Introduction
Pancreatic cancer is a highly invasive malignant
tumor. Its incidence is roughly close to the mortality rates. At
present, surgical resection is still the first treatment choice by
which to attempt to cure pancreatic cancer. However, only 20% of
patients at the time of diagnosis are resectable; 30–40% of
patients with pancreatic cancer are unresectable, even though the
tumor is confined to the pancreatic area (1). Tumorigenesis is the result of the
interaction between multiple genetic and epigenetic factors and
abnormal gene methylation is one of the significant factors. High
methylation of tumor-suppressor genes results in transcriptional
suppression and even loss of expression; while low methylation may
result in extremely active transcription and uncontrolled
expression that may lead to abnormal differentiation and
proliferation, and the development of cancer (2).
Epigenetic modifications have attracted much
attention in the research on the development of tumors. Epigenetic
modifications mainly include DNA methylation and
hydroxylmethylation, histone methylation and acetylation, chromatin
remodeling, genomic imprinting and RNA interference. The formation
of DNA methylation is catalyzed by DNA methyltransferases (DNMTs),
mainly including DNMT1, DNMT3a and DNMT3b, and 5mC is formed under
catalysis of these enzymes (3). DNA
methylation modification is closely related to transcriptional
inactivation, gene imprinting and X chromosome inactivation
(4). Research evidence has shown
that DNA methylation plays an important role in the development of
many malignant tumors.
Similar to other malignancies, the development and
progression of pancreatic carcinoma is a multistage process that
involves a series of genetic alterations and gene methylation
changes. Research has demonstrated that tumor-suppressor genes P16
and RASSF1A are highly methylated in pancreatic cancer, Ueki et
al (5) and Fukushima (6) et al reported that ppENK gene
methylation levels were increased in more than 90% of pancreatic
cancer cases. Schutte et al (7) reported that the P16 gene was
inactivated in 95% of cases, and 15% of these cases were correlated
with methylation. Moore et al (8) reported that the P16 gene was
methylated in 27% of pancreatic cancer cell lines. Dammann et
al (9) reported RASSF1A gene
methylation levels were increased in 64% of primary pancreatic
ductal carcinoma cells, 83% of pancreatic endocrine tumors and 88%
of pancreatic cancer cell lines. When pancreatic cancer cells were
treated with the demethylation drug 5-Aza-CdR, expression levels of
ppENK, P16 and RASSF1A which were downregulated by methylation
respectively had different degrees of re-expression to exert an
antitumor efficiency. This provided the theoretical basis for
antitumor treatment using demethylation drugs in clinical
study.
5-Aza-CdR is currently one of the most commonly used
demethylation nucleoside analogues (10), and plays a role in methylation
mainly by inhibiting the expression and activity of DNMT under a
low concentration. 5-Aza-CdR was approved by the food and drug
administration (FDA) to be mainly used for the treatment of blood
system tumors. Zhang et al (11) reported that, when pancreatic cancer
PANC-1 cells treated with 1 µmol/l 5-Aza-CdR for 72 h, the
tumor-suppressor genes RASSF1A, P16 and ppENK had different levels
of demethylation. In recent years, Chinese herbal medicines have
been recognized as having antitumor efficacy. Emodin
(1,3,8-trihydroxy-6-methylanthraquinone), one such Chinese herbal
medicine, has extensive pharmacological effects such as immune
regulation, antibacterial, anti-inflammatory and antitumor
activities (12). Liu et al
(13) reported that emodin
inhibited pancreatic cancer cell growth through different modes of
action, yet the detailed mechanism remained unclear. It was
reported that emodin caused a certain degree of demethylation in
pancreatic cancer PANC-1 cells, but the demethylation intensity was
weaker when compared with 5-Aza-CdR.
The etiology of tumors include multiple factors.
Comprehensive treatment is the main treatment mode for tumors at
present. Drug combinations are currently an important strategy for
antitumor treatment. The aim of the present study was to
investigate the demethylation efficacy of emodin in combination
with 5-Aza-CdR on pancreatic cancer PANC-1 cells. It was
demonstrated that emodin combined with 5Aza-CdR enhanced the
demethylation by 5-Aza-CdR alone on tumor-suppressor genes RASSF1A,
P16 and ppENK in pancreatic cancer cells by reducing the expression
of methyltransferases DNMT1 and DNMT3a. This finding provides a new
strategy for the clinical treatment of pancreatic cancer.
Materials and methods
Chemicals and reagents
Emodin (purity ≥98%), 5-Aza-CdR and
dimethylsulfoxide (DMSO) were purchased from Sigma (St. Louis, MO,
USA). Emodin was dissolved in DMSO to create a stock solution at
concentrations of 10 and 20 mmol/l, which were stored at −70°C. The
DMSO concentration was maintained below 0.1% in all of the cell
cultures and did not exert any detectable effect on cell growth or
cell death. The Cell Counting Kit-8 (CCK-8) was purchased from
Gibco. A cell and tissue genomic DNA extraction kit, methylation
and FQ-PCR primers were purchased from Fastagen Biotech (Shanghai,
China). The EpiTect® Bisulfite and EpiTect®
methylation-specific PCR (MSP) kits were purchased from Qiagen. The
RNA extraction kit was purchased from Tiangen (Beijing, China).
SYBR-Green fluorescent dye was purchased from Invitrogen. The
antibodies, anti-RASSF1A and anti-ppENK were purchased from Abcam.
The anti-P16/INK4a antibody and anti-β-actin were purchased from
Epitomics.
Cell line and culture
Human pancreatic cancer cell line PANC-1 was
obtained from the American Type Culture Collection (ATCC; Manassas,
VA, USA). The cells were cultured in Dulbecco's modified Eagle's
medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100
U/ml penicillin and 100 µg/ml streptomycin. Cells were
maintained at 37°C in a humidified atmosphere of 5% CO2.
The medium was changed every 2–3 days, and the cells were
subcultured when confluency reached 70–80% in 0.25% trypsin at
37°C.
Cell proliferation assay
Cell survival was determined using the CCK-8 kit.
Briefly, logarithmic phase PANC-1 cells were plated into 96-well
culture plates (~5×103 cells/well). After 24 h of
incubation, the cells were treated with the vehicle alone (0.1%
DMSO) and various concentrations (10, 20, 40 and 80 µM) of
emodin, followed by a 24-, 48- and 72-h cell culture. Each group
had 6-wells. A total of 10 µl CCK-8 was added to each well
for 1 h before the end of the incubation period. The absorbance at
450 nm was read using the Bio-Tek ELx800 absorbance microplate
reader. The experiment was repeated three times. To determine the
inhibition rate of the drugs on the cells, the following
calculation was performed: Relative % inhibition = 1 − (dosing
absorbance - blank absorbance)/(control absorbance − blank
absorbance) × 100%.
Dot-blot assay
The PANC-1 cells were treated with the optimal
concentration of emodin (40 µM) and 5-Aza-CdR (1 µM),
either alone or in combination for 72 h. The control cells were
treated with 0.1% DMSO only. The total DNA was isolated from the
cultured cells using the cell/tissue genomic DNA extraction kit
according to the manufacturer's instructions. The concentrations of
DNA were determined by fluorometry using the Qubit®
dsDNA HS kit and fluorom-eter (both from Invitrogen). The procedure
for the Dot-blot assay was performed with reference to a previous
study (14). First, DNA in each
group was placed on a nylon membrane (Hybond-N+; GE), and put in
ultraviolet light-emitting instruments for 5 min, after being
blocked for 1.5 h in 5% skim milk and washed with ice-cold
phosphate-buffered saline (PBS) for 5 min. The membrane was then
incubated with the desired primary antibody overnight at 4°C. The
membrane was then treated with the appropriate
peroxidase-conjugated secondary antibody for 1 h at room
temperature, and the immune complexes were observed using an
enhanced chemiluminescence reagent followed by analysis of the grey
value of the dot with ImageJ software.
Methylation-specific PCR (MSP)
The PANC-1 cells were treated with the optimal
concentration of emodin (40 µM) and 5Aza-CdR (1 µM),
either alone or in combination for 72 h. The control cells were
treated with 0.1% DMSO only. Genomic DNA was extracted using the
cell/tissue genomic DNA extraction kit according to the
manufacturer's instructions. When the DNA was concentrated to 1
µg, bisulfite modification of genomic DNA was performed
using the EpiTect® Bisulfite kit. The methylation and
unmethylation primer sequences for P16, RASSF1A and ppENK are shown
in Table I. Bisulfite-modified DNA
(4 µl), the methylation (3 µl) and unmethylation
primer (3 µl), 2X Taq PCR Master Mix (25 µl)
and RNase-free water (17 µl) were added to achieve a final
volume of 50 µl. PCR amplification conditions were as
follows: 95°C for 10 min, 94°C for 30 sec, annealing for 30 sec,
and extension at 72°C for 45 sec; a total of 35 cycles; followed by
a final extension at 72°C for 10 min. A total of 10 µl of
the PCR product was separated using 2.5% agarose gel
electrophoresis including GoldView I type of nucleic acid stain for
45 min, and the results were photographed and analyzed.
 | Table IPrimer sequences for PCR, MSP and
bisulfite sequencing. |
Table I
Primer sequences for PCR, MSP and
bisulfite sequencing.
| Genes | | Primer pairs
(5′-3′) | Product size
(bp) |
|---|
| P16 | F |
GCCGATCCAGGTCATGATGAT | |
| R |
GCATCTATGCGGGCATGGTTA | 300 |
| RASSF1A | F |
TGGGGAGGTGAACTGGGAC | |
| R |
ACACGGCACGCACTTGG | 217 |
| ppENK | F |
GCGGTTCCTGACACTTTGC | |
| R |
GGGTGCTGGTGCCATCTT | 245 |
| DNMT1 | F |
GACCCATCTCTTGAAGGTGGTGTT | |
| R |
CCTCGTCATAACTCTCCACCTGCT | 164 |
| DNMT3a | F |
AGGTGGACCGCTACATTGCC | |
| R |
GAGATGTCCCTCTTGTCACTAACG | 143 |
| P16 | MF |
TTATTAGAGGGTGGGGCGGATCGC | |
| MR |
GACCCCGAACCGCGACCGTAA | 150 |
| UF |
TTATTAGAGGGTGGGGTGGATTGT | |
| UR |
CAACCCCAAACCACAACCATAA | 151 |
| RASSF1A | MF |
GTGTTAACGCGTTGCGTATC | |
| MR |
AACCCCGCGAACTAAAAACGA | 93 |
| UF |
TTTGGTTGGAGTGTGTTAATGTG | |
| UR |
CAAACCCCACAAACTAAAAACAA | 105 |
| ppENK | MF |
TGTGGGGAGTTATCGAGC | |
| MR |
GCCTTCGCGAAAAAAATCG | 96 |
| UF |
TTGTGTGGGGAGTTATTGAGT | |
| UR |
CACCTTCACAAAAAAAATCAATC | 100 |
| P16 | BS-F |
TTGTTGTTTAGGTTGGAGTGTAGTG | |
| BS-R |
TCAAAAACATATATTAATAACAACCATCAA | 256 |
| ppENK | BS-F |
AAAGAGTTTTTGGAAATAGGGGATA | |
| BS-R |
CATCAACAATTTCCCACTAAAAAAT | 241 |
| RASSF1A | BS-F |
GTATGTAAGGGTTGGATGTGTAGAGA | |
| BS-R |
CCCCAAATAAAATCTCCACAAAAATC | 298 |
Bisulfite sequencing PCR (BSP)
The PANC-1 cells were treated with the optimal
concentration of emodin (40 µM) and 5Aza-CdR (1 µM),
either alone or in combination for 72 h. The control cells were
treated with 0.1% DMSO only. Genomic DNA was extracted using the
cell/tissue genomic DNA extraction kit according to the
manufacturer's instructions, which were respectively modified by
sulfite. The modified DNA was used for BSP. The sequences of the
primers for P16, RASSF1A and ppENK are shown in Table I. Template DNA (4 µl),
primers (each 1.5 µl), 2X Taq PCR Master Mix (12.5
µl) and DEPC-H2O (5.5 µl) were added to
achieve a final volume of 25 µl. PCR amplification
conditions were as follows: 95°C for 5 min, 94°C for 30 sec,
annealing for 45 sec, and extension at 72°C for 45 sec; a total of
40 cycles; followed by a final extension at 72°C for 10 min. A
total of 10 µl of the PCR product was separated using 2%
agarose gel electrophoresis, and the results were photographed. BSP
products were extracted from agarose gel including GoldView I type
of nucleic acid stain for 45 min, then purified and sequenced
(ShangHai Maipu Biotechnology Co., Ltd., China). The methylation of
the sample was analyzed using BiQ Analyzer software.
Fluorescent quantitative PCR
(FQ-PCR)
The PANC-1 cells were treated with the optimal
concentration of emodin (40 µM) and 5Aza-CdR (1 µM),
either alone or in combination for 72 h. The control cells were
treated with 0.1% DMSO only. Total RNA was isolated from the cells
using TRIzol reagent according to the manufacturer's instructions.
Quantitative analysis of RNA was carried out by ELISA and the
integrity of the RNA was verified by agarose gel electrophoresis.
For reverse transcriptase analysis, 2 µg of total RNA was
reversely transcribed using the RevertAid™ First Strand cDNA
Synthesis kit. Cell cDNA was diluted three times with DEPC. P16,
RASSF1A, ppENK and GAPDH primers were diluted with 1X TBE buffer at
10 pmol/ml. PCR amplification with 10 µl of the reaction
system [prepared cDNA 1 µl and diluent primers (upstream and
downstream primers each 1 µl), fluorescence PCR water,
SYRB-Green 5 µl] was performed with SYBR-Green PCR Master
Mix-Plus. Results were analyzed with LightCycler 480 software. All
samples were performed in triplicate, and the relative amount of
the target gene was normalized to GAPDH.
Western blot analysis
The PANC-1 cells were treated with the optimal
concentration of emodin (40 µM) and 5Aza-CdR (1 µM),
either alone or in combination for 72 h. The control cells were
treated with 0.1% DMSO only. Total proteins were extracted from the
cells using precooling lysis buffer (RIPA:PMSF=100:1), and placed
on ice for 20 min, and treated with ultrasound (150 W) for 5 sec
for an average of three times. After centrifugation at 120,000 x g
for 5 min at 4°C, the supernatant was collected and the protein
concentration was determined using the BCA protein assay kit
according to the manufacturer's instructions. The protein lysates
(20 µg/lane) were separated on 10% SDS polyacrylamide gel
and transferred onto a nitrocellulose membrane. Each membrane was
blocked with 5% skim milk and then incubated with the indicated
primary antibodies against P16, RASSF1A, ppENK, DNMT1, DNMT3a,
DNMT3b and β-actin overnight at 4°C. Subsequently, the membrane was
incubated with the secondary antibodies, goat anti-rabbit and
anti-mouse IgG conjugated with HRP, for 1 h at room temperature,
and the formed immunocomplex was visualized by enhanced
chemiluminescence reagent and exposed to X-ray film. Quantitative
data are expressed as a percentage of the mean ± standard deviation
(SD) of the relative levels of the objective protein and control
β-actin of each group of cells from three independent
experiments.
Statistical analysis
All results were repeated in at least three separate
experiments. The data are expressed as the means ± SD. Statistical
comparisons were carried out using one-way analysis of variance,
which revealed significant differences between groups, and a
Student's t-test which revealed significant differences between two
sample means. Statistical analyses were carried out using SPSS
version 17.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
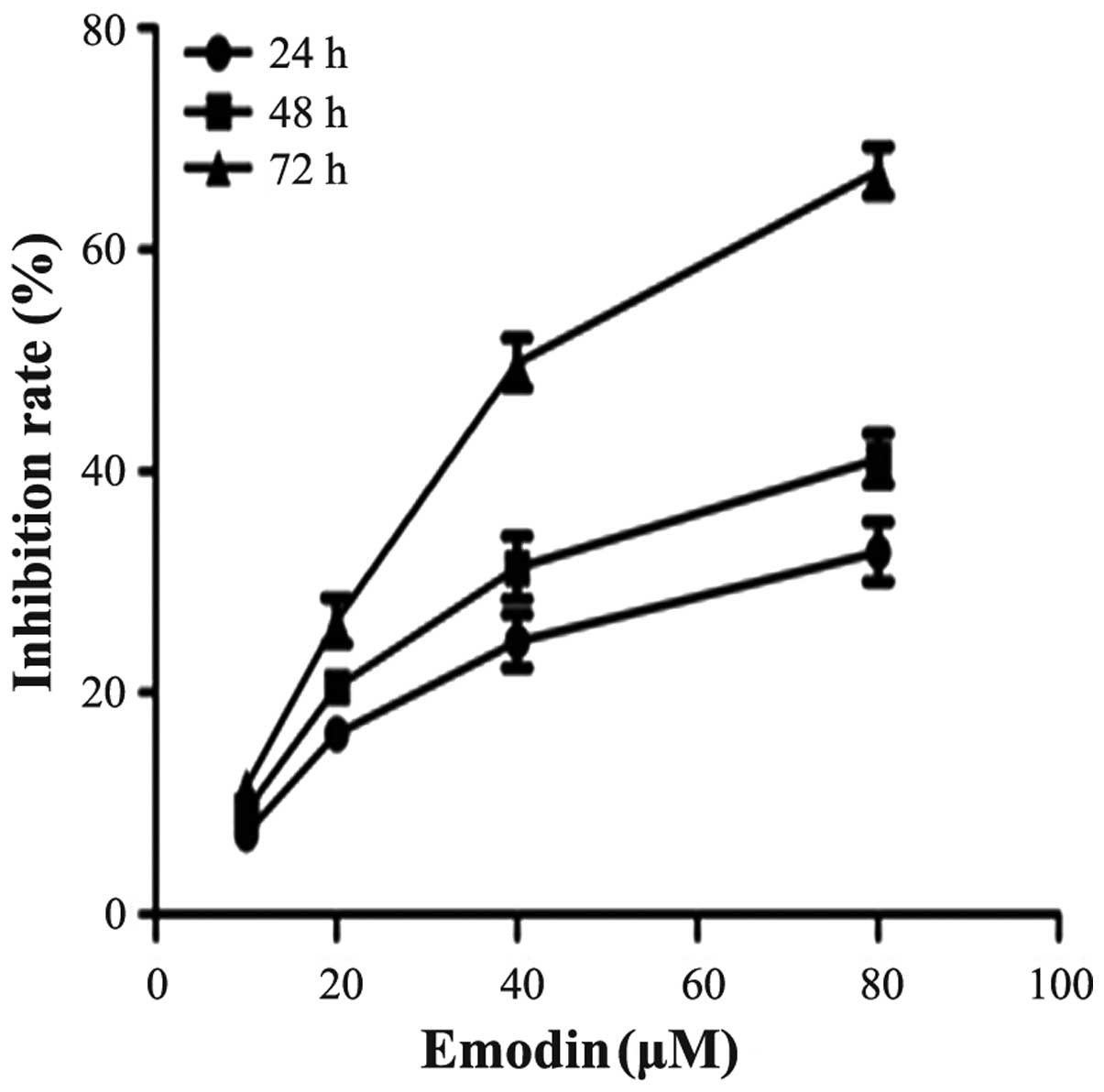
Effect of emodin on PANC-1 cell
proliferation
To investigate the effect of emodin on cell growth,
PANC-1 cells were cultured with 0, 10, 20, 40 and 80 µM
emodin for 24, 48 and 72 h. Cell proliferation was determined by
the CCK-8 assay. As demonstrated in Fig. 1, emodin was shown to inhibit the
growth of the cells in a dose- and time-dependent manner. The
inhibition rate of emodin at a concentration of 40 µM for 72
h was 50.6%, thus 40 µM was close to the half maximal
inhibitory concentration (IC50). This result was similar
to a previous study (11).
Therefore, emodin at a concentration of 40 µM was used for
the following experimental research.
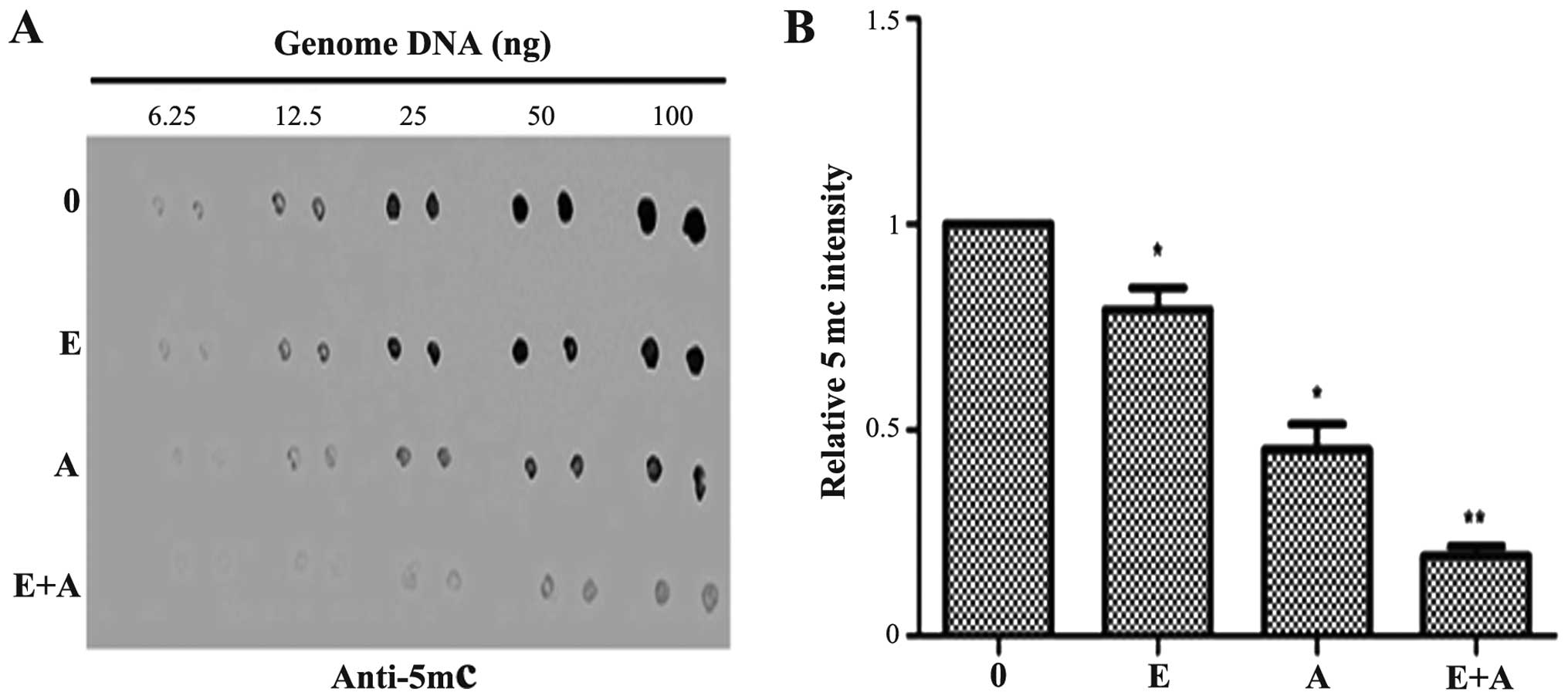
Effects of emodin and 5-Aza-CdR on the
level of genomic 5mC
Dot-blot assay was performed to determine the
effects of emodin (40 µM) and 5-Aza-CdR (1 µM), alone
or in combination on the genomic DNA methylation level in PANC-1
cells. As shown in Fig. 2, emodin
(40 µM) and 5-Aza-CdR (1 µM) exhibited an inhibitory
effect, respectively and reduced the 5mC level as compared with the
level in the control group. The effects observed in the 5-Aza-CdR
group were more evident than that in the emodin group. The 5mC
level was significantly decreased when the PANC-1 cells were
treated with a combination of emodin and 5-Aza-CdR.
Effects of emodin and 5-Aza-CdR on the
methylation of tumor-suppressor genes P16, RASSF1A and ppENK
It was reported that methylation of tumor-suppressor
genes P16, RASSF1A and ppENK played an important role in the
pathogenesis of pancreatic cancer. In order to further clarify the
regulatory effect of emodin alone and in combination with 5-Aza-CdR
on the methylation of these genes, the MSP assay was used to detect
the methylation status of these genes. As shown in Fig. 3, P16, RASSF1A and ppENK presented a
high degree of methylation and a faint demethylation state in the
pancreatic cancer PANC-1 cells. When the PANC-1 cells were treated
with emodin and 5-Aza-CdR alone, the methylation showed different
degrees of decline. Simultaneously, demethylation was enhanced at
different degrees. In addition, the effect of 5-Aza-CdR was
stronger than the effect of emodin. When the PANC-1 cells were
treated with emodin in combination with 5-Aza-CdR, methylation of
the three tumor-suppressor genes P16, RASSF1A and ppENK was
significantly reduced to a greater degree and the demethylation was
significantly enhanced when compared with the effect of emodin or
5-Aza-CdR alone. These data showed that emodin or 5-Aza-CdR alone
exhibited a certain methylation effect on the P16, RASSF1A and
ppENK genes, while the combination of emodin and 5-Aza-CdR had a
significantly enhanced methylation effect on these genes.
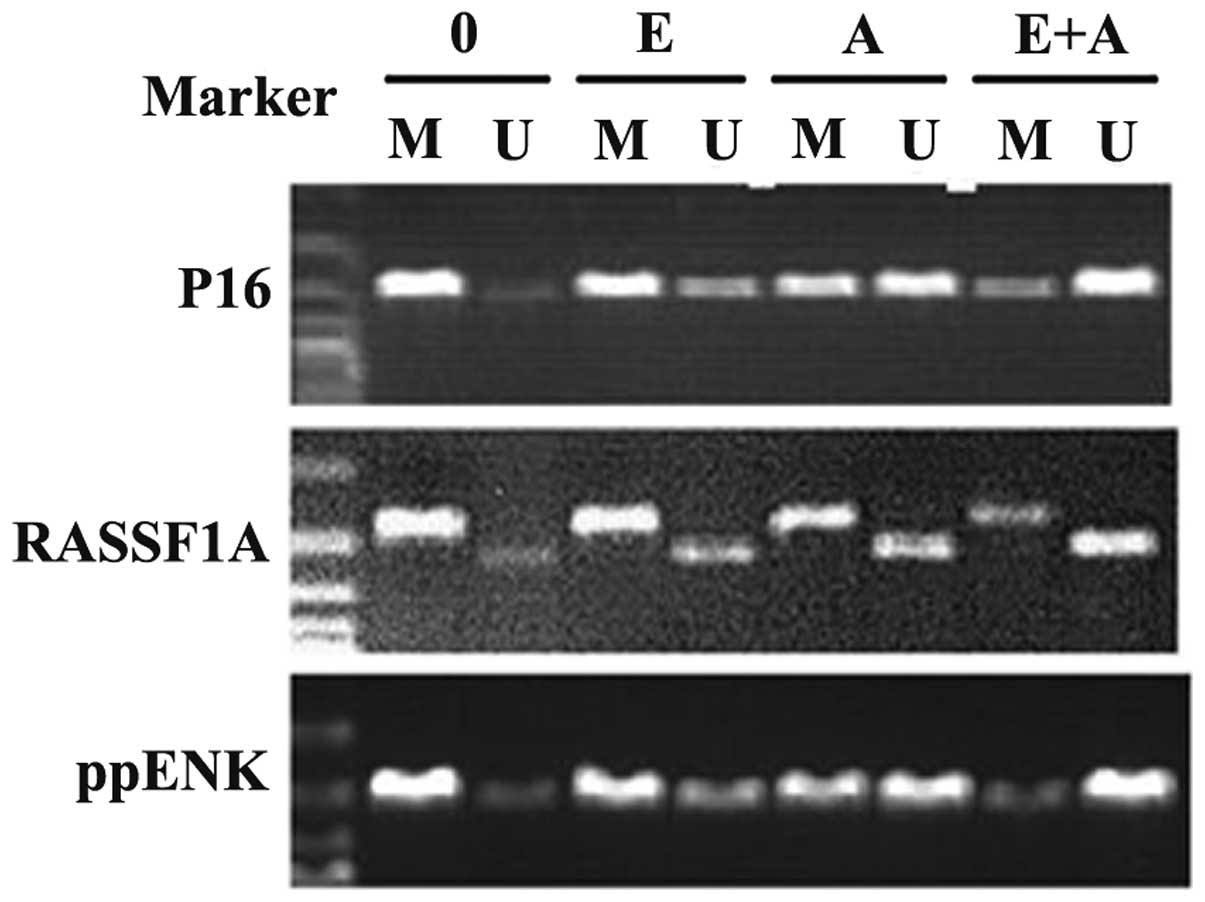 | Figure 3Effect of emodin and 5-Aza-CdR on the
methylation of tumor-suppressor genes P16, RASSF1A, ppENK in PANC-1
cells. The PANC-1 cells were treated with emodin (40 µM) and
5-Aza-CdR (1 µM) alone or in combination for 72 h, and the
methylation of these genes was detected by MSP assay. Emodin and
5-Aza-CdR alone or in combination was observed to weaken the
methylation band and gradually strengthened the demethylation band
of P16, RASSF1A and ppENK. M, methylation; U, demethylation; 0,
control group; E, 40 µM emodin group; A, 1 µM
5-Aza-CdR group; E+A, combination group. |
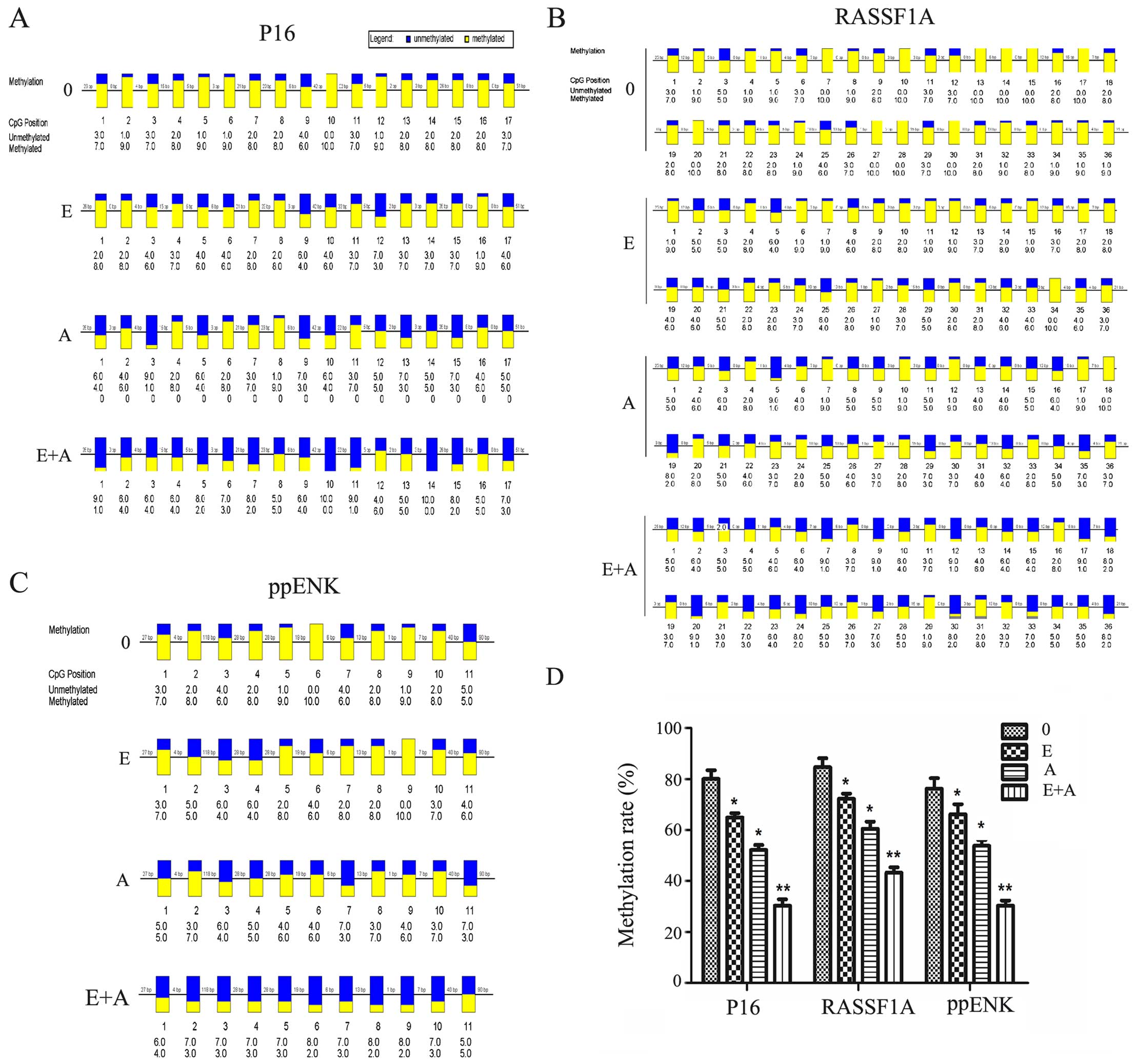
Effects of emodin and 5-Aza-CdR on the
methylation of tumor-suppressor genes P16, RASSF1A and ppENK
In order to verify the reliability of the above
results, the BSP method was used to verify the methylation levels
of the three tumor-suppressor genes P16, RASSF1A and ppENK. As
shown in Fig. 4, when the PANC-1
cells were treated with emodin or 5-Aza-CdR, alone or in
combination for 72 h, the methylation levels of the three
tumor-suppressor genes P16, RASSF1A and ppENK were reduced and the
demethylation levels were increased compared with the controls.
Scopes of P16, ppENK and RASSF1A gene sequencing were respectively
composed of 17, 11 and 36 CpG islands, 10 of which were randomly
selected to clone and sequence. The methylation rates of the P16
gene were 80, 66.5, 51.8 and 30%, respectively; the meth-ylation
rates of the ppENK gene were 76.4, 66.4, 53.6 and 30%,
respectively; the methylation rates of the RASSF1A gene were 85,
72.7, 60.3 and 43.6%, respectively. These results further confirmed
the above results that emodin and 5-Aza-CdR decreased the
methylation levels and increased the demethylation levels of the
promoter region of key tumor-suppressor genes RASSF1A, P16 and
ppENK in pancreatic cancer. The effect was more apparent when a
combination of both were used, indicating that emodin enhanced the
demethylation efficiency of 5-Aza-CdR on tumor-suppressor genes
RASSF1A, P16 and ppENK.
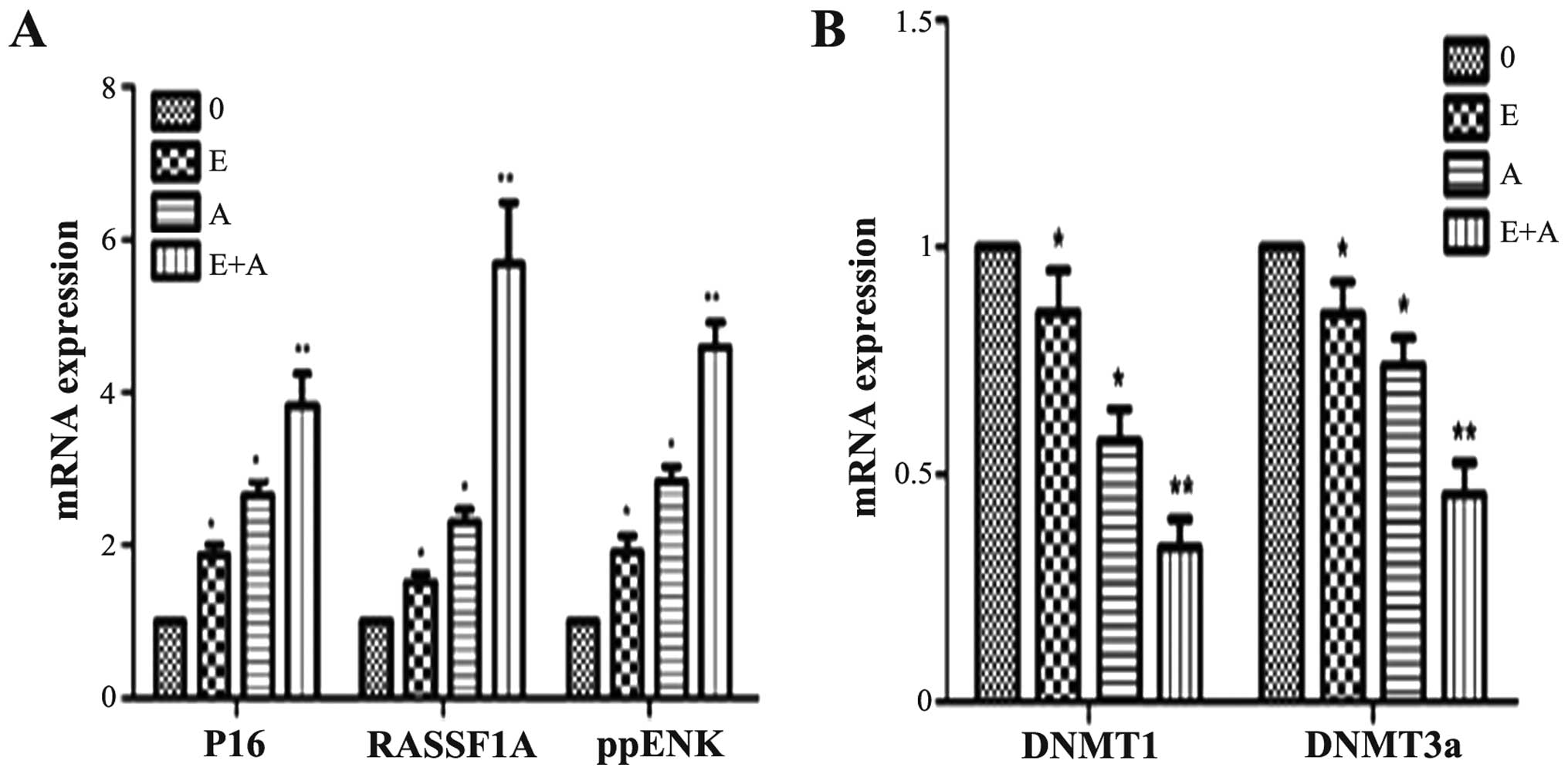
Effects of emodin and 5-Aza-CdR on the
mRNA expression levels of tumor-suppressor genes P16, RASSF1A,
ppENK and DNMTs
In order to demonstrate whether emodin and 5Aza-CdR
have an effect at the transcription level, PCR was used to detect
gene transcription levels of tumor-suppressor genes P16, RASSF1A,
ppENK and DNMTs. As shown in Fig.
5, when the PANC-1 cells were treated with emodin or 5Aza-CdR
alone or in combination for 72 h, compared with the control group,
the expression levels of P16, RASSF1A and ppENK were increased to
different degrees. In addition, the effect of 5-Aza-CdR was
stronger than the effect of emodin. When the PANC-1 cells were
treated with emodin in combination with 5-Aza-CdR, the mRNA
expression levels of P16, RASSF1A and ppENK were more significantly
enhanced when compared with the levels following treatment with
emodin or 5-Aza-CdR alone. In contrast, mRNA expression levels of
DNMT1 and DNMT3a were reduced at different degree. The effect was
the most obvious when a combination was used. However, the mRNA
expression levels of DNMT3b had no change (data not shown).
Effects of emodin and 5-Aza-CdR on the
protein expression levels of tumor-suppressor genes P16, RASSF1A,
ppENK and DNMTs
In order to further demonstrate the effects of
emodin and 5-Aza-CdR at the protein level. western blotting was
used to detect protein levels of P16, RASSF1A, ppENK and DNMTs. As
shown in Fig. 6, when the PANC-1
cells were treated with emodin or 5-Aza-CdR alone or in combination
for 72 h, compared with control group, the protein expression of
P16, RASSF1A and ppENK was significantly increased. When the PANC-1
cells were treated with emodin in combination with 5-Aza-CdR, the
protein expression levels of P16, RASSF1A and ppENK were more
significantly enhanced than these levels following treatment with
emodin or 5-Aza-CdR alone. At the same time, the expression levels
of DNMT1 and DNMT3a were reduced; the expression levels of DNMT1
and DNMT3a were more significantly decreased when a combination of
emodin and 5-Aza-CdR was used when compared to treatment with each
agent alone. These findings were consistent with the FQ-PCR
results.
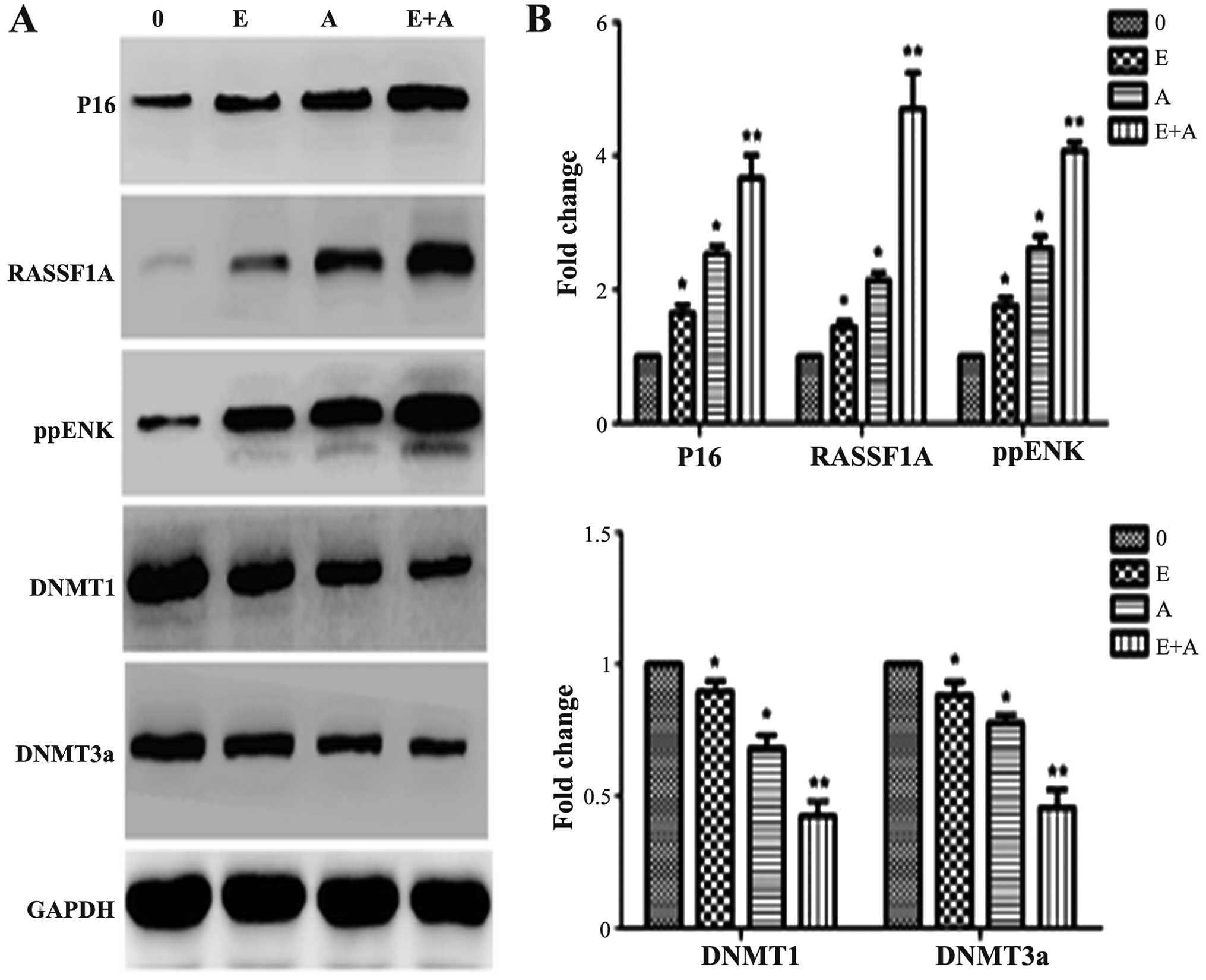 | Figure 6Effects of emodin and 5-Aza-CdR on
the protein expression levels of P16, RASSF1A, ppENK, DNMT1 and
DNMT3a in the PANC-1 cells. (A) Protein expression levels were
detected by western blot analysis. Protein expression levels of
P16, RASSF1A and ppENK were increased, while protein expression
levels of DNMT1 and DNMT3a were decreased. The effect of emodin in
combination with 5-Aza-CdR was stronger than the effect of either
agent alone in the above experiments. (B) The quantification was
performed assigning a value of 1 to the control group. The results
obtained from three separate experiments are expressed as mean ±
SD. *P<0.05, **P<0.01 vs. control or
cells treated with 5-Aza-CdR alone. 0, the control group; E, 40
µM emodin group; A, 1 µM 5-Aza-CdR group; E+A,
combination group. |
Discussion
In recent years, the incidence of pancreatic cancer
has increased yearly. Most patients have advanced disease at the
time of diagnosis and hence a dismal prognosis. Gemcitabine has
been the standard systemic therapy for the palliative treatment of
pancreatic cancer over the last decade, although the 1-year
survival rate of ~20% remains unsatisfactory. Evidence suggests
that many types of cancer are associated with epigenetic changes
and gene mutations, which initiate the process of tumor
development. Epigenetic changes refer to heritable changes in gene
expression (active versus inactive genes) that do not involve
changes to the underlying DNA sequence; a change in phenotype
without a change in genotype. DNA methylation is one of the
important gene epigenetic modifications. DNA methylation is
catalyzed by DNA methyltransferase (DNMT) and S-adenosine
methionine (SAM) as a methyl donor which is transferred to a
specific base, that does not change the DNA sequences; the genetic
code is reversible. DNMTs mainly consist of DNMT1, DNMT3a and
DNMT3b. New synthetic single-strand DNA is methylated by DNMT1, and
methylation information is transmitted to daughter cells; while
DNMT3a and DNMT3b are new methyltransferases for which DNA
methylation patterns are established in the process of embryonic
development, and are involved in the generation of methylation
(15). High expression of DNMTs in
tumors results in the high methylation of tumor-suppressor genes
(TSGs) and inactivation, and eventually promotes tumorigenesis
(16). In the oncogenome, the
demethylation of gene promoter CpG islands promote the expression
of TSGs, thus they are likely to become new targets for the gene
therapy of pancreatic cancer.
The P16 gene can encode the inhibitional proteins of
cell cyclin-dependent kinase 4 (CDK4) and plays an important role
in cell cycle regulation. P16 is a type of negative regulatory
factor, that inhibits the expression of transcription factors, DNA
replication and tumorigenesis. In nude mouse transfection
experiments, high CpG island of P16 gene was heavily methylated,
with loss of expression (17). Peng
et al (18) reported that
the P16 and APC genes were highly methylated in patients with
pancreatic ductal adenocarcinoma. Yang et al (19) analyzed and evaluated the expression
and methylation of the P16 gene in 46 cases of human pancreatic
cancer and adjacent tissues. The results showed that the rate of
P16 protein expression in pancreatic cancer was 41.3% (19/46), and
that in the adjacent tissues was 95.7% (44/46); the difference was
statistically significant. Moreover, cytosine methylation was not
detected in 19 cases of P16 protein-positive pancreatic cancer
specimens, while cytosine methylation was detected in 18 cases of
27 cases of p16 protein-negative specimens; the methylation rate
was 39.1%. This was in accordance with a previous study (20). When 25 cases of pancreatic cancer
were compared with normal tissues, P16 expression was decreased in
80%, 65% of which was caused by the high methylation of the
promoter. RASSF1A, whose encoded protein mainly acts on the cell
signaling transduction pathways related to Ras proteins, induces
cell apoptosis and blocks the accumulation of endogenous cyclin D1
and D3 and inhibits cell malignant transformation via combined with
the Ras protein. Shimizu et al (21) reported that in the tumor tissue of
hamster pancreatic ductal adenocarcinoma, the expression of RASSF1A
was significantly less than that in the normal tissue. When 4
groups of significantly decreased tissues were used to study the
methylation status, it was found that the 5′-terminal CpG island of
the RASSF1A gene was almost highly methylated, and not mutated.
Dammann et al (9) found that
in 64% of primary pancreatic cancers and in 83% of pancreatic
endocrine tumor cell lines, the RASSF1A promoter was highly
methylated. Ueki et al (5)
reported that the ppENK gene contained 42 CpG islands, and the
methylation rate of seven CpG islands in a pancreatic cancer cell
line was 93%, while no methylation was noted in normal pancreatic
tissues, showing that abnormal methylation and transcription
inhibition of ppENK are ordinary incidents in the development of
pancreatic cancer. Fukushima et al (6) found 14 cases of ppENK methylation in
15 cases of invasive ductal adenocarcinoma, while no methylation
was noted in the nontumor pancreas epithelium. In Panc-l cells, the
ppENK methylation ratio increased with higher classification,
suggesting that ppENK methylation was a middle or late event in the
development of pancreatic cancer. However, compared with other
diseases, ppENK methylation was more easily detected in pancreatic
cancer. Thus it can be used as a potential malignant biomarker of
the pancreatic epithelial cells.
5-Aza-CdR is currently one of the most commonly used
demethylation nucleoside analogues, and plays a role in
demethylation mainly through inhibiting the expression and
activation of DNMTs at a low concentration, yet resulted in many
side-effects. Drug toxicity and bone marrow suppression are the
most obvious side-effects, which limit its application in the
clinic. Thus, the identification of drugs with good specificity,
high safety, few side-effects and demethylation effects has become
an urgent task. According to Zhang et al (11), emodin caused a certain degree of
methylation in pancreatic cancer PANC-1 cells, while the degree of
demethylation was weaker than that of 5-Aza-CdR. At present, drug
combinations are the main mode for the treatment of tumors. Thus,
we aimed to ascertain whether the effect of emodin combined with
5-Aza-CdR on pancreatic cancer cells would be stronger than each
agent alone. In the present study, we found that emodin inhibited
the growth of pancreatic cancer cells in a time- and dose-dependent
manner. When the PANC-1 cells were treated with 80 µM emodin
for 72 h, the growth inhibition rate was 65.2%, and there was a
significant change in cell morphology. These findings were
consistent with a previous study by Lin et al (22). When the PANC-1 cells were treated
with 40 µM emodin for 72 h, the growth inhibition rate was
50.6%, and there was no significant change in cell morphology.
Thus, emodin at a concentration of 40 µM was used in the
following experiment. Dot-blot assay showed that emodin at a
concentration of 40 µM or 5Aza-CdR at 1 µM reduced
the 5mC level when compared with the 5mC level in the control
group. Yet, emodin (40 µM) in combination with 5Aza-CdR (1
µM) significantly reduced the 5mC level, showing that emodin
enhanced the demethylation effect by 5-Aza-CdR on pancreatic cancer
cells, further causing the downregulation of 5mC expression. BSP
assay was used to further verify the result of our experiment and
showed that emodin combined with 5-Aza-CdR caused demethylation of
the P16, RASSF1A and ppENK genes; the drug combination was more
effective than either drug alone, consistent with the results of
the MSP assay. The loss of expression of tumor-suppressor genes is
often caused by methylation. The expression is inversely
proportional to the methylation density of the CpG island, and the
loss of expression of 67–90% of tumor-suppressor genes are caused
by low levels of methylation. Moreover, complete loss of expression
of tumor-suppressor genes are caused by high density methylation of
CpG islands (23). The FQ-PCR
results also confirmed that the re-expression of P16, RASSF1A and
ppENK genes, inactivated by methylation, was stronger when emodin
was used in combination with 5-Aza-CdR. Moreover, the western
blotting results were consistent with the FQ-PCR results, further
demonstrating that emodin in combination with 5-Aza-CdR enhanced
the demethylation effect of 5-Aza-CdR on pancreatic cancer cell
tumor-suppressor genes RASSF1A, P16 and ppENK.
In vivo, methylation is mainly catalyzed by
methyltransferases (DNMT1, DNMT3a and DNMT3b), and inhibition of
methyltransferase activity and reduction in methyltransferase
expression are the two main strategies for the demethylation of
tumor suppressor genes. According to the experimental results of
FQ-PCR and WB, emodin combined with 5-Aza-CdR significantly reduced
the expression of DNMT1 and DNMT3a, and the effect was stronger
than either agent alone. Therefore, we speculated that emodin
combined with 5-Aza-CdR could enhance the demethylation by
5-Aza-CdR of pancreatic cancer cell tumor-suppressor genes P16 and
RASSF1A by reducing the expression of methyltransferase DNMT1 and
DNMT3a. In addition, whether emodin and 5-Aza-CdR have effects on
methyltransferase activity needs further investigation. Our
research is mainly confined to the cellular level, and whether
emodin and 5-Aza-CdR also play a role in demethylation in
vivo also requires further research.
In summary, Dot-blot assay confirmed that emodin
combined with 5-Aza-CdR reduced the genomic 5mC level and the
effect was stronger than either agent used alone. Emodin combined
with 5-Aza-CdR had a demethylation effect on tumor-suppressor genes
RASSF1A, P16 and ppENK in pancreatic cancer PANC-1 cells as
confirmed by the results of MSP and BSP assays. FQ-PCR and western
blotting results further confirmed that emodin combined with
5-Aza-CdR increased the expression of p16, RASSF1A and ppENK,
showing that emodin enhanced the demethylation by 5Aza-CdR on
pancreatic cancer Panc-1 cells by inhibiting the expression of
DNMT1 and DNMT3a. These findings provide new insight into the
clinical treatment of pancreatic cancer with the understanding that
the epigenetic demethylation pathway is an important mechanism of
action.
Acknowledgments
We are grateful for the financial support from the
Zhejiang Medical Science and Technology Project (grant no.
2016KYB286), the key subject of Jiaxing Medicine (General Surgery,
grant no. 04-F-15), the Jiaxing Science and Technology Projects
(grant no. 2013AY21042-5), the Jiaxing Science and Technology
Innovation Team Project (grant no. 2013-03), the Administration of
Traditional Chinese Medicine of Zhejiang, China (grant no.
2013ZQ026), the Science and Technology Development Program of
Hangzhou (grant no. 20140733Q34), and the Jiaxing Science and
Technology Projects (grant no. 2015C23012).
References
|
1
|
Saif MW: Pancreatic neoplasm in 2011: An
update. JOP. 12:316–321. 2011.PubMed/NCBI
|
|
2
|
Miranda TB and Jones PA: DNA methylation:
The nuts and bolts of repression. J Cell Physiol. 213:384–390.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cheng X and Blumenthal RM: Mammalian DNA
methyltransferases: A structural perspective. Structure.
16:341–350. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bird A: DNA methylation patterns and
epigenetic memory. Genes Dev. 16:6–21. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ueki T, Toyota M, Skinner H, Walter KM,
Yeo CJ, Issa JP, Hruban RH and Goggins M: Identification and
characterization of differentially methylated CpG islands in
pancreatic carcinoma. Cancer Res. 61:8540–8546. 2001.PubMed/NCBI
|
|
6
|
Fukushima N, Sato N, Ueki T, Rosty C,
Walter KM, Wilentz RE, Yeo CJ, Hruban RH and Goggins M: Aberrant
methylation of preproenkephalin and p16 genes in pancreatic
intraepithelial neoplasia and pancreatic ductal adenocarcinoma. Am
J Pathol. 160:1573–1581. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schutte M, Hruban RH, Geradts J, Maynard
R, Hilgers W, Rabindran SK, Moskaluk CA, Hahn SA, Schwarte-Waldhoff
I, Schmiegel W, et al: Abrogation of the Rb/p16 tumor-suppressive
pathway in virtually all pancreatic carcinomas. Cancer Res.
57:3126–3130. 1997.PubMed/NCBI
|
|
8
|
Moore PS, Sipos B, Orlandini S, Sorio C,
Real FX, Lemoine NR, Gress T, Bassi C, Klöppel G, Kalthoff H, et
al: Genetic profile of 22 pancreatic carcinoma cell lines. Analysis
of K-ras, p53, p16 and DPC4/Smad4. Virchows Arch. 439:798–802.
2001. View Article : Google Scholar
|
|
9
|
Dammann R, Schagdarsurengin U, Liu L, Otto
N, Gimm O, Dralle H, Boehm BO, Pfeifer GP and Hoang-Vu C: Frequent
RASSF1A promoter hypermethylation and K-ras mutations in pancreatic
carcinoma. Oncogene. 22:3806–3812. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brueckner B, Kuck D and Lyko F: DNA
methyltransferase inhibitors for cancer therapy. Cancer J.
13:17–22. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang H, Chen L, Bu HQ, Yu QJ, Jiang DD,
Pan FP, Wang Y, Liu DL and Lin SZ: Effects of emodin on the
demethylation of tumor-suppressor genes in pancreatic cancer PANC-1
cells. Oncol Rep. 33:3015–3023. 2015.PubMed/NCBI
|
|
12
|
Li-Weber M: Targeting apoptosis pathways
in cancer by Chinese medicine. Cancer Lett. 332:304–312. 2013.
View Article : Google Scholar
|
|
13
|
Liu A, Chen H, Tong H, Ye S, Qiu M, Wang
Z, Tan W, Liu J and Lin S: Emodin potentiates the antitumor effects
of gemcitabine in pancreatic cancer cells via inhibition of nuclear
factor-κB. Mol Med Rep. 4:221–227. 2011.PubMed/NCBI
|
|
14
|
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim
SH, Ito S, Yang C, Wang P, Xiao MT, et al: Oncometabolite
2-hydroxyglutarate is a competitive inhibitor of
α-ketoglutarate-dependent dioxygenases. Cancer Cell. 19:17–30.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bestor TH: The DNA methyltransferases of
mammals. Hum Mol Genet. 9:2395–2402. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Baylin SB and Herman JG: DNA
hypermethylation in tumorigenesis: Epigenetics joins genetics.
Trends Genet. 16:168–174. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hanaoka M, Shimizu K, Shigemura M, Kato A,
Fujii H, Honoki K and Tsujiuchi T: Cloning of the hamster p16 gene
5′ upstream region and its aberrant methylation patterns in
pancreatic cancer. Biochem Biophys Res Commun. 333:1249–1253. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Peng DF, Kanai Y, Sawada M, Ushijima S,
Hiraoka N, Kitazawa S and Hirohashi S: DNA methylation of multiple
tumor-related genes in association with overexpression of DNA
methyltransferase 1 (DNMT1) during multistage carcinogenesis of the
pancreas. Carcinogenesis. 27:1160–1168. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang WH, Wang CY, Zhu QS, et al: Gene
methylation changes and protein expression analysis of Pl6 in
pancreatic cancer. Chinese J of General Surgery. 16:46–450. 2007.In
Chinese.
|
|
20
|
Attri J, Srinivasan R, Majumdar S, Radotra
BD and Wig J: Alterations of tumor suppressor gene
pl6INK4a in pancreatic duetal carcinoma. BMC
Gastroenterol. 5:222005. View Article : Google Scholar
|
|
21
|
Shimizu K, Itsuzaki Y, Fujii H, Honoki K
and Tsujiuchi T: Reduced expression of the Rassf1a gene and its
aberrant DNA methylation in pancreatic duct adenocarcinomas induced
by N-nitrosobis(2-oxopropyl)amine in hamsters. Mol Carcinog.
47:80–87. 2008. View
Article : Google Scholar
|
|
22
|
Lin SZ, Wei WT, Chen H, Chen KJ, Tong HF,
Wang ZH, Ni ZL, Liu HB, Guo HC and Liu DL: Antitumor activity of
emodin against pancreatic cancer depends on its dual role:
Promotion of apoptosis and suppression of angiogenesis. PLoS One.
7:e421462012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tahiliani M, Koh KP, Shen Y, Pastor WA,
Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et
al: Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in
mammalian DNA by MLL partner TET1. Science. 324:930–935. 2009.
View Article : Google Scholar : PubMed/NCBI
|