1. Introduction
Chronic myeloid leukemia (CML) has a yearly
incidence of approximately 1 in 50,000 individuals and accounts for
15% of all adult leukemias. The onset of this disease is ~45-55
years, with the majority of patients being asymptomatic at
diagnosis which is commonly made after routine blood tests. The
evolution of CML occurs via a biphasic or triphasic course. The
majority of the cases (~85%) are diagnosed during the asymptomatic
chronic phase (CP) where the cells are mainly differentiated,
minimally invasive and maintain their functionality (1). If left untreated, the disease
inevitably progresses after three to five years to an intermediate
accelerated phase (AP) and to blast crisis (BC) (2). Nevertheless, up to a quarter of
patients progress directly to the BC phase (3). In 1960, Rudkin et al detected a
consistent chromosomal abnormality characteristic of CML, which
later was named the Philadelphia (Ph) chromosome (4). The Ph chromosome results from a
reciprocal translocation, which involves the Abelson (ABL)
proto-oncogene on chromosome 9 and breakpoint cluster region (BCR)
on chromosome 22, t(9;22)(q34;q11) (5). This translocation creates the BCR-ABL
fusion gene, which is believed to be the principal cause of CML and
is considered as a hallmark of this disease. Depending on the
breakpoint in the BCR gene, three main types of fusion proteins are
formed: p210BCR/ABL (M-bcr breakpoint), which is the most common in
CML, p230BCR/ABL (µ-bcr breakpoint) and p190BCR/ABL (m-bcr
breakpoint) (5). The BCR-ABL
protein is exclusively localized to the cytoplasm and it is able to
trigger multiple downstream pathways leading to enhanced cell
proliferation and transformation, reduced growth factor dependence,
resistance to apoptosis, perturbed adhesion to bone marrow and
stroma, and genetic instability (6). This results in the expansion of the
leukemic cell population, initially characterized by overproduction
of mature myeloid cells with normal morphology (7). Many BCR-ABL substrates and binding
partners have been identified, and current efforts are directed at
linking these pathways to specific defects for CML (5,8,9).
Selective therapies are aimed for the treatment of CML since its
target is well defined in contrast to other cancers (10). Hundreds of protein kinases are known
in the human genome and a drug is required that targets a single
adenosine triphosphate (ATP) binding site of protein kinase. By
blocking the binding of ATP, phosphorylation is prevented and
BCR-ABL-expressing cells either have a growth disadvantage or they
undergo apoptosis (10). Imatinib
(IM) (STI571) is the first BCR-ABL tyrosine kinase inhibitor (TKI)
that prevents ATP from binding by itself to the ABL domain via
interaction with six hydrogen bonds (10). Hydrogen bonds involve the pyridine-N
and backbone-NH of Met-318, the aminopyrimidine and side chain
hydroxyl of Thr-315, the amide-NH and side chain carboxylate of
Glu-285, the carbonyl and backbone-NH of Asp-381, the protonated
methylpiperazine with the backbone-carbonyl atoms of Ile-360 and
His-361. Additionally, a number of van der Waals interactions
contribute to binding (11–13). Resistance faced by imatinib can be
subdivided into BCR-ABL-independent and dependant mechanisms
(14). The dependent mechanism is
associated with the duplication of the BCR-ABL tyrosine kinase gene
in the DNA sequence leading to higher expression of the protein
(10). A point mutation in the
kinase domain of BCR-ABL leads to disruption in the binding site of
imatinib on the tyrosine kinase, resulting in the loss of
sensitivity of the drug (14).
T315I is a unique mutation due to its resistance to all approved
BCR-ABL inhibitors, prior to ponatinib (15). It may be due to the displacement of
cytosine to thiamine (C->T) base pair at 944 of the ABL gene. It
causes the elimination of critical O2 molecules needed
for hydrogen bonding between imatinib and BCR-ABL kinases (10). The most common mutation occurs in
ATP binding and the activation loop. It causes the derangement of
loops as a result of which the kinase domain cannot assume inactive
conformation required for imatinib binding (14). BCR-ABL-independent resistance occurs
either due to overexpression of the P-glycoprotein efflux pump,
activation of rous sarcoma oncogene cellular homolog (Src) family
kinase or may be due to low expression, activity or polymorphism of
organic cation transporter 1 (OCT1) (10–16).
Nilotinib (AMN107) and dasatinib (BMS-345825) are
second generation drugs that were aimed to have less resistance and
intolerance than imatinib (10).
Nilotinib is a selective inhibitor and binds to the inactive
conformation of the ABL kinase domain, largely through lipophilic
interactions and thus blocks its catalytic activity, being 10- to
30-fold more potent than imatinib (17–19).
It is effective against all type of resistance except the T315I
mutation. Its failure against T315I is due to the loss of an H-bond
interaction between threonine-O and aniline-NH on nilotinib
and a steric clash between the isoleucine-methyl and 2-methylphenyl
phenyl groups of nilotinib (17,18).
Dasatinib is a multi-targeted inhibitor of wild-type BCR-ABL and
Src family kinases having additional inhibitory activity against
downstream kinases (19). Contrary
to most TKIs, dasatinib binds to active conformation of ABL kinase
(13). First and second generation
inhibitors have provided promising results, but new mutations are
continuously being encountered that requires discovery of more
drugs.
Bosutinib is based on a quinolone scaffold and it
also has the ability to inhibit the mutation of T3151 (18). One of the most promising TKIs able
to inhibit the T315I mutation is ponatinib (AP24534). In addition,
it inhibits Src, vascular endothelial growth factor receptor
(VEGFR), fibroblast growth factor receptor (FGFR) and
platelet-derived growth factor receptor (PDGFR) family kinases
(20). Clinical trials with
ponatinib are ongoing and initial results from a phase II trial,
called the Ponatinib P-positive Acute Lymphocytic Leukemia (ALL)
and CML Evaluation (PACE), involves CML and Ph-positive ALL
patients with dasatinib and/or nilotinib intolerance or resistance
(including T315I). However, this drug was correlated with a nearly
12% incidence of cardiovascular events such as serious arterial
thrombotic events in adults, resulting in withdrawal from the
market (21,22).
Bafetinib (INNO-406), an oral dual ABL/LYN tyrosine
kinase inhibitor, demonstrates specific LYN kinase activity with no
or limited activity against other Src-family member kinases.
Several BCR-ABL kinase domain mutations are sensitive to INNO-406
in vitro, including the F317L and F317V mutations.
Kantarjian et al found that INNO-406 can be used in
Ph-positive CML or ALL post-imatinib resistance or intolerance.
Such a drug with efficacy against various point mutations in the
BCR-ABL kinase, with few adverse effects and with narrower kinase
spectra, is also in phase II clinical trials (23). Befitinib and imatinib have
structural and binding similarities, the notable difference being
hydrophobic interaction between the trifluoromethyl group and the
hydrophobic pocket created by Ile-293, Leu-298, Leu-354 and Val-379
(24).
Although more potent BCR TKIs are available,
imatinib still remains the frontline TKI. Nilotinib, dasatinib,
bosutinib and ponatinib are approved for the treatment of imatinib
resistance or intolerant CML. The availability of highly potent
TKIs, such as nilotinib, has broadened the treatment armamentarium
in CML. Nilotinib appears to overcome imatinib resistance in
patients with chronic, accelerated and blastic phase CML, producing
sustained cytogenetic and hematological responses (25). The first line data for these drugs
are encouraging and suggest that some or all of them may replace
imatinib as a frontline standard BCR-ABL tyrosine kinase inhibitor
in the near future.
In recent years, miRNAs have received wide attention
as important regulators of gene expression in leukemogenesis and
they are associated with resistance to BCR-ABL TKIs.
2. MicroRNAs (miRNAs)
miRNAs are conserved non-coding RNAs with a short
sequence (~20–23 nucleotides) that participate in the traslational
regulation of many genes involved in the control of important
biological processes by selectively binding to their messenger RNAs
(mRNAs) and generally blocking their protein expression (26,27).
Most miRNA genes are coded in the intergenic regions of the genome
that are distant from other genes and they have their regulatory
elements derived from independent transcription units (28). Nevertheless, genomic studies have
revealed that 30% of miRNAs are included in the introns of the
pre-mRNA of protein-coding genes and frequently in the same
orientation, implying that they are transcribed in the same mRNA
primary transcript (although they could also be independently
transcribed through an alternative promoter) (28). The majority of human miRNA genes are
isolated from each other within the genome, but others can be
clustered together and transcribed as a polycistronic primary
transcript (28). Since they were
discovered and characterized on 1993 by Lee et al (29), the number of miRNA sequences
deposited in miRBase databases is continuously growing, and they
have been proven to play an important role in cellular regulatory
pathways (30).
miRNA biological relevance: Involvement
in gene expression control
Considering that a single miRNA can target several
mRNAs, a single mRNA may contain in the 3′-untranslated region
(UTR) sequence several signals for miRNA recognition and it has
been determined that at least 10–40% of human mRNAs are a target
for miRNAs (31). In fact, a single
miRNA may bind to as many as 200 gene targets and these targets can
be diverse in their function; they include transcription and
secreted factors, receptors and transporters. Thus, miRNAs
potentially control the expression of about one-third of human
mRNAs (32). Therefore, great
interest is concentrated on the identification of validated targets
of miRNAs. This specific field of miRNA research has confirmed that
the complex networks constituted by miRNAs and RNA targets coding
for structural and regulatory proteins lead to the control of
highly regulated biological functions, such as differentiation
(33), cell cycle (34) and apoptosis (35). Since a single 3′UTR of a given mRNA
contains signal sequences for several miRNAs, which miRNA should be
targeted in order to achieve alteration of the expression of the
gene should be experimentally evaluated. With respect to the
possible effects of the expression of other mRNA targets, it should
be clearly stated that alteration of a single miRNA may retain
multiple effects. In contrast, miRNAs usually bind to their targets
with incomplete complementarity; for this reason, the
identification of gene targets with only a simple BLAST search is
impossible. However, current bioinformatic approaches have taken
advantage of the fact that miRNAs within families have highest
homology at the 5′-end of the mature miRNA, which is crucial for
the stability and proper loading of the miRNA into the
miRNA-mediated silencing complex (miRISC) (36).
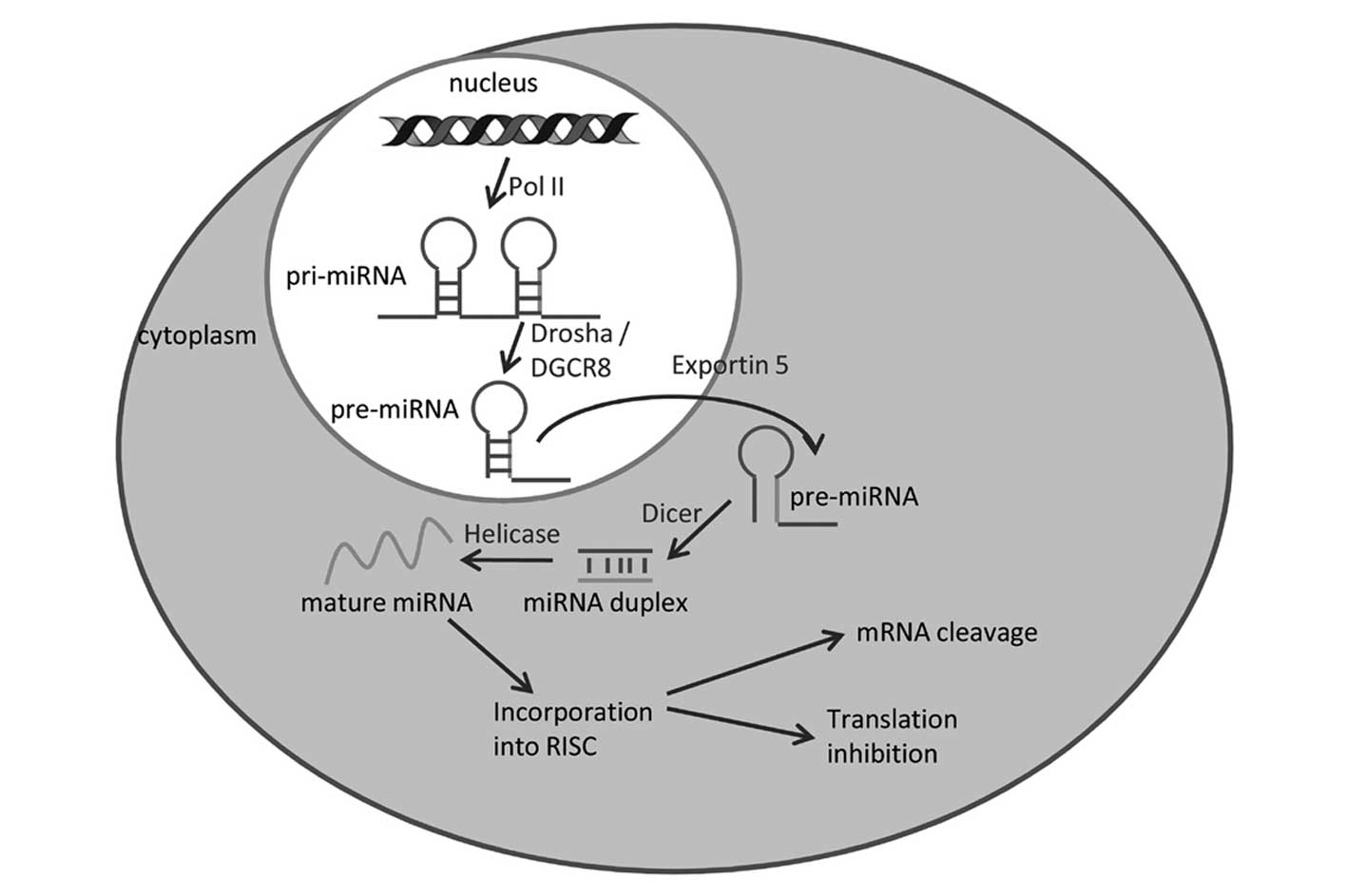
Biogenesis of miRNAs
Various miRNAs are encoded by unique genes
(intergenic miRNAs) (37) and
others are embedded into the intronic regions of protein-coding
genes (intragenic miRNAs) (38).
The transcription by RNA polymerase II of these miRNA genes gives
rise to long primary miRNAs (pri-miRNAs) with typical stem-loop
structures. These are rapidly processed by the nuclear RNase
endonuclease-III Drosha, which, removing the branches, gives rise
to precursor miRNAs of ~60–100 nucleotides in length. In both cases
of intergenic miRNAs and intragenic miRNAs, the pre-miRNAs are
transported from the nucleus to the cytoplasm by exportin-5. In the
cytoplasm, pre-miRNAs are further processed by another RNase
endonuclease-III (Dicer) to generate mature miRNAs ~22-nt long,
which generate the RNA-induced silencing complex (RISC) (Fig. 1).
miRNA and gene regulation
Structurally, miRNAs are small non-coding regulatory
RNAs; these small RNAs post-transcriptionally repress gene
expression by recognizing complementary target sites most often in
the 3′UTR of target mRNAs (39).
However, animal miRNAs may also target 5′UTR and coding regions of
mRNAs, as documented by experiments involving both artificial and
natural mRNAs and also by bioinformatic predictions (40). miRNAs silence the expression of
target mRNAs, either by mRNA cleavage or by translational
repression. Nevertheless, it has been described that miRNAs can
also increase the expression of a target mRNA (41). Perfect miRNA:mRNA complementarity
leads to cleavage of the mRNA by Argonaute protein (AGO2); this is
the small interfering RNA (siRNA) pathway, which while important
experimentally, is not thought to occur with endogenous mammalian
miRNAs. Instead, the imperfect pairing with mRNA causes a
downregulation of translation, even if the mechanism by which this
occurs is still not clear. Whatever the precise nature of the
mechanism, affected mRNAs accumulate in granular cytoplasmic
'P-bodies' along with RISC proteins (42). Generally, mRNA abundance is
ultimately also reduced (43). This
is important, as it means that the impact of miRNA activity can be
assessed (at least to some extent) by measures of mRNA abundance
such as expression profiling.
The basic mechanism leading to alteration of gene
expression is based on the recruitment of mature miRNAs at the
level of the RISC silencing complex. This process occurs in the
cytoplasm, where the pre-miRNA hairpin is cleaved by the RNase III
enzyme Dicer, which interacts with the 3′-end of the hairpin and
cuts away the loop joining the 3′ and 5′ arms, yielding an
imperfect miRNA/miRNA duplex. One of the strands is incorporated
into the RISC, where it binds to the target mRNA sequence. Perfect
or near perfect base pairing with the target RNA promotes cleavage
of the RNA (44). It is proposed
that in the case of partial complementarity, miRNAs, in order to
recognize their targets, nucleotides 2–7 of the miRNA (the 'seed
sequence') are important (45).
This is the key process permitting mature miRNAs to exert their
effects in gene regulation. The final effect of miRNA activity is
the inhibition of the synthesis of the protein(s) encoded by the
target mRNA(s). This has of course important biological
implications depending on the role of the protein in the cellular
network. Since a single 3′UTR of a given mRNA contains signal
sequences for several miRNAs, applied biological studies are needed
to determine which miRNA should be targeted to achieve alteration
of gene expression. Possible effects on the expression of other
mRNA targets should be considered. An alteration of a single miRNA
may exhibit multiple effects, possibly in combination with the
targeting activity of other miRNAs, enabling the achievement of a
strong biological effect (46,47).
3. The roles of microRNAs in the
pathogenesis and drug resistance of CML
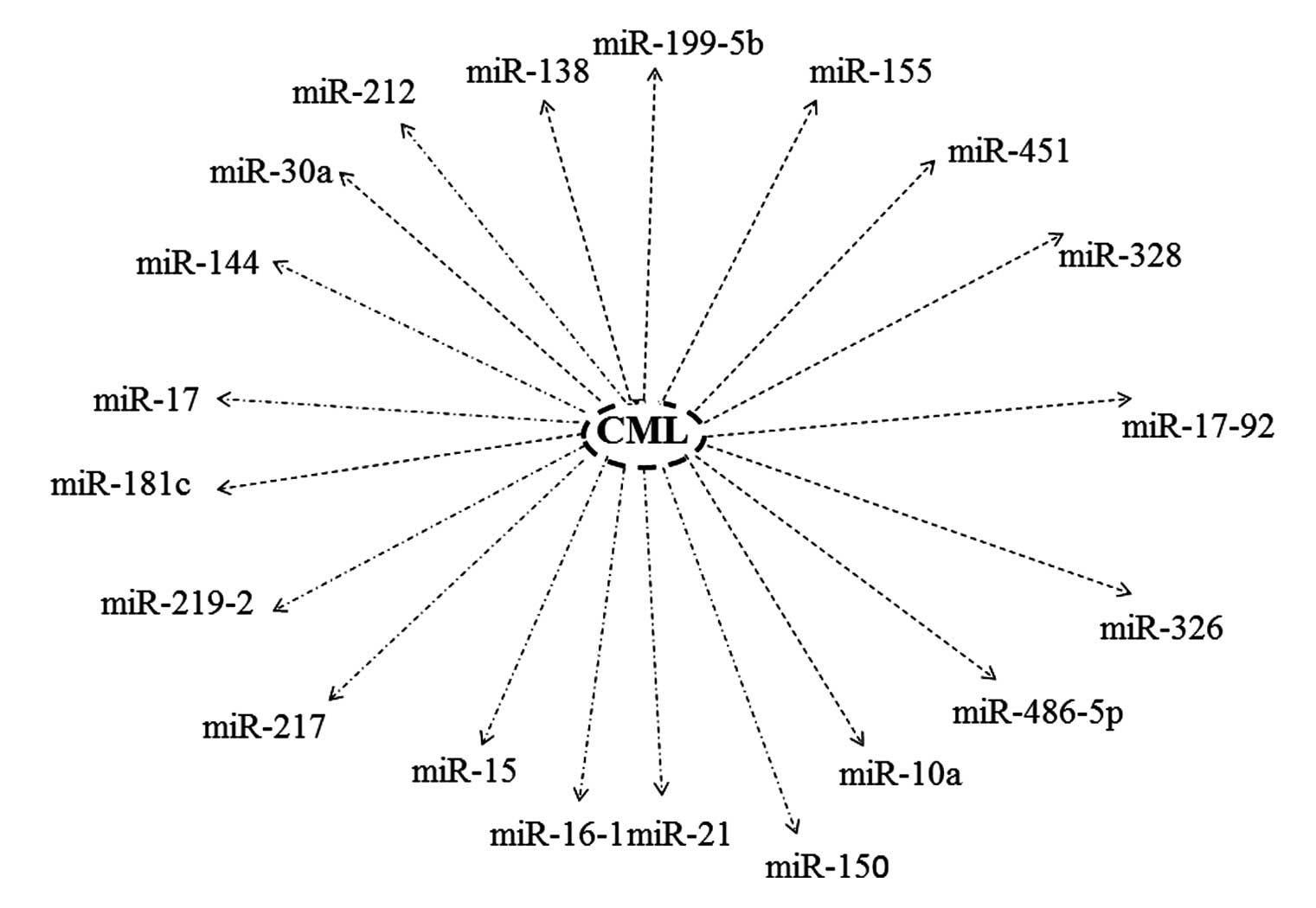
Aberrant expression of miRNAs has been observed in
hematological cancers, exhibiting unique expression signatures in
comparison to normal counterparts. Furthermore, numerous studies
have solely identified a loose association between certain
hematological cancers and aberrant miRNA expression, while others
have been able to illustrate their role in clinical diagnosis,
prognosis and cancer therapy (Fig.
2) (32,48–56).
The first investigation discovered that miR-15 and miR-16-1 have a
deleted region, 13q14, in chronic lymphocytic leukemia (CLL)
(57) and they act as tumor
suppressors, and their expression is inversely correlated with
anti-apoptotic B-cell CLL/lymphoma 2 (Bcl-2) expression (58). Additionally, Bcl-2 and myeloid cell
leukemia sequence 1 (Mcl-1) have been predicted as potential target
genes of the miR-29 family and both belong to the Bcl-2 family
which plays a central role in cell death regulation (26). A recent study found that a
significant low expression of miR-29a/29b is related to a high
expression level of both Bcl-2 and Mcl-1 in peripheral blood
mononuclear cells (PBMCs) derived from acute myeloid leukemia (AML)
and CML patients compared with healthy subjects (59). Overexpression of miR-21 has also
been implicated in directly modulating the expression of several
apoptotic-related proteins such as Bcl-2. Seca et al
observed that its downregulation caused a decrease in the
expression levels of Bcl-2 protein in leukemic cells. In addition,
treatment with anti-miR-21 caused both an increase in the
autophagy-related proteins (Beclin-1 and LC3-II) and a cellular
sensitivity to etoposide or doxorubicin (K562 and KYO-1 cells)
(60). Another previous study found
that over 50% of miRNA genes are located within regions of loss of
heterozygosity, amplification, fragile sites and viral integration
sites, and other cancer-related genomic regions (61). Using quantitative reverse
transcription-polymerase chain reaction (qRT-PCR), upregulation of
miR-96 and downregulation of miR-151, miR-150, miR-125a and miR-10a
were detected in CD34+ cells derived from CML patients
when compared with the same cells derived from healthy donors.
Additionally, transcription factor upstream stimulatory factor 2
(USF2) has been considered as an miR-10a target and it appears to
be upregulated in CML patients (62). Wang et al observed a
miR-486-5p overexpression in CML CD34 cells compared with normal
CD34 cells (63). In addition, the
investigators showed that miR-486-5p levels were increased after
erythroid differentiation in normal CD34 cells and its inhibition
reduced such differentiation of both normal CD34 and CML CD34
cells. These results indicate an important role for miR-486-5p in
modulating normal and leukemic hematopoietic progenitor growth and
erythroid differentiation (63).
Furthermore, this miRNA was able both to enhance
phosphatidylinositol-3 kinase (PI3K)/AKT (protein kinase B)
signaling and to reduce phosphatase and tensin homolog (PTEN) and
Forkhead box O1 (FoxO1) levels in hematopoietic cells. Meanwhile,
downregulation of miR-326 was a possible mechanism for the
unrestricted activation of smoothened (Smo) signal transducer of
the oncogenic Hedgehog (Hh) pathway and resulted in elevated cell
proliferation and decreased rate of apoptosis in CD34+
CML cells (64). One study
suggested an oncogenic role for the miR-17-92 cluster (which
encodes seven miRNAs: miR-17-5p, miR-17-3p, miR-18a, miR-19a,
miR-20a, miR-19b-1 and miR-92-1) in CML. Chromosomal amplification
at 13q31-q32 led to overexpression of the miR-17-92 cluster encoded
by the chromosome 13 open reading frame 25 (C13orf25) gene in
CD34+ CML. Its overexpression was found to be associated
with CP-CML but not BC-CML (65). A
novel role of miRNAs has been identified during blast crisis.
Aberrant activity of RNA binding proteins (RBPs) is related to
increased BCR-ABL activity (66).
One RBP affected is heterogeneous nuclear ribonucleoprotein E2
(hnRNP E2) which arrests myeloid differentiation through
interaction with CCAAT/enhancer-binding protein-α (C/EBP-α)
(67,68). miR-328 downregulation has been shown
in BC-CML secondary to BCR-ABL activity both in vitro and
in vivo using microarray, northern blotting and qRT-PCR
methods (66). Postulating that
miRNA and RBPs may interact, Eiring et al demonstrated that
hnRNP E2 mediated the loss of miR-328 in BC-CML and re-expression
of miR-328 was able to restore differentiation and block blast
survival by simultaneously interacting with the hnRNP E2 and the
mRNA encoding the survival factor provirus integration site for
Moloney murine leukemia virus 1 (Pim 1) (66). Another study found that the
heterogeneous nuclear RNP (hnRNP) A1 protein is also upregulated in
BC-CML and it is linked to pri-miR-17-92 at the same time (68). Another study provided support for
the idea that the lack of miR-17-92 expression can play a pivotal
role during BC stage in CML (67).
Another hallmark of CML is miR-150 whose reduced expression is
found in CD34+ cells derived from CML patients at
diagnosis (62) and also in total
leukocytes of PBMCs derived from CML patients in AP and BC phases
(69). Moreover, increased
expression of miR-150 and miR-146a and decreased expression of
miR-142-3p and miR-199b-5p were observed in PBMCs derived from 140
patients newly diagnosed with CML and treated with imatinib for two
weeks. Expression levels of these miRNAs also tended to normalize
after imanitib treatment (69).
Another recent study, conducted on 17 patients, demonstrated that
the miR-451 expression level increased after imanitib therapy and
it was inversely correlated with the BCR-ABL transcript in some CML
patients. However, this study suggests that expression of miR-451,
being heterogeneous among patients, can be regulated by other
mediators (70). Recently, Rokah
et al characterized the miRNA expression profile of CML cell
lines and patients compared to a normal counterpart derived from
healthy donors, using miRNA microarrays and miRNA real-time PCR.
The expression levels of miR-31, miR-155 and miR-564 were decreased
in CML and influenced by BCR-ABL activity. After 30 days of BCR-ABL
inhibition (by imatinib), upregulation was observed in all three
miRNAs indicating that BCR-ABL does not play a role in repressing
these miRNAs. Notably, the analysis identified CML disease as
possibly associated with major deregulation of these miRNAs
(71). In addition, miR-130a
expression was also found to be regulated by BCR-ABL in K562 cells.
Small interference (si)RNA knockdown of BCR-ABL in K562 cells,
decreased miR-130a and miR-130b, and increased the expression of
their putative target, the growth negative regulator CCN3 (72). Another study identified miR-96
upregulation, and miR-120, miR-151 and miR-10 downregulation in
CML. Conversely, miR-10 downregulation was shown to be independent
to BCR-ABL signalling (62).
Consistent with these data, a recent study demonstrated that the
expression of miR-424 is markedly low in CML cell lines and patient
samples at the time of diagnosis. In addition, with the aid of
bioinformatic analysis and via luciferase assays, the authors
revealed a conserved target site for miR-424 in the 3′UTR of the
ABL gene, showing that this miRNA directly targets BCR-ABL. In
fact, its overexpression was able to induce apoptosis of K562 cells
as well as sensitize these cells to imatinib treatment. These data
suggest that miR-424 acts as a tumor suppressor by downregulating
BCR-ABL expression (73).
Furthermore, Fallah et al found differential expression of
miRNAs, derived from leukocytes in the peripheral blood of 50 newly
diagnosed CML patients in chronic phase using stem-loop reverse
transcription-polymerase chain reaction. Some onco-miRNAs were
found to be downregulated (miR-155 and miR-106), and some
tumor-suppressor miRs (miR-16-1, miR-15a, miR-101 and miR-568) were
upregulated. These results showed that few miRNAs alone are good
candidates for CML diagnosis independently of conflicting results,
but together could be an additional tool for CML diagnosis
(74).
CML treatment has been revolutionized by TKIs most
notably imatinib, which acts by inhibiting BCR-ABL. A significant
number of patients, however, suffer from imatinib resistance
(75). For example, miR-146
upregulation targets members of the nuclear-factor-κ-B (NF-κB)
pathway [interleukin-1-associated kinase 1
(IRAK1)/tumor-necrosis-factor -receptor-associated factor 6
(TRAF6)] that are found to be constitutively activated in CML by
BCR-ABL (76). Furthermore,
inhibition of the NF-κB pathway in CML results in apoptosis,
suggesting that the upregulation of miR-146 post-imatinib treatment
makes CML cells more sensitive to apoptotic signaling (77). miR-199-5b was found to be a
regulator of the Notch pathway through its targeting of the
transcription factor Hes1 and it may promote cancer growth in CML,
inhibiting Hes1 (70). Xu et
al showed the feedback regulation among BCR-ABL, GATA1
transcription factor and miR-138. In fact, these authors
demonstrated that overexpression of miR-138 represses BCR-ABL and
cyclin D3 (CCND3) by binding to the coding and 3′UTR regions,
respectively (78). Notably,
miR-138 expression is increased by GATA1, and repressed by BCR-ABL
in addition to imatinib resistance in CML (78). Turrini et al found that
miR-212 increased the ATP-binding cassette subfamily G member 2
(ABCG2) expression upon treatment with imatinib in CML (79). Another recent study showed that
imatinib markedly inhibits expression of miR-30a in human CML
cells. This miRNA is a potent inhibitor of autophagy by
downregulating Beclin-1 and autophagy protein 5 (ATG5) expression
and miR-30a mimic enhances imatinib-induced cytotoxicity and
promotes mitochondrial-dependent intrinsic apoptosis. In contrast,
knockdown of miR-30a by antagomir-30a increases the expression of
Beclin-1 and ATG5, and inhibits imatinib-induced cytotoxicity.
These findings indicate that dysregulation of miR-30a may interfere
with the effectiveness of imatinib-mediated apoptosis by an
autophagy-dependent pathway, thus representing a novel potential
therapeutic target in CML (80).
Different studies were performed to find a relationship between
epigenetic dysregulation of miRNAs and CML. A recent study found
that 48 miRNAs of CpG-rich 212 miRNAs were upregulated after
imatinib treatment via microarray analysis. In particular, imatinib
induced the demethylation of the miR-203 promoter region, resulting
in low expression of targeted BCR-ABL gene, and loss of
proliferation of leukemic cells (81). Recent evidence indicates that the
miRNA expression could be linked to the onset of resistance to
TKIs. In this context, a study demonstrated the relationship among
expression changes of specific miRNAs and resistance to imatinib or
responsiveness to imatinib after treatment in CML patients.
Nineteen miRNAs differentially expressed between resistant and
responder patients (imatinib) were identified: 18 of them were
downregulated (hsa-miR-7, hsa-miR-23a, hsa-miR-26a, hsa-miR-29a,
hsa-miR-29c, hsa-miR-30b, hsa-miR-30c, hsa-miR-100, hsa-miR-126#,
hsa-miR-134, hsa-miR-141, hsa-miR-183, hsa-miR-196b, hsa-miR-199a,
hsa-miR-224, hsa-miR-326, hsa-miR-422b and hsa-miR-520a) and only
one was upregulated (hsa-miR-191) in resistant CML patients
(82). Lopotová et al
(83) and Scholl et al
(84) reported that CML patients
with imatinib-resistance showed lower levels of miR-451 compared
with responders. In particular, the increased levels of 13 miRNAs
(miR-19a, miR-19b, miR-17, miR-20, miR-92a, miR-106a, miR-221,
miR-222, miR-126, miR-146a, miR-181a, miR-181b, let7c and miR-55)
and decreased levels of 4 miRNAs (miR-103, miR-150, miR-451 and
miR-144) in PBMCs from Blast Crisis (BC)-CML patients were used to
uncover signaling pathways and their role in CML (83,84).
Several miRNAs (e.g. miR-191, miR-29a, miR-422b, miR-100, miR-326
and miR-26a) are promising predictors of imatinib resistance in
newly diagnosed CML patients. A study suggested that miR-181b and
miR181d are associated with myeloid cell leukemia-1 (Mcl 1) in
Lyn-mediated imatinib-resistant CML cells. Incubation of these
cells with a Lyn inhibitor (dasatinib) increased miR-181b and
miR-181d expression (85). In line
with this study, Liu et al (86) described a reciprocal regulatory
correlation between c-Myc and miRNA-144/451. In particular, Myc is
upregulated in imatinib-resistant K562 cells, where it inhibited
miRNA-144/451 transcription. Additionally, You et al
analyzed the circulating miRNA profile in the culture supernatant
of imatinib-resistant K562 CML cells (K562-R) by microarray chip
analysis. They found that specific miRNAs are associated with
nilotinib sensitivity by comparison of the miRNA expression
patterns from the culture supernatant of nilotinib-treated K562-R
cells with the culture supernatant of untreated K562-R cells
(miRNA-221, -379, -548, -603 and -648). The information obtained
from this study may have the potential to identify a novel
biomarker to predict drug response in the future (87). In another study, the authors
examined the expression levels of miR-17 which possesses oncogenic
activities through downregulation of cyclin-dependent kinase N1A
(CDKN1A), p21 and E2 transcription factor 1 (E2F1) tumor suppressor
genes, in imatinib-sensitive and -resistant K562 cells compared to
PBMCs derived from healthy donors by stem-loop RT-PCR. A
significant decrease was observed in miR-17 levels in response to
imatinib, dasatanib and nilotinib in K562 IM-R cells. These data
proved that miR-17 may be a crucial target for the treatment of CML
(88). Consistent with these data,
Mosakhani et al performed miRNA microarray (followed by
qRT-PCR verification) on available diagnostic bone marrow core
biopsies derived from CML patients including 4 imatinib-resistant
and 5 imatinib-responder CML patients. They found a significant
downregulation of miR-181c in imatinib-resistant vs.
imatinib-responders and some miR-181c target genes such as pre-B
cell leukemia homeobox 3 (pre-PBX3), heat shock protein 90 kDa β
member 1 (HSP90B1), N-myristoyltransferase 2 (NMT2) and RAD21 were
associated with the drug response (89). Finally, Ohyashiki et al
identified downregulation of miR-148b in patients of the STOP-IM
group and in a subset of the imatinib group (90). In this context, a recent study found
both new and differential expression of miRNAs in CD34+
CML stem/progenitor cells obtained at diagnosis from bone marrow of
CML patient IM-responders, IM-non-responders after imatinib
therapy, and of healthy donors. Bioconductor Illumina deep
sequencing (DESeq) analysis revealed 63 differentially expressed
miRNAs in the CD34+ cells from CML and healthy donor
samples. Notably, 12 of these were differentially expressed in
CD34+ cells from the IM-responders and non-responders.
Most of the 63 differentially expressed miRNAs identified were
present at reduced levels in the CD34+ CML cells as
compared to cells derived from healthy donors, while 17 miRNAs were
increased. In addition, 34 novel miRNAs were identified in the
CD34+ CML stem/progenitor cells. The authors, next,
validated the sequencing data in CD34+ cells from
IM-responders and IM-non-responders and normal individuals using a
high-throughput quantitative microfluidic device. This study
confirmed the differential expression in CD34+ CML cells
of 32 of the 63 identified miRNAs, including an increased level of
oncomiRs miR-155 and miR-17-92, and a decreased level of the tumor
suppressors, miR-145, miR-151 and miR-452. Importantly, the authors
detected significant changes in some of these miRNAs in
CD34+ cells from CML patients after three months of
nilotinib treatment (23 normalized after three months of nilotinib
treatment, whereas 10 showed little change). To further correlate
miRNA profiles with corresponding mRNA expression changes and to
identify potential target genes, RNA-sequencing was performed on
the same RNA samples. Differentially expressed mRNAs (1,210) were
identified that were predicted targets of the deregulated miRNAs in
the comparison of CD34 cells derived from CML patients and normal
individuals. Strikingly, only seven differentially expressed mRNAs
were identified from comparison of the IM-responders and
non-responders. Most of these were predicted to have roles in
regulation of the cell cycle, mitogen-activated protein kinase
(MAPK) signaling and transforming growth factor-β (TGF-β) signaling
pathways. Thus, aberrant, differentially expressed miRNAs and
target genes identified in primitive CML stem/progenitor cells may
serve as useful biomarkers to predict clinical response of CML
patients to TKI therapy (91).
Additionally, since the miR-219-2 and miR-199b (centromeric to the
ABL1 gene) are frequently lost in CML patients, a study analyzed
150 CML patients in order to identify 9q deletion. Fluorescent
in situ hybridization (FISH) (BCR-ABL dual color) revealed
9q34.1 deletion in 34 (23%) CML patients. The expression level of
miRNA, analyzed by real-time polymerase chain reaction (RT-PCR),
showed downregulation of miR-199b and miR-219-2 in the 9q deleted
patients (34 CML) as compared to a pool of patients without
deletion. However, miR-199b (9q34.11) was significantly
downregulated compared to miR-219-2. The follow-up study showed
that miR-199b was found to be strongly associated with imatinib
resistance, as 44.11% patients showed resistance to imatinib
therapy. Hence, the deletion in the 9q34.1 region (ABL) plays an
important role in disease pathogenesis (92). Finally, Nishioka et al found
that long-term exposure of K562 cells to BCR-ABL TKI caused
drug-resistance in association with an increase in levels of DNA
methyltransferases (DNMTs) and a decrease in levels of miR-217. In
addition, an increase in levels of DNMT3A in association with
downregulation of miR-217 were observed in leukemia cells isolated
from individuals with BCR-ABL TKI-resistant Philadelphia-positive
ALL and CML. Further studies with TKI-resistant K562 cells found
that forced expression of miR-217 inhibited expression of DNMT3A
through a miR-217-binding site within the 3′ untranslated region of
DNMT3A and sensitized these cells to growth inhibition mediated by
the TKI. Notable, long-term exposure of K562 cells to dasatinib in
combination with 5-Aza-2′-deoxycytidine (5-Aza-dC) potently
inhibited proliferation of these cells in association with
upregulation of miR-217 and downregulation of DNMT3A in
vitro. Furthermore, a decrease in levels of DNMT3A and an
increase in levels of miR-217 were noted in K562 tumors in
immune-deficient mice that were treated with the combination of
5-Aza-dC and dasatinib. Taken together, Ph+ leukemia
cells acquired TKI resistance via downregulation of miR-217 and
upregulation of DNMT3A (93).
4. Conclusion
A decade after its discovery, miRNAs are rapidly
entering the clinic as biomarkers and therapeutic tools. The fact
that miRNAs can regulate the expression of multiple genes makes
them attractive drug targets, as multiple oncogenes can be
inhibited at the same time. However, the fact that miRNAs can
regulate multiple genes is also a major drawback, due to the risk
of unwanted side-effects. To reduce side-effects, tumor-specific
delivery may be necessary. miRNAs, that can act as oncogenes or
tumor suppressor genes in CML, contribute to the pathogenesis,
disease progression, and response to therapy of CML and resistance
to TKIs. The potential of using these small RNAs as therapeutic
targets opens up new opportunities for leukemia therapy by either
inhibiting or augmenting their activity. In conclusion, with
miRNA-based therapeutics entering early phase human clinical
trials, hope for a novel class of effective anticancer agents may
be realized.
References
|
1
|
Faderl S, Talpaz M, Estrov Z, O'Brien S,
Kurzrock R and Kantarjian HM: The biology of chronic myeloid
leukemia. N Engl J Med. 341:164–172. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kantarjian HM, Dixon D, Keating MJ, Talpaz
M, Walters RS, McCredie KB and Freireich EJ: Characteristics of
accelerated disease in chronic myelogenous leukemia. Cancer.
61:1441–1446. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kantarjian HM, Deisseroth A, Kurzrock R,
Estrov Z and Talpaz M: Chronic myelogenous leukemia: A concise
update. Blood. 82:691–703. 1993.PubMed/NCBI
|
|
4
|
Rudkin CT, Hungerford DA and Nowell PC:
DNA contents of chromosome Ph1 and chromosome 21 in
human chronic granulocytic leukemia. Science. 144:1229–1231. 1964.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Druker BJ: Translation of the Philadelphia
chromosome into therapy for CML. Blood. 112:4808–4817. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Holyoake DT: Recent advances in the
molecular and cellular biology of chronic myeloid leukaemia:
Lessons to be learned from the laboratory. Br J Haematol.
113:11–23. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Asimakopoulos FA, Shteper PJ, Krichevsky
S, Fibach E, Polliack A, Rachmilewitz E, Ben-Neriah Y and
Ben-Yehuda D: ABL1 methylation is a distinct molecular event
associated with clonal evolution of chronic myeloid leukemia.
Blood. 94:2452–2460. 1999.PubMed/NCBI
|
|
8
|
Mancini M, Veljkovic N, Leo E, Aluigi M,
Borsi E, Galloni C, Iacobucci I, Barbieri E and Santucci MA:
Cytoplasmatic compartmentalization by Bcr-Abl promotes TET2
loss-of-function in chronic myeloid leukemia. J Cell Biochem.
113:2765–2774. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang Q, Yang Y, Li X and Huang S:
Transcription suppression of SARI (suppressor of AP-1, regulated by
IFN) by BCR-ABL in human leukemia cells. Tumour Biol. 32:1191–1197.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bixby D and Talpaz M: Mechanisms of
resistance to tyrosine kinase inhibitors in chronic myeloid
leukemia and recent therapeutic strategies to overcome resistance.
HHematology Am Soc Hematol Educ Program. 1:461–476. 2009.
View Article : Google Scholar
|
|
11
|
Mughal A, Aslam HM, Khan AM, Saleem S,
Umah R and Saleem M: Bcr-Abl tyrosine kinase inhibitors - current
status. Infect Agent Cancer. 8(23)2013. View Article : Google Scholar
|
|
12
|
Asaki T, Sugiyama Y, Hamamoto T,
Higashioka M, Umehara M, Naito H and Niwa T: Design and synthesis
of 3-substituted benzamide derivatives as Bcr-Abl kinase
inhibitors. Bioorg Med Chem Lett. 16:1421–1425. 2006. View Article : Google Scholar
|
|
13
|
Eck MJ and Manley PW: The interplay of
structural information and functional studies in kinase drug
design: Insights from BCR-Abl. Curr Opin Cell Biol. 21:288–295.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
An X, Tiwari AK, Sun Y, Ding P-R, Ashby CR
Jr and Chen ZS: BCR-ABL tyrosine kinase inhibitors in the treatment
of Philadelphia chromosome positive chronic myeloid leukemia: A
review. Leuk Res. 34:1255–1268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Stein B and Smith BD: Treatment options
for patients with chronic myeloid leukemia who are resistant to or
unable to tolerate imatinib. Clin Ther. 32:804–820. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Thomas J, Wang L, Clark RE and Pirmohamed
M: Active transport of imatinib into and out of cells: Implications
for drug resistance. Blood. 104:3739–3745. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jabbour E, Cortes J and Kantarjian H:
Nilotinib for the treatment of chronic myeloid leukemia: An
evidence-based review. Core Evid. 4:207–213. 2009. View Article : Google Scholar
|
|
18
|
Manley PW, Cowan-Jacob SW and Mestan J:
Advances in the structural biology, design and clinical development
of Bcr-Abl kinase inhibitors for the treatment of chronic myeloid
leukaemia. Biochim Biophys Acta. 1754:3–13. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Olivieri A and Manzione L: Dasatinib: A
new step in molecular target therapy. Ann Oncol. 18(Suppl 6):
vi42–vi46. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
O'Hare T, Shakespeare WC, Zhu X, Eide CA,
Rivera VM, Wang F, Adrian LT, Zhou T, Huang WS, Xu Q, et al:
AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia,
potently inhibits the T315I mutant and overcomes mutation-based
resistance. Cancer Cell. 16:401–412. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
FDA Drug Safety Communication: FDA asks
manufacturer of the leukemia drug Iclusig (ponatinib) to suspend
marketing and sales. U.S. Food and Drug Administration. 2013,
http://www.fda.gov/Drugs/DrugSafety/ucm373040.htm.
Accessed Nov 26, 2013.
|
|
22
|
Lipshultz SE, Diamond MB, Franco VI,
Aggarwal S, Leger K, Santos MV, Sallan SE and Chow EJ: Managing
chemotherapy-related cardiotoxicity in survivors of childhood
cancers. Paediatr Drugs. 16:373–389. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kimura S, Naito H, Segawa H, Kuroda J,
Yuasa T, Sato K, Yokota A, Kamitsuji Y, Kawata E, Ashihara E, et
al: NS-187, a potent and selective dual Bcr-Abl/Lyn tyrosine kinase
inhibitor, is a novel agent for imatinib-resistant leukemia. Blood.
106:3948–3954. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Horio T, Hamasaki T, Inoue T, Wakayama T,
Itou S, Naito H, Asaki T, Hayase H and Niwa T: Structural factors
contributing to the Abl/Lyn dual inhibitory activity of
3-substituted benzamide derivatives. Bioorg Med Chem Lett.
17:2712–2717. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Valent P: Standard treatment of
Ph+ CML in 2010: How, when and where not to use what
BCR/ABL1 kinase inhibitor? Eur J Clin Invest. 40:918–931. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Peláez N and Carthew RW: Biological
robustness and the role of microRNAs: A network perspective. Curr
Top Dev Biol. 99:237–255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nam JW, Rissland OS, Koppstein D,
Abreu-Goodger C, Jan CH, Agarwal V, Yildirim MA, Rodriguez A and
Bartel DP: Global analyses of the effect of different cellular
contexts on microRNA targeting. Mol Cell. 53:1031–1043. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Di Leva G, Garofalo M and Croce CM:
MicroRNAs in cancer. Annu Rev Pathol. 9:287–314. 2014. View Article : Google Scholar :
|
|
29
|
Lee R, Feinbaum R and Ambros V: The
heterochronic gene lin-4 of C. elegans encodes small RNAs with
antisense complementarity to lin-14. Cell. 75:843–854. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kozomara A and Griffiths-Jones S: miRBase:
Integrating microRNA annotation and deep-sequencing data. Nucleic
Acids Res. 39:D152–D157. 2011. View Article : Google Scholar :
|
|
31
|
Tsai LM and Yu D: MicroRNAs in common
diseases and potential therapeutic applications. Clin Exp Pharmacol
Physiol. 37:102–107. 2010. View Article : Google Scholar
|
|
32
|
Esquela-Kerscher A and Slack FJ: Oncomirs
- microRNAs with a role in cancer. Nat Rev Cancer. 6:259–269. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Masaki S, Ohtsuka R, Abe Y, Muta K and
Umemura T: Expression patterns of microRNAs 155 and 451 during
normal human erythropoiesis. Biochem Biophys Res Commun.
364:509–514. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang Y and Blelloch R: Cell cycle
regulation by microRNAs in embryonic stem cells. Cancer Res.
69:4093–4096. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Subramanian S and Steer CJ: MicroRNAs as
gatekeepers of apoptosis. J Cell Physiol. 223:289–298.
2010.PubMed/NCBI
|
|
36
|
Schwarz DS, Hutvágner G, Du T, Xu Z,
Aronin N and Zamore PD: Asymmetry in the assembly of the RNAi
enzyme complex. Cell. 115:199–208. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Berezikov E, Chung WJ, Willis J, Cuppen E
and Lai EC: Mammalian mirtron genes. Mol Cell. 28:328–336. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hinske LC, Galante PA, Kuo WP and
Ohno-Machado L: A potential role for intragenic miRNAs on their
hosts' interactome. BMC Genomics. 11:533–541. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bagga S, Bracht J, Hunter S, Massirer K,
Holtz J, Eachus R and Pasquinelli AE: Regulation by let-7 and lin-4
miRNAs results in target mRNA degradation. Cell. 122:553–563. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ørom UA, Nielsen FC and Lund AH:
MicroRNA-10a binds the 5′UTR of ribosomal protein mRNAs and
enhances their translation. Mol Cell. 30:460–471. 2008. View Article : Google Scholar
|
|
41
|
Vasudevan S, Tong Y and Steitz JA:
Switching from repression to activation: microRNAs can up-regulate
translation. Science. 318:1931–1934. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pillai RS, Bhattacharyya SN, Artus CG,
Zoller T, Cougot N, Basyuk E, Bertrand E and Filipowicz W:
Inhibition of translational initiation by let-7 MicroRNA in human
cells. Science. 309:1573–1576. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hendrickson DG, Hogan DJ, McCullough HL,
Myers JW, Herschlag D, Ferrell JE and Brown PO: Concordant
regulation of translation and mRNA abundance for hundreds of
targets of a human microRNA. PLoS Biol. 7:e10002382009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Choe J, Cho H, Lee HC and Kim YK:
microRNA/Argonaute 2 regulates nonsense-mediated messenger RNA
decay. EMBO Rep. 11:380–386. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cuccato G, Polynikis A, Siciliano V,
Graziano M, di Bernardo M and di Bernardo D: Modeling RNA
interference in mammalian cells. BMC Syst Biol. 5:19–24. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hemida MG, Ye X, Thair S and Yang D:
Exploiting the therapeutic potential of microRNAs in viral
diseases: Expectations and limitations. Mol Diagn Ther. 14:271–282.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bader AG, Brown D and Winkler M: The
promise of microRNA replacement therapy. Cancer Res. 70:7027–7030.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen CZ: MicroRNAs as oncogenes and tumor
suppressors. N Engl J Med. 353:1768–1771. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lu J, Getz G, Miska EA, Alvarez-Saavedra
E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA,
et al: MicroRNA expression profiles classify human cancers. Nature.
435:834–838. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang H, Luo XQ, Zhang P, Huang LB, Zheng
YS, Wu J, Zhou H, Qu LH, Xu L and Chen YQ: MicroRNA patterns
associated with clinical prognostic parameters and CNS relapse
prediction in pediatric acute leukemia. PLoS One. 4:e78262009.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Labbaye C and Testa U: The emerging role
of MIR-146A in the control of hematopoiesis, immune function and
cancer. J Hematol Oncol. 5(13)2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Calin GA, Ferracin M, Cimmino A, Di Leva
G, Shimizu M, Wojcik SE, Iorio MV, Visone R, Sever NI, Fabbri M, et
al: A MicroRNA signature associated with prognosis and progression
in chronic lymphocytic leukemia. N Engl J Med. 353:1793–1801. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Garzon R, Volinia S, Liu CG,
Fernandez-Cymering C, Palumbo T, Pichiorri F, Fabbri M, Coombes K,
Alder H, Nakamura T, et al: MicroRNA signatures associated with
cytogenetics and prognosis in acute myeloid leukemia. Blood.
111:3183–3189. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gimenes-Teixeira HL, Lucena-Araujo AR, Dos
Santos GA, Zanette DL, Scheucher PS, Oliveira LC, Dalmazzo LF,
Silva-Júnior WA, Falcão RP and Rego EM: Increased expression of
miR-221 is associated with shorter overall survival in T-cell acute
lymphoid leukemia. Exp Hematol Oncol. 2(10)2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Jongen-Lavrencic M, Sun SM, Dijkstra MK,
Valk PJ and Löwenberg B: MicroRNA expression profiling in relation
to the genetic heterogeneity of acute myeloid leukemia. Blood.
111:5078–5085. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fernando TR, Rodriguez-Malave NI and Rao
DS: MicroRNAs in B cell development and malignancy. J Hematol
Oncol. 5(7)2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Calin GA, Dumitru CD, Shimizu M, Bichi R,
Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, et al:
Frequent deletions and down-regulation of micro-RNA genes miR15 and
miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci
USA. 99:15524–15529. 2002. View Article : Google Scholar
|
|
58
|
Cimmino A, Calin GA, Fabbri M, Iorio MV,
Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, et
al: miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl
Acad Sci USA. 102:13944–13949. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Xu L, Xu Y, Jing Z, Wang X, Zha X, Zeng C,
Chen S, Yang L, Luo G, Li B, et al: Altered expression pattern of
miR-29a, miR-29b and the target genes in myeloid leukemia. Exp
Hematol Oncol. 3(17)2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Seca H, Lima RT, Lopes-Rodrigues V,
Guimaraes JE, Almeida GM and Vasconcelos MH: Targeting miR-21
induces autophagy and chemosensitivity of leukemia cells. Curr Drug
Targets. 14:1135–1143. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Calin GA, Sevignani C, Dumitru CD, Hyslop
T, Noch E, Yendamur S, Shimizu M, Rattan S, Bullrich F, Negrini M,
et al: Human microRNA genes are frequently located at fragile sites
and genomic regions involved in cancers. Proc Natl Acad Sci USA.
101:2999–3004. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Agirre X, Jiménez-Velasco A, San
José-Enériz E, Garate L, Bandrés E, Cordeu L, Aparicio O, Saez B,
Navarro G, Vilas-Zornoza A, et al: Down-regulation of hsa-miR-10a
in chronic myeloid leukemia CD34+ cells increases
USF2-mediated cell growth. Mol Cancer Res. 6:1830–1840. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wang LS, Li L, Li L, Chu S, Shiang KD, Li
M, Sun HY, Xu J, Xiao FJ, Sun G, et al: MicroRNA-486 regulates
normal eryth-ropoiesis and enhances growth and modulates drug
response in CML progenitors. Blood. 125:1302–1313. 2015. View Article : Google Scholar :
|
|
64
|
Babashah S, Sadeghizadeh M, Hajifathali A,
Tavirani MR, Zomorod MS, Ghadiani M and Soleimani M: Targeting of
the signal transducer Smo links microRNA-326 to the oncogenic
Hedgehog pathway in CD34+ CML stem/progenitor cells. Int
J Cancer. 133:579–589. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Venturini L, Battmer K, Castoldi M,
Schultheis B, Hochhaus A, Muckenthaler MU, Ganser A, Eder M and
Scherr M: Expression of the miR-17-92 polycistron in chronic
myeloid leukemia (CML) CD34+ cells. Blood.
109:4399–4405. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Eiring AM, Harb JG, Neviani P, Garton C,
Oaks JJ, Spizzo R, Liu S, Schwind S, Santhanam R, Hickey CJ, et al:
miR-328 functions as an RNA decoy to modulate hnRNP E2 regulation
of mRNA translation in leukemic blasts. Cell. 140:652–665. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Chang JS, Santhanam R, Trotta R, Neviani
P, Eiring AM, Briercheck E, Ronchetti M, Roy DC, Calabretta B,
Caligiuri MA, et al: High levels of the BCR/ABL oncoprotein are
required for the MAPK-hnRNP E2 dependent suppression of
C/EBPalpha-driven myeloid differentiation. Blood. 110:994–1003.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Perrotti D, Cesi V, Trotta R, Guerzoni C,
Santilli G, Campbell K, Iervolino A, Condorelli F,
Gambacorti-Passerini C, Caligiuri MA, et al: BCR-ABL suppresses
C/EBPalpha expression through inhibitory action of hnRNP E2. Nat
Genet. 30:48–58. 2002. View
Article : Google Scholar
|
|
69
|
Machová Poláková K, Lopotová T, Klamová H,
Burda P, Trněný M, Stopka T and Moravcová J: Expression patterns of
microRNAs associated with CML phases and their disease related
targets. Mol Cancer. 10(41)2011. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Flamant S, Ritchie W, Guilhot J, Holst J,
Bonnet ML, Chomel JC, Guilhot F, Turhan AG and Rasko JE: Micro-RNA
response to imatinib mesylate in patients with chronic myeloid
leukemia. Haematologica. 95:1325–1333. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Rokah OH, Granot G, Ovcharenko A, Modai S,
Pasmanik-Chor M, Toren A, Shomron N and Shpilberg O: Downregulation
of miR-31, miR-155, and miR-564 in chronic myeloid leukemia cells.
PLoS One. 7:e355012012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Suresh S, McCallum L, Lu W, Lazar N,
Perbal B and Irvine AE: MicroRNAs 130a/b are regulated by BCR-ABL
and down-regulate expression of CCN3 in CML. J Cell Commun Signal.
5:183–191. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Hershkovitz-Rokah O, Modai S,
Pasmanik-Chor M, Toren A, Shomron N, Raanani P, Shpilberg O and
Granot G: Restoration of miR-424 suppresses BCR-ABL activity and
sensitizes CML cells to imatinib treatment. Cancer Lett.
360:245–256. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Fallah P, Amirizadeh N, Poopak B, Toogeh
G, Arefian E, Kohram F, Hosseini Rad SM, Kohram M, Teimori Naghadeh
H and Soleimani M: Expression pattern of key microRNAs in patients
with newly diagnosed chronic myeloid leukemia in chronic phase. Int
J Lab Hematol. 37:560–568. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Bhamidipati PK, Kantarjian H, Cortes J,
Cornelison AM and Jabbour E: Management of imatinib-resistant
patients with chronic myeloid leukemia. Ther Adv Hematol.
4:103–117. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Taganov KD, Boldin MP, Chang KJ and
Baltimore D: NF-kappaB-dependent induction of microRNA miR-146, an
inhibitor targeted to signaling proteins of innate immune
responses. Proc Natl Acad Sci USA. 103:12481–12486. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Duncan EA, Goetz CA, Stein SJ, Mayo KJ,
Skaggs BJ, Ziegelbauer K, Sawyers CL and Baldwin AS: IkappaB kinase
beta inhibition induces cell death in imatinib-resistant and T315I
Dasatinib-resistant BCR-ABL+ cells. Mol Cancer Ther.
7:391–397. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Xu C, Fu H, Gao L, Wang L, Wang W, Li J,
Li Y, Dou L, Gao X, Luo X, et al: BCR-ABL/GATA1/miR-138 mini
circuitry contributes to the leukemogenesis of chronic myeloid
leukemia. Oncogene. 33:44–54. 2014. View Article : Google Scholar
|
|
79
|
Turrini E, Haenisch S, Laechelt S, Diewock
T, Bruhn O and Cascorbi I: MicroRNA profiling in K-562 cells under
imatinib treatment: Influence of miR-212 and miR-328 on ABCG2
expression. Pharmacogenet Genomics. 22:198–205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Yu Y, Yang L, Zhao M, Zhu S, Kang R,
Vernon P, Tang D and Cao L: Targeting microRNA-30a-mediated
autophagy enhances imatinib activity against human chronic myeloid
leukemia cells. Leukemia. 26:1752–1760. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Shibuta T, Honda E, Shiotsu H, Tanaka Y,
Vellasamy S, Shiratsuchi M and Umemura T: imatinib induces
demethylation of miR-203 gene: An epigenetic mechanism of
anti-tumor effect of imatinib. Leuk Res. 37:1278–1286. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
San José-Enériz E, Román-Gómez J,
Jiménez-Velasco A, Garate L, Martin V, Cordeu L, Vilas-Zornoza A,
Rodríguez-Otero P, Calasanz MJ, Prósper F, et al: MicroRNA
expression profiling in imatinib-resistant chronic myeloid leukemia
patients without clinically significant ABL1-mutations. Mol Cancer.
8(69)2009. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Lopotová T, Záčková M, Klamová H and
Moravcová J: MicroRNA-451 in chronic myeloid leukemia:
miR-451-BCR-ABL regulatory loop? Leuk Res. 35:974–977. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Scholl V, Hassan R and Zalcberg IR:
miRNA-451: A putative predictor marker of imatinib therapy response
in chronic myeloid leukemia. Leuk Res. 36:119–121. 2012. View Article : Google Scholar
|
|
85
|
Zimmerman EI, Dollins CM, Crawford M,
Grant S, Nana-Sinkam SP, Richards KL, Hammond SM and Graves LM: Lyn
kinase-dependent regulation of miR181 and myeloid cell leukemia-1
expression: Implications for drug resistance in myelogenous
leukemia. Mol Pharmacol. 78:811–817. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Liu L, Wang S, Chen R, Wu Y, Zhang B,
Huang S, Zhang J, Xiao F, Wang M and Liang Y: Myc induced
miR-144/451 contributes to the acquired imatinib resistance in
chronic myelogenous leukemia cell K562. Biochem Biophys Res Commun.
425:368–373. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
You RI, Ho CL, Hung HM and Chao TS:
Identification of nilotinib-altered microRNA expression patterns in
imatinib-resistant chronic myeloid leukemia cells. Biomark Genomic
Med. 5:71–73. 2013. View Article : Google Scholar
|
|
88
|
Firatligil B, Biray Avci C and Baran Y:
miR-17 in imatinib resistance and response to tyrosine kinase
inhibitors in chronic myeloid leukemia cells. J BUON. 18:437–441.
2013.PubMed/NCBI
|
|
89
|
Mosakhani N, Mustjoki S and Knuutila S:
Down-regulation of miR-181c in imatinib-resistant chronic myeloid
leukemia. Mol Cytogenet. 6(27)2013. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Ohyashiki JH, Ohtsuki K, Mizoguchi I,
Yoshimoto T, Katagiri S, Umezu T and Ohyashiki K: Downregulated
microRNA-148b in circulating PBMCs in chronic myeloid leukemia
patients with undetectable minimal residual disease: A possible
biomarker to discontinue imatinib safely. Drug Des Devel Ther.
8:1151–1159. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Lin H, Rothe K, Ruschmann J, Petriv O,
O'Neill K, Knapp D, Brinkman RR, Birol I, Forrest DL, Hansen C, et
al: Identification of new microRNA biomarkers and candidate target
genes in primitive CML cells using global comparative RNA analyses.
3133 Poster AHA; 2014
|
|
92
|
Joshi D, Chandrakala S, Korgaonkar S,
Ghosh K and Vundinti BR: Down-regulation of miR-199b associated
with imatinib drug resistance in 9q34.1 deleted BCR/ABL positive
CML patients. Gene. 542:109–112. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Nishioka C, Ikezoe T, Yang J, Nobumoto A,
Tsuda M and Yokoyama A: Downregulation of miR-217 correlates with
resistance of Ph+ leukemia cells to ABL tyrosine kinase
inhibitors. Cancer Sci. 105:297–307. 2014. View Article : Google Scholar
|