emodin (0, 10, 20 and 40 μM) and 5-Aza-CdR
(1 μM) for 72 h, the methylation rates of the P16 gene were
93.5, 71.8, 59.4 and 33.5%, respectively. While the methylation
rate in the 5-Aza-CdR group was decreased to 25%. The effect in the
5-Aza-CdR group was stronger than that in the emodin-treated group.
The methylation rates of the ppENK gene were 86.4, 82.7, 67.3 and
52.7%, respectively, while that in the 5-Aza-CdR group was 39.1%.
The methylation rates of the RASSF1A gene were 65.3, 60, 27.2 and
16.4%, respectively, while that in the 5-Aza-CdR group was 15.3%.
These results demonstrated that emodin reduced the methylation
levels of P16, RASSF1A and ppENK in the promoter region in
pancreatic cancer cells.
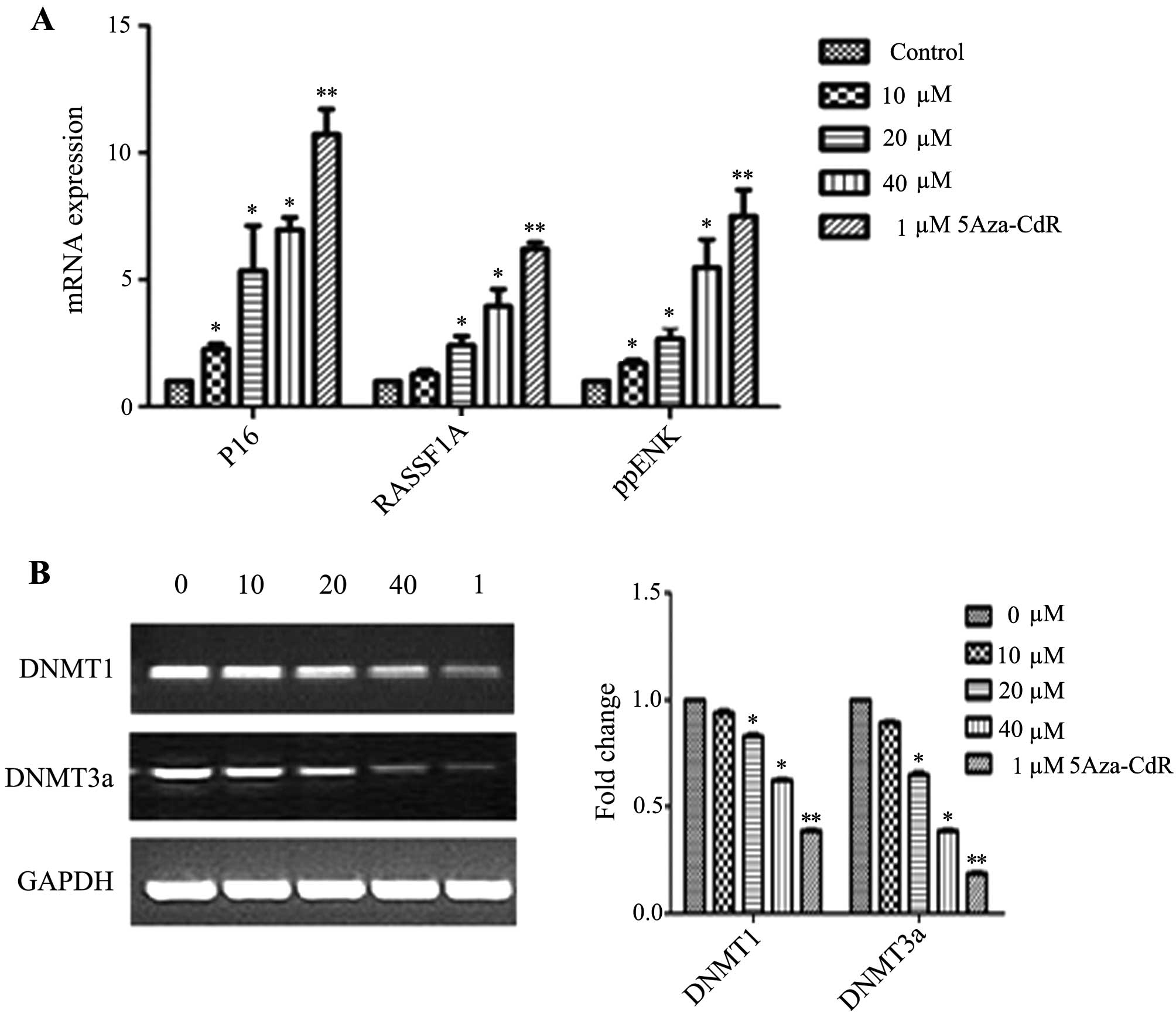
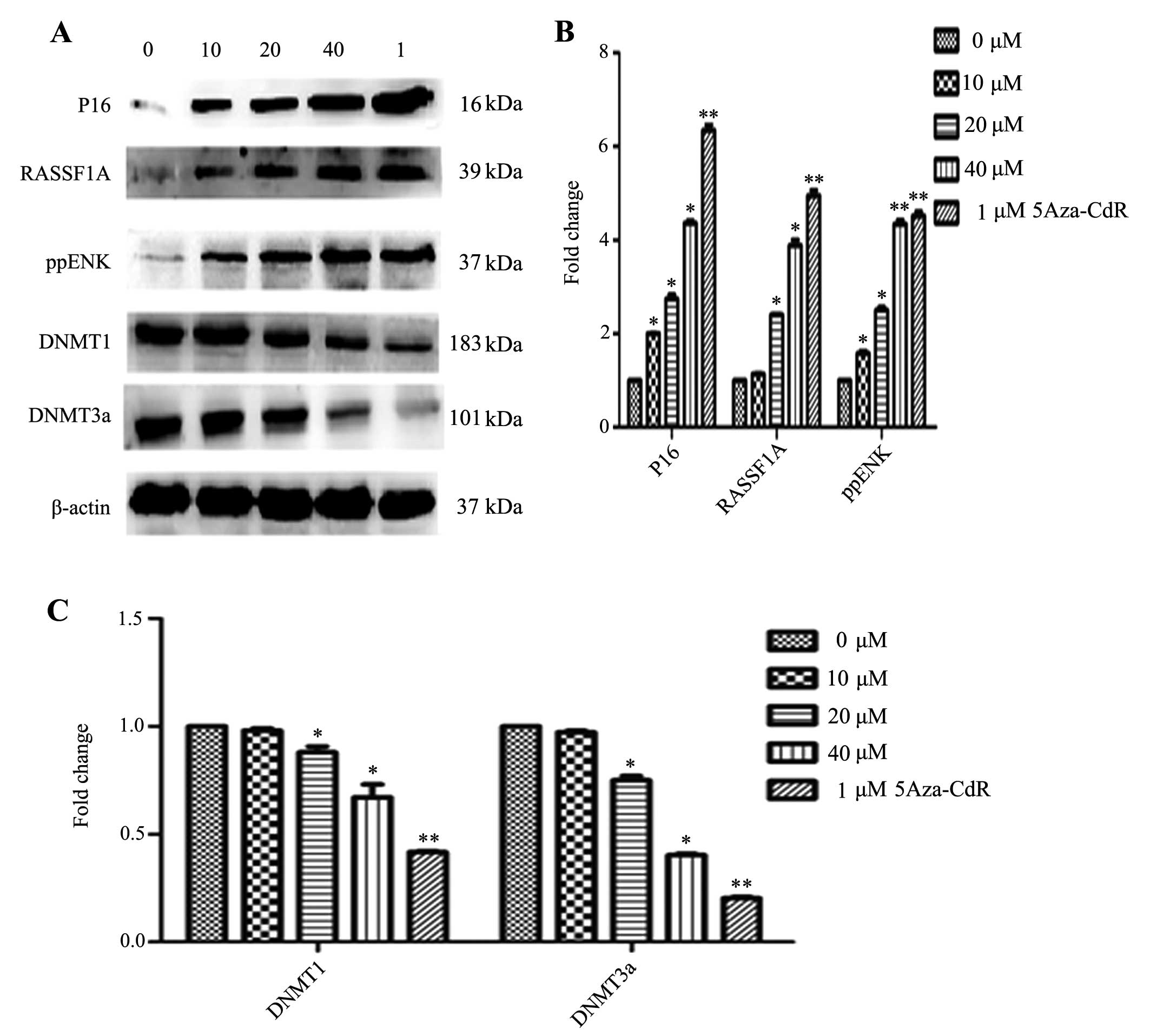
Effects of emodin on mRNA and protein
expression of P16, RASSF1A, ppENK and DNA methyltransferases
(DNMTs)
The levels of methylation of P16, RASSF1A and ppENK
in the promoter region were decreased in the PANC-1 cells. However,
whether the transcription and protein levels were affected in the
emodin-treated PANC-1 cells was unknown. Thus, we investigated the
mRNA and protein expression of P16, RASSF1A, ppENK and DNMTs. As
shown in Fig. 5, emodin increased
the mRNA expression levels of P16, RASSF1A and ppENK in a
dose-dependent manner. 5-Aza-CdR also increased the mRNA expression
levels of P16, RASSF1A and ppENK. The effect was stronger than that
of emodin. As shown in Fig. 6,
emodin increased the protein expression levels of P16, RASSF1A and
ppENK in a dose-dependent manner. 5-Aza-CdR also increased the
protein expression levels of P16, RASSF1A and ppENK. The effect was
stronger than that of emodin. In Figs.
5B and 6, while emodin
decreased the mRNA and protein expression of methyltransferase
DNMT1 and DNMT3a, it had no effect on DNMT3b (data not shown). The
results suggest that emodin may decrease overall genomic
methylation levels by the down-regulation of DNMT1 and DNMT3a at
the transcription and protein levels, thereby affecting the
transcriptional level of the whole genome, upregulating the mRNA
and protein levels of the tumor-suppressor genes, P16, RASSF1A and
ppENK, and consequently inhibiting tumor growth.
Discussion
The incidence of pancreatic cancer is increasing
yearly, and the fatality rate is high. According to reports in
2008, 37,680 new patients in the US were diagnosed with pancreatic
cancer and 34,290 of them (91%) succumbed to the disease (30). The pathogenesis of pancreatic cancer
remains unclear and discovering a treatment for this cancer has
been a challenge. Emodin has shown curative effects on pancreatic
cancer in the clinic. Emodin was found to trigger apoptosis in
cancer pancreatic cells (31), to
inhibit the formation of new vessels (29) and to improve the resistance to
gemcitabine in pancreatic cancer cells (30). However, the specific mechanisms of
these curative effects remain unclear. Our data showed that emodin
affected the whole genome expression by demethylation, which
especially decreased methylation levels of the tumor-suppressor
genes P16, RASSF1A and ppENK, thus playing an important role in
cancer treatment.
In the present study, the data indicated that
varying concentrations of emodin effectively inhibited the
proliferation of the PANC-1 cell line at different time points.
When PANC-1 cells were treated with 80 μM emodin for 72 h,
there were significant changes in the growth inhibition rate and
the cell morphology, which is consistent with previous literature
(29). Notably, when PANC-1 cells
were treated with 40 μM emodin for 72 h, a decrease in the
growth inhibition rate was noted, but no significant changes in the
cell morphology were noted. Therefore, emodin at concentrations of
0, 10, 20 and 40 μM were selected for the experiments. Our
results revealed that emodin caused various degrees of
demethylation of tumor-suppressor genes P16, RASSF1A and ppENK in
PANC-1 cells. Methylation is often the cause of tumor-suppressor
gene inactivation, and its expression was found to be inversely
proportional to the density of CpG island methylation. Low levels
of methylation caused a 67–90% inactivation of gene expression,
while methylation of high density CpG islands completely
extinguished gene expression (32).
PCR also confirmed that emodin caused the re-expression of P16,
RASSF1A and ppENK inactivated by methylation, and whose expression
levels increased in a dose- and time-dependent manner. The results
of the western blot analysis further confirmed our research, which
provides further evidence that emodin causes the demethylation of
tumor-suppressor genes in PANC-1 cells.
In order to further investigate the role of emodin
in the demethylation at the epigenetic level in PANC-1 cells,
5-Aza-CdR, which was proven to have demethylation effects in
clinical trials, was used in the present study. The results showed
that 5-Aza-CdR significantly reduced the methylation levels of the
tumor-suppressor genes P16, RASSF1A and ppENK by enhancing the
expression of mRNA and protein. Although emodin also plays a
demethylation role in PANC-1 cells, the effect was weaker than that
of 5-Aza-CdR. In vivo, the methylation was mainly catalyzed
by methyltransferases (DNMT1, DNMT3a and DNMT3b). There are two
ways to demethylate tumor-suppressor genes: inhibition of
methyltransferase activity and reduction in methyltransferase
expression. RT-PCR and western blot analysis showed that both 40
μM emodin and 1 μM 5-Aza-CdR significantly reduced
the expression of DNMT1 and DNMT3a, suggesting that demethylation
of emodin may be linked with the inhibition of methyltransferase
expression. Our study confirmed that emodin caused a certain degree
of demethylation in the tumor-suppressor genes P16, RASSF1A, ppENK
in the PANC-1 cells. However, it remains to be discovered whether
emodin plays a role in the demethylation of different suppressor
genes in other pancreatic cancer cell lines. In addition, whether
emodin enhances the demethylation of 5-Aza-CdR warrants further
research.
The methylation of DNA CpG islands (5mC) plays an
integral role in gene transcriptional silencing, genomic imprinting
and X chromosome inactivation. Currently, 5hmC is the main focus of
research. There are 10 to 11 translocation enzymes which catalyze
the oxidation of 5mC to produce 5hmC; the prominent translocation
enzymes include TET1, TET2 and TET3 (32). The specific function of 5hmC is not
very clear; 5hmC is considered to be an intermediate from 5mC to 5C
(33). The expression of 5hmC in
many tumor tissues was found to be significantly reduced compared
with that in normal tissues. According to a recent report (34), the expression of 5hmC in pancreatic,
liver, lung, prostate and breast cancer tissues was lower than that
in normal tissues, thus acting as a biological indicator for the
detection of tumors. In the present study, a dot-blot assay
indicated that 40 μM emodin and 1 μM 5-Aza-CdR
significantly reduced the 5mC expression, but showed no significant
change in the expression of 5hmC. Therefore, our study indicates
that emodin can reduce the generation of 5mC by inhibiting the
expression of DNMT. These results are consistent with the above
results from PCR and western blotting. However, as 5mC is catalyzed
by TET to generate 5hmC, emodin may have no effect on the
expression and activity of TET enzymes.
In summary, BSP demonstrated that emodin, to a
certain degree, affected the demethylation of tumor-suppressor
genes P16, RASSF1A, and ppENK in the pancreatic cancer cell line
PANC-1. RT-PCR and western blot results showed that emodin caused
the re-expression of P16, RASSF1A and ppENK which were previously
not expressed or weakly expressed in the PANC-1 cells. Emodin also
reduced the expression of DNMT1 and DNMT3a. The dot-blot results
confirmed that emodin reduced the expression of the 5mC genome,
likely by inhibiting the expression of DNMT. This discovery
provides a novel strategy for the treatment of pancreatic cancer.
Clinical treatment of emodin in pancreatic cancer occurs not only
through apoptosis or inhibition of angiogenesis but also through
demethylation in epigenetics.
Acknowledgments
We are grateful for the financial support from the
Jiaxing Science and Technology Projects (grant no. 2013AY21042-5)
and Jiaxing Science and Technology innovation team project (grant
no. 2013-03).
References
|
1
|
Reske SN: PET and PET-CT of malignant
tumors of the exocrine pancreas. Radiologe. 49:131–136. 2009.In
German. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hariharan D, Saied A and Kocher HM:
Analysis of mortality rates for pancreatic cancer across the world.
HPB Oxf. 10:58–62. 2008. View Article : Google Scholar
|
|
3
|
Guo X and Cui Z: Current diagnosis and
treatment of pancreatic cancer in China. Pancreas. 31:13–22. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cheng X and Blumenthal RM: Mammalian DNA
methyltransferases: a structural perspective. Structure.
16:341–350. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bird A: DNA methylation patterns and
epigenetic memory. Genes Dev. 16:6–21. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gambichler T, Sand M and Skrygan M: Loss
of 5-hydroxymeth-ylcytosine and ten-eleven translocation 2 protein
expression in malignant melanoma. Melanoma Res. 23:218–220. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matsuoka S, Edwards MC, Bai C, Parker S,
Zhang P, Baldini A, Harper JW and Elledge SJ: p57KIP2, a
structurally distinct member of the p21CIP1 Cdk
inhibitor family, is a candidate tumor suppressor gene. Genes Dev.
9:650–662. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sato N, Fukushima N, Maehara N,
Matsubayashi H, Koopmann J, Su GH, Hruban RH and Goggins M:
SPARC/osteonectin is a frequent target for aberrant methylation in
pancreatic adenocarcinoma and a mediator of tumor-stromal
interactions. Oncogene. 22:5021–5030. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Attri J, Srinivasan R, Majumdar S, Radotra
BD and Wig J: Alterations of tumor suppressor gene P16INK4a in
pancreatic ductal carcinoma. BMC Gastroenterol. 5:222005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dammann R, Schagdarsurengin U, Liu L, Otto
N, Gimm O, Dralle H, Boehm BO, Pfeifer GP and Hoang-Vu C: Frequent
RASSF1A promoter hypermethylation and K-ras mutations in pancreatic
carcinoma. Oncogene. 22:3806–3812. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ueki T, Toyota M, Skinner H, Walter KM,
Yeo CJ, Issa JP, Hruban RH and Goggins M: Identification and
characterization of differentially methylated CpG islands in
pancreatic carcinoma. Cancer Res. 61:8540–8546. 2001.PubMed/NCBI
|
|
12
|
Fukushima N, Sato N, Ueki T, Rosty C,
Walter KM, Wilentz RE, Yeo CJ, Hruban RH and Goggins M: Aberrant
methylation of preproenkephalin and P16 genes in pancreatic
intraepithelial neoplasia and pancreatic ductal adenocarcinoma. Am
J Pathol. 160:1573–1581. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schutte M, Hruban RH, Geradts J, Maynard
R, Hilgers W, Rabindran SK, Moskaluk CA, Hahn SA, Schwarte-Waldhoff
I, Schmiegel W, et al: Abrogation of the Rb/P16 tumor-suppressive
pathway in virtually all pancreatic carcinomas. Cancer Res.
57:3126–3130. 1997.PubMed/NCBI
|
|
14
|
Moore PS, Sipos B, Orlandini S, Sorio C,
Real FX, Lemoine NR, Gress T, Bassi C, Klöppel G, Kalthoff H, et
al: Genetic profile of 22 pancreatic carcinoma cell lines. Analysis
of K-ras, p53, P16 and DPC4/Smad4. Virchows Arch. 439:798–802.
2001. View Article : Google Scholar
|
|
15
|
Brueckner B, Kuck D and Lyko F: DNA
methyltransferase inhibitors for cancer therapy. Cancer J.
13:17–22. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Szyf M: DNA methylation and demethylation
as targets for anticancer therapy. Biochemistry (Mosc). 70:533–549.
2005. View Article : Google Scholar
|
|
17
|
Goffin J and Eisenhauer E: DNA
methyltransferase inhibitors - state of the art. Ann Oncol.
13:1699–1716. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lyko F and Brown R: DNA methyltransferase
inhibitors and the development of epigenetic cancer therapies. J
Natl Cancer Inst. 97:1498–1506. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xiang N, Zhao R, Song G and Zhong W:
Selenite reactivates silenced genes by modifying DNA methylation
and histones in prostate cancer cells. Carcinogenesis.
29:2175–2181. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brueckner B, Garcia Boy R, Siedlecki P,
Musch T, Kliem HC, Zielenkiewicz P, Suhai S, Wiessler M and Lyko F:
Epigenetic reactivation of tumor suppressor genes by a novel
small-molecule inhibitor of human DNA methyltransferases. Cancer
Res. 65:6305–6311. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cui X, Wakai T, Shirai Y, Yokoyama N,
Hatakeyama K and Hirano S: Arsenic trioxide inhibits DNA
methyltransferase and restores methylation-silenced genes in human
liver cancer cells. Hum Pathol. 37:298–311. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lin SZ, Chen KJ, Tong HF, Jing H, Li H and
Zheng SS: Emodin attenuates acute rejection of liver allografts by
inhibiting hepatocellular apoptosis and modulating the Th1/Th2
balance in rats. Clin Exp Pharmacol Physiol. 37:790–794.
2010.PubMed/NCBI
|
|
23
|
Li HL, Chen HL, Li H, Zhang KL, Chen XY,
Wang XW, Kong QY and Liu J: Regulatory effects of emodin on NF-κB
activation and inflammatory cytokine expression in RAW 264.7
macrophages. Int J Mol Med. 16:41–47. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li-Weber M: Targeting apoptosis pathways
in cancer by Chinese medicine. Cancer Lett. 332:304–312. 2013.
View Article : Google Scholar
|
|
25
|
Liu A, Chen H, Tong H, Ye S, Qiu M, Wang
Z, Tan W, Liu J and Lin S: Emodin potentiates the antitumor effects
of gemcitabine in pancreatic cancer cells via inhibition of nuclear
factor-κB. Mol Med Rep. 4:221–227. 2011.PubMed/NCBI
|
|
26
|
Liu Z, Xie Z, Jones W, Pavlovicz RE, Liu
S, Yu J, Li PK, Lin J, Fuchs JR, Marcucci G, et al: Curcumin is a
potent DNA hypomethylation agent. Bioorg Med Chem Lett. 19:706–709.
2009. View Article : Google Scholar
|
|
27
|
Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H,
Welsh W and Yang CS: Tea polyphenol (−)-epigallocatechin-3-gallate
inhibits DNA methyltransferase and reactivates methylation-silenced
genes in cancer cell lines. Cancer Res. 63:7563–7570.
2003.PubMed/NCBI
|
|
28
|
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim
SH, Ito S, Yang C, Wang P, Xiao MT, et al: Oncometabolite
2-hydroxyglutarate is a competitive inhibitor of
α-ketoglutarate-dependent dioxy-genases. Cancer Cell. 19:17–30.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lin SZ, Wei WT, Chen H, Chen KJ, Tong HF,
Wang ZH, Ni ZL, Liu HB, Guo HC and Liu DL: Antitumor activity of
emodin against pancreatic cancer depends on its dual role:
promotion of apoptosis and suppression of angiogenesis. PLoS One.
7:e421462012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu A, Luo J and Zhang JH: Emodin combined
gemcitabine inhibited the growth of pancreatic cancer in vitro and
in vivo and its mechanisms study. Zhongguo Zhong Xi Yi Jie He Za
Zhi. 32:652–656. 2012.In Chinese. PubMed/NCBI
|
|
31
|
Chen H, Wei W, Guo Y, Liu A, Tong H, Wang
Z, Tan W, Liu J and Lin S: Enhanced effect of gemcitabine by emodin
against pancreatic cancer in vivo via cytochrome C-regulated
apoptosis. Oncol Rep. 25:1253–1261. 2011.PubMed/NCBI
|
|
32
|
Tahiliani M, Koh KP, Shen Y, Pastor WA,
Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et
al: Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in
mammalian DNA by MLL partner TET1. Science. 324:930–935. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu SC and Zhang Y: Active DNA
demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol.
11:607–620. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang H, Liu Y, Bai F, Zhang JY, Ma SH, Liu
J, Xu ZD, Zhu HG, Ling ZQ, Ye D, et al: Tumor development is
associated with decrease of TET gene expression and
5-methylcytosine hydroxylation. Oncogene. 32:663–669. 2013.
View Article : Google Scholar
|