Introduction
Lung cancer is the leading cause of cancer death
worldwide. Only 16.6% of lung cancer patients live 5 years or more
after diagnosis (1). In addition to
chemotherapy, radiotherapy, and molecular targeted therapy,
adjuvant therapies targeting the tumor microenvironment have
received a great deal of attention in recent years (2). The inflammatory milieu comprises
infiltrated immune cells, cytokines, chemokines, and growth
factors, which contribute to tumor proliferation, angiogenesis,
metastasis, and chemoresistance (3). Increasing evidence emphasizes that
chronic inflammation is crucial for the progression of a tumor
(4). Thus, cancer-related
inflammation has been recognized as the 'seventh hallmark of
cancer' (5).
Inflammasome, an intracellular multi-protein
complex, switches on the inflammatory response of tissues to
various danger signals. Among the inflammasomes, NLRP3 inflammasome
is the most characterized activated by a diverse range of 'danger
signals'. It is formed by the assembly of NOD-like receptor NLRP3,
adaptor protein apoptosis-associated speck-like protein containing
a caspase recruitment domain (ASC), and pro-caspase-1. Once
activated, NLRP3 inflammasome triggers the proteolytic processing
of pro-caspase-1 into its active form, caspase-1 (p10 or p20),
which subsequently cleaves pro-IL-1β and pro-IL-18 to mature
bioactive forms. It has been well documented that two signals are
required for the formation and activation of NLRP3 inflammasome.
Firstly, pathogen-associated molecular patterns (PAMPs) or
danger-associated molecular patterns (DAMPs) including viruses,
bacteria, or lipopolysaccharide (LPS), prime the expressions of
NLRP3, pro-IL-1β and pro-IL-18. Then, a second stimulus such as
adenosine 5′-triphosphate (ATP), silica or monosodium urate (MSU)
activates the NLRP3 inflammasome (6).
Mounting evidence indicates that NLRP3 inflammasome
plays an important role in the development and progression of
gastrointestinal cancer, skin cancer, breast cancer and
hepatocellular carcinoma (7).
However, the effects of NLRP3 inflammasome on the progression of
various cancers are complex. For instance, overexpressed and
constitutively activated NLRP3 inflammasome contributed to the
progression of late stage of melanoma (8), while significant downregulation of
NLRP3 inflammasome was observed in human hepatocellular carcinoma
(9). Currently, it remains unclear
whether NLRP3 inflammasome has a beneficial or detrimental effect
on the development of lung cancer. As the most frequent cause of
major cancer incidence and mortality worldwide, lung cancer are
included in two main histological categories: small cell lung
cancer (SCLC) and non-small cell lung cancer (NSCLC). NSCLC
comprises approximately 80% of all diagnosed lung cancer, and lung
adenocarcinoma is the most common subtype of NSCLC. Thus, among
lung cancer variants, adenocarcinoma is the most common
histological subtype (1).
Therefore, using human lung adenocarcinoma A549 cells, we
investigated the role of NLRP3 inflammasome in the proliferation
and migration of lung cancer cells and the potential underlying
mechanisms.
Materials and methods
Reagents and cell culture
The A549 human alveolar epithelial adenocarcinoma
cell line was purchased from the Institute of Biochemistry and Cell
Biology of the Chinese Academy of Sciences (Shanghai, China). Cells
were cultured in RPMI-1640 medium supplemented with 10% fetal
bovine serum (FBS) (ScienCell, CA, USA) at 37°C in a humidified
atmosphere of 5% CO2. LPS and ATP were purchased from
Sigma-Aldrich (St. Louis, MO, USA). The caspase-1 inhibitor
benzyloxycar-bonyl-tyrosine-valine-alanine-aspartate-fluoromethyl
ketone (Z-YVAD-FMK) was purchased from PromoCell (Heidelberg,
Germany). IL-18 binding protein (IL-18BP) was purchased from
GenScript (Nanjing, China), and IL-1 receptor antagonist (IL-1Ra)
was obtained from Fitzgerald Industries (MA, USA). Cells were
stimulated by 1 µg/ml LPS for 8 h with or without 5 mmol/l
ATP for the last half an hour. To inhibit the activation of
caspase-1 or block the IL-18 and IL-1β signal transduction, cells
were pre-treated with 10 µmol/l Z-YVAD-FMK (10), 1 µg/ml IL-18BP (11), and 2 µg/ml IL-1Ra (12) for half an hour prior to LPS
stimulation, respectively.
Immunofluorescence assay
Cells were fixed with 4% paraformaldehyde for 30
min, permeabilized with 0.5% Triton X-100 for 15 min, and incubated
with 5% bovine serum albumin (BSA) in phosphate-buffered saline
(PBS) for 1 h at room temperature (RT). Subsequently, cells were
incubated with primary antibodies against NLRP3 (1:200, Abcam,
London, UK) and ASC (1:200, Cell Signaling Technologies, MA, USA)
overnight at 4°C. After three washes with PBS (5 min per wash),
cells were incubated with Alexa Fluor 488-conjugated donkey
anti-goat (1:1,000, Invitrogen, CA, USA) and Alexa Fluor
555-conjugated donkey anti-rabbit (1:500, Invitrogen) for 1 h at
RT. After three washes with PBS, nuclei were stained with Hoechst
33342 (Beyotime, Nantong, China) for 30 min. Immunofluorescence
images were captured using a Zeiss LSM5 Live confocal laser
scanning microscope (Carl Zeiss, Jena, Germany).
Enzyme-linked immunosorbent assay
(ELISA)
Cells were seeded in 24-well plates overnight. For
NLRP3 inflammasome activation, cells were stimulated by 1
µg/ml LPS for 8 h with or without 5 mmol/l ATP for the last
half an hour. After that, cell supernatants were collected, and
centrifuged at 300 g for 8 min at 4°C for elimination of cells
detached due to cell death, to generate cell-free medium
preparations. Then supernatants were concentrated by centrifugation
at 12,000 g for 30 min at 4°C through a column with a cut-off of 10
kDa (Microcon; Merck-Millipore, Darmstadt, Germany). The levels of
IL-1β and IL-18 in cell culture supernatants were measured by human
eLISA kits from R&D Systems (Minneapolis, MN, USA) and MBL
(Nagoya, Japan), according to the manufacturer's instructions.
Cell proliferation assay
5-ethynyl-2′-deoxyuridine (EdU) incorporation
proliferation assay was performed to investigate the proliferation
of A549 cells using a Cell-Light™ EdU Imaging detecting kit
(RiboBio, Guangzhou, China). Cells were seeded in 6-well plates,
and incubated for 24 h after different treatments. All of the EdU
incorporation experiments were performed according to the
manufacturer's protocol (13). The
ratio of EdU-positive nuclei to total nuclei was calculated as the
proliferation rate of cells in six random high-power fields per
well. The cells were visualized by fluorescence microscopy (DM2500,
Leica, Wetzlar, Germany).
Apoptosis detection
Cells were harvested after different treatments and
washed with PBS. Then cells were resuspended in 500 µl
buffer, and stained with 5 µl Annexin V FITC and 5 µl
PI (BD Biosciences, NJ, USA) for 15 min in the dark. The stained
cells were detected by flow cytometry (BD Biosciences).
Cell migration assays
Cell migration was examined using scratch assay and
Transwell chamber migration assay. For scratch assay, cells were
seeded in 6-well plates. Until the cells reached 100% confluence
forming a monolayer, a sterile 50-µl pipette tip was used to
create a scratch on cell monolayer after different treatments. The
wounds were photographed at baseline and 24 h later, using a phase
contrast microscope (Nikon, Tokyo, Japan). For Transwell chamber
migration assay, 5×104 cells were resuspended in 200
µl of serum-free medium in the upper chamber, and 600
µl medium with different treatments was placed in the bottom
chamber. Then, 10% FBS was added to the lower chamber to act as a
chemoattractant. The Transwell system (24-well, 8 µm,
Corning, NY, USA) was incubated for 4 h at 37°C and 5%
CO2. Non-migrated cells on the top of the membrane were
gently removed with a cotton swab. The cells that migrated through
the pores onto the lower side of the membrane were fixed with 4%
paraformaldehyde for 30 min. Finally, the cells were stained with
crystal violet (Beyotime) and counted using a phase contrast
microscope (Nikon).
Downregulation of NLRP3 expression by
short hairpin RNA (shRNA) interference
To knock down the NLRP3 expression, the A549 cells
were transfected with lentiviral-mediated shRNA. The cells were
transfected by lentiviral particles at a multiplicity of infection
of 20–30 with control shRNA (shCtrl) or NLRP3 shRNAs (shNLRP3)
(GenePharma, Shanghai, China), according to the manufacturer's
instructions. Downregulation of NLRP3 expression was verified by
reverse transcriptase polymerase chain reaction (RT-PCR) and
western blotting (data not shown).
RNA isolation and RT-PCR
Total RNA was extracted from cells using TRIzol
(Invitrogen). Reverse transcription was performed using a reverse
transcription kit (Takara, Shiga, Japan) according to the
manufacturer's instructions. Standard real-time quantitative PCR
was performed using the following primers: NLRP3: forward
5′-AAGGGCCATGGACTATTTCC-3′ and reverse 5′-GACTCCACCCGATGACAGTT-3′;
GAPDH: forward 5′-GAAGGTGAAGGTCGGAGTC-3′ and reverse
5′-GAAGATGGTGATGGGATTTC-3′. All primers were synthesized by
Invitrogen Life Technologies. The mRNA levels were quantified by
SYBR Green Technology. Fold changes in gene expression were
calculated using the 2−ΔΔCT method (14).
Western blot analysis
Cells were lysed on ice in lysis buffer with 1% EDTA
and 1% protease inhibitor cocktail (Thermo Fisher Scientific, MA,
USA). The total protein concentrations were quantified by BCA
method (Beyotime). Proteins (60–100 µg) were resolved by
SDS-PAGE and transferred to PVDF membranes (Millipore, MA, USA).
After blocking with 5% milk for 1 h at RT, the membranes were
incubated with primary antibodies against total and phosphorylated
protein kinase B (Akt), extracellular regulated protein kinase 1/2
(eRK1/2), cyclic adenosine monophosphate (cAMP) response element
binding protein (CREB), NLRP3, E-cadherin, GAPDH (Cell Signaling
Technologies), caspase-1 (Santa Cruz, CA, USA), and Snail (Abcam)
at 4°C overnight. After three 10-min washes with Tris-buffered
saline containing 0.1% Tween-20 (TBST), the blots were incubated
with HRP-conjugated secondary antibodies (Bioworld, Nanjing,
China), and signals were detected by enhanced chemiluminescent
reagents and analyzed with Bio-Rad Gel Doc/Chemi Doc Imaging System
and Quantity One software (Hercules, CA, USA).
Statistical analysis
All the data are expressed as the mean ± SEM.
Differences between means were analyzed using one-way analysis of
variance (ANOVA) or two-way ANOVA followed by Student-Newman-Keuls
tests for multiple comparisons. Statistical significance was
defined as P<0.05. All analyses were performed using the
Statistical Package for Social Sciences statistical software
(SPSS), version 20.0 (SPSS Inc., Chicago, IL, USA).
Results
LPS + ATP activates NLRP3 inflammasome in
A549 cells
To investigate whether LPS and ATP trigger the
formation of NLRP3 inflammasome in A549 cells, the association
between NLRP3 and ASC was determined by immunofluorescence. As
shown in Fig. 1A, many
intracellular NLRP3 and ASC colocalizations were found in punctate
spot style after the stimulation of LPS+ATP. However, less NLRP3
and ASC colocalizations were detected in the control group and the
LPS or ATP single-treated group. Consistently, western blot
analysis showed that the expression of active caspase-1 p10
significantly increased by stimulation of LPS+ATP (Fig. 1B). Moreover, the extracellular
levels of IL-18 and IL-1β significantly increased in the LPS+ATP
group compared to the other groups (Fig. 1C). Together, these data demonstrate
that LPS+ATP induced the assembly and activation of NLRP3
inflammasome in A549 cells.
Activation of NLRP3 inflammasome enhances
A549 cell proliferation
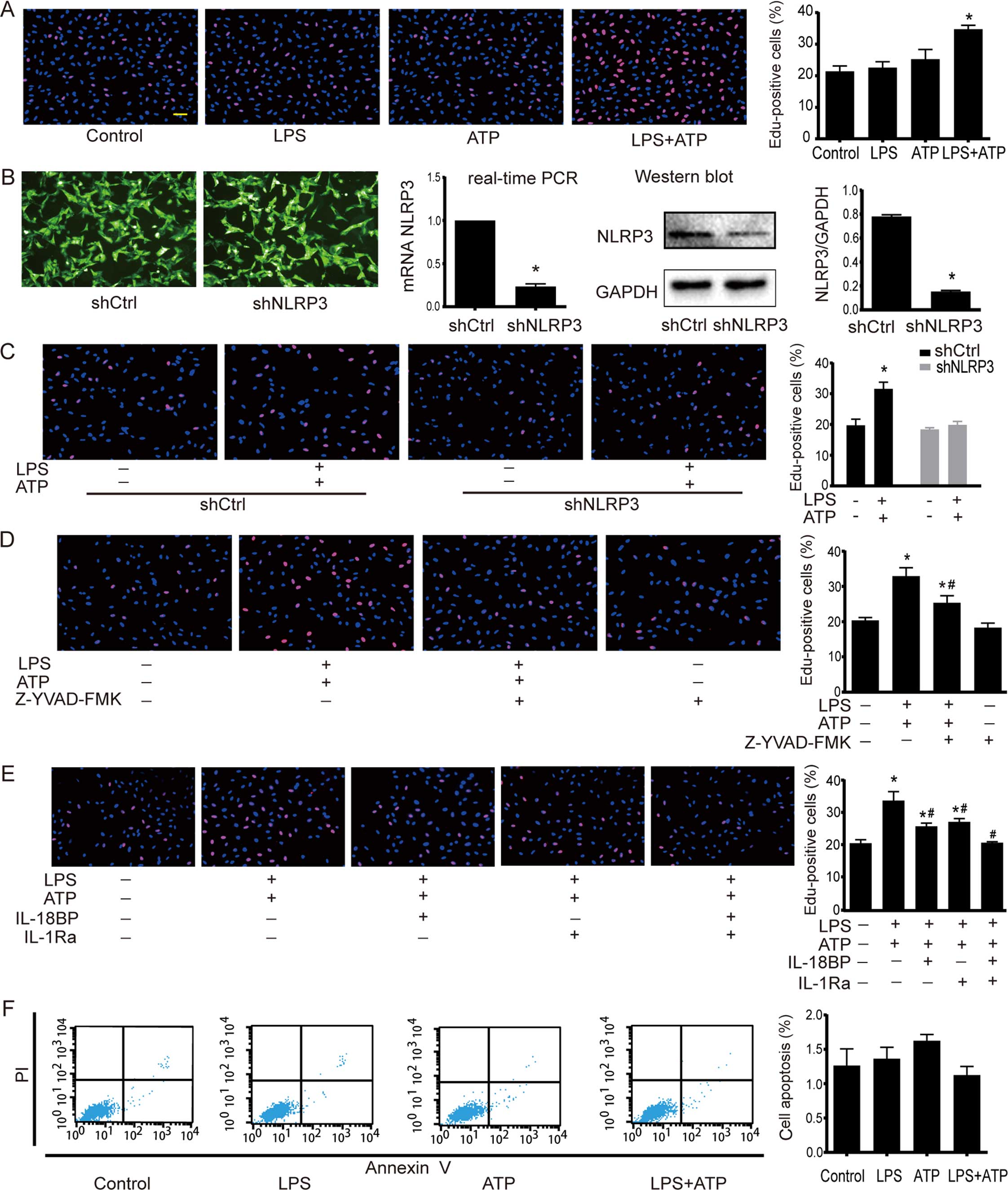
EdU incorporation assay was used to determine the
cell proliferation. As shown in Fig.
2A, a significant increase in EdU-positive cells was found
after the treatment of LPS+ATP, but not LPS or ATP alone. NLRP3
inflammasome activation involves several stages, including NLRP3
inflammasome assembly, pro-IL-1β and pro-IL-18 enzymatic cleavage
by active caspase-1, and subsequent effects following the release
of mature IL-1β and IL-18. Therefore, inhibiting NLRP3 inflammasome
by NLRP3 downregulation, caspase-1 inhibitor Z-YVAD-FMK, IL-18BP
and IL-1Ra was used to further confirm the effects of NLRP3
inflammasome activation on A549 cells. The downregulation of NLRP3
by shRNA interference was confirmed by RT-PCR and western blotting.
The NLRP3 expression in shNLRP3 was downregulated to 18% of shCtrl
(Fig. 2B). As shown in Fig. 2C, LPS+ATP increased the number of
EdU-positive cells in the shCtrl cells, but not in the shNLRP3
cells. The caspase-1 inhibitor Z-YVAD-FMK had no effect on A549
proliferation under base condition, but it attenuated
LPS+ATP-induced cell proliferation (Fig. 2D). Moreover, neutralization of IL-18
activity with IL-18BP or inhibition of IL-1β activity with IL-1Ra
attenuated the LPS+ATP-induced enhancement of proliferation, while
the combined use of IL-18BP and IL-1Ra abolished the
pro-proliferative effect of LPS+ATP (Fig. 2E). As shown in Fig. 2F, flow cytometry analysis of Annexin
V-FITC/PI-staining revealed that LPS, ATP, and LPS+ATP did not
alter apoptosis of A549 cells.
Activation of NLRP3 inflammasome enhances
A549 cell migration
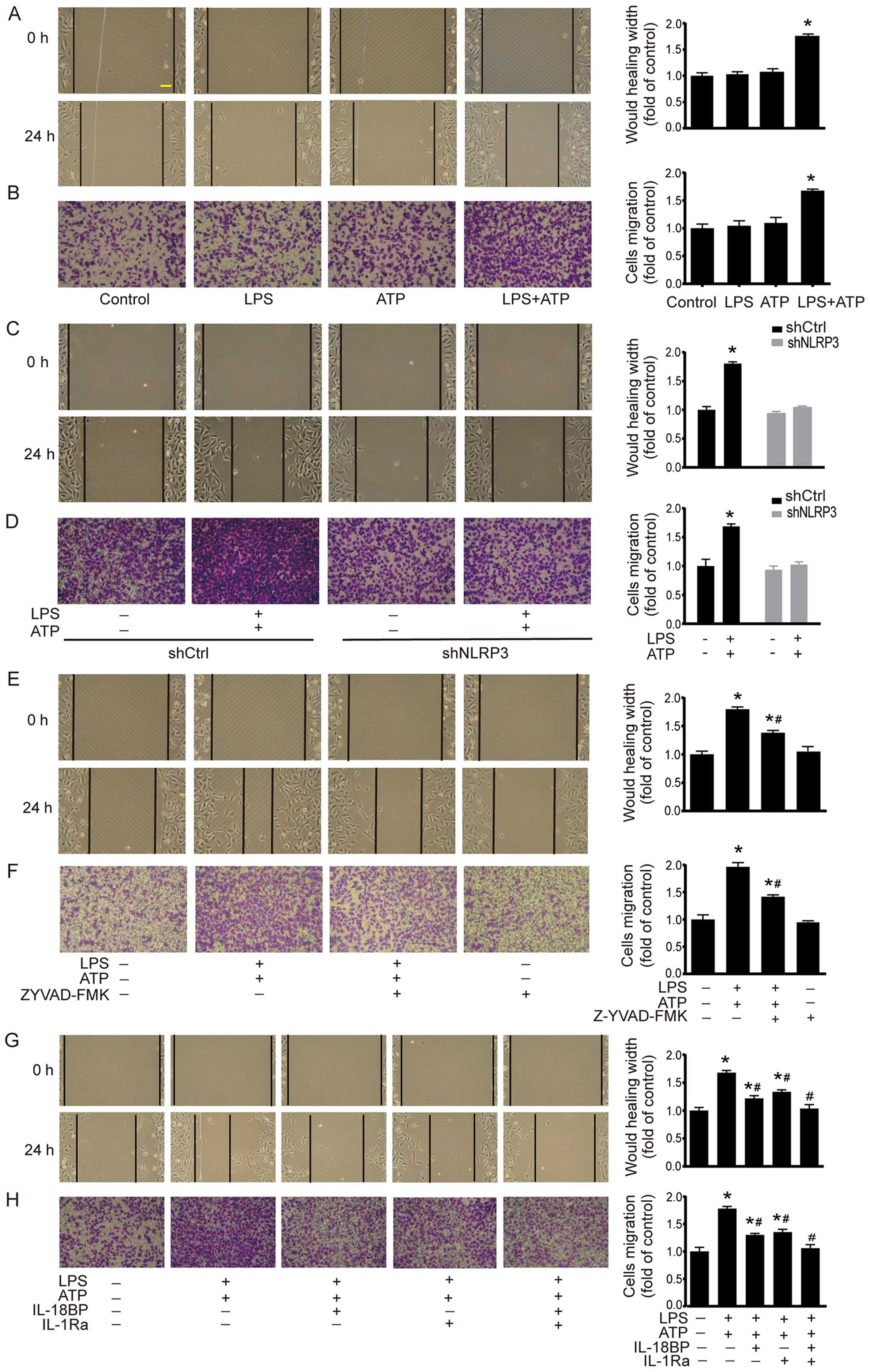
Cell migration was investigated by using scratch
assay and Transwell chamber migration assay. As shown in Fig. 3A, scratch assay demonstrated that
LPS+ATP elicited a significant increase in the migrated distance
compared with the control group. The results of Transwell chamber
migration assay were consistent with the scratch assay (Fig. 3B). In shCtrl A549 cells, LPS+ATP
also enhanced cell migration in scratch assay and Transwell chamber
migration assay. However, the LPS+ATP-induced enhancement of cell
migration was abolished by NLRP3 knockdown (Fig. 3C and D). In addition, caspase-1
inhibitor Z-YVAD-FMK attenuated LPS+ATP-induced migration in both
scratch assay and Transwell chamber migration assay (Fig. 3E and F). IL-18BP or IL-1Ra also
attenuated LPS+ATP-induced migration, while the combined treatment
of IL-18BP and IL-1Ra reversed the LPS+ATP-induced enhancement of
cell migration (Fig. 3G and H).
Activation of NLRP3 inflammasome enhances
the phosphorylation of Akt, ERK1/2, and CREB
To investigate the potential molecular mechanisms of
activated NLRP3 inflammasome on A549 cell proliferation, the
phosphorylation of two intracellular kinases (Akt and ERK1/2) and a
transcription factor (CREB) involved in cell proliferation was
determined by western blotting. As shown in Fig. 4, activation of NLRP3 inflammasome by
LPS+ATP increased the phosphorylation of Akt, ERK1/2, and CREB.
However, downregulation of NLRP3 reversed the phosphorylation of
Akt, ERK1/2, and CREB induced by the treatment of LPS+ATP (Fig. 4A). Caspase-1 inhibitor Z-YVAD-FMK
abolished LPS+ATP-induced Akt phosphorylation, and attenuated
LPS+ATP-induced ERK1/2 and CREB phosphorylation (Fig. 4B). IL-18BP abolished LPS+ATP-induced
Akt phosphorylation, and attenuated ERK1/2 and CREB
phosphorylation, while IL-1Ra abolished ERK1/2 phosphorylation, and
attenuated Akt and CREB phosphorylation. Moreover, the combination
of IL-18BP and IL-1Ra reversed the phosphorylation of Akt, ERK1/2
and CREB induced by the activation of NLRP3 inflammasome (Fig. 4C).
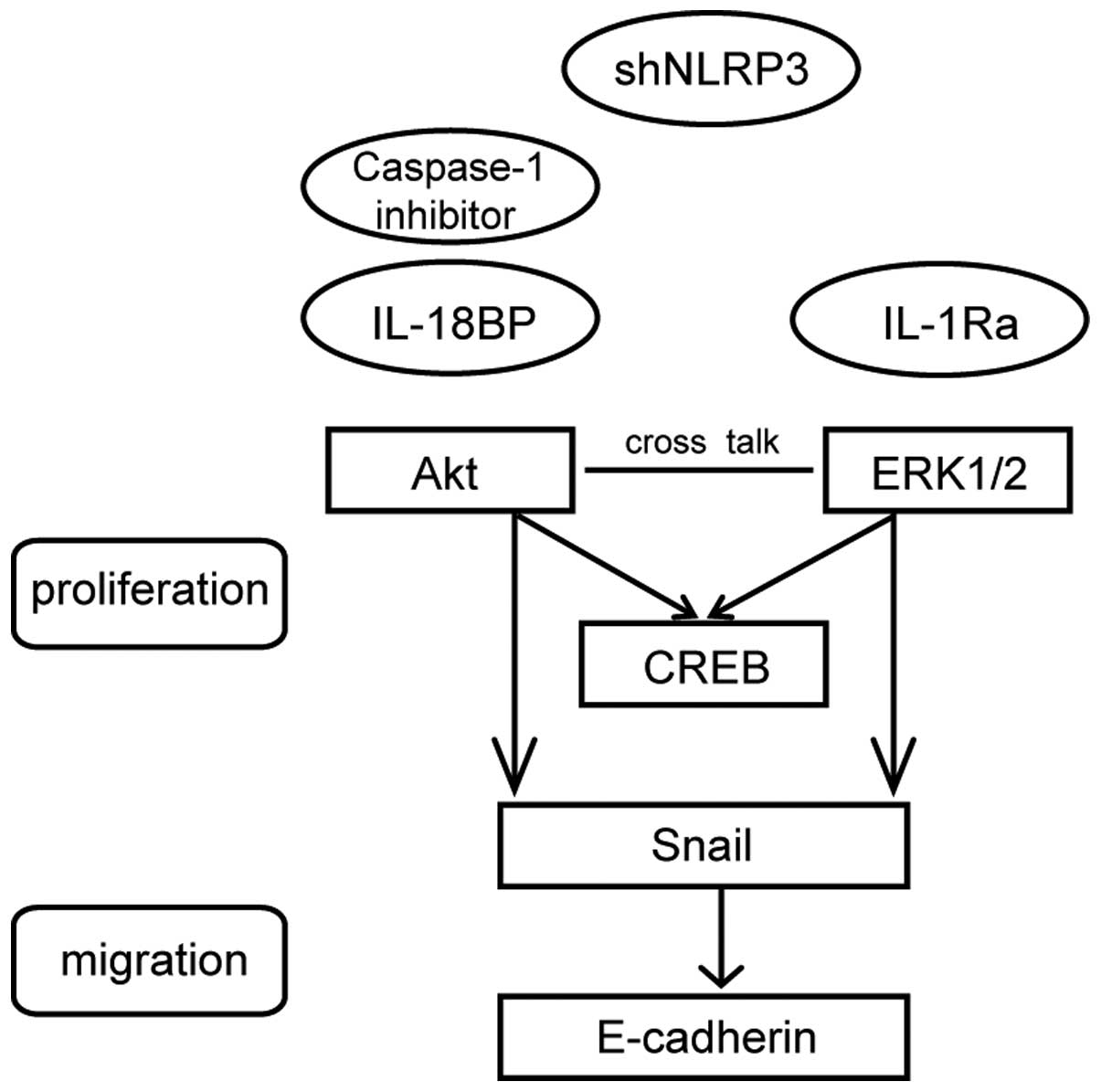
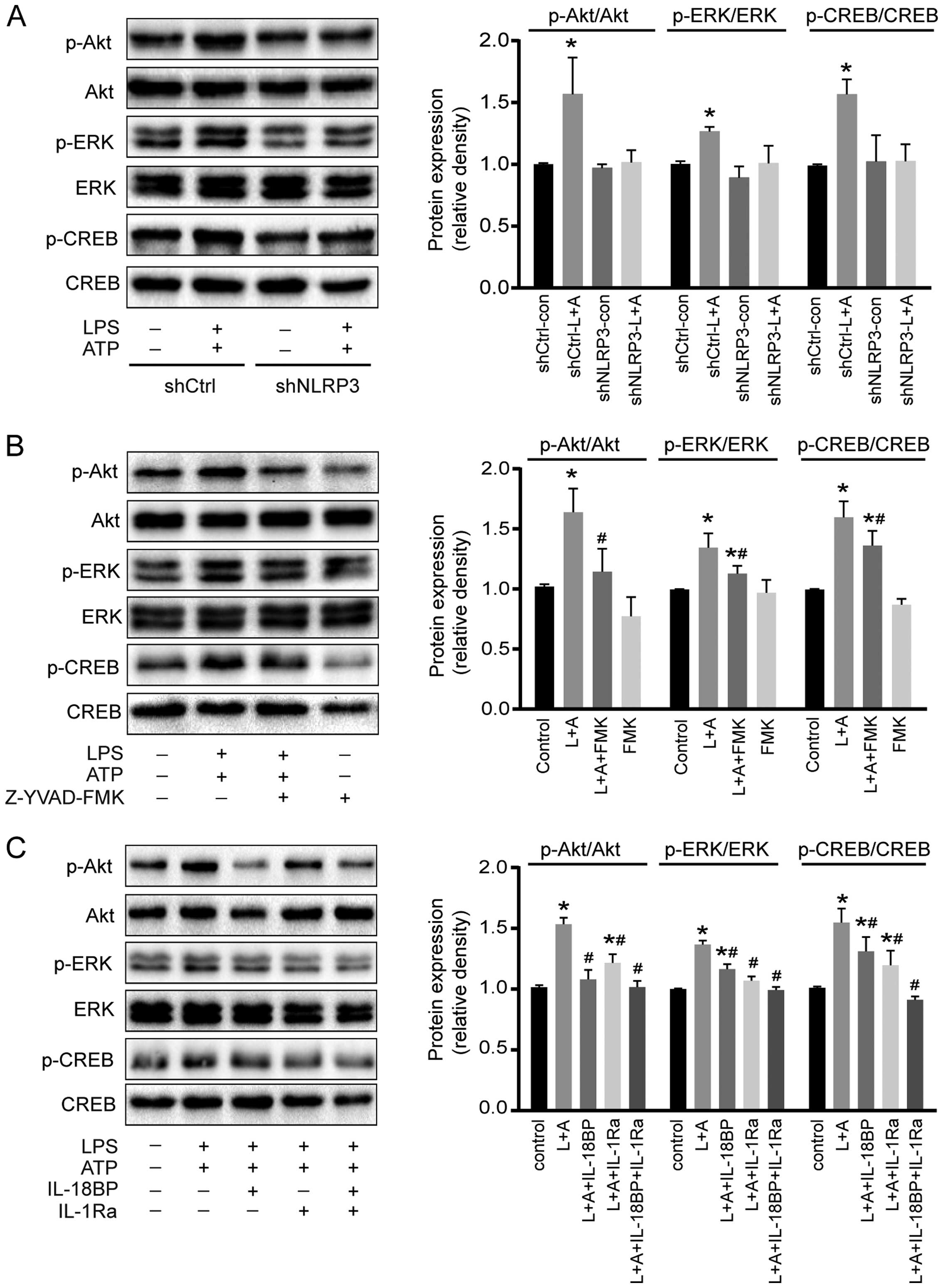 | Figure 4Effects of NLRP3 inflammasome
activation on Akt, ERK1/2, and CREB phosphorylation. (A)
Downregulation of NLRP3 reversed the LPS+ATP-induced
phosphorylation of Akt, ERK1/2 and CREB. (B) Caspase-1 inhibitor
Z-YVAD-FMK abolished LPS+ATP-induced Akt phosphorylation and
inhibited ERK1/2 and CREB phosphorylation. (C) L-18BP, or IL-1Ra
inhibited LPS+ATP-induced phosphorylation of Akt, ERK1/2 and CREB,
and combined use of IL-18BP and IL-1Ra abolished the
LPS+ATP-induced phosphorylation of Akt, ERK1/2 and CREB. L, LPS; A,
ATP; FMK, Z-YVAD-FMK. Four independent experiments were examined.
*P<0.05 compared with the control group;
#P<0.05 compared with the LPS+ATP group. |
Activation of NLRP3 inflammasome
downregulates E-cadherin expression and upregulates Snail
expression
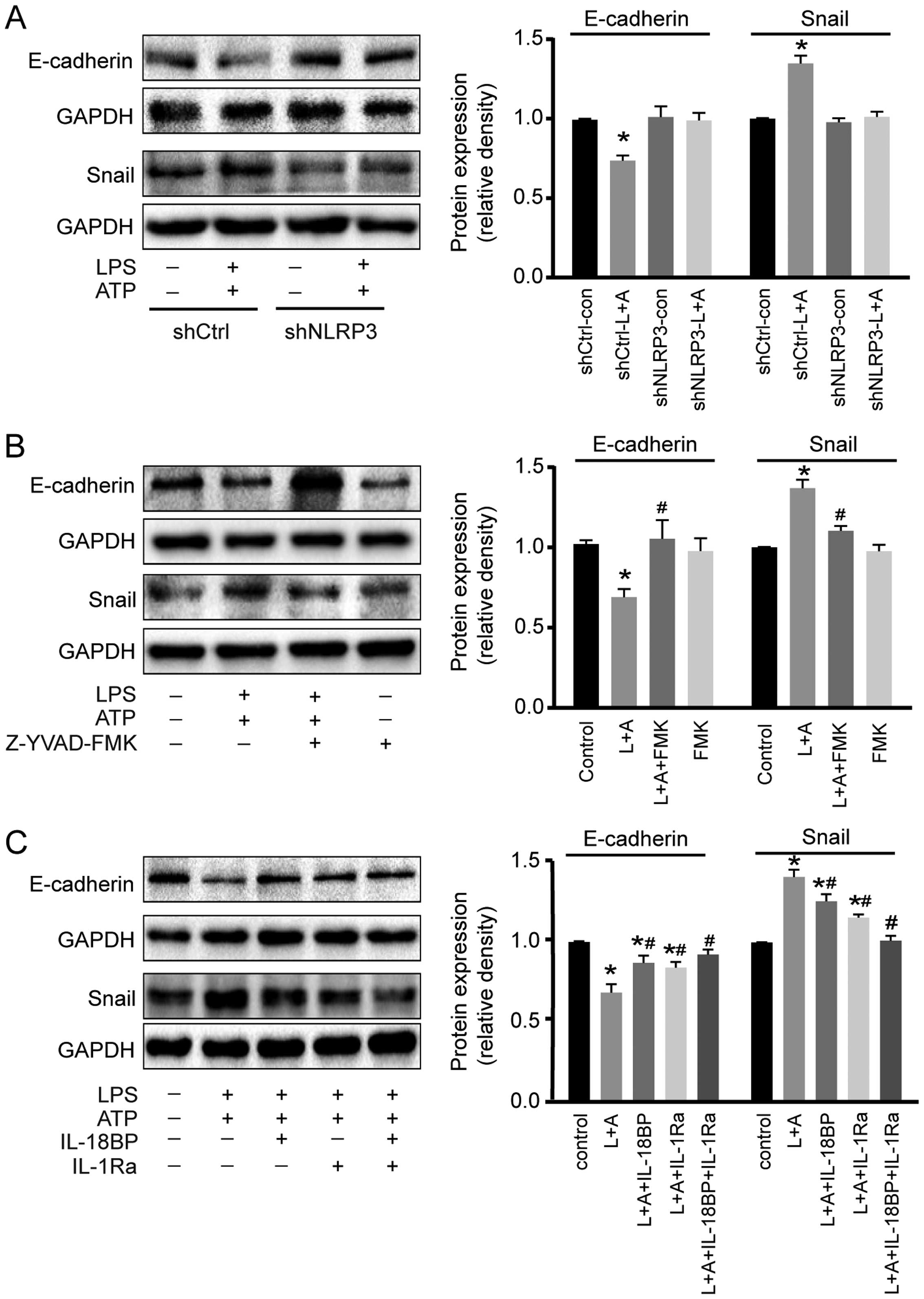
The cellular adhesion molecule E-cadherin and the
transcription factor Snail are two important factors involved in
inflammation-induced cancer cell migration (15). As shown in Fig. 5A and 5B, LPS+ATP treatment resulted in
E-cadherin down-regulation and Snail upregulation in A549 cells,
while these effects were abolished by NLRP3 interference or
caspase-1 inhibitor. Moreover, LPS+ATP-induced E-cadherin
down-regulation and Snail upregulation were inhibited by IL-18BP or
IL-1Ra, and reversed by the combined use of IL-18BP and IL-1Ra
(Fig. 5C).
Discussion
Cancer-related inflammation supplies cytokines,
chemokines, and extracellular matrix, to promote tumor growth and
metastasis. Currently, inflammasomes, which sense exogenous and
endogenous dangers and initiate inflammatory responses, are
considered to contribute the development of cancer-related
inflammation and have complex functions in carcinogenesis (16). Among the inflammasomes, NLRP3 is
widely expressed in a number of cells including epithelial cells,
macrophages, dendritic cells and keratinocytes. NLRP3 inflammasome
can be activated by a number of stimuli (17). It has been well documented that
NLRP3 inflammasome plays important roles in inflammatory disease,
autoimmune disease, and in particular, several types of cancer
(18). However, the roles of NLRP3
inflammasome in different cancers are cell- and tissue-specific
(8,19). Thus, in-depth understanding the
effects of NLRP3 inflammasome in lung cancer cells and the
underlying mechanisms may offer new insights into tumor therapy. In
the present study, we demonstrated that NLRP3 inflammasome
activation exacerbated the proliferation and migration of A549
cells. These effects were attenuated by inhibiting the activity of
NLRP3 inflammasome by different ways such as downregulating NLRP3
expression, inhibiting the activity of caspase-1, or blocking IL-18
and/or IL-1β signal transduction. Thus, NLRP3 inflammasome could be
an important molecule mediating the proliferation and migration of
A549 cells.
Activation of NLRP3 inflammasome commonly includes
priming with a TLR agonist (such as LPS) and activating with a
second stimulus (such as ATP) (20). Once activated, NLRP3 inflammasome
triggers proteolytic processing of pro-caspase-1 into its active
form, caspase-1 (p10 or p20), which subsequently cleaves pro-IL-1β
and pro-IL-18 to mature bioactive forms. Our results showed that
the down-regulation of NLRP3 abolished the effects of LPS+ATP on
the proliferation and migration of A549 cells. However, inhibiting
the activity of caspase-1, which is downstream of NLRP3, only
partially attenuated the effects of LPS+ATP. Furthermore, the
combined use of IL-1Ra and IL-18BP to block the IL-1β and IL-18
signaling pathways, which are downstream of caspase-1, reversed the
effects of LPS+ATP. These results suggest that: i) NLRP3
inflammasome activation-induced IL-1β and IL-18 may work through
mechanisms other than the caspase-1 pathway; ii) the effects of
activated NLRP3 inflammasome on A549 cell proliferation and
migration should be mediated by releasing IL-1β and IL-18 in an
autocrine or paracrine manner. Although caspase-1 activation is the
major downstream event of NLRP3 inflammasome assembly, recent
studies report that NLRP3 inflammasome can also activate caspase-8
(21). In addition, caspase-8 is
also involved in the maturation of IL-1β and IL-18 by inducing a
non-canonical process of releasing IL-1β (22–24)
and IL-18 (25). In this regard, we
consider that NLRP3 inflammasome can mediate the release of IL-1β
and IL-18 through a caspase-1-dependent or -independent
pathway.
In the present study, blockage of IL-18 or IL-1β
signaling alleviated the effects of activated NLRP3 inflammasome on
A549 cell proliferation and migration. This result indicates that
both IL-18 and IL-1β could contribute to the progression of lung
adenocarcinoma. In a mouse model, Lewis lung carcinoma cells
engineered to produce IL-1β exhibited increased tumor growth and
expression of angiogenic factors (26). In non-small cell lung cancer (NSCLC)
patients, serum IL-1β is significantly elevated compared with
healthy donors (27). Moreover,
in vitro studies show that IL-1β promotes the proliferation
and migration of NSCLC cells by repressing the expression of
microRNA-101 through the COX2-HIF1α pathway (27). In other cancers, it has been
reported that IL-1β-mediated inflammation contributes to the
development and progression of melanoma (8). IL-18 also belongs to the IL-1
superfamily. The biologic activity of IL-18 is mainly regulated by
enzymatic processing, whereas IL-1β is controlled at the
transcriptional level (28).
Although there are some differences between IL-18 and IL-1β, IL-18
also has a tumor-promoting activity similar to that of IL-1β. In
lung cancer patients, IL-18 level in induced sputum is
significantly higher than that in healthy controls (29). Moreover, in gastric cancer, IL-18
increases tumor cell proliferation (30), promotes cell migration (31), and enhances angiogenesis (32). IL-18 regulates hepatic melanoma
metastasis, favoring the adhesion of melanoma cells to liver
(33). These effects of IL-1β and
IL-18 on the proliferation and migration of A549 cells were also
confirmed in our study. Moreover, the combined use of IL-1 Ra and
IL-18BP reversed the effects of activated NLRP3 inflammasome.
Therefore, we assume that NLRP3 inflammasome activation can
regulate the proliferation and migration of A549 cells by releasing
IL-1 superfamily cytokines in an autocrine or paracrine manner
(34).
Uncontrolled tumor cell growth and metastasis mainly
result from the disruptions of regulatory signaling pathways. The
Akt and ERK1/2 are two important signaling molecules in tumor
proliferation. Our data demonstrated that activation of NLRP3
inflammasome could enhance the phosphorylation of Akt, ERK1/2 and
CREB. The Akt pathway plays a pivotal role in fundamental cellular
functions by phosphorylating a variety of substrates. Another
important signal transduction pathway involves ERK1/2, which forms
major cell-proliferation signaling pathways from the cell surface
to the nucleus. Recent studies have demonstrated crosstalk between
Akt and ERK1/2 signaling (35,36).
It is well documented that Akt and ERK1/2 kinase can activate CREB,
which is a key transcription factor for cell proliferation
(37). In the present study,
caspase-1 inhibitor Z-YVAD-FMK suppressed the LPS+ATP-induced
phosphorylation of Akt more effectively than that of eRK1/2.
Moreover, IL-18BP functioned more effectively on the
phosphorylation of Akt while IL-1Ra functioned more effectively on
the phosphorylation of ERK1/2. Previous studies have indicated that
IL-18 promotes proliferation via Akt pathway (38,39) in
various cell types, but has few effects on ERK1/2 activation
(40). ERK1/2 pathway participates
in IL-1β-induced sensitization of nociception in rats (41). However, it is also demonstrated that
Akt and ERK1/2, which are simultaneously involved in IL-18 or IL-1β
signaling, act equally (42,43).
Therefore, both Akt and ERK1/2 signaling are involved in activated
NLRP3 inflammasome-induced A549 cell proliferation. We provide a
schematic model (Fig. 6) to
illustrate clearly the mechanisms. However, the precise signal
transduction pathways need to be further investigated by using
kinase inhibitors.
Tumor metastasis is one of the most common events
that lead to mortality of cancer. There are several molecules
involved in tumor metastasis, such as E-cadherin (44), matrix metalloproteinases (MMPs)
(45,46) and chemokine (C-X-C motif) ligand
(CXCL) (47). E-cadherin, which is
a prime adhesion molecule on cell membrane of epithelial cells and
epithelia-derived cancer cells, participates in the architectural
maintenance of epithelial tissues (48). Snail is a zinc-finger transcription
factor which represses E-cadherin transcription (49). Our results showed that NLRP3
inflammasome activation could downregulate the expression of
E-cadherin and upregulate the expression of Snail. In human NSCLC
patients, reduced E-cadherin expression correlates with lymph nodes
metastasis (50) and reduced
overall survival (44).
Upregulation of Snail is associated with poor prognosis and
promotes the progression of NSCLC in vivo (51). Downregulation of Snail expression
not only inhibits TNFα-induced cancer cell migration and invasion
in vitro but also suppresses LPS-mediated metastasis in
vivo (49). The present study
also showed that blockage of IL-1β and IL-18 signaling attenuated
the effects of activation of NLRP3 inflammasome on the expressions
of E-cadherin and Snail. In oral cancer (52) and gastric cancer (53), it is documented that IL-1β
downregulates E-cadherin expression and upregulates Snail
expression, and subsequently enhances migration and invasion.
Besides IL-1β, IL-18 also decreases the expression of E-cadherin in
epithelial cells (54). Thus, IL-1
family cytokine-mediated regulation of E-cadherin and Snail may
contribute to the A549 cell migration induced by activated NLRP3
inflammasome.
The final purpose of experimental research is to
provide useful information for the clinical application. Literature
shows that caspase-1 inhibitor, IL-1Ra and IL-18BP, which inhibit
the NLRP3 inflammasome, are used in the clinical or in animal model
research. For instance, the human recombinant IL1ra (hrIL1ra) has
been used in phase II or III randomized control trial for stroke,
severe sepsis and traumatic brain injury (55). The caspase-1 inhibitor ameliorates
experimental autoimmune myasthenia gravis by innate DC IL-1-IL-17
pathway (56). IL-18BP ameliorates
cardiac ischemia/reperfusion injury in a murine model (57). There are two limitations in our
present study: firstly, the study was conducted only in one cell
line; some data in other cell lines would be more powerful to
verify the conclusion. Secondly, this study was conducted in
vitro, however, we plan further experiments in vivo to
confirm our results to supply more valuable information for the
translational application.
In conclusion, our data show that NLRP3 inflammasome
activation can accelerate the proliferation and migration of A549
lung cancer cells by releasing IL-1β and IL-18 in an autocrine or
paracrine manner. These results suggests that molecules
participating in NLRP3 inflammasome signaling may be promising
therapeutic targets for the treatment of lung adenocarcinoma.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (NSFC) under grant 81273571, the
National Technological Special Project for 'Significant New Drugs
Development' (2011ZX09302-003-02), a Jiangsu Clinical Research
Center for Respiratory Diseases project under grant BL2012012 and a
project funded by the Priority Academic Program Development of
Jiangsu Higher education Institutions (PAPD).
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tarassishin L, Casper D and Lee SC:
Aberrant expression of interleukin-1β and inflammasome activation
in human malignant gliomas. PLoS One. 9:e1034322014. View Article : Google Scholar
|
|
3
|
Landskron G, De la Fuente M, Thuwajit P,
Thuwajit C and Hermoso MA: Chronic inflammation and cytokines in
the tumor microenvironment. J Immunol Res. 2014:1491852014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Coussens LM and Werb Z: Inflammation and
cancer. Nature. 420:860–867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mantovani A: Cancer: Inflaming metastasis.
Nature. 457:36–37. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ozaki E, Campbell M and Doyle SL:
Targeting the NLRP3 inflammasome in chronic inflammatory diseases:
Current perspectives. J Inflamm Res. 8:15–27. 2015.PubMed/NCBI
|
|
7
|
Kolb R, Liu GH, Janowski AM, Sutterwala FS
and Zhang W: Inflammasomes in cancer: A double-edged sword. Protein
Cell. 5:12–20. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Okamoto M, Liu W, Luo Y, Tanaka A, Cai X,
Norris DA, Dinarello CA and Fujita M: Constitutively active
inflamma-some in human melanoma cells mediating autoinflammation
via caspase-1 processing and secretion of interleukin-1beta. J Biol
Chem. 285:6477–6488. 2010. View Article : Google Scholar :
|
|
9
|
Zhu Z, Zhong S and Shen Z: Targeting the
inflammatory pathways to enhance chemotherapy of cancer. Cancer
Biol Ther. 12:95–105. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Meng XF, Wang XL, Tian XJ, Yang ZH, Chu
GP, Zhang J, Li M, Shi J and Zhang C: Nod-like receptor protein 1
inflammasome mediates neuron injury under high glucose. Mol
Neurobiol. 49:673–684. 2014. View Article : Google Scholar
|
|
11
|
Fortin CF, Ear T and McDonald PP:
Autocrine role of endogenous interleukin-18 on inflammatory
cytokine generation by human neutrophils. FASEB J. 23:194–203.
2009. View Article : Google Scholar
|
|
12
|
Rivollier A, Mazzorana M, Tebib J, Piperno
M, Aitsiselmi T, Rabourdin-Combe C, Jurdic P and Servet-Delprat C:
Immature dendritic cell transdifferentiation into osteoclasts: A
novel pathway sustained by the rheumatoid arthritis
microenvironment. Blood. 104:4029–4037. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yu Y, Arora A, Min W, Roifman CM and
Grunebaum E: EdU incorporation is an alternative non-radioactive
assay to [(3)H] thymidine uptake for in vitro measurement of mice
T-cell proliferations. J Immunol Methods. 350:29–35. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
15
|
Tania M, Khan MA and Fu J: epithelial to
mesenchymal transition inducing transcription factors and
metastatic cancer. Tumour Biol. 35:7335–7342. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zitvogel L, Kepp O, Galluzzi L and Kroemer
G: Inflammasomes in carcinogenesis and anticancer immune responses.
Nat Immunol. 13:343–351. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Janowski AM, Kolb R, Zhang W and
Sutterwala FS: Beneficial and Detrimental Roles of NLRs in
Carcinogenesis. Front Immunol. 4:3702013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Strowig T, Henao-Mejia J, Elinav E and
Flavell R: Inflammasomes in health and disease. Nature.
481:278–286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Allen IC, TeKippe EM, Woodford RM, Uronis
JM, Holl EK, Rogers AB, Herfarth HH, Jobin C and Ting JP: The NLRP3
inflammasome functions as a negative regulator of tumorigenesis
during colitis-associated cancer. J exp Med. 207:1045–1056. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang A, Wang P, Ma X, Yin X, Li J, Wang
H, Jiang W, Jia Q and Ni L: Mechanisms that lead to the regulation
of NLRP3 inflammasome expression and activation in human dental
pulp fibroblasts. Mol Immunol. 66:253–262. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sagulenko V, Thygesen SJ, Sester DP, Idris
A, Cridland JA, Vajjhala PR, Roberts TL, Schroder K, Vince JE, Hill
JM, et al: AIM2 and NLRP3 inflammasomes activate both apoptotic and
pyroptotic death pathways via ASC. Cell Death Differ. 20:1149–1160.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maelfait J, Vercammen E, Janssens S,
Schotte P, Haegman M, Magez S and Beyaert R: Stimulation of
Toll-like receptor 3 and 4 induces interleukin-1beta maturation by
caspase-8. J exp Med. 205:1967–1973. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Antonopoulos C, El Sanadi C, Kaiser WJ,
Mocarski ES and Dubyak GR: Proapoptotic chemotherapeutic drugs
induce noncanonical processing and release of IL-1beta via
caspase-8 in dendritic cells. J Immunol. 191:4789–4803. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gringhuis SI, Kaptein TM, Wevers BA,
Theelen B, van der Vlist M, Boekhout T and Geijtenbeek TB: Dectin-1
is an extracellular pathogen sensor for the induction and
processing of IL-1β via a noncanonical caspase-8 inflammasome. Nat
Immunol. 13:246–254. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li S, Sun R, Chen Y, Wei H and Tian Z:
TLR2 controls the development of hepatocellular carcinoma by
reducing interleukin-18-mediated immunosuppression. Cancer Res. Jan
19–2015.Epub ahead of print. View Article : Google Scholar
|
|
26
|
Saijo Y, Tanaka M, Miki M, Usui K, Suzuki
T, Maemondo M, Hong X, Tazawa R, Kikuchi T, Matsushima K, et al:
Proinflammatory cytokine IL-1 beta promotes tumor growth of Lewis
lung carcinoma by induction of angiogenic factors: In vivo analysis
of tumor-stromal interaction. J Immunol. 169:469–475. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang L, Zhang LF, Wu J, Xu SJ, Xu YY, Li
D, Lou JT and Liu MF: IL-1β-mediated repression of microRNA-101 is
crucial for inflammation-promoted lung tumorigenesis. Cancer Res.
74:4720–4730. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fabbi M, Carbotti G and Ferrini S:
Context-dependent role of IL-18 in cancer biology and
counter-regulation by IL-18BP. J Leukoc Biol. 97:665–675. 2015.
View Article : Google Scholar
|
|
29
|
Rovina N, Hillas G, Dima E, Vlastos F,
Loukides S, Veldekis D, Roussos C, Alhanatis M and Bakakos P: VEGF
and IL-18 in induced sputum of lung cancer patients. Cytokine.
54:277–281. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim KE, Song H, Hahm C, Yoon SY, Park S,
Lee HR, Hur DY, Kim T, Kim CH, Bang SI, et al: expression of ADAM33
is a novel regulatory mechanism in IL-18-secreted process in
gastric cancer. J Immunol. 182:3548–3555. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim KE, Song H, Kim TS, Yoon D, Kim CW,
Bang SI, Hur DY, Park H and Cho DH: Interleukin-18 is a critical
factor for vascular endothelial growth factor-enhanced migration in
human gastric cancer cell lines. Oncogene. 26:1468–1476. 2007.
View Article : Google Scholar
|
|
32
|
Kim J, Kim C, Kim TS, Bang SI, Yang Y,
Park H and Cho D: IL-18 enhances thrombospondin-1 production in
human gastric cancer via JNK pathway. Biochem Biophys Res Commun.
344:1284–1289. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Vidal-Vanaclocha F, Fantuzzi G, Mendoza L,
Fuentes AM, Anasagasti MJ, Martín J, Carrascal T, Walsh P, Reznikov
LL, Kim SH, et al: IL-18 regulates IL-1beta-dependent hepatic
melanoma metastasis via vascular cell adhesion molecule-1. Proc
Natl Acad Sci USA. 97:734–739. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Apte RN, Dotan S, Elkabets M, White MR,
Reich E, Carmi Y, Song X, Dvozkin T, Krelin Y and Voronov E: The
involvement of IL-1 in tumorigenesis, tumor invasiveness,
metastasis and tumor-host interactions. Cancer Metastasis Rev.
25:387–408. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Martín MJ, Calvo N, de Boland AR and
Gentili C: Molecular mechanisms associated with PTHrP-induced
proliferation of colon cancer cells. J Cell Biochem. 115:2133–2145.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Will M, Qin AC, Toy W, Yao Z,
Rodrik-Outmezguine V, Schneider C, Huang X, Monian P, Jiang X, de
Stanchina E, et al: Rapid induction of apoptosis by PI3K inhibitors
is dependent upon their transient inhibition of RAS-ERK signaling.
Cancer Discov. 4:334–347. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Johannessen M and Moens U: Multisite
phosphorylation of the cAMP response element-binding protein (CREB)
by a diversity of protein kinases. Front Biosci. 12:1814–1832.
2007.
|
|
38
|
Reddy VS, Valente AJ, Delafontaine P and
Chandrasekar B: Interleukin-18/WNT1-inducible signaling pathway
protein-1 signaling mediates human saphenous vein smooth muscle
cell proliferation. J Cell Physiol. 226:3303–3315. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hosotani Y, Kashiwamura S, Kimura-Shimmyo
A, Sekiyama A, Ueda H, Ikeda T, Mimura O and Okamura H:
Interleukin-18 prevents apoptosis via PI3K/Akt pathway in normal
human keratinocytes. J Dermatol. 35:514–524. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fix C, Bingham K and Carver W: Effects of
interleukin-18 on cardiac fibroblast function and gene expression.
Cytokine. 53:19–28. 2011. View Article : Google Scholar :
|
|
41
|
Yang KY, Bae WS, Kim MJ, Bae YC, Kim YJ,
Kim HJ, Nam SH and Ahn DK: Participation of the central p38 and
ERK1/2 pathways in IL-1β-induced sensitization of nociception in
rats. Prog Neuropsychopharmacol Biol Psychiatry. 46:98–104. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Venkatesan B, Valente AJ, Reddy VS, Siwik
DA and Chandrasekar B: Resveratrol blocks interleukin-18-EMMPRIN
cross-regulation and smooth muscle cell migration. Am J Physiol
Heart Circ Physiol. 297:H874–H886. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chang MC, Hung HP, Lin LD, Shyu YC, Wang
TM, Lin HJ, Chan CP, Huang CC and Jeng JH: Effect of interleukin-1β
on ICAM-1 expression of dental pulp cells: Role of PI3K/Akt,
MEK/ERK, and cyclooxygenase. Clin Oral Investig. 19:117–126. 2015.
View Article : Google Scholar
|
|
44
|
Yan B, Zhang W, Jiang LY, Qin WX and Wang
X: Reduced E-cadherin expression is a prognostic biomarker of
non-small cell lung cancer: A meta-analysis based on 2395 subjects.
Int J Clin exp Med. 7:4352–4356. 2014.
|
|
45
|
Wang R, Ke ZF, Wang F, Zhang WH, Wang YF,
Li SH and Wang LT: GOLPH3 overexpression is closely correlated with
poor prognosis in human non-small cell lung cancer and mediates its
metastasis through upregulating MMP-2 and MMP-9. Cell Physiol
Biochem. 35:969–982. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Fiorentini C, Bodei S, Bedussi F, Fragni
M, Bonini SA, Simeone C, Zani D, Berruti A, Missale C, Memo M, et
al: GPNMB/OA protein increases the invasiveness of human metastatic
prostate cancer cell lines DU145 and PC3 through MMP-2 and MMP-9
activity. Exp Cell Res. 323:100–111. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bandapalli OR, Ehrmann F, Ehemann V, Gaida
M, Macher-Goeppinger S, Wente M, Schirmacher P and Brand K:
Down-regulation of CXCL1 inhibits tumor growth in colorectal liver
metastasis. Cytokine. 57:46–53. 2012. View Article : Google Scholar
|
|
48
|
Gogali A, Charalabopoulos K, Zampira I,
Konstantinidis AK, Tachmazoglou F, Daskalopoulos G, Constantopoulos
SH and Dalavanga Y: Soluble adhesion molecules E-cadherin,
inter-cellular adhesion molecule-1, and E-selectin as lung cancer
biomarkers. Chest. 138:1173–1179. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wu Y and Zhou BP: Inflammation: A driving
force speeds cancer metastasis. Cell Cycle. 8:3267–3273. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kase S, Sugio K, Yamazaki K, Okamoto T,
Yano T and Sugimachi K: Expression of E-cadherin and beta-catenin
in human non-small cell lung cancer and the clinical significance.
Clin Cancer Res. 6:4789–4796. 2000.
|
|
51
|
Grant JL, Fishbein MC, Hong LS, Krysan K,
Minna JD, Shay JW, Walser TC and Dubinett SM: A novel molecular
pathway for Snail-dependent, SPARC-mediated invasion in non-small
cell lung cancer pathogenesis. Cancer Prev Res (Phila). 7:150–160.
2014. View Article : Google Scholar
|
|
52
|
Lee CH, Chang JS, Syu SH, Wong TS, Chan
JY, Tang YC, Yang ZP, Yang WC, Chen CT, Lu SC, et al: IL-1β
promotes malignant transformation and tumor aggressiveness in oral
cancer. J Cell Physiol. 230:875–884. 2015. View Article : Google Scholar
|
|
53
|
Jee YS, Jang TJ and Jung KH: Prostaglandin
E(2) and interleukin-1β reduce E-cadherin expression by enhancing
snail expression in gastric cancer cells. J Korean Med Sci.
27:987–992. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bani-Hani AH, Leslie JA, Asanuma H,
Dinarello CA, Campbell MT, Meldrum DR, Zhang H, Hile K and Meldrum
KK: IL-18 neutralization ameliorates obstruction-induced
epithelial-mesenchymal transition and renal fibrosis. Kidney Int.
76:500–511. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Helmy A, Guilfoyle MR, Carpenter KLH,
Pickard JD, Menon DK and Hutchinson PJ: Recombinant human
interleukin-1 receptor antagonist in severe traumatic brain injury:
A phase II randomized control trial. J Cereb Blood Flow Metab.
34:845–851. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang CC, Li H, Zhang M, Li XL, Yue LT,
Zhang P, Zhao Y, Wang S, Duan RN, Li YB, et al: Caspase-1 inhibitor
ameliorates experimental autoimmune myasthenia gravis by innate
dendric cell IL-1-IL-17 pathway. J Neuroinflammation. 12:1182015.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Gu H, Xie M, Xu L, Zheng X, Yang Y and Lv
X: The protective role of interleukin-18 binding protein in a
murine model of cardiac ischemia/reperfusion injury. Transpl Int.
28:1436–1444. 2015. View Article : Google Scholar : PubMed/NCBI
|