Introduction
AMP-activated protein kinase (AMPK) is a pleiotropic
enzyme playing a role in the control of numerous functions both
centrally and peripherally (1).
AMPK has now emerged as a potential therapeutic target for cancer
as a consequence of being a substrate and a mediator of the tumour
suppressor gene LKB1 (2). The
tumour suppressing effects associated with AMPK activation include
the ability of the enzyme to repress the lipogenic enzymes, fatty
acid synthase and acetyl coA carboxylase (both enzymes are
upregulated in cancer cells), to inhibit Akt phosphorylation and to
phosphorylate p53 and p21, thereby triggering apoptotic and cell
cycle arresting signals. Protein kinase B (PKB), also known as Akt,
is a serine threonine kinase which belongs to the same family of
protein kinases A, G and C (AGC family). Three human isoforms have
been identified (PKBα or Akt1, PKBβ/Akt2 and PKBγ or Akt3). Akt
influences several biological functions including metabolism,
growth and proliferation by phosphorylating and modulating the
activity of numerous cellular substrates, such as glycogen synthase
kinase 3, the mammalian target of rapamycin (mTOR), breast cancer
susceptibility gene 1 (BRCA1) and others (3). Understanding the molecular mechanism
of action and regulation of this enzyme is crucial as a result of
its aberrant regulation in life-threatening disorders, such as
cancer (4). Akt is activated
downstream of phosphatidyl inositol 3-kinase (PI3K), which is
activated upon interaction of growth factors with their receptors
(5,6). Akt isoform specific upregulation in
various types of human cancers has been reported. Increased
expression of AKT1 was reported in gastric cancers (7), while AKT2 has been reported to be
amplified in ovarian carcinomas (8)
and cancers of pancreas (9) and
AKT3 in cancerous melanocytes (10). Akt1 has been reportedly activated in
breast cancer (11), Akt2 in
hepatic (12) and colorectal
(13) cancers and finally, Akt3 is
upregulated in ER-negative breast cancers (14). The involvement of both AMPK and Akt
in metabolic and survival control suggests that, in cancer, there
is crosstalk between Akt and AMPK. This study has been undertaken
to investigate the effect of AMPK activation on Akt,
phosphorylation and Akt inhibition on AMPK phosphorylation and
proliferation in three human breast cancer cell lines each with
differing genetic backgrounds. The adipose tissue-related hormone,
leptin, is encoded by the Ob (obese) gene (7q31). Leptin is a
pleiotropic hormone having numerous peripheral and central effects
(15,16). Leptin is an angiogenic, mitogenic
and survival factor and is also an important metabolic regulator
(17). Leptin binds to OB-R
receptor and activates the JAK/STAT signalling pathway, which in
turn activates MAPK and PI3K/Akt signalling pathways. So in
addition, the effect of leptin on AMPK phosphorylation and on cell
proliferation was also investigated.
Materials and methods
5-aminoimidazole carboxamide ribonucleotide
(AICAR) was from Sigma-Aldrich, Poole, Dorset, UK. InSolution™ Akt
Inhibitor VIII (Isozyme-Selective, Akt-1/2), was purchased from
Calbiochem®, Watford, UK. Dulbecco's minimal essential
medium (DMEM) was obtained from Gibco BRL, Paisley, UK and fetal
calf serum (FCS) was from Autogenbioclean, UK Ltd.
Phosphate-buffered saline and trypsin/EDTA were obtained from
Sigma-Aldrich, Irvine, Ayrshire, UK. CellTiter 96®
Aqueous One Solution cell proliferation assay was purchased from
Promega, Madison, WI, USA. T25 and T75 tissue culture flasks were
from Nunc™, Denmark and tissue culture sterile 6-, 24- and 96-well
plates were from Costar®, USA. Anti-phospho-AMPK
(Thr172) and anti-phospho-Akt (Ser473) antibodies were purchased
from Cell Signaling Technology; Danvers, MA, USA. All blotting
reagents were purchased from Bio-Rad® (CA, USA and
München, Germany) except TBST and transfer buffer, which were
prepared from their components. Leptin, human, recombinant
expressed in E. coli (#L4146) was purchased from
Sigma-Aldrich, GmbH Germany.
Breast cancer cell lines MCF-7 (p53+ and
ER+), T47D (p53 mutant and ER+) and
MDA-MB-231 (p53 mutant and ER−) were purchased from the
European Collection of Cell Cultures (ECACC, Porton Down, UK).
Cell culture
Breast cancer cell lines were grown in a continuous
monolayer culture in T75 top filtered sterile tissue culture flasks
inside a sterile humidified incubator at 37°C and 5% CO2
in air. Cells were then sub-cultured as necessary for maintenance.
Cells were used between passages 57–70, 9–22 and 12–24 for MCF-7,
MDA-MB-231 and T47D cells respectively.
Western blotting
Cell lines were sub-cultured, counted and plated at
1×106 cells in appropriate tissue culture plates and
incubated at 37°C and 5% CO2 in a humidified atmosphere
overnight. Cells were then incubated with AICAR (0.83 mM) or with
corresponding vehicle for 3 or 24 h. Equivalent sets of cells were
incubated and subjected to the same procedure with Akt inhibitor
VIII in the presence or absence of AICAR for 1 h with corresponding
controls. This was to investigate the effect of Akt inhibition on
AMPK phosphorylation. To study the effect of leptin on AMPK
phosphorylation, cells were incubated with AICAR ± leptin and
leptin alone. Cell lysates from control and treated cells were
collected, followed by protein assay. Equal protein concentrations
(w/v) of control and stimulated cells were premixed with Laemmli
sample buffer (Bio-Rad) containing β-mercaptoethanol and loaded in
a maximum volume of 30 µl/well into wells in pre-formed
Tris-HCl-SDS gels (Bio-Rad). Proteins were then separated by
electrophoresis and then transferred to a PDF membrane. The
membrane was blocked overnight in 10% dry milk before being
incubated overnight at 4°C with anti-phospho-Akt or
anti-phospho-AMPK antibody, directed against p-Ser473 or p-Thr172
amino acid residues, respectively. The membrane was washed in TBST
four times (15 min each) then incubated with polyclonal anti-rabbit
IgG antibody then washed again four times before adding the
horseradish peroxidase (HRP)-linked substrate and generating
signals that were detected on X-ray film. Band densitometry was
performed (as an equivalent percentage) using ImageJ software.
Statistical analysis was investigated by Graphpad Prism.
Cell proliferation
Cells were sub-cultured, counted and plated at
7×103 cells/well in sterile 96-well plates and incubated
overnight at 37°C and 5% CO2 in air. The next day,
serial concentrations of AICAR (0-16.65 µM), Akt inhibitor
VIII alone (0.00–4.16 µM) or in combination with AICAR
(0.415 mM), or leptin (0–300 ng/ml) were added. The number of
viable cells was then estimated after 72 h using the MTS method
(Aqueous One assay kit, Promega), which assesses the ability of
viable cells to reduce the tetrazolium salt,
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS), producing brownish red coloured formazan, which was measured
at 490 and 630 nm using a Biotek ELX800 Microplate reader (Biotek
Instruments, Winooski, VT, USA). Cell proliferation was then
calculated as percentages of control and plotted as negative dose
response curves using Graphpad Prism and statistical analysis by
ANOVA.
Results
We have reported previously on the significant
effects of AICAR on AMPK in all three cell types (18), so to simplify presentation, we do
not present statistical data on this effect alone.
Effects of AMPK activation on Akt Ser473
phosphorylation
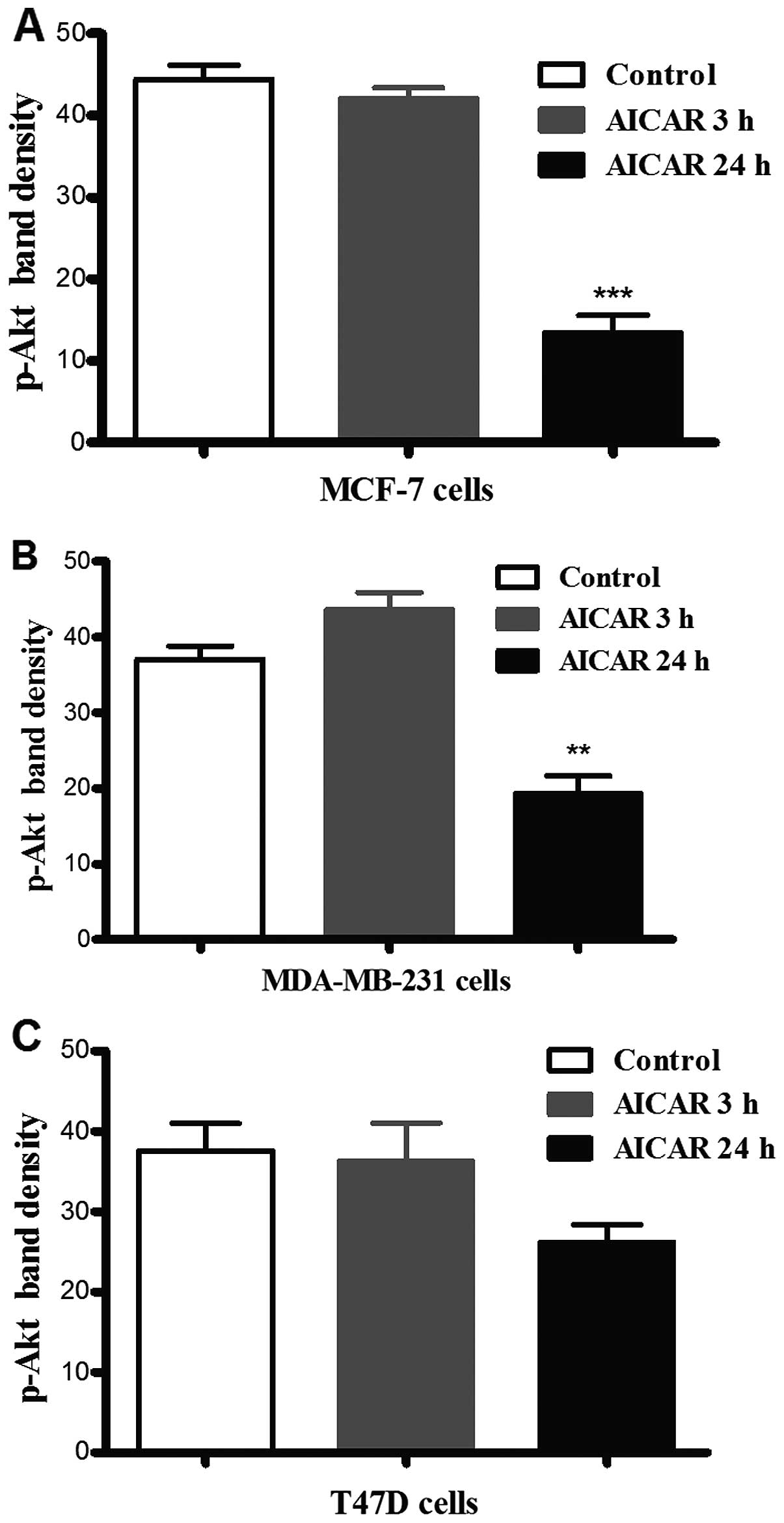
Incubation of each of the cell lines with AICAR for
3 or 24 h resulted in a significant reduction in p-Akt (Ser473)
levels in MCF-7 and MDA-MB-231 cells after 24 h as compared to the
corresponding controls (there was no observable difference after 3
h). In T47D cells, however, there was no significant difference in
Akt phosphorylation in response to activation of AMPK at either
time-point (Fig. 1).
Effects of inhibition of Akt on AMPK
phosphorylation
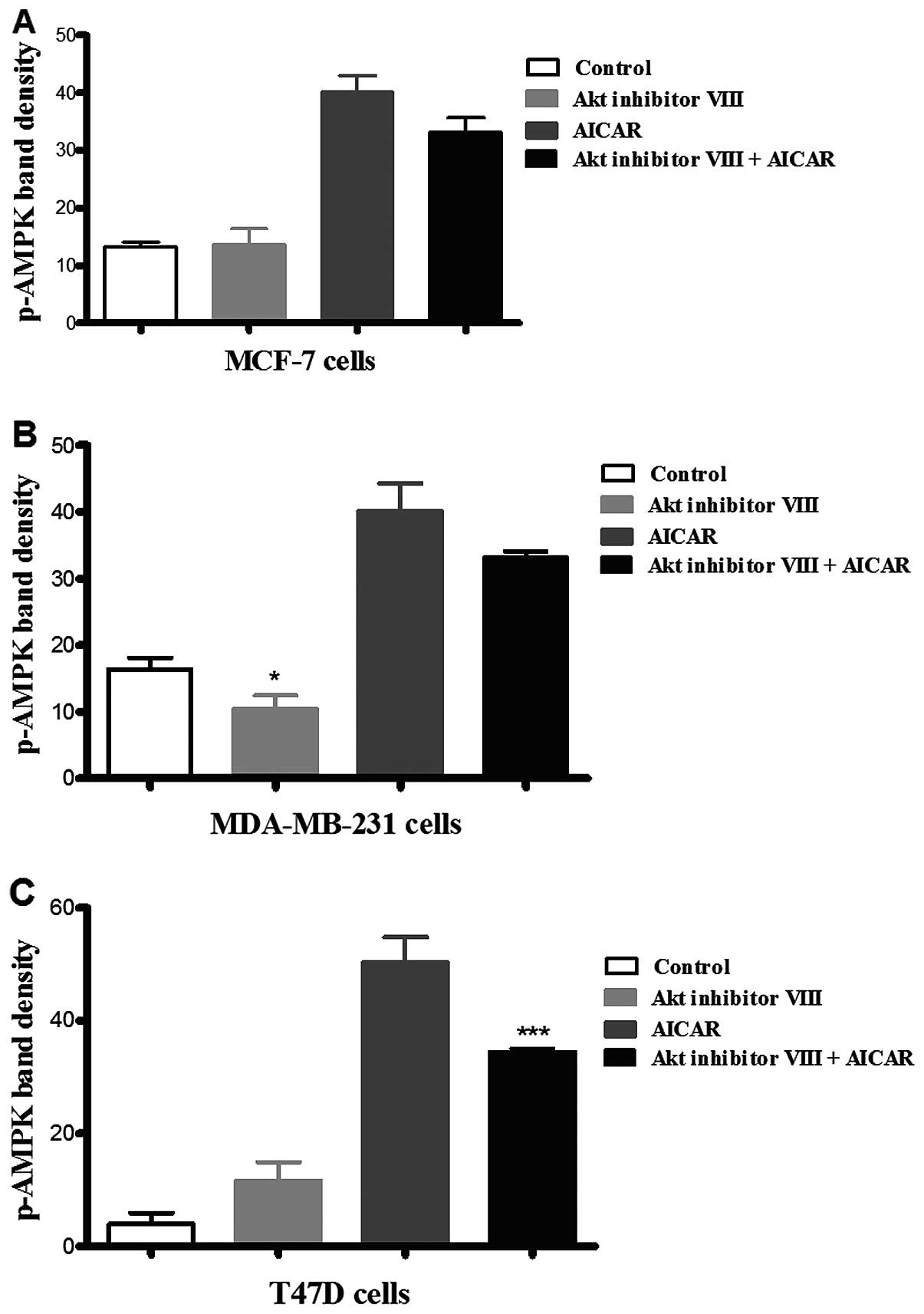
Incubation of the three cell types with Akt
inhibitor VIII revealed that it did not have a significant effect
on AMPK phosphorylation in MCF-7 and T47D cells in comparison with
the corresponding control (Fig. 2).
In contrast, in MDA-MB-231 cells, treatment with Akt inhibitor VIII
resulted in an apparent decrease in AMPK phosphorylation when
compared to the corresponding control of medium alone. In the
presence of AICAR, inhibition of Akt resulted in a significant
reduction in AMPK phosphorylation in T47D cells, but did not
suppress the phosphorylating effect of AICAR on AMPK in the other
cell lines investigated. These results suggested that Akt
inhibition increased AMPK phosphorylation in T47D cells, but not
the other two cell types (Fig.
2).
Cell proliferation
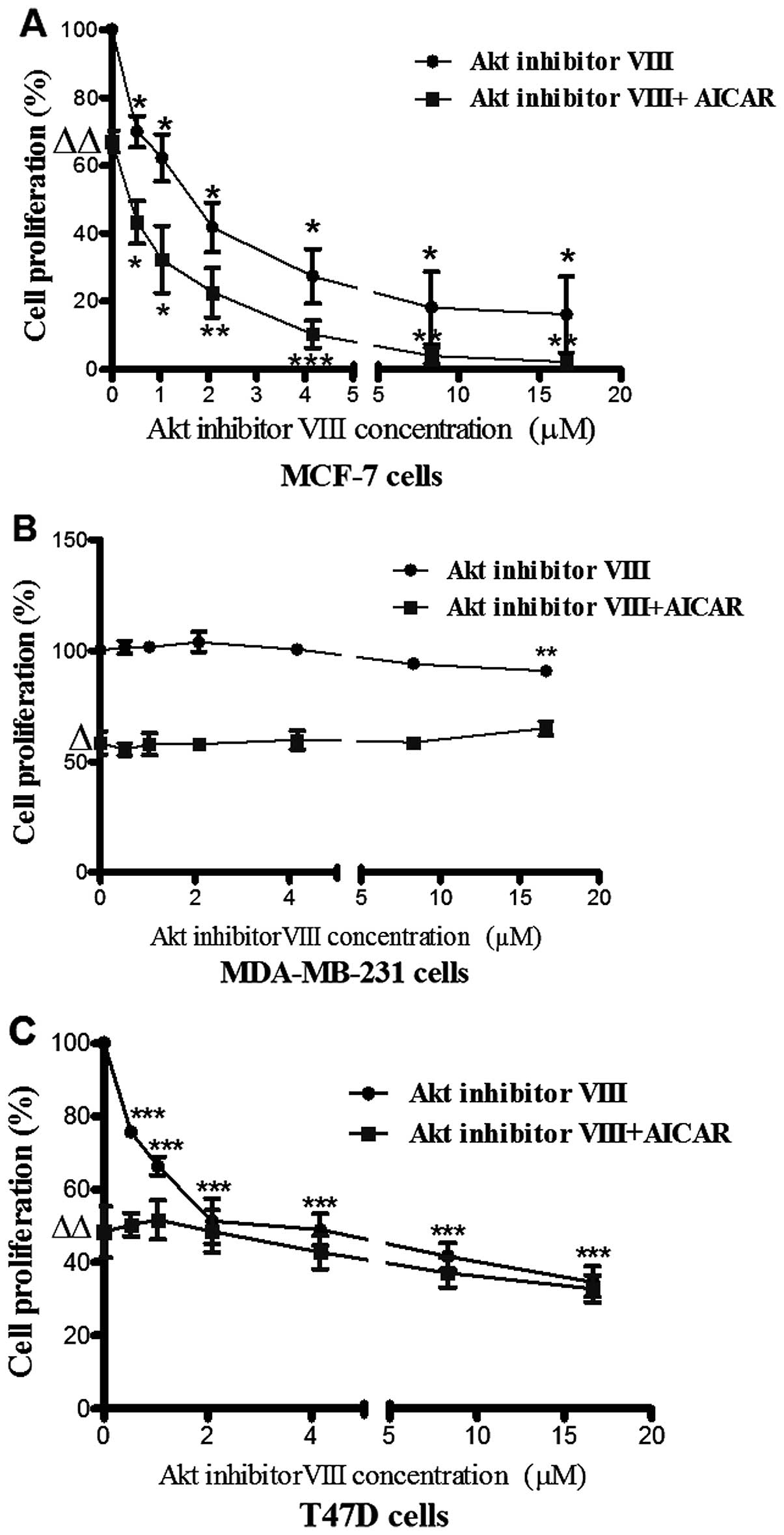
Akt inhibition resulted in differential
anti-proliferative effects in the 3 breast cancer cell types.
a) MCF-7 cells
The activating effect of AICAR on AMPK was
previously reported by the present group to be associated with
inhibition of breast cancer cell proliferation (18). In the present study, Akt inhibitor
VIII (0–16.65 µM) caused a dose-dependent reduction in MCF-7
cell proliferation. Co-administration of AICAR (0.415 mM) and Akt
inhibitor VIII indicated an additive effect in this cell line
(Fig. 3A).
b) MDA-MB-231 cells
The MDA-MB-231 cell line was not responsive to Akt
inhibitor VIII and on combination with AICAR there was no change in
the anti-proliferative action of AICAR suggesting that MDA-MB-231
cells might express an Akt isoform which is less responsive to Akt
inhibitor VIII (Fig. 3B).
c) T47D cells
Although incubation of T47D cells with increasing
concentrations of Akt inhibitor VIII resulted in a dose responsive
inhibition of cell proliferation, co-administration of AICAR (0.415
mM) with Akt inhibitor VIII appeared to have no influence on cell
proliferation (Fig. 3C).
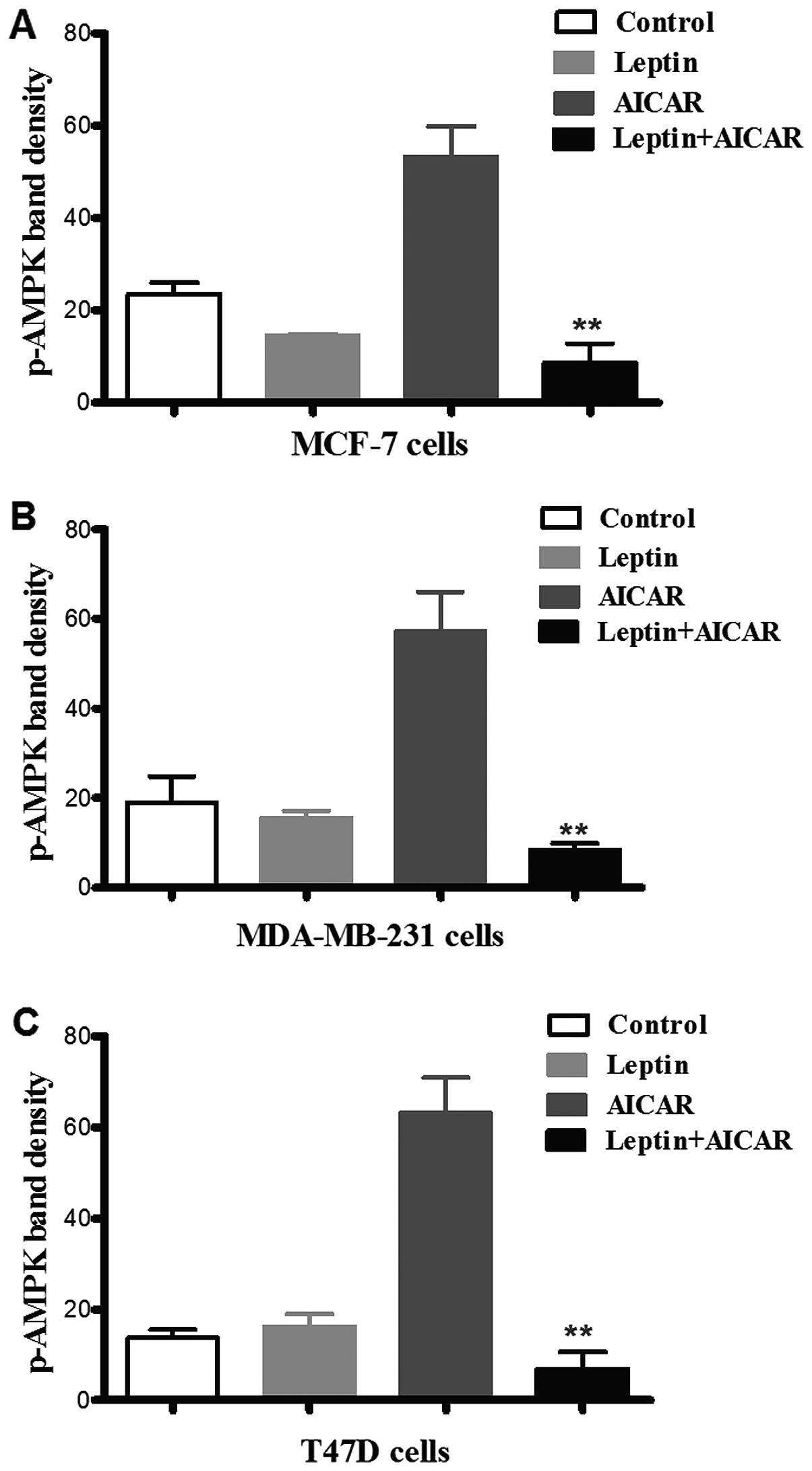
Leptin suppressed activated AMPK
phosphorylation and cell proliferation
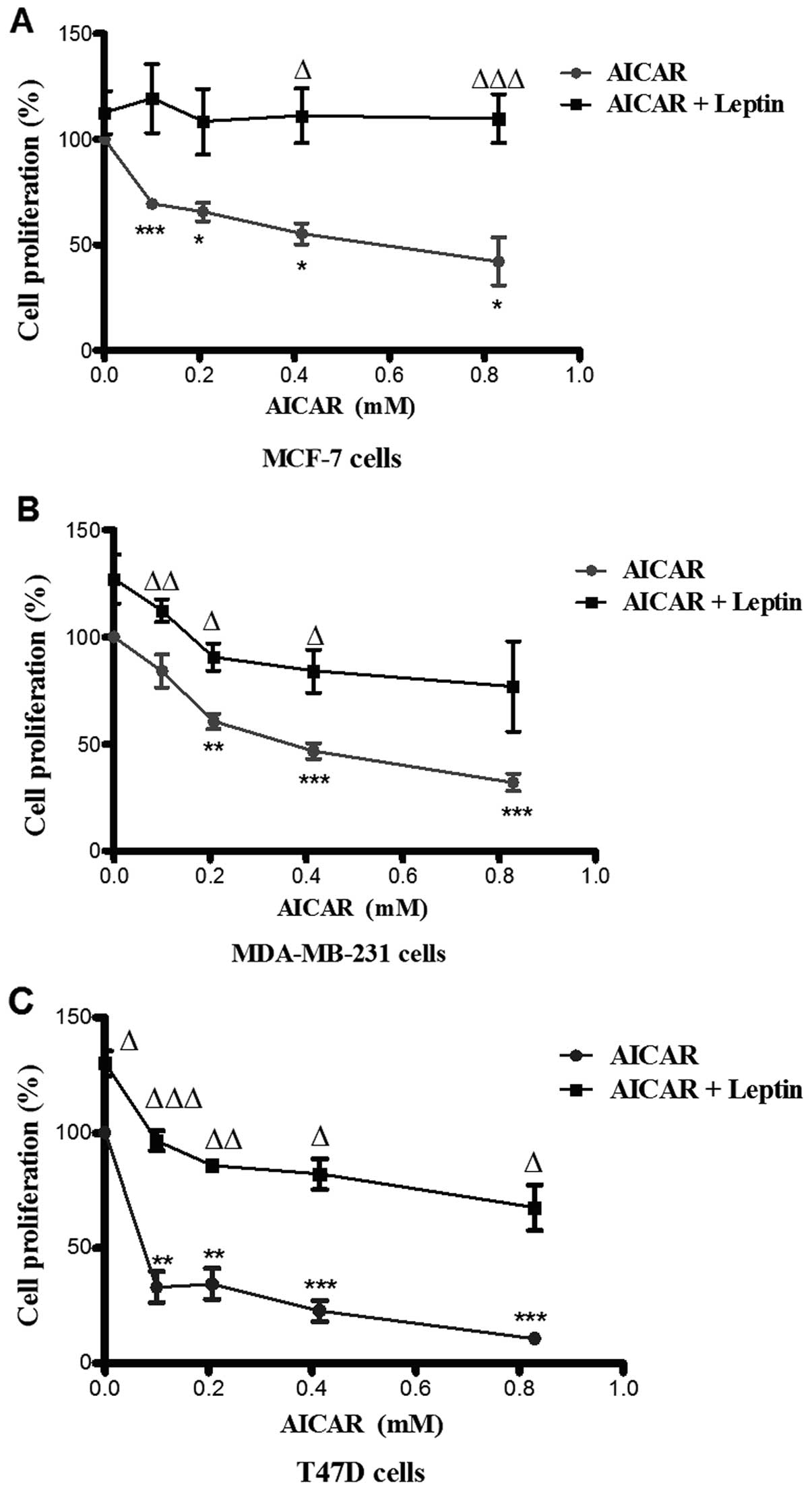
AICAR (0.83 mM)-induced AMPK phosphorylation was
suppressed by co-administration with leptin in all three cell
types. There was no evidence of a significant effect of leptin
alone (Fig. 4). Leptin also
differentially attenuated breast cancer cell proliferation. Indeed,
the effects of leptin were also apparent at lower doses of AICAR.
In MCF-7 cells, leptin abolished the effect of AMPK activation.
However, in the other two cell types, this blockade was partial,
but still significant (Fig. 5).
Discussion
Both the PI3K/Akt/mTOR and LKB/AMPK signalling
pathways have been implicated in cancer (19). We have previously reported that
activation of AMPK has differential effects on cell proliferation
in MCF-7, MDA-MB-231 and T47D cells (18). The present study reports an
investigation into the potential crosstalk between these pathways
in breast cancer cell lines with differing p53 and estrogen
receptor status. Activation of AMPK by AICAR exerted differential
inhibitory effects on Akt phosphorylation in the three cell types
investigated (Fig. 1). There was a
clear inhibitory effect in MCF-7 and MDA-MB-231 cells, which was
not significant in T47D cells. The inhibitory effect of AICAR on
PI3K pathway components has also been reported in C6 glioma cells;
a reduction in Akt phosphorylation observed both in vivo and
in vitro (20,21). It has also been reported that AICAR
inhibited the phosphorylation of mTOR at serine 1448, a crucial
event in mTOR activation (22). In
contrast, in the present study, inhibition of Akt had only minimal
or no significant inhibitory effects on AMPK phosphorylation in all
three cell types.
There were clear differences between the cell types
in response to co-administration of AICAR and Akt inhibitor VIII
when cell proliferation was assessed. In MCF-7 cells, the
co-administration resulted in an apparent additive inhibitory
effect on cell proliferation (Fig.
3A). In MDA-MB-231 cells, however, Akt inhibitor VIII failed to
produce a noticeable effect on cell proliferation (Fig. 3B) perhaps as a result of
overexpression of Akt3 (the least responsive isoform to Akt
inhibitor VIII) in ER-negative breast cancer cells (14). In T47D cells, simultaneous
activation of AMPK by AICAR and inhibition of Akt by Akt inhibitor
VIII did not produce a significant decrease in cell proliferation
when compared to the effect of Akt inhibitor VIII alone (Fig. 3C). The absence of significant
differences between the effects of Akt inhibitor VIII combined with
AICAR might suggest that any potential additive effect of the drug
combination is attenuated. This could be related to the reduction
in AICAR-mediated AMPK phosphorylation upon treating cells with Akt
inhibitor VIII and AICAR together (Fig.
2C).
In line with our results, another report showed that
metformin-activated AMPK in MCF-7 cells resulted in
de-phosphorylation of Akt, an action that helped to improve the
sensitivity to anti-HER2 receptor drugs and decrease the risk of
cardiomyopathy (23). It was also
reported that AMPK activation downregulates the mTOR pathway in
breast cancer following its activation by metformin (24).
There is growing literature linking obesity to
cancer incidence (25). In this
context, it was reported that leptin switches the metabolic profile
of breast cancer cells from fatty acid β oxidation to the aerobic
glycolytic pathway (26). This
effect was found to be accompanied by Akt activation and
downregulation of the LKB1 gene with a subsequent decrease in AMPK
phosphorylation (26). In the
present study, treatment of MCF-7, MDA-MB-231 and T47D cells with
leptin resulted in blockade of the phosphorylating effect of AICAR
on AMPK (Fig. 4). This was
associated with attenuation of the inhibitory effect of AMPK
activation on cell proliferation in MDA-MB-231 and T47D cells and
complete blockade in MCF-7 cells. In this regard, it has been
reported that leptin stimulated Akt phosphorylation at Ser473
(27,28) and induced PI3K-regulted PKC-α gene
expression suggesting that leptin activates the PI3K pathway
(29). A 6-fold reduction in
apoptosis was observed in MCF-7 cells treated with leptin for
>24 h (30). This was in line
with the ability of leptin to reduce the levels of p53 and Bax
(31). These effects of leptin on
MCF-7 cells support the concept established in the present study
that leptin could oppose the antitumour effects of P-AMPK not only
in this cell line but also to some extent in the other two. It has
also been reported that leptin receptor is expressed on mammary
cancer stem cells suggesting that leptin may act as an initiation
factor for the primary mammary tumour which, once established,
proliferates, migrates and invades local tissues also with the aid
of leptin. Moreover, leptin could sustain angiogenesis by acting on
endothelial cells to enhance the pro-inflammatory processes that
are required to support tumour growth by exerting its effects on
other stromal cells, such as immune cells and fibroblasts (32).
This study suggests that crosstalk between Akt and
AMPK in breast cancer cells is dependent on cellular genetics.
Consequently further investigation is required before deciding on
the use of particular Akt inhibitors with or without an activator
of AMPK. Therefore, in the context of therapeutics, administration
of an Akt inhibitor with AMPK activators may be effective in some,
but not all, breast cancers depending on cellular characteristics.
These findings support indications that genetic profiling could be
very important in defining the most effective treatment regimes
across a range of therapeutic targets. Indeed, it is clear that
administration of a drug targeting an intracellular process has to
take account of crosstalking pathways that might interfere with
treatment effectiveness. Of particular importance, from the present
study, is that attempts to employ any new generation AMPK
activators in the treatment of breast cancer could be less
effective in patients with high circulating levels of leptin, again
dependent on the genetic background of the tumour.
Acknowledgments
We wish to thank The Egyptian Cultural Agency for
part funding. We also thank Professor David Hornby for his
generosity in accommodating part of this project in the Department
of Molecular Biology and Biotechnology. We are grateful to Drs
Janice Royds and Stuart Johnson for useful discussions.
References
|
1
|
Hardie DG: The AMP-activated protein
kinase pathway - new players upstream and downstream. J Cell Sci.
117:5479–5487. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lizcano JM, Göransson O, Toth R, Deak M,
Morrice NA, Boudeau J, Hawley SA, Udd L, Mäkelä TP, Hardie DG, et
al: LKB1 is a master kinase that activates 13 kinases of the AMPK
subfamily, including MARK/PAR-1. EMBO J. 23:833–843. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lawlor MA and Alessi DR: PKB/Akt: A key
mediator of cell proliferation, survival and insulin responses? J
Cell Sci. 114:2903–2910. 2001.PubMed/NCBI
|
|
4
|
Calleja V, Laguerre M, Parker PJ and
Larijani B: Role of a novel PH-kinase domain interface in PKB/Akt
regulation: structural mechanism for allosteric inhibition. PLoS
Biol. 7:e172009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Calleja V, Alcor D, Laguerre M, Park J,
Vojnovic B, Hemmings BA, Downward J, Parker PJ and Larijani B:
Intramolecular and intermolecular interactions of protein kinase B
define its activation in vivo. PLoS Biol. 5:e952007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Calleja V, Laguerre M and Larijani B: 3-D
structure and dynamics of protein kinase B-new mechanism for the
allosteric regulation of an AGC kinase. J Chem Biol. 2:11–25. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Staal SP: Molecular cloning of the akt
oncogene and its human homologues AKT1 and AKT2: Amplification of
AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci
USA. 84:5034–5037. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cheng JQ, Godwin AK, Bellacosa A, Taguchi
T, Franke TF, Hamilton TC, Tsichlis PN and Testa JR: AKT2, a
putative oncogene encoding a member of a subfamily of
protein-serine/threonine kinases, is amplified in human ovarian
carcinomas. Proc Natl Acad Sci USA. 89:9267–9271. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cheng JQ, Ruggeri B, Klein WM, Sonoda G,
Altomare DA, Watson DK and Testa JR: Amplification of AKT2 in human
pancreatic cells and inhibition of AKT2 expression and
tumorigenicity by antisense RNA. Proc Natl Acad Sci USA.
93:3636–3641. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stahl JM, Sharma A, Cheung M, Zimmerman M,
Cheng JQ, Bosenberg MW, Kester M, Sandirasegarane L and Robertson
GP: Deregulated Akt3 activity promotes development of malignant
melanoma. Cancer Res. 64:7002–7010. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stål O, Pérez-Tenorio G, Akerberg L,
Olsson B, Nordenskjöld B, Skoog L and Rutqvist LE: Akt kinases in
breast cancer and the results of adjuvant therapy. Breast Cancer
Res. 5:R37–R44. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu X, Sakon M, Nagano H, Hiraoka N,
Yamamoto H, Hayashi N, Dono K, Nakamori S, Umeshita K, Ito Y, et
al: Akt2 expression correlates with prognosis of human
hepatocellular carcinoma. Oncol Rep. 11:25–32. 2004.
|
|
13
|
Roy HK, Olusola Bf, Clemens DL, Karolski
WJ, Ratashak A, Lynch HT and Smyrk TC: AKT proto-oncogene
overexpression is an early event during sporadic colon
carcinogenesis. Carcinogenesis. 23:201–205. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nakatani K, Thompson DA, Barthel A, Sakaue
H, Liu W, Weigel RJ and Roth RA: Up-regulation of Akt3 in estrogen
receptor-deficient breast cancers and androgen-independent prostate
cancer lines. J Biol Chem. 274:21528–21532. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Houseknecht KL and Spurlock ME: Leptin
regulation of lipid homeostasis: dietary and metabolic
implications. Nutr Res Rev. 16:83–96. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Myers MG Jr: Leptin receptor signaling and
the regulation of mammalian physiology. Recent Prog Horm Res.
59:287–304. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cirillo D, Rachiglio AM, la Montagna R,
Giordano A and Normanno N: Leptin signaling in breast cancer: An
overview. J Cell Biochem. 105:956–964. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
El-Masry OS, Brown BL and Dobson PR:
Effects of activation of AMPK on human breast cancer cell lines
with different genetic backgrounds. Oncol Lett. 3:224–228.
2012.PubMed/NCBI
|
|
19
|
Hardie DG and Alessi DR: LKB1 and AMPK and
the cancer-metabolism link - ten years after. BMC Biol. 11(36)2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brazil DP, Yang ZZ and Hemmings BA:
Advances in protein kinase B signalling: AKTion on multiple fronts.
Trends Biochem Sci. 29:233–242. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rattan R, Giri S, Singh AK and Singh I:
5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits
cancer cell proliferation in vitro and in vivo via AMP-activated
protein kinase. J Biol Chem. 280:39582–39593. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kimball SR: Interaction between the
AMP-activated protein kinase and mTOR signaling pathways. Med Sci
Sports Exerc. 38:1958–1964. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Vazquez-Martin A, Oliveras-Ferraros C, del
Barco S, Martin-Castillo B and Menendez JA: The antidiabetic drug
metformin: a pharmaceutical AMPK activator to overcome breast
cancer resistance to HER2 inhibitors while decreasing risk of
cardiomyopathy. Ann Oncol. 20:592–595. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hadad SM, Fleming S and Thompson AM:
Targeting AMPK: a new therapeutic opportunity in breast cancer.
Crit Rev Oncol Hematol. 67:1–7. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sundaram S, Johnson AR and Makowski L:
Obesity, metabolism and the microenvironment: Links to cancer. J
Carcinog. 12(19)2013.PubMed/NCBI
|
|
26
|
Ando S and Catalano S: The multifactorial
role of leptin in driving the breast cancer microenvironment. Nat
Rev Endocrinol. 8:263–275. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Frankenberry KA, Skinner H, Somasundar P,
McFadden DW and Vona-Davis LC: Leptin receptor expression and cell
signaling in breast cancer. Int J Oncol. 28:985–993.
2006.PubMed/NCBI
|
|
28
|
Garofalo C and Surmacz E: Leptin and
cancer. J Cell Physiol. 207:12–22. 2006. View Article : Google Scholar
|
|
29
|
Okumura M, Yamamoto M, Sakuma H, Kojima T,
Maruyama T, Jamali M, Cooper DR and Yasuda K: Leptin and high
glucose stimulate cell proliferation in MCF-7 human breast cancer
cells: reciprocal involvement of PKC-alpha and PPAR expression.
Biochim Biophys Acta. 1592:107–116. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Perera CN, Spalding HS, Mohammed SI and
Camarillo IG: Identification of proteins secreted from leptin
stimulated MCF-7 breast cancer cells: A dual proteomic approach.
Exp Biol Med (Maywood). 233:708–720. 2008. View Article : Google Scholar
|
|
31
|
Nkhata KJ, Ray A, Schuster TF, Grossmann
ME and Cleary MP: Effects of adiponectin and leptin co-treatment on
human breast cancer cell growth. Oncol Rep. 21:1611–1619.
2009.PubMed/NCBI
|
|
32
|
Park J and Scherer PE: Leptin and cancer:
from cancer stem cells to metastasis. Endocr Relat Cancer.
18:C25–C29. 2011. View Article : Google Scholar : PubMed/NCBI
|