Introduction
Acute myeloid leukemia (AML) is defined by an
increase in undifferentiated myeloid cells in bone marrow (BM) with
abnormal genetic changes, resulting in hematopoietic insufficiency
(1). The cells of most leukemias,
including AML, interact with the BM microenvironment (BMM), which
influences their survival (2). In
particular, leukemic blasts after chemotherapy are protected by
factors in tumor environments and this may result in a relapse in
leukemia. Because alterations in the BMM promote retention and
survival of leukemic cells, targeting this niche is regarded as an
advanced strategy to eradicate drug-resistant leukemic blasts
(3,4). The interaction between hematopoietic
cells and the BMM through various factors is critical for
regulation of cell homing, proliferation, and differentiation.
Several studies have reported that BM stromal cells protect
leukemic cells from chemotherapy-induced apoptosis (5,6). Among
many factors in the BMM, immune cells participate in crosstalk with
hematopoietic stem cells (HSC) (7).
The chemokine CXC motif ligand 12 (CXCL12), also known as
stromal-derived factor-1 (SDF-1), is a strong attractant that
recruits CXC receptor 4 (CXCR4)-expressing hematopoietic cells to
BM. The interaction of SDF-1/CXCR4 plays pivotal roles in the
cross-interaction between blasts and the BMM to prevent retention
and mobilization of leukemic cells (8), as well as in normal hematopoiesis
including the development of immune cells (9,10).
Plerixafor (Mozobil™) rapidly induces mobilization of hematopoietic
stem cells from BM into peripheral circulation by blocking
SDF-1/CXCR4 (11,12). Because most primary AML retains a
dependency on the BMM, plerixafor can function as a
microenvironmental factor in leukemia, decreasing the resistance of
leukemic cells by modulating the BMM and directly suppressing
malignancies through modulating the expression of CXCR4 (13,14).
The correlation between upregulation of CXCR4 after chemotherapy
and poor outcomes has been shown by Sison et al (15). In addition, the inhibition of
SDF-1/CXCR4 has been shown to enhance the sensitivity of leukemic
cells to chemotherapeutic agents, as well as partly abolish their
protection by the BMM (13,16). Based on these studies, we examined
whether CXCR4 inhibition by plerixafor could activate the killing
function of immune cells, resulting in the suppression of blasts
in vivo. To answer this question, an established syngeneic
leukemic mouse model using C1498 cells (a murine myelogenous
leukemia cell line) was used (17).
We found that plerixafor did not eradicate the leukemic blasts
in vitro; however, it made leukemic blasts more sensitive to
cytotoxic chemotherapy with cytosine arabinoside (Ara-C) in
vivo, suggesting biological effects on the microenvironment
through immune cell activation. Furthermore, we report an
additional role for CXCR4 inhibition in tumors as a stimulator of
immune cells, except migration capacity. The present study shows
that CXCR4 inhibition induces the suppression of AML blasts with
chemotherapy by the upregulation of cytokines to kill the cancerous
cells. Further, it provides some clues to develop therapeutic
strategies involving immune cell activation in the leukemic
microenvironment.
Materials and methods
Human primary cells and cell lines
All experiments were performed with authorization
from the Institutional Review Board for Human Research at the
Catholic University of Korea. AML blood samples were obtained from
the Catholic Blood and Marrow Transplantation Center at Seoul St.
Mary's Hospital. A total of 19 AML samples were prospectively
collected and examined. Mononuclear cells were separated from
leukapheresed peripheral blood (LPB) by density gradient
centrifugation using Ficoll Paque™ Plus (17-1440-03; GE Healthcare
Life Sciences, Piscataway, NJ, USA). The clinical characteristics
and experimental information of the AML patients enrolled in the
present study are listed in Table
I. The human AML cell line, Jurkat (TIB-152™; American Type
Culture Collection, ATCC), and the murine AML cell line, C1498 (a
murine myelogenous leukemia cell line, TIB-49™; ATCC), were used.
All cells were cultured in the proper media at 37°C in a humidified
atmosphere of 5% CO2, according to the supplier's
suggestions.
 | Table IClinical and laboratory features of
the primary AML cells. |
Table I
Clinical and laboratory features of
the primary AML cells.
| Patients | FAB subtype | Age at
diagnosis | Gender | Cell source | WBC/mm3
at diagnosis | Cytogenetic
anomalies |
|---|
| 1 | M3 | 53 | M | PB | 114,980 |
46,XY,t(15;17)(q22;q12)[20] |
| 2 | M4 | 65 | M | PB | 185230 | 46,XY[20] |
| 3 | M4 | 41 | F | PB | 100260 |
46,XX,t(6;11)(q27;q23)[30] |
| 4 | M4 | 23 | F | PB | 177420 |
48~49,XX,+1,der(1;14)(q10;q10),
t(7;11)(q32;p15),+8,+8,t(9;11) (p22;q23),t(12;20)(q12;q13.1),
del(19)(p13.1),+mar[cp2]/46,XX[28] |
| 5 | M1 | 48 | M | PB | 115550 |
47,XY,+11[29]/46,XY[1] |
| 6 | M5 | 27 | M | PB | 40 | 46,XY[20] |
| 7 | M3 | 32 | F | PB | 53.98 |
46,XX,t(15;17)(q22;q12)[28]/46,XX[2] |
| 8 | M3 | 28 | M | PB | 121900 | 46,XY[20] |
| 9 | M1 | 50 | F | PB | 123140 |
46,XX,15ps+[20] |
| 10 | M5b | 41 | F | PB | 143720 |
46,XX,t(6;11)(q27;q23)[20] |
| 11 | M5 | 82 | F | PB | 109540 | 46,XX[20] |
| 12 | M4 | 39 | M | PB | 141200 |
47,XY,+mar[3]/46,XY[22] |
| 13 | M2 | 25 | M | PB | 185000 | 46,XY[20] |
| 14 | M4 | 45 | M | PB | 71820 | 46,XY,inv(16)(p13.1q22)[20] |
| 15 | M5b | 28 | F | PB | 128020 |
46,XX,t(6;11)(q27;q23)[19]/46,XX[1] |
| 16 | M4 | 36 | F | PB | 240640 | 46,XX[20] |
| 17 | M0 | 58 | M | PB | 317620 | 46,XY[20] |
| 18 | M1 | 79 | F | PB | 207970 |
46,XX,t(11;12)(p15;q13),del(3)(q12q22)[20] |
| 19 | M2 | 31 | F | PB | 129700 | 46,XX[20] |
Leukemic mouse model
C57Bl/6J mice were purchased from the Jackson
Laboratory (Bar Harbor, ME, USA) and bred under pathogen-free
conditions in the Department of Laboratory Animals at the Catholic
University of Korea. All of the animal experiments were approved by
the Institutional Animal Care and Use Committee of the Catholic
University of Korea. For the syngeneic model, 2×106
C1498 cells were suspended in 200 µl phosphate-buffered
saline (PBS) and were intravenously injected into the 7-week-old
mice. For the treatment, plerixafor (Mozobil™, 2.5 mg/kg; Sanofi
Oncology) were subcutaneously injected 2 h before and 2 h after
intraperitoneal injection of Ara-C (100 mg/kg; Sigma) into the mice
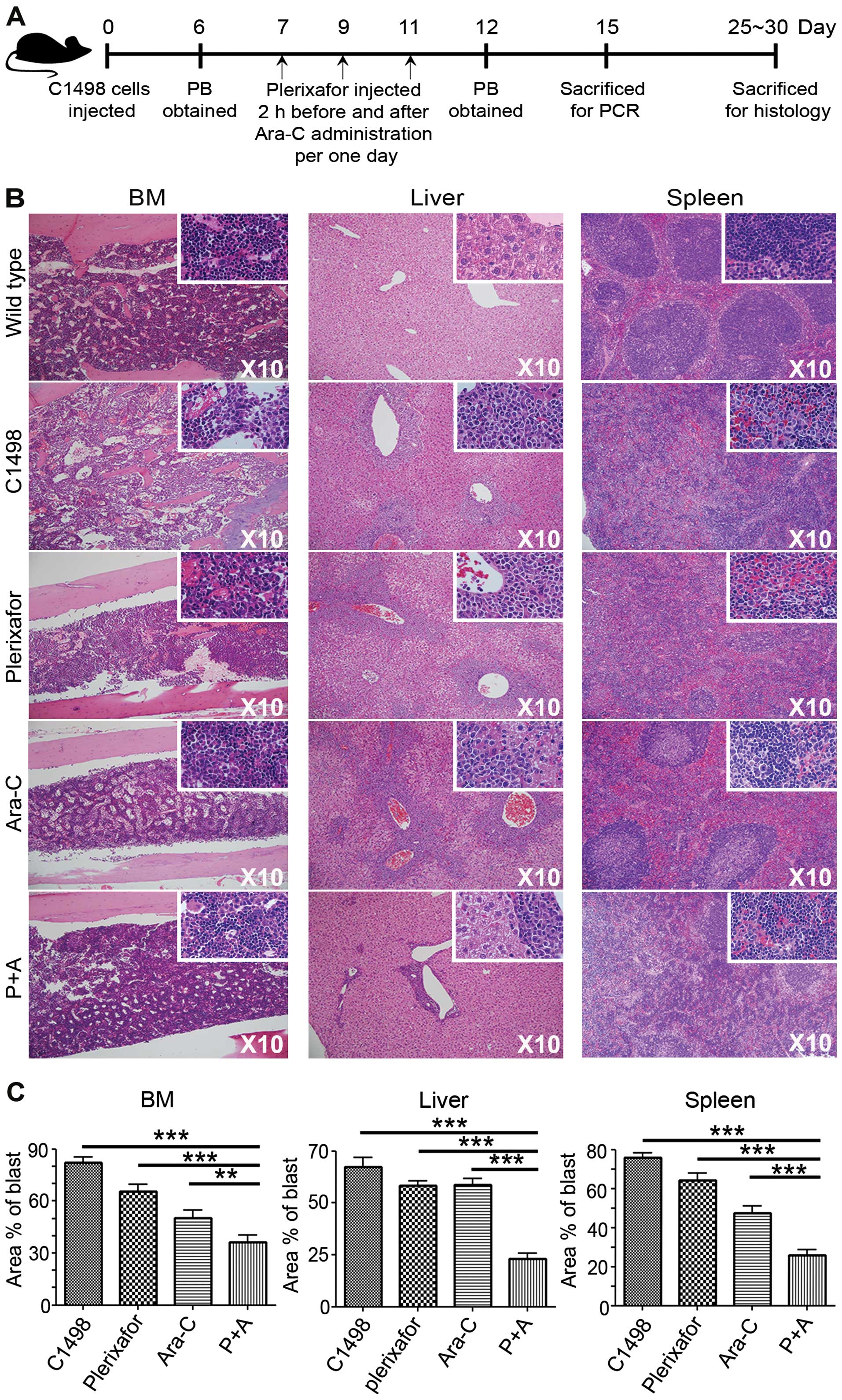
administered C1498 cells, as described in Fig. 3A (schematic diagram). Peripheral
blood (PB) was obtained from a facial vein and samples from organs
were obtained from the sacrificed mice until day 30
post-injection.
Cell migration assay
Approximately 3×105 C1498 cells and human
primary cells were pretreated with serially diluted plerixafor in
DMEM with 10% fetal bovine serum (FBS) for 2 h. After washing,
total cells were resuspended in 500 µl of Dulbecco's
modified Eagle's medium (DMEM) with 1% FBS and placed in the upper
chamber of Transwell plates (3 µm membrane pore size;
Corning). Inserts were placed in the lower chamber containing the
same medium, with or without SDF-1α (100 ng/ml; R&D Systems,
Minneapolis, MN, USA). Migration assays were performed at 37°C for
4 h. The migrated cells in the lower chamber were counted by an
automated cell counter (Luna™, LB-L10001).
Cell apoptosis assay
C1498 cells (2×105) were pretreated in 0
or 5 µM CXCR4, plerixafor, in DMEM with 10% FBS at 37°C for
2 h. Then, cells were cultured for 24 h at 37°C in DMEM with 10%
FBS with or without Ara-C (40 ng/ml) and SDF-1α (100 ng/ml).
Annexin V+ apoptotic cells were stained (ApoScan;
BioBud) and were counted by flow cytometry.
Flow cytometry
PB, spleen and BM cells were flushed from mouse
femurs, suspended in 200 µl of PBS, and incubated with
antibodies. After washing, cells were analyzed using a FACSCalibur
flow cytometer equipped with CellQuest® software (BD
Biosciences, San Diego, CA, USA). The antibodies used to detect
mouse cells included FITC-conjugated anti-mouse CD4 (clone: GK1.5,
553729), PE-Cy™5-conjugated anti-mouse CD8 (clone: 53-6.7, 553034),
and PE-conjugated anti-mouse NK1.1 (clone: PK136, 553165) (all from
BD Pharmingen™), biotin-conjugated anti-mouse CXCR4 (clone: REA107,
130-102-021; Miltenyi Biotec), APC-conjugated streptavidin
(17-4317-82; eBioscience), for human cells, APC-conjugated
anti-human CXCR4 (clone: 12G5, 555976; BD Pharmingen™). Flow
cytometric data were analyzed using appropriate controls with
proper isotype-matched IgG and unstained controls.
Histology
Liver, spleen, and BM from the each group were fixed
in 4% paraformaldehyde. BM samples were fixed in paraformadehyde,
decalcified with 5% formic acid, and embedded in paraffin. Prepared
slides were counterstained with Meyer's hematoxylin. Hematoxylin
and eosin (H&E) staining was used after fixation to confirm
leukemic blast infiltration in tissues including BM, spleen, and
liver. For immunohistochemistry, after antigen retrieval, prepared
slides were blocked for endogenous peroxidase activity and were
incubated with primary antibody anti-mouse IFN-γ (clone: DB-1,
NB100-78214; Novus Biologicals). Proper secondary antibody
(IH-8056-50; Gentaur, Brussels, Belgium) was used for
immunohistochemistry and detected using the DAB chromogen/substrate
system (HistoMouse™-MAX kit, 89-9551; Invitrogen, Camarillo, CA,
USA). Slides were counterstained with Meyer's hematoxylin.
Quantitative real-time PCR (RT-qPCR)
Total RNA was extracted from BM cells, liver, and
spleen of mice as described in the schematic diagram (Fig. 3A) at day 15. RNA (1 µg) was
reverse transcribed into cDNA at 42°C for 60 min in a 20-µl
reaction mixture using a Transcriptor First Strand cDNA Synthesis
kit (04 897 030 001; Roche, Mannheim, Germany). The primers used in
the study are listed in Table II.
The RT-qPCR was performed with TaqMan probes by
LightCycler® 480 (Roche). All data were normalized to
the amount of GAPDH expression, with samples run in triplicate.
| Table IIPrimer sequences for RT-PCR and
RT-qPCR. |
Table II
Primer sequences for RT-PCR and
RT-qPCR.
| Gene | Sequence | Method |
|---|
| Mouse CXCR4 | F:
TACCTCGCTATTGTCCACGC
R: GTGCACGATGCTCTCGAAGT | RT-PCR |
| Mouse GAPDH | F:
CGTGTTCCTACCCCCAATGT
R: GGCCCTCAGATGCCTGCTTCAC | |
| Mouse IFN-γ | F:
CAGCCGATGGGTTGTACCTT
R: GGCAGCCTTGTCCCTTGA
P: TGAGCTCATCCGAGTGGTCC | RT-qPCR |
| Mouse perforin | F:
GACTGCTGCCCACGACAGA
R: TGCCCGGAAATTGCTTACC
P: CTTGGCCCATTTGG | |
| Mouse granzyme
B | F:
CCCAGGCGCAATGTCAAT
R: CCCCAACCAGCCACATAGC
P: TGAAGCCAGGAGATGTG | |
| Mouse GAPDH | F:
CGTGTTCCTACCCCCAATGT
R: TGTCATCATACTTGGCAGGTTTCT
P: TCGTGGATCTGACGTGCCGC | |
Statistical analysis
The results are presented as the mean ± standard
error (SE). Data were compared by the Mann-Whitney U test, and
GraphPad Prism ver. 4 software (GraphPad Software, La Jolla, CA,
USA) was used for the analyses. Image J was used for analysis of
stained cells in slides. Percentage of stained area was calculated
as the ratio of the stained area to the total area detected in the
image. Values of P<0.05 were considered statistically
significant.
Results
Levels of CXCR4 are decreased in
plerixafor-treated leukemic cells
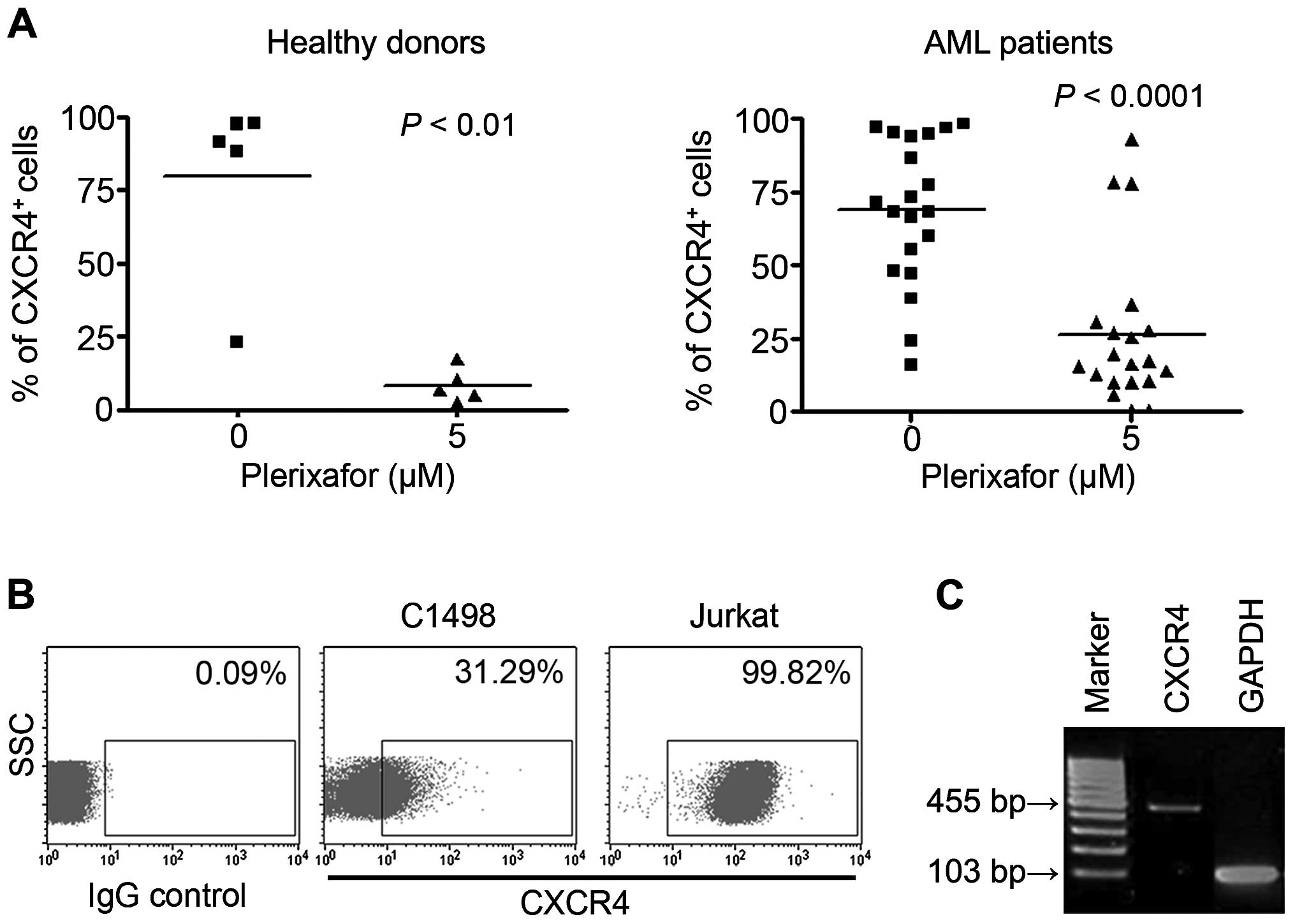
To examine the level of CXCR4 expressed by AML
blasts and murine cells, 19 AML samples and murine C1498 and human
Jurkat cells were subjected to FACS analysis. Data revealed that
both normal human samples and AML mono-nuclear cells highly
expressed CXCR4. CXCR4 expression in both groups was significantly
decreased by CXCR4 inhibition, when treated with 5 µM
plerixafor (normal, 79.7±14.2%; normal + plerixafor, 8.4±2.6%; AML,
68.6±5.9%; AML + plerixafor, 30.8±7.2%; Fig. 1A). In addition, CXCR4 expression in
C1498 cells was ~31.29%, compared to 99.7% in the CXCR4 + Jurkat
cell line and RNA expression was also confirmed (Fig. 1B and C).
Plerixafor inhibits SDF-1α-induced
migration of leukemic cells, but has no effect on cell apoptosis in
vitro
The role of CXCR4 in migration is well known.
Osteoblasts and mesenchymal stromal cells (MSCs) in the BM niche
produce cytokine SDF-1α, which is an attractant for CXCR4 +
hematopoietic cells and encourages their migration. Because tumor
cell migration into BM caused the recurrence of cancer in patients,
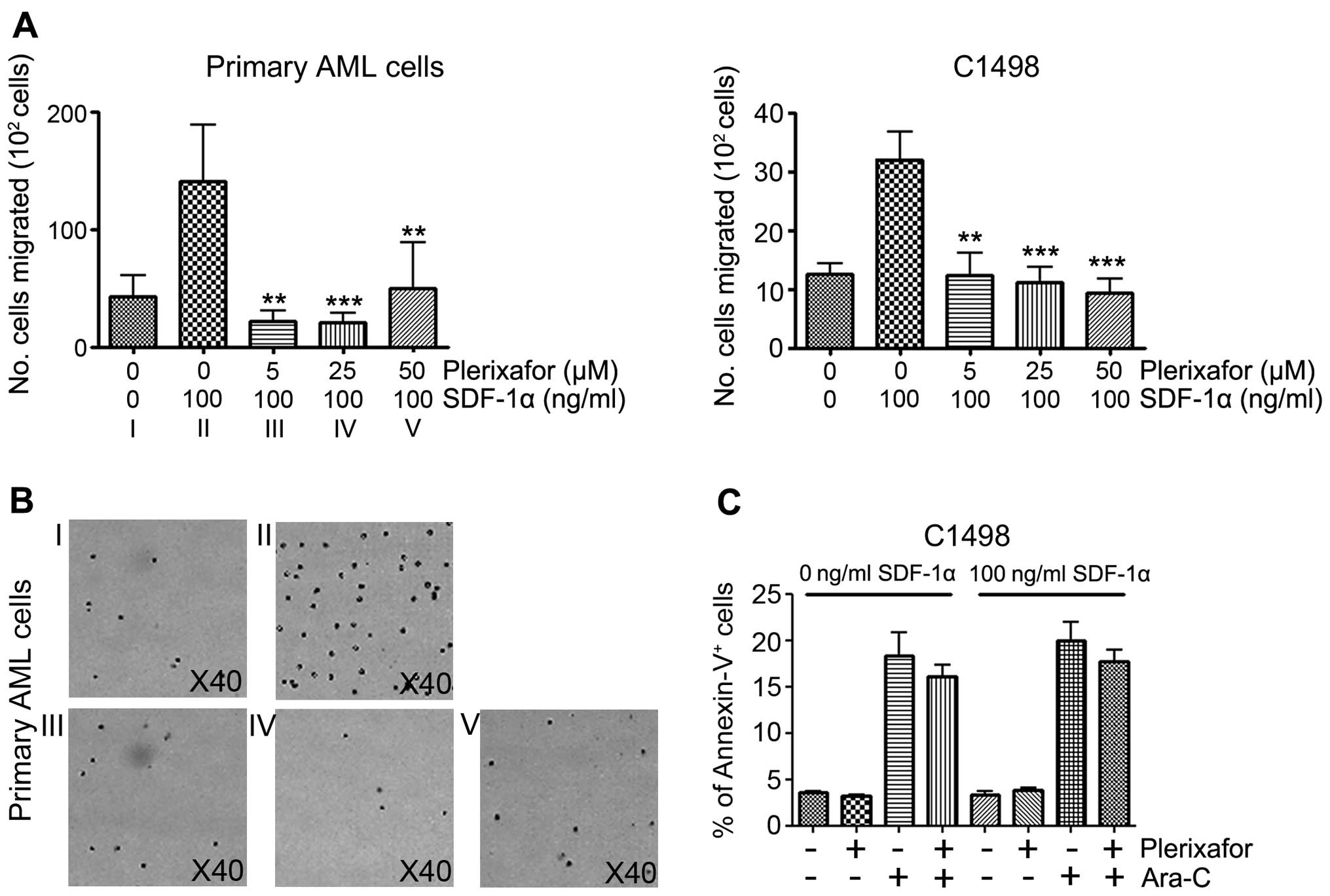
we tested whether CXCR4 inhibition can block migration of C1498
cells under SDF-1α exposure. Consistent with previous studies
(14), our migration assay clearly
showed the inhibitory effect of plerixafor on SDF-1α-induced
migration of primary AML and C1498 cells. Under SDF-1α conditions
(100 ng/ml), C1498 cells were co-cultured with the plerixafor at
various concentrations. Migrations of both C1498 and primary AML
cells were similarly inhibited. While
42.8×102±18.1×102 migrated cells were
detected without SDF-1α, SDF-1α induced
140.6×102±48×102 cells to migrate, showing an
~3.28-fold increase. Although there is individual variation in
primary AML cells, plerixafor significantly inhibited migration at
all indicated doses. Similarly, in C1498 cells, cell migration was
significantly inhibited by plerixafor in a dose-dependent manner
(Fig. 2A). Primary AML cells were
counted to avoid the induction of apoptosis during the experiments
(Fig. 2B). As expected, plerixafor
effectively inhibited tumor cell migration in vitro,
confirming the functional role of the SDF-1/CXCR4 interaction in
AML migration. Next, although the anti-leukemic effects by the
plerixafor are still debatable, studies continue to show that
SDF-1/CXCR4 can protect AML blasts from chemotherapy and induce the
apoptotic machinery of leukemic blasts under BMM disruption
(8,18). Thus, to test the direct role of
plerixafor in apoptosis, C1498 cells were cultured with or without
Ara-C. At 24 h after co-culture, a FACS analysis was performed to
evaluate the frequency of dead cells. A high number of apoptotic
cells were observed following Ara-C treatment, compared to no
apoptotic cells without Ara-C treatment. However, the Ara-C and
plerixafor dual-treated group (termed P+A group) displayed no
significant difference in apoptosis when compared to the Ara-C only
group (without SDF-1α group: Plerixafor−
Ara-C− cells, 3.6±0.1%; plerixafor+
Ara-C− cells, 3.2±0.2%; plerixafor−
Ara-C+ cells, 18.4±2.5%; plerixafor+
Ara-C+ cells, 16.0±1.3%; and with SDF-1α group:
plerixafor− Ara-C− cells, 3.3±0.3%;
plerixafor+ Ara-C− cells, 3.8±0.3%;
plerixafor− Ara-C+ cells, 20.0±2.0%;
plerixafor+ Ara-C+ cells, 17.7±1.3%; Fig. 2C). Dual treatment, therefore, does
not increase leukemic blast deaths, suggesting that apoptosis is
exclusively controlled by Ara-C, but not plerixafor, in
vitro.
Significant suppression of blasts was
detected in the plerixafor and Ara-C combination group in vivo
Because an in vitro system cannot
recapitulate in vivo conditions, a syngeneic mouse model was
used to further investigate the effects of plerixafor on blast
suppression in a leukemic microenvironment. The protocol shown in
Fig. 3A was used in experiments
in vivo with tissues including liver, BM, and spleen from
each group that were prepared and subjected to immunohistochemistry
one month after C1498 injection. Results clearly displayed blast
suppression in all tissues. Aberrant spindle-shaped C1498 cells
distinguished these cells from normal HSC in BM, and leukemic
clusters in the liver and spleen were detected. Leukemic blasts
synergistically and significantly decreased in the P+A group,
compared to those of the other groups (Fig. 3B). It also provided quantificational
significance in P+A group, comparing the results to other groups
(Fig. 3C) showing the following: In
the BM control: 82.1±3.5%; plerixafor, 65.7±3.8%; Ara-C, 50.4±4.5%;
and P+A, 36.1±4.4%. In the liver control, 67.4±4.6%; plerixafor,
58.2±2.5%; Ara-C, 58.7±2.9%; and P+A, 23.1±2.8%. In the spleen
control, 75.9±2.1%; plerixafor, 64.4±3.4%; Ara-C, 47.4±3.5%; and
P+A, 26.0±2.7%. These data suggested an unexpected role for
plerixafor in leukemic blast suppression specifically in the AML
niche.
The frequency of immune cells was not
increased; however, cytotoxicity-related factors were significantly
increased in the Ara-C and plerixafor dual-injected group in
leukemia
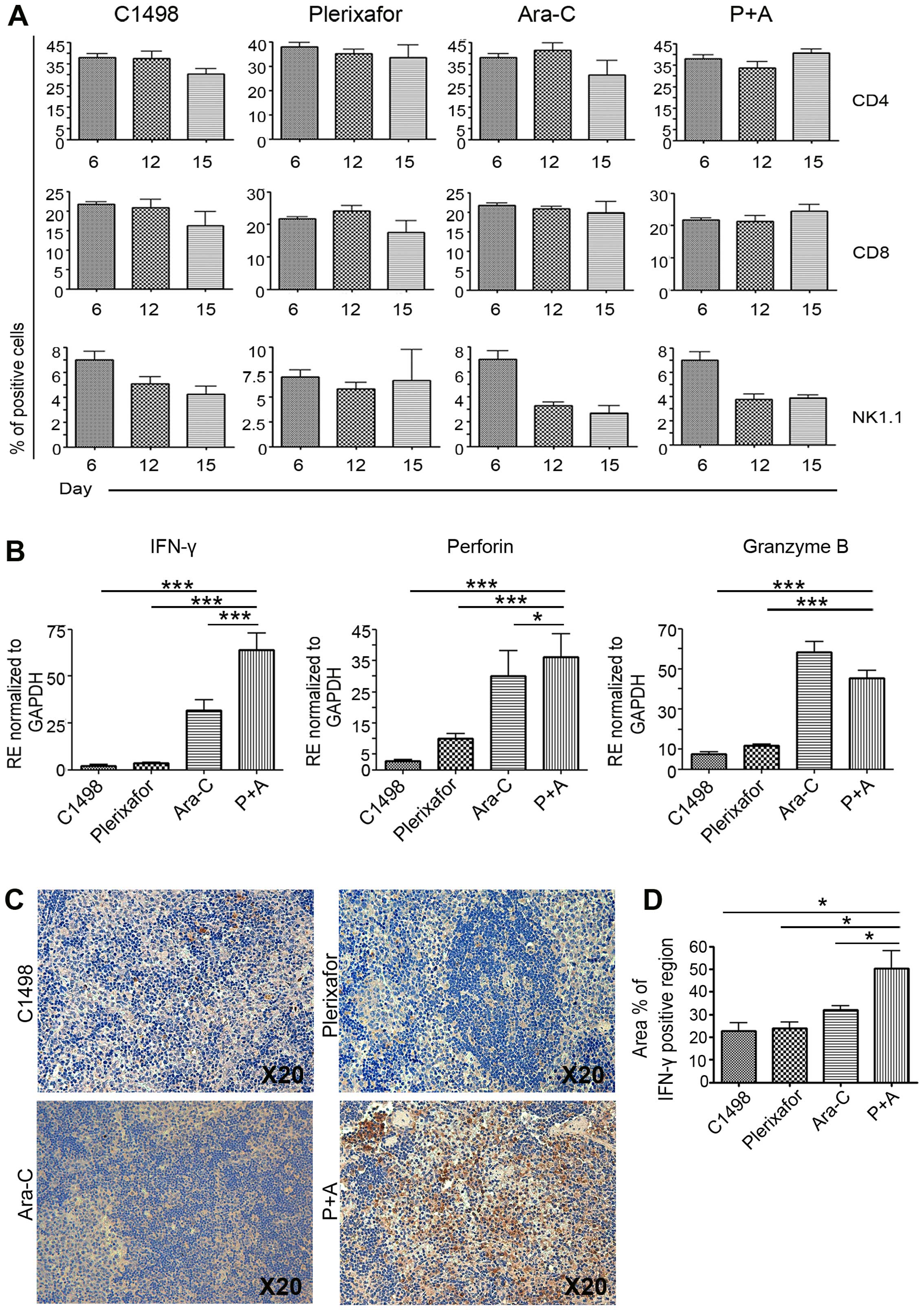
To further examine whether immune cell inhibition by
CXCR4 could diminish the number of leukemic blasts, we investigated
CD3 leukocytes and NK cells, which express CXCR4 in functional
cells distributed in the spleen and BM. PB cells from the Ara-C-
and plerixafor-only groups and the P+A group were isolated and
subjected to FACS analysis. The untreated C1498 injection only
group and plerixafor treated group showed that CD4 and CD8 cells
gradually decreased in a day-dependent manner, implying that
plerixafor cannot independently alter the frequency of immune
cells. However, the frequency of CD4 and CD8 cells was maintained
in the P+A group, compared to the Ara-C-only and C1498 injected
groups (Fig. 4A). To examine their
functional capacity, real-time PCR was performed using primers for
IFN-γ and cytotoxic-related factors. PCR results revealed a
significant upregulation of IFN-γ expression and
cytotoxic-related factors including perforin and granzyme
B. IFN-γ, a main cytokine produced by cytotoxic T and NK
cells, can help to kill target cells by immune system activation.
As shown in Fig. 4B, the expression
levels of IFN-γ, perforin, and granzyme B in
the spleens of the P+A group were significantly increased, compared
to those of the control (untreated C1498 injection only) group and
the plerixafor only group. These results showed high level of genes
in P+A group, compared to leukemia and single treated group (for
IFN-γ; C1498 injected group vs. 28.0-fold, plerixafor vs.
17.7-fold, Ara-C vs. 2.0-fold, for perforin; C1498 injected
group vs. 13.1-fold, plerixafor vs. 3.6-fold, Ara-C vs. 1.2-fold,
for granzyme B; C1498 injected group vs. 6.0-fold,
plerixafor vs. 3.9-fold), suggesting transcriptional activation in
response to leukemic blasts (Fig.
4B). With the Ara-C treated group, a significant difference
between the treated and P+A groups in expression of the
IFN-γ and perforin gene, but not granzyme B,
was detected. In protein level of spleen, high level of
IFN-γ was detected in P+A group, compared to control, Ara-C-
and plerixafor-only groups (Fig. 4C and
D). Taken together, those results imply a role of functional
relevance in immune cells for plerixafor, especially when combined
with Ara-C.
Discussion
Previous studies suggested that CXCR4 is directly
involved in tumor promotion and clinical outcomes (19,20).
CXCR4 modulation is regarded as a promising strategy to eliminate
tumor promotion in tumor environments as well as at the cellular
level. Although many studies have shown the effect of plerixafor,
the role of CXCR4 in the immune system still remains unclear. To
test the antitumoral effects of plerixafor, we investigated whether
plerixafor contributed to the suppression of leukemic cells both
in vitro and in vivo. In contrast to our expectation,
no direct antitumoral effect was found in vitro, regardless
of whether the treatment was plerixafor alone or in combination
with Ara-C. However, a remarkable suppression of leukemic blasts
in vivo was detected in the P+A combination group,
suggesting an unknown role for CXCR4 in the tumor microenvironment.
Direct killing from chemotherapy and radiotherapy or through immune
cells such as cytotoxic T cells and NK cells is usually regarded as
the main strategy to eliminate tumor cells. Because there was no
effective suppression of tumor cells in vitro but effective
inhibition of blasts in vivo, we postulate that plerixafor
has indirect effects, and that it can trigger factors such as
immune cells to support the decrease in blasts.
Recently, de Oliveira et al reported the
correlation between IFN-γ and high CXCR4 expression in
immunopathogenesis and suggested that IFN-γ induces high
levels of CXCR4 and its ligand, resulting in the migration of tumor
cells and metastasis, but not exerting antitumor effects (21). In contrast, our data showed a high
level of IFN-γ mRNA expression in the P+A group. Moreover,
cytotoxic factors, perforin and granzyme B, were
highly expressed in the P+A group, and their expression was
significantly different from that of the C1498 injection only group
as a control and plerixafor injection only group, suggesting
enhanced killing of immune cells by inhibition of CXCR4 with
chemotherapy in leukemia. The function and frequency of immune
cells and the level of IFN-γ overall has been shown to be
decreased in leukemia (17). To
exclude pathologic conditions, we performed a FACS analysis using
normal mononuclear cells (MNCs) with the Ara-C and plerixafor
combination treatment in vitro and examined the expression
of IFN-γ, perforin, and granzyme B. However,
no significant difference in immune cell activation was detected in
the P+A group (data not shown), implying that immune activation by
CXCR4 inhibition may require another mediator in vivo. For a
better understanding of the correlation between CXCR4 and immune
cells, further functional studies on the precise mechanism induced
at the immune cell level as well as in tissues should be
undertaken. Niche-focused in vivo studies of CXCR4 and
immune cells are needed to clarify this interaction in
leukemia.
The BMM is important for the progression of AML and
maintenance of minimal residual disease through soluble factors
that are associated with leukemic cell resistance to chemotherapy
(22). The centrally located
vascular niche of BM is a site that induces differentiation and
eventually mobilization of hematopoietic cells to the peripheral
blood (23). Remarkably, mature
megakaryocytes are located adjacent to sinusoidal endothelial cells
(SEC) within BM and migrate to the peripheral circulation through
SEC (24,25). We also found high numbers of
megakaryocytes around the SEC and decreased circulating leukemic
blasts inside the BM sinusoidal capillary (data not shown). In
addition, the SEC capillary morphology returned to that of
wild-type when plerixafor was injected together with Ara-C (data
not shown). This suggests that plerixafor can also support the
reconstitution of BM architecture with conventional chemotherapy.
While this is not directly relevant to leukemia elimination, we
cannot rule out the possibility that niche rearrangement by
plerixafor can inhibit engraftment of leukemia.
Not all pathological conditions need proper CXCR4
expression to develop and activate lymphocytes. In WHIM (warts,
hypogammaglobulinemia, infections, and myelokathexis) syndrome with
rare combined immunodeficiency disorder in BM, a strong response to
CXCL12 and displayed chronic non-cyclic leukopenia was shown.
Specifically, in the case of CXCR+/1013 mutant mice
presenting WHIM syndrome, CXCR4 desensitization with plerixafor
reversed lymphoneutropenia that had disturbed leukocyte homeostasis
(26). This indicates that the
microenvironment is governed by diverse pathologic conditions that
cause it to differ from an in vitro system.
In summary, we provide clues for the CXCR4 function
accompanied with high level of cytotoxicity related factors in
leukemic microenvironments that may suggest an advanced therapeutic
strategy for leukemia through the modulation of immune cells or
their niche by plerixafor.
Acknowledgments
This study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education (2014R1A1A2053407).
References
|
1
|
Löwenberg B, Downing JR and Burnett A:
Acute myeloid leukemia. N Engl J Med. 341:1051–1062. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liesveld JL, Bechelli J, Rosell K, Lu C,
Bridger G, Phillips G II and Abboud CN: Effects of AMD3100 on
transmigration and survival of acute myelogenous leukemia cells.
Leuk Res. 31:1553–1563. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lane SW, Wang YJ, Lo Celso C, Ragu C,
Bullinger L, Sykes SM, Ferraro F, Shterental S, Lin CP, Gilliland
DG, et al: Differential niche and Wnt requirements during acute
myeloid leukemia progression. Blood. 118:2849–2856. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Krause DS, Fulzele K, Catic A, Sun CC,
Dombkowski D, Hurley MP, Lezeau S, Attar E, Wu JY, Lin HY, et al:
Differential regulation of myeloid leukemias by the bone marrow
microenvironment. Nat Med. 19:1513–1517. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Moshaver B, van der Pol MA, Westra AH,
Ossenkoppele GJ, Zweegman S and Schuurhuis GJ: Chemotherapeutic
treatment of bone marrow stromal cells strongly affects their
protective effect on acute myeloid leukemia cell survival. Leuk
Lymphoma. 49:134–148. 2008. View Article : Google Scholar
|
|
6
|
Konopleva M, Konoplev S, Hu W, Zaritskey
AY, Afanasiev BV and Andreeff M: Stromal cells prevent apoptosis of
AML cells by up-regulation of anti-apoptotic proteins. Leukemia.
16:1713–1724. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mercier FE, Ragu C and Scadden DT: The
bone marrow at the crossroads of blood and immunity. Nat Rev
Immunol. 12:49–60. 2012. View
Article : Google Scholar
|
|
8
|
Peled A and Tavor S: Role of CXCR4 in the
pathogenesis of acute myeloid leukemia. Theranostics. 3:34–39.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tokoyoda K, Egawa T, Sugiyama T, Choi BI
and Nagasawa T: Cellular niches controlling B lymphocyte behavior
within bone marrow during development. Immunity. 20:707–718. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Foudi A, Jarrier P, Zhang Y, Wittner M,
Geay JF, Lecluse Y, Nagasawa T, Vainchenker W and Louache F:
Reduced retention of radioprotective hematopoietic cells within the
bone marrow microenvironment in CXCR4−/− chimeric mice.
Blood. 107:2243–2251. 2006. View Article : Google Scholar
|
|
11
|
Fricker SP, Anastassov V, Cox J, Darkes
MC, Grujic O, Idzan SR, Labrecque J, Lau G, Mosi RM, Nelson KL, et
al: Characterization of the molecular pharmacology of AMD3100: A
specific antagonist of the G-protein coupled chemokine receptor,
CXCR4. Biochem Pharmacol. 72:588–596. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Burger JA and Peled A: CXCR4 antagonists:
Targeting the microenvironment in leukemia and other cancers.
Leukemia. 23:43–52. 2009. View Article : Google Scholar
|
|
13
|
Zhang Y, Patel S, Abdelouahab H, Wittner
M, Willekens C, Shen S, Betems A, Joulin V, Opolon P, Bawa O, et
al: CXCR4 inhibitors selectively eliminate CXCR4-expressing human
acute myeloid leukemia cells in NOG mouse model. Cell Death Dis.
3:e3962012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zeng Z, Shi YX, Samudio IJ, Wang RY, Ling
X, Frolova O, Levis M, Rubin JB, Negrin RR, Estey EH, et al:
Targeting the leukemia microenvironment by CXCR4 inhibition
overcomes resistance to kinase inhibitors and chemotherapy in AML.
Blood. 113:6215–6224. 2009. View Article : Google Scholar :
|
|
15
|
Sison EA, McIntyre E, Magoon D and Brown
P: Dynamic chemotherapy-induced upregulation of CXCR4 expression: A
mechanism of therapeutic resistance in pediatric AML. Mol Cancer
Res. 11:1004–1016. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nervi B, Ramirez P, Rettig MP, Uy GL, Holt
MS, Ritchey JK, Prior JL, Piwnica-Worms D, Bridger G, Ley TJ, et
al: Chemo-sensitization of acute myeloid leukemia (AML) following
mobilization by the CXCR4 antagonist AMD3100. Blood. 113:6206–6214.
2009. View Article : Google Scholar :
|
|
17
|
Lee JY, Park S, Min WS and Kim HJ:
Restoration of natural killer cell cytotoxicity by VEGFR-3
inhibition in myelogenous leukemia. Cancer Lett. 354:281–289. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kremer KN, Peterson KL, Schneider PA, Meng
XW, Dai H, Hess AD, Smith BD, Rodriguez-Ramirez C, Karp JE,
Kaufmann SH, et al: CXCR4 chemokine receptor signaling induces
apoptosis in acute myeloid leukemia cells via regulation of the
Bcl-2 family members Bcl-XL, Noxa, and Bak. J Biol Chem.
288:22899–22914. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mannelli F, Cutini I, Gianfaldoni G,
Bencini S, Scappini B, Pancani F, Ponziani V, Bonetti MI, Biagiotti
C, Longo G, et al: CXCR4 expression accounts for clinical phenotype
and outcome in acute myeloid leukemia. Cytometry B Clin Cytom.
86:340–349. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Müller A, Homey B, Soto H, Ge N, Catron D,
Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, et al:
Involvement of chemokine receptors in breast cancer metastasis.
Nature. 410:50–56. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
de Oliveira KB, Guembarovski RL,
Guembarovski AM, da Silva do Amaral Herrera AC, Sobrinho WJ, Ariza
CB and Watanabe MA: CXCL12, CXCR4 and IFNγ genes expression:
Implications for proinflammatory microenvironment of breast cancer.
Clin Exp Med. 13:211–219. 2013. View Article : Google Scholar
|
|
22
|
Meads MB, Hazlehurst LA and Dalton WS: The
bone marrow microenvironment as a tumor sanctuary and contributor
to drug resistance. Clin Cancer Res. 14:2519–2526. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kopp HG, Avecilla ST, Hooper AT and Rafii
S: The bone marrow vascular niche: Home of HSC differentiation and
mobilization. Physiology (Bethesda). 20:349–356. 2005. View Article : Google Scholar
|
|
24
|
Psaila B, Lyden D and Roberts I:
Megakaryocytes, malignancy and bone marrow vascular niches. J
Thromb Haemost. 10:177–188. 2012. View Article : Google Scholar :
|
|
25
|
Yin T and Li L: The stem cell niches in
bone. J Clin Invest. 116:1195–1201. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Balabanian K, Brotin E, Biajoux V,
Bouchet-Delbos L, Lainey E, Fenneteau O, Bonnet D, Fiette L, Emilie
D and Bachelerie F: Proper desensitization of CXCR4 is required for
lymphocyte development and peripheral compartmentalization in mice.
Blood. 119:5722–5730. 2012. View Article : Google Scholar : PubMed/NCBI
|