Tumorigenesis is a complicated and multistep
process, in which successive mutations in oncogenes and
tumor-suppressor genes virtually result in enhanced proliferation
and resistance to cell death. Most human tumor types share various
hallmarks, which include sustainment of proliferative signals,
evasion of growth suppressors, resistance to cell death,
replicative immortality, induction of angiogenesis, activation of
invasion and metastasis, energy metabolism, evasion of immune
destruction, genome instability and mutation, and tumor-promoting
inflammation (1,2). During the past decades of cancer
research, our focus on cancer research has shifted from the
malignant cancer cell itself to the tumor microenvironment and the
complex interactions. The tumor microenvironment, which consists of
resident fibroblasts, endothelial cells, pericytes, leukocytes and
extracellular matrix, also contributes to the progression of cancer
(3).
Studies have provided some evidence that human
tumors are more than a mass of accumulating malignant cancer cells.
Actually, tumor cells can efficiently recruit stromal cells
(4), immune cells (5) and vascular cells (6) by secreting stimulatory growth factors,
chemokines and cytokines. In turn, these recruited cells release
growth-promoting signals and intermediate metabolites as well as
remodel tissue structure to build the microenvironment. The
reciprocal communication between cancer cells and the
microenvironment eventually leads to enhanced proliferation and
metastatic capability, and finally death.
As the tumor microenvironment actively participates
in tumor progression and metastasis rather than acting as a
by-stander, therapeutic strategies targeting the tumor
microenvironment hold great potential. It is known that non-tumor
cells are presumably and genetically more stable than tumor cells,
thus, therapies targeting the tumor microenvironment are less
likely to cause adaptive mutations and rapid metastasis. Yet,
considering the complex interactions (stromal cells can both
promote and inhibit tumor cell growth), therapies targeting the
tumor microenvironment for cancer therapy should be highly
selective. Therefore, further studies must provide new insight into
the tumor microenvironment for better cancer therapeutic
strategies. We will review how tumor cells recruit stromal cells to
the primary tumor site and build the microenvironment. Moreover, we
will highlight the role of the tumor microenvironment in the
regulation of tumor progression and discuss the potential value for
cancer therapy.
A tumor is a highly complex tissue composed of
neoplastic and stromal cells. It is widely known that stromal cells
contain a variety of mesenchymal cells, particularly fibroblasts,
myofibroblasts, endothelial cells, pericytes and inflammatory cells
associated with the immune system. Accumulating evidence has
confirmed that tumor cells must recruit and reprogram the
surrounding normal cells to serve as contributors to tumor
progression. Tumor cells and the supporting normal cells form an
organ-like structure and make concerted efforts for rapid
proliferation, local invasion and metastases. These normal cells in
the tumor microenvironment mainly consist of fibroblasts, immune
cells and vascular cells. These cells are recruited to the primary
tumor site and build the tumor microenvironment for tumor
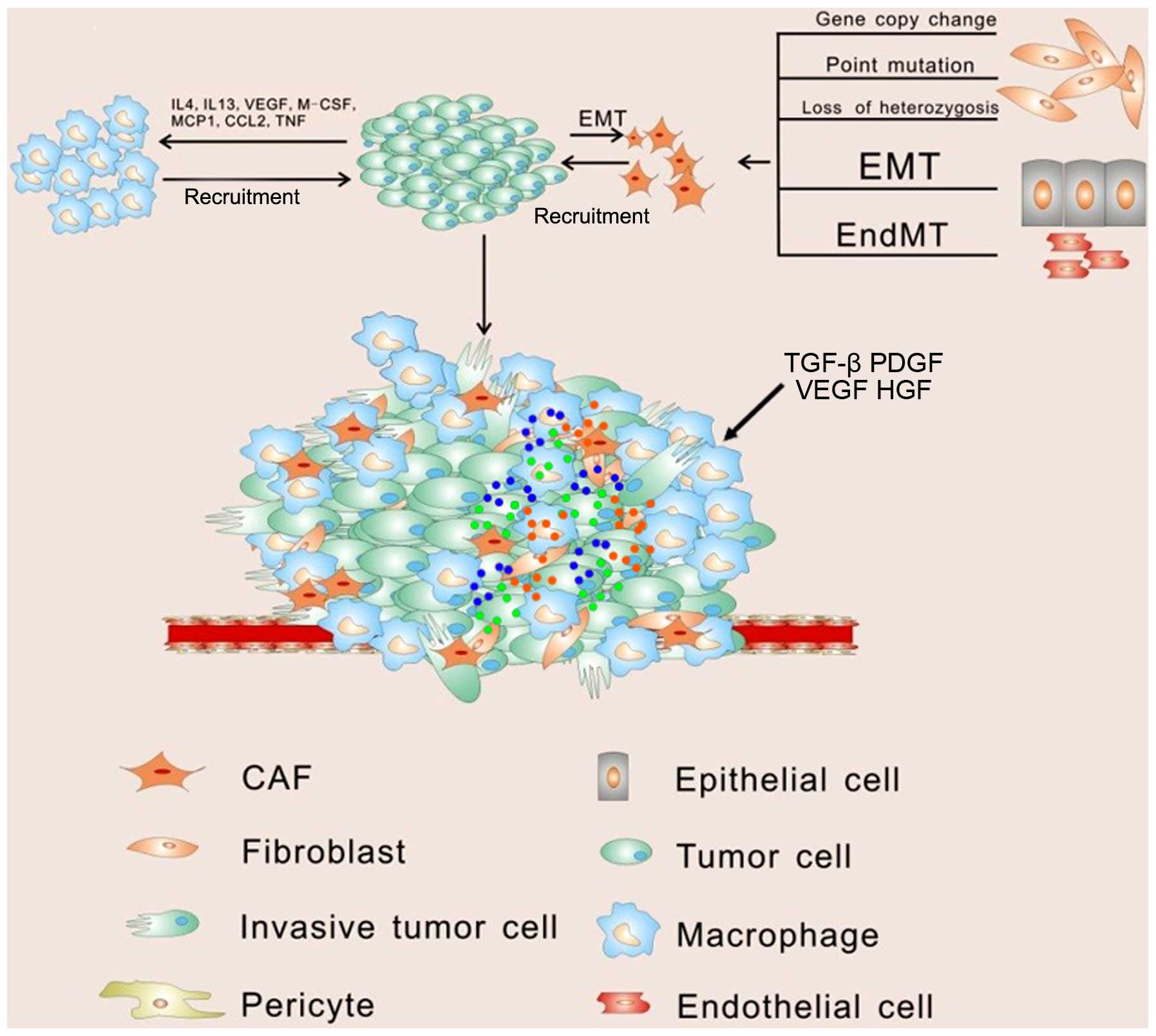
progression in soluble paracrine signals (Fig. 1).
Among the supporting cells, fibroblasts represent
the majority of the stromal cells in various types of human
cancers. Initially, activated fibroblasts inhibit the early stages
of tumor progression (7), and this
effect is carried out through simple gap junctions between
fibroblasts and IL-6 production (8,9).
Fibroblasts can then be modulated by tumor cells and develop into
cancer-associated fibroblasts (CAFs), which are identified by
expression of different biomarks, such as α-smooth muscle actin,
vimentin, desmin and fibroblast-activation protein. Although
research has made great contributions in this field, the original
source of CAFs remains controversial. CAFs are critically involved
in promoting growth and angiogenesis, remolding of the
extracellular matrix (ECM) and directing cell-cell interaction
(10). Clinical and experimental
data indicate that tumor cells secrete a high level of transforming
growth factor β (TGF-β), which is strongly chemotactic for
fibroblasts and transdifferentiates fibroblasts into CAFs (11,12).
The main source of CAFs is thought to be derived from normal
fibroblasts through genetic alteration. It has been observed that
expression of genes in fibroblasts may be altered via point
mutation, loss of heterozygosis, and the number of gene copy
changes. The mutation or inactivation of
phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase (PTEN) and
p53 is frequently detected in CAFs around the primary tumor lesion
(13). However, the evidence for
genetic alterations as a factor to induce CAFs is still
unconvincing. Normal dermal fibroblasts can also be orchestrated
indirectly dependent on immune cells by carcinoma cells to express
pro-inflammatory genes (14).
Except for normal fibroblasts, CAFs are thought to be generated
from epithelial cells, endothelial cells and, interestingly, cancer
cells (Fig. 1). The myofibroblast,
an essential cell type, participates in wound healing (15) and was also found to be a major
source of CAFs (16). Laminin,
which critically contributes to cell attachment and
differentiation, is downregulated in myofibroblasts in cancer
regions, providing an additional evidence that CAFs can be directly
differentiated from myofibroblasts (17). In addition, vascular cells such as
vascular smooth muscle cells show similar markers and morphology
with myofibroblasts, providing another probability that CAFs may be
derived from mural cells (18).
Platelet-derived growth factor (PDGF) can also indirectly recruit
myofibroblasts by stimulation of TGF-β release from macrophages
(19). Another potential source of
CAFs is human bone marrow-derived mesenchymal stem cells (hMSCs).
hMSCs, which are thought to be multipotent cells, are present in
adult marrow and have the potential to differentiate into lineages
of mesenchymal tissues (20). Under
hypoxic conditions, tumor cells secrete IL-6 and activate both
Stat3 and MAPK signaling pathways to enhance the migratory
potential of hMSCs (21,22). The recruited hMSCs have the
potential to develop into CAFs. Notable, surrounding normal
epithelial cells can be another source of CAFs by undergoing
epithelial-to-mesenchymal transition (EMT) in response to stimuli
from the microenvironment. A previous study reported that
proliferating endothelial cells induced by TGF-β can undergo a
phenotypic conversion into fibroblast-like cells (23). Another recent study confirmed that
EndMT (endothelial-to-mesenchymal transition) frequently appears in
a variety of cancers. Zeisberg et al found that endothelial
cells are a source of CAFs by undergoing EndMT at the invasive
front of tumors in transgenic mice (24). This suggests that EndMT is an
important process for the accumulation of CAFs. Interestingly, CAFs
can also be derived from cancer cells directly which shows
dangerous signaling. Cancer cells are obstinate and are not
eradicated easily. A previous study revealed that under the proper
conditions, breast tumor cells can transdifferentiate into
myoepithelial cells and finally become myofibroblasts, which are
the ancestors of CAFs (25).
Meanwhile, recent genetic analysis found that CAFs isolated from
human breast tumor biopsies were indeed derived from epithelial
cancer cells (26). However,
genetic alterations present in both CAFs and cancer cells are not
identical, suggesting that only a small part of stromal cells and
cancer cells may share a common origin (27). Therefore, it is worthwhile to
consider the consequences of tumor cell-derived CAFs in tumor
progression, the indirect action of nonmalignant CAFs on associated
tumor cells as a mechanism of facilitating tumor growth. The
current paradigm would appear then to be that some CAFs encourage
their neighbors to become more malignant rather than performing
this function themselves.
Once CAFs are stimulated, they can secrete stromal
cell-derived factor 1 (SDF-1), which recruits circulating
endothelial progenitor cells (EPCs) into the tumor mass to
stimulate angiogenesis (28).
Importantly, a recent report shed new light on the roles of miRNAs
in tumor microenvironment. Downregulation of miR-320 and
upregulation of ETS2 (v-ets erythroblastosis virus E26 oncogene
homolog 2, one of the direct targets of miR-320), were found to
contribute to tumor angiogenesis and tumor-cell invasion in
PTEN-deleted stromal fibroblasts (29). Another report revealed that CAFs
mediate tamoxifen resistance through IL-6-induced degradation of
ER-α in luminal breast cancer (30). This study demonstrated that CAFs
also play a role in drug resistance. Studies designed to ascertain
how CAFs provide a suitable tumor microenvironment may facilitate
the development of new therapeutic strategies against tumor
progression.
Oncogenic mutations and transcription factor
activation induce high levels of inflammatory mediators, including
cytokines and chemokines. Chemokines and cytokines are critical
autocrine and paracrine factors in tumor development, which are
secreted into the tumor microenvironment to recruit and activate
various inflammatory cells. In turn, these 'educated' inflammatory
cells produce more inflammatory signals and form a cancer-related
inflammatory microenvironment to induce cancer cell evasion from
immune destruction. Finally these inflammatory cells promote tumor
progression. Among these immune cells, macrophages represent the
majority and play leading roles in cancer-related inflammation.
Macrophages can polarize into two different types of macrophages
upon different stimulation. Classically activated macrophages (M1),
following exposure to interferon, have antitumor activity and
elicit tissue destructive reactions. However in response to IL-4 or
IL-13, macrophages undergo alternative activation (M2) (31). Tumor-associated macrophages (TAMs)
closely resemble alternative (M2) macrophages, which produce high
amounts of interleukin IL-10. Moreover, these cells exhibit
anti-inflammatory and tissue repair functions (32). Vascular endothelial growth factor
(VEGF), macrophage-colony stimulating factor (M-CSF) and monocyte
chemotactic protein 1 (MCP-1) produced by tumor cells efficiently
recruit macrophages to the tumor microenvironment by promoting
migration and survival (33).
Interestingly, a low level of MCP-1 induces modest monocyte
infiltration resulting in tumor formation, whereas a high level is
associated with massive monocyte/macrophage infiltration into the
tumor mass, leading to tumor destruction (34). Experimental evidence suggests that
signaling molecules produced by both tumor cells and these
macrophages, work together to activate integrin α4β1, with
subsequent stimulation of myeloid cells entering the tumor
microenvironment (35). Among these
signaling molecules, chemokine and chemokine receptors make up a
complex network, which influence the development of primary tumors
and metastases (36). Recent data
showed that tumor cells and host organ-derived chemokine chemokine
(C-C motif) ligand 2 (CCL2) recruit inflammatory monocytes, which
differentiate into macrophages and facilitate efficient tumor cell
metastasis seeding and growth in distant meta-static sites of the
lung (37). CCL2 can also increase
prostate tumor growth and bone metastasis through macrophage and
osteoclast recruitment (38). These
studies have made great contributions to our understanding of the
microphage recruitment to the tumor site (Fig. 1). Cancer cells that overexpress
chemokine (C-X-C motif) ligand 2 (CXCL1/2) can also attract
CD11b+Gr1+ myeloid cells into the primary
tumor site, which produce chemokines, including S100A8/9, that
enhance cancer cell survival (39).
CCL21, expressed by melanoma tumors, shifts the host immune
response from immunogenic to tolerogenic, and facilitates tumor
progression (40). Other soluble
factors, such as prostaglandin E2 (PGE2) and TGF-β, also play an
active role in facilitating tumor progression by limiting natural
killer (NK) cells (41). Tumor
necrosis factor (TNF) signaling can drive myeloid-derived
suppressor cell accumulation, and promote tumor cell escape from
the immune system (42). In a
spontaneous murine model of patent ductus arteriosus (PDA), Bayne
et al demonstrated that tumor-derived granulocyte-macrophage
colony-stimulating factor (GM-CSF) drives the accumulation of
Gr-1+CD11b+ myeloid cells as part of the
cancer-associated inflammatory reaction, which in turn suppresses
antitumor T cell immunity and promotes tumor growth (43). In particular, CXCL12 can regulate
cancer cell survival, proliferation and migration, and, indirectly,
via angiogenesis or recruiting immune cells to affect tumor
progression (44). These 'educated'
immune cells work together with local tumor cells and CAFs to
produce more inflammatory factors forming an inflammatory
microenvironment and protecting tumor cells from immune
destruction. Finally, this cooperation promotes tumor progression
and metastasis. However, studies on the relationship between
inflammation and cancer are sparse. Progress in the identification
of inflammation-dependent mechanisms that affect tumor cell
survival, trafficking and chemo-attractive functions are valuable
to new drug development. Understanding of the biological and
molecular mechanisms of carcinogenesis may provide more
opportunities for clinical therapy.
Tumors require the formation of a complex vascular
network to meet the metabolic and nutritional needs for growth.
Recent evidence suggests that endothelial cells and pericytes,
which play essential roles in the 'turn on' of the angiogenic
switch (45,46), can also be modulated by tumor cells.
Several studies indicate that VEGF is highly expressed in a variety
of human tumors, including lung (47), breast (48), ovarian (49), bladder and kidney (50). VEGF elicits a pronounced angiogenic
response in a variety of in vivo models. VEGF directly
activates enterochromaffin cells (Ecs) through mitogenic and
promigratory effects, and also mobilizes endothelial progenitor
cells (EPCs) from the bone marrow, modulates EPC kinetics and
promotes EPC differentiation (51).
Interestingly, miR-126 regulates endothelial recruitment and
metastatic colonization through IGFBP2, PITPNC1 and MERTK targeting
(52). Protein kinase C (PKC)
inhibition plays a crucial role in the extracellular
signal-regulated kinase (ERK) phosphorylation that mediates
proliferation of pulmonary vascular endothelial cells (53). Another important signaling pathway
PI3K/AKT/mTOR (phosphatidylinositol 3-kinase/AKT/mammalian target
of rapamycin) is activated in most human cancers and is closely
related with the production of VEGF. It has been reported that VEGF
secretion is increased both in hypoxia-inducible factor 1
(HIF-1)-dependent and -independent manners in response to PI3K/AKT
activation (54). Activation of
mTOR was detected in head and neck squamous cell carcinoma (HNSCC)
patients with metastasis and inhibition of mTOR decreased vascular
formation and lymph node metastasis (55,56).
Inhibitors targeting signaling and molecules involved in
angiogenesis may be a viable strategy for the treatment of
cancer.
Growth-promoting signals in the microenvironment
play a critical role in both normal and pathological tissues.
Normal tissues require that growth factors be induced from a
quiescent state into an active proliferation state. They tightly
control the production and the release of growth-promoting signals
that instruct themselves or the entry of other cells into the cell
growth and division cycle. At the same time, growth-promoting
signals contribute to cancer-sustaining proliferation, which has
been confirmed as a hallmark of cancer. Cancer cells obtain growth
signals through autocrine and paracrine pathways. Analyzing
previous research, we conclude that tumor stromal cells provide
cancer cells with growth-promoting signals, including growth
factors, cytokines and chemokines. Table I lists these various tumor-promoting
molecules.
Growth factors, secreted by stromal cells into the
microenvironment, promote tumor progression via stimulation of
cellular growth, proliferation and cellular differentiation. For
instance, TGFβ is known to enhance EMT and invasiveness in primary
carcinomas (57). Blocking the TGFβ
signaling pathway can reduce intravasation and metastatic seeding
in the lung as well as bone (58–62). A
recent report by Labelle et al suggests that platelets
secrete TGFβ-1 to activate the TGFβ/Smad pathway in tumor cells,
and enhance invasiveness and metastasis (63). Hepatocyte growth factor (HGF), which
was originally cloned as a mitogenic protein in hepatocytes
(64), can specifically activate
MET receptor tyrosine kinase as well as stimulate mitogenesis, cell
motility and matrix invasion (65,66).
Two studies found that the higher a patient's HGF level, the less
likely he/she was to remain in remission. The study found that
stromal cells secreted HGF resulting in activation of the MET,
reactivation of the mitogen-activated protein kinase (MAPK) and
phosphatidylinositol-3-OH kinase (PI3K)-AKT signaling pathways.
These cellular signaling changes in tumor cells immediately induce
resistance to RAF inhibition and confer resistance to BRAF
inhibitor in BRAF-mutant melanoma cells (67,68).
In addition to chemical inhibitors for the inhibition of growth
factors, antibodies that target receptors have been developed.
Cetuximab, an EGFR monoclonal antibody, acts as an efficient
antitumor drug in many types of cancer. The efficiency of cetuximab
against chemo- or radio-resistant HNSCC was demonstrated (69). Other growth factors secreted by
stromal cells can also promote cancer cell growth. For instance,
VEGF, PDGF, bFGF and IGF1 can facilitate tumor progression by
stimulating angiogenesis (table
I).
In addition to growth factors, chemokines are also
important for enhancing tumor growth. Chemokines are chemotactic
cytokines, which are induced by inflammatory cytokines, growth
factors and pathogenic stimuli (70–72).
Chemokine signaling plays a major role in cellular transformation,
inflammation, and wound healing; as well as tumor growth,
angiogenesis, tumorigenesis and metastasis (73) (Table
I). Currently, the research of cancer-associated chemokines has
mainly focused on CXC chemokines and CC chemokines. Some CXC
chemokines promote cancer development mainly by promoting
angiogenesis and enhancing tumor metastasis. CXCL1, a small
cytokine belonging to the CXC chemokine family, is overexpressed in
70% of human melanomas and is involved in CRC tumor growth and
angiogenesis (74). Overexpression
of CXCl1/2 in cancer cells attracts
CD11b+Gr1+ myeloid cells to the primary tumor
site, and finally, enhances the viability of cancer cells through
S100A8/9 factors (39). The subset
of cc chemokine families, which are secreted by stromal cells have
multiple functions in the progression of cancer. For example, CCL2
released from stromal cells can promote tumor growth, facilitate
macrophage infiltration and induce metastasis (75–78).
Meanwhile, other molecules, such as osteopontin (OPN), galectin-3
and brain-derived neutrophic factor (BDNF), are also involved in
the tumor microenvironment and promote cancer progression.
Furthermore, some molecules in the vasculature, such as LPA, S1P,
and prothrombin, play crucial roles in tumor development (Table I).
Small and non-coding RNAs (miRNAs)
post-transcriptionally control the translation and stability of
mRNAs. These RNAs participate in the regulation of metabolism and
tumorigenesis (79–81). RNAs cannot function as extracellular
signaling molecules because they are vulnerable to be degraded by
ribonucleases (82). But
interestingly, recent evidence shows that miRNAs contained in
exosomes act as signal transducers and play important roles in the
tumor microenvironment acting as a bridge between cancer cells and
stromal cells (82–85). Kosaka et al showed that
miR-143 expression in normal prostate cells was higher and
transferred growth-inhibitory signals to prostate cancer cells both
in vitro and in vivo. They highlighted that secretory
tumor-suppressive miRNAs may be a death signal from winners to
losers in the context of cell competition (85). Macrophages also regulate the
invasiveness of breast cancer cells through exosome-mediated
delivery of oncogenic miRNAs (86).
Notably, tumor-secreted miR-21 and miR-29a also can function by an
unexpected mechanism, by binding as ligands to receptors of the
Toll-like receptor (TLR) family, murine TLR7 and human TLR8 in
immune cells, triggering a TLR-mediated prometastatic inflammatory
response that ultimately may lead to tumor growth and metastasis
(87). However, the detailed
mechanisms of the role of secretory miRNAs in the tumor
microenvironment are still poorly understood. Some studies only
report that microRNAs can be stable blood-based markers for cancer
detection. For example, a significant increase in miR10b, miR34a
and miR155 concentrations in the peripheral blood of breast cancer
patients and the observed correlation with tumor progression
suggest a potential clinical utility of circulating miRNAs as a new
class of future biomarkers (88).
Serum levels of miR-141 (an miRNA expressed in prostate cancer) can
distinguish patients with prostate cancer from healthy controls,
and may be used as an important approach for the blood-based
detection of human cancer (89).
Although the understanding of the role of miRNAs in the tumor
microenvironment remains poorly understood, it has been proposed
that miRNAs in the tumor microenvironment may potentially serve as
paracrine signaling molecules having both tumor-promoting as well
as tumor-suppressing effects.
Over the past 10 years of cancer research, the
reprogramming of energy metabolism has been considered as a
hallmark of cancer. Metabolic reprogramming is always considered to
be intrinsic to cancer cells, such as oncogene activation,
inactivation of tumor-suppressor genes as well as the mutation of
glycolytic enzymes (90). Yet,
recent research has shifted our focus on the regulation of the
tumor microenvironment in tumor metabolism. Compared with normal
differentiated cells, cancer cells mainly rely on aerobic
glycolysis rather than mitochondrial oxidative phosphorylation to
gain the energy needs for rapid proliferation even under normal
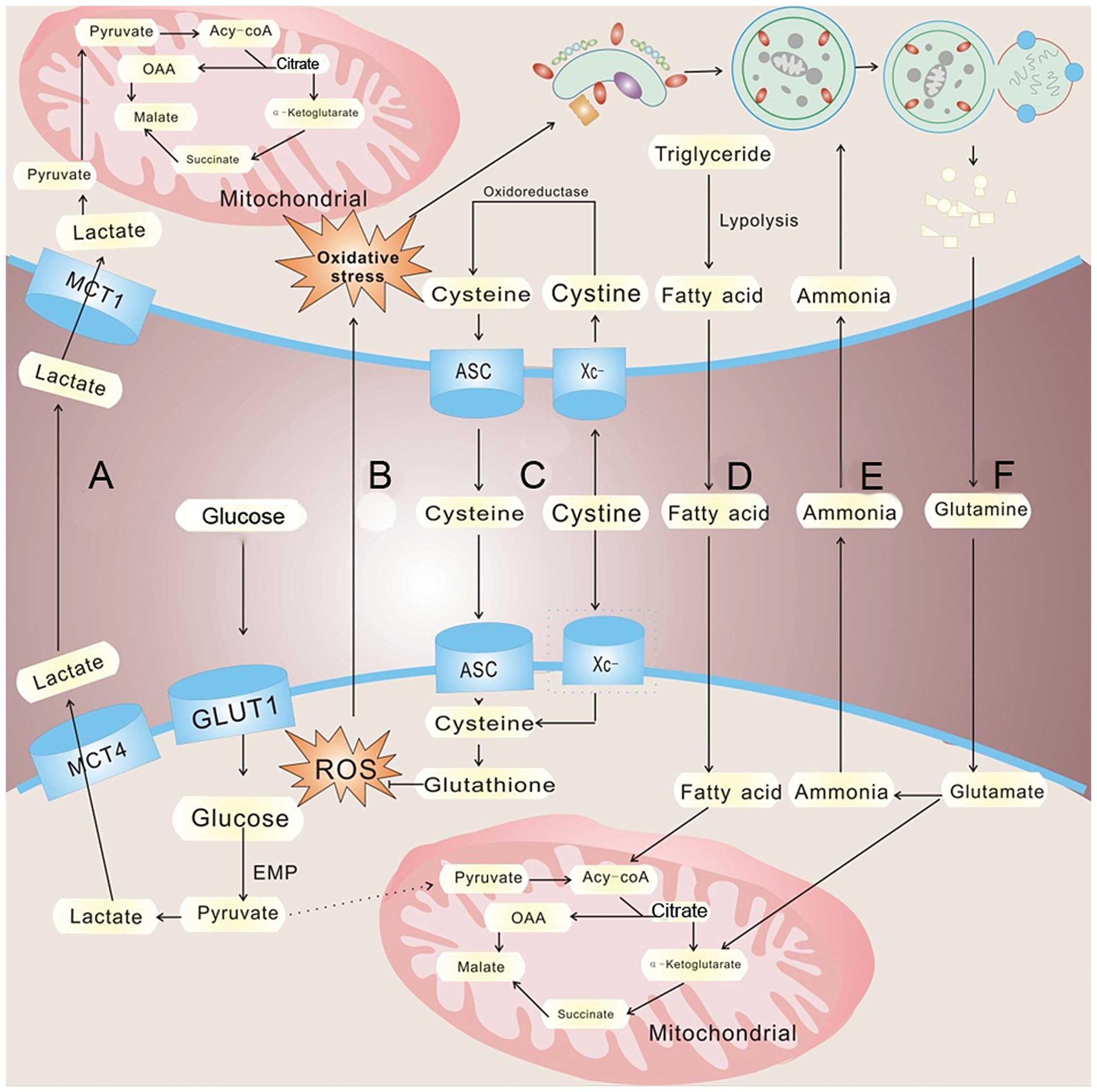
conditions, which is called the 'Warburg effect' (91) (Fig.
2). Yet, aerobic glycolysis is less efficient than oxidative
phosphorylation for generating ATP; 4 mol ATP per mol glucose
compared with 36 mol ATP per mol glucose when under oxidative
phosphorylation. To meet the energy needs and high levels of
glycolytic intermediates supporting anabolic reactions, tumor cells
maintain enhanced glucose uptake through high levels of glucose
transporters, lactate dehydrogenase and other glycolytic enzymes
(92). Due to the high level of
aerobic glycolysis, much lactate is generated by cancer cells,
which contributes to an acidic condition, ROS production and MAPK
signaling activation (93). High
incidence of distant metastasis is related to the suppressed
proliferation and cytokine production of human cytotoxic T
lymphocytes (94,95). But an elevated level of lactate
accumulation in the tumor cells or the microenvironment also leads
to an inhibitory effect of glycolysis and restriction of cell
growth and proliferation. Tumor cells must secrete lactate into the
surroundings via increased expression of lactate transporter
monocarboxylate transporters 4 (MCT4). In response, MCT1 expression
increases in CAFs resulting in the uptake of tumor-extruded
lactate. CAFs increase the expression level of lactate
dehydrogenase (LDH-B), resulting in the conversion of the influxed
lactate to pyruvate. The pyruvate is shunted to the tricarboxylic
acid cycle for ATP generation via oxidative phosphorylation,
thereby satisfying the energy needs of the CAFs (96) (Fig.
2A). The released lactate can also be consumed by endothelial
cells and stimulate angiogenesis through NF-κB/IL-8 signaling
(97). Interestingly, lactate
produced by hypoxic tumor cells may indeed diffuse and be taken up
by oxygenated tumor cells (98,99).
Sonveaux et al suggest that hypoxic tumor cells depend on
glucose and glycolysis to produce energy and secrete lactate. The
lactate is diffused along its concentration gradient and is taken
up by oxygenated tumor cells and used to meet the energy needs
(100). A previous study examined
the expression of major proteins involved in cellular aerobic and
anaerobic metabolism, PDH, PDK1, LDH1, LDH5, MCT1, MCT2 and GLUT1,
between normal tissues, cancer cells and tumor-associated stromal
cells. The results suggest that the newly formed stroma and
vasculature express complementary metabolic pathways, buffering and
recycling products of anaerobic metabolism to sustain cancer cell
survival (101). Nieman et
al provide strong evidence that adipocytes promote the initial
homing of tumor cells to the omentum through adipokine secretion.
Subsequently, adipocytes provide fatty acids to the cancer cells,
fueling rapid tumor growth (102)
(Fig. 2D). These studies highlight
how tumors can survive and grow in hypoxia and an energy crisis.
They are capable of organizing the regional stromal cells into a
harmoniously collaborating metabolic domain living in 'the same
boat'.
In addition to glucose, glutamine is the other
molecule catabolized in appreciable quantities in most mammalian
cell in vitro cultures (103); metabolic profiling of the colon
and stomach cancer microenvironment by capillary electrophoresis
time-of-flight mass spectrometry identified a significant
accumulation of all amino acids except glutamine in the tumors
(104) (Fig. 2F). CAFs undergo an autophagic
program, leading to the generation and secretion of high glutamine
levels into the tumor microenvironment to meet the glutamine needs
of cancer cells. Cancer cells accumulate glutamine and convert it
to glutamate which is further catabolized to α-ketoglutarate,
enters the TCA cycle and increases the mitochondrial activity of
cancer cells. The by-product, ammonia, freely diffuses into the
microenvironment, and then induces autophagy and glutamine
production in CAFs, which confirm a cascade between CAFs and cancer
cell interactions (105) (Fig. 2E). Not only can glutamine be used in
the TCA flux to meet the energy needs of cancer cells, but is also
involved in the synthesis of non-essential amino acids alanine,
serine, arginine, and proline (106). Jain et al suggested a key
role for glycine in rapid cancer cell proliferation using metabolic
profiling approaches. Interestingly, they found that rapidly
proliferating non-transformed cells, including human bronchial
epithelial cells and lymphocytes, release rather than consume
glycine (107). These reports
found that normal cells near tumor cells may provide glycine for
the rapid proliferation of cancer cells. Treatments that target
tumors and surrounding cells should be considered, rather than
targeting only the tumor cells. This concept may provide us with
novel strageties for tumor treatment.
Cellular metabolism is critical for the generation
of energy in biological systems, however, as a result of electron
transfer reactions, reactive oxygen species (ROS) are generated in
aerobic cells. ROS and cellular oxidant stress have long been
associated with cancer (108–111). Previous evidence suggests that
cancer cells normally produce a higher ROS level than normal cells
(112). Hypoxia, mitochondrial
dysfunction, and inflammation, ionizing radiation, chemotherapeutic
agents, hyperthermia, inhibition of antioxidant enzymes, or
depletion of cellular reductants such as NADPH and glutathione, can
all lead to the accumulation of ROS in cancer cells (113,114). Although a low level of ROS is
easily managed by cancer cells, abnormally high levels of ROS
induce oxidative stress. A low level of ROS promotes cell
proliferation and differentiation, while a high level of ROS can
cause oxidative damage to lipids, proteins, DNA and finally cause
cell death (115). Recent evidence
highlights the role of ROS in the tumor microenvironment and
provides new insight into metabolic associations between cancer
cells and non-malignant neighbors in the stroma. Accumulated ROS
are dispersed from cancer cells to adjacent fibroblasts (116), leading to the oxidative stress of
stromal cells, which, in turn, drives autophagy via HiF1 induction
and NF-κB activation; meanwhile providing recycled nutrients via
catabolism and aerobic glycolysis to feed the appetite of adjacent
cancer cells. In addition, stromal ROS production induced by cancer
cells leads to local DNA damage as well as DNA damage in adjacent
cancer cells via a 'bystander effect'. As a consequence, stromal
ROS promote aneuploidy and genomic instability in cancer cells,
driving tumor-stroma coevolution. These studies proposed a simple
solution to the autophagy paradox [both promote cell death and
survival (117–119)], which is called 'The Autophagic
Tumor Stroma Model of Cancer' (120–126). In this simplistic model, it is
proposed that autophagy acts as a tumor suppressor when it occurs
in epithelial cancer cells; conversely, autophagy acts as a tumor
promoter when it occurs in CAFs. Zhang et al provide another
mechanism of tumor-stroma interactions to avoid ROS accumulation in
cancer cells. They showed that bone marrow stromal cells can
expressed a high level of Xc- transporter and effectively take up
cystine to synthesize GSH (127).
It is known that a high level of cellular GSH can both release
oxidative stress and promote cell survival. Metastatic stress and
ROS both are crucial for tumor survival and growth. It is important
for us to understanding how tumor cells conquer the energy crisis
and oxidative stress. When we discuss invasion and metastasis, it
must be remembered that tumor cells have accomplices.
Like normal tissues, tumors need to sustain a
nutrient supply and evacuate metabolic wastes. Blood vessels
nourish nearly every organ of the body, and deviations from normal
vessel growth can contribute to numerous diseases. Angiogenesis
allows tumors to obtain nutrients and evacuate metabolic waste with
no difficulty (128).
Tumor-associated angiogenesis is currently known as a hallmark of
cancer. It is now widely accepted that the 'angiogenic switch' is
under the tight control of pro-angiogenic molecules and
anti-angiogenic molecules, and the 'angiogenic switch' is on only
when the net balance between pro-angiogenic molecules and
anti-angiogenic molecules is tipped in favor of angiogenesis
(128–130). Mounting evidence suggests that
stromal cells in the tumor microenvironment play critical roles in
switching on and sustaining chronic angiogenesis in many tumor
types. Among the various types of stromal cells, TAMs are one of
the most important cell types involved in promoting
tumor-associated angiogenesis. Leek et al found that the
number of TAMs is positively correlated with tumor angiogenesis in
breast carcinomas (131).
Subsequent studies have confirmed such a link in a wide array of
tumor types. TAMs can induce angiogenesis through different
pathways, which can be divided into three categories. First, TAMs
release pro-angiogenic factors directly activating endothelial
cells. In early 1984, TAMs were demonstrated to be potent
stimulators of neovascularization and endothelial cell
proliferation, and that depletion of macrophages from tumor cell
suspensions significantly decreased their angiogenic potential
(132). VEGF, bFGF, TGF-α/β, EGF
and IL-1β secreted by macrophages control tumor angiogenesis
(133–138). These factors induce endothelial
cell activation and differentiation into tumor neovessels.
Recently, Chen et al confirmed that m2 phenotype macrophages
can promote angiogenesis in a paracrine manner via the endothelial
nitric oxide synthase (eNOS) signaling pathway (139). Additionally, tIe2-expressing
macrophages (TEMs) can directly interact with ECs, is important for
tumor angiogenesis and can be targeted to induce effective
antitumor responses (140).
Secondly, TAMs recruit other pro-angiogenic cells. TAMs can attract
mononuclear macrophages (MONO) and TEMs into the tumor
microenvironment. Recruited TEMs can also recruit endothelial and
myeloid progenitors capable of directly incorporating into the
tumor vasculature (141). Thirdly,
TAMs modulate ECM. Matrix metalloproteinases, which are ECM
remodeling enzymes, regulate signaling pathways that control cell
growth, inflammation and angiogenesis. Mounting evidence suggests
that TAMs also produce and secrete MMP2, MMP7, MMP9 and MMP12 to
the tumor microenvironment (142–144). These MMPs can interact with ECM
and increase the bioavailability of pro-angiogenic factors,
including VEGFA and bFGF and promote angiogenesis.
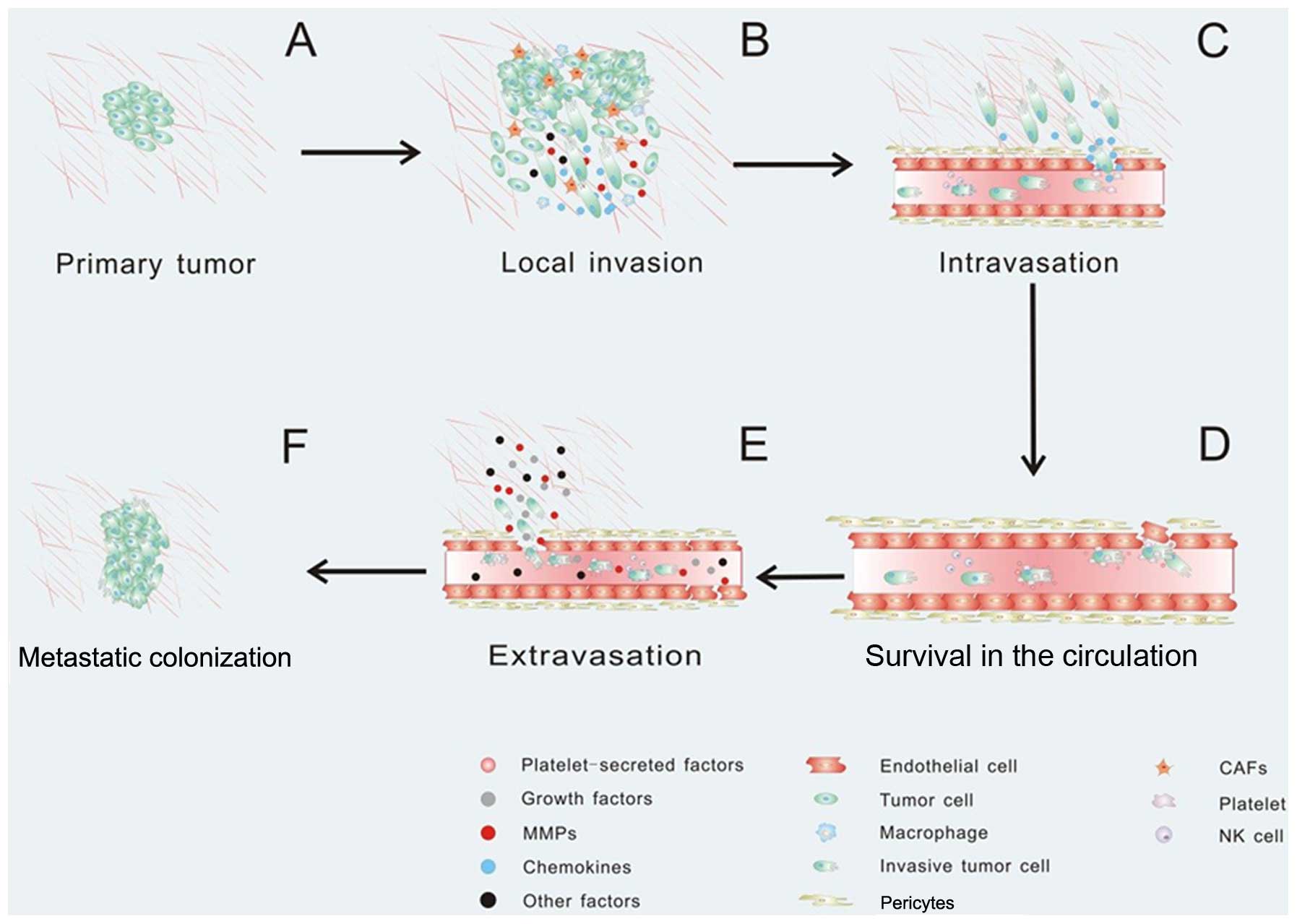
Tumor metastasis always consists of a series of
discrete biological processes that move tumor cells from the
primary neoplasm to a distant organ. This process involves local
invasion, intravasation, survival in the circulation, arrest at a
distant organ site, extravasation, micrometastasis formation, and
then metastatic colonization, and finally clinically detectable
macroscopic metastases are formed (147–149) (Fig.
3). These events have been considered to be induced by genetic
and/or epigenetic alterations within tumor cells, but accumulating
evidence supports that tumor metastasis is also mediated by tumor
stromal cells. Recent publications have confirmed that the tumor
microenvironment contributes to every stage of tumor metastasis.
Once the tumor cells need to escape from the primary tumor, they
must interact with preexisting host basement membranes and the ECM
(Fig. 3A). In early 1986, Liotta
et al proposed a three-step hypothesis describing the
sequence of biochemical events during tumor cell local invasion: i)
tumor cell attachment via cell surface receptors which specifically
bind to components of the matrix, ii) secretion of hydrolytic
enzymes and iii) tumor cell locomotion into the region of the
matrix (150,151). MMP-7 expressed by breast
epithelial cancer cells not only cleaves matrix components in the
tumor microenvironment, but also cleaves the cell surface adhesion
molecule E-cadherin, leading to disruption of basement membrane
structures and breast epithelial cell-cell junctions (152,153). It has been confirmed that CAFs and
macrophages promote cancer cell invasion by secreting matrix
metalloproteases which cause proteolysis of the ECM leading to the
promotion of cancer cell invasion (154–156). Fibroblasts also facilitate tumor
cell invasion through force- and protease-mediated ECM remodeling
(157–159), and intrinsic fibroblast caveolin-1
enhances tumor cell invasion by force-dependent remodeling of the
surrounding environment via Rho GTPase activation (160). Activated macrophages and TAMs
produce CCL18 and promote the invasiveness of breast cancer cells
via the functional receptor for CCL18, PITPNM3 (161) (Fig.
3B). Interestingly, the ECM in the path of the invading cell
can be remodeled by invadopodia (162), which are actin-rich membrane
protrusions with a matrix degradation activity formed by invasive
carcinoma cells (163). Podosomes,
which are similar to invadopodia in molecular composition with
ventral membrane protrusions and invaginations formed by
macrophages and other type of cells (164–166), are proposed to play a role in ECM
remodeling and then promote carcinoma cell invasion. For example,
v-src-transformed 3Y1 rat fibroblast (3Y1-src) cells cultured on
fibronectin degrade the fibronectin mainly at the podosomes, which
is thought to underlie the invasive phenotype of 3Y1-src cells
(167). Yamaguchi et al
showed that macrophage podosomes have a matrix degradation activity
and that colony-stimulating factor-1 (CSF-1) regulates the
formation and organization of macrophage podosomes (162). These observations highlight the
critical role of these specialized protrusive structures,
invadopodia and podosomes, in tumor invasion (Fig. 3B). EMT is a hypothesized program of
development of biological cells characterized by loss of cell
adhesion, repression of E-cadherin expression, and increased cell
mobility. EMT is essential for numerous developmental processes
including mesoderm formation and neural tube formation and is
regulated by many transcription factors, including zinc finger
protein snail 1 (SNAI1), SNAI2, zinc finger E-box-binding homeobox
1 (ZEB1), ZEB2, TWIST, FOXC1 (forkhead box protein 1), FOXC2, TCF3
(transcription factor 3 - also known as E47), and GSC (homeobox
protein goosecoid) (168). During
tumor cell invasion, tumor cells co-opt EMT and the basement
membrane becomes fragmented. The tumor cells can intravasate into
lymph or blood vessels, allowing their passive transport to distant
organs (169). It has been
reported that tumor-associated fibroblasts (TAFs) induce the
significant overexpression of FgFr4 in colorectal cancer cell lines
and play a critical role in colorectal cancer EMT and metastasis
(170). The stromal cells also
stimulate EMT and promote tumor cell invasion. Pericytes,
associated with endothelial cells, promote tumor angiogenesis, and
promote tumor progression (171,172). By using genetic mouse models or
pharmacological inhibitors, Cooke et al showed that pericyte
depletion suppressed tumor growth but resulted in
hypoxia-associated EMT (173).
Meanwhile, inflammation plays an important role in inducing EMT.
Snail, which plays an essential role in inducing EMT and cancer
metastasis by repressing expression of E-cadherin (174–176), can be stabilized by the
inflammatory cytokine TNFα through the activation of the NF-κB
pathway (177). It has been
revealed that EMT is a dynamic process controlled by an
inflammatory microenvironment.
After local invasion, the tumor cells infiltrate
into the vascular spaces and establish direct contact with the
blood. In this step, the tumor cells invade across the endothelial
basal lamina and migrate between the endothelial cells lining the
capillaries that service the tumor (178). Intravasation is always critical
and is the rate limiting step of tumor metastasis (148,179). Recent evidence suggests that the
tumor microenvironment can promote cancer cell intravasation and
metastasis. For instance, CCL2-expressing tumor cells attract
monocytes and activate endothelial cells through CCR2, showing that
a tumor cell-derived chemokine induces vascular permeability and
enables efficient tumor cell intravasation (37,180)
(Fig. 3C). In particular, platelets
influence vascular integrity and play an important part in tumor
metastasis (181). Notably,
platelets secrete TGFβ1 to activate the TGFβ/Smad pathway, which
synergize with the NF-κB pathway, and enhance invasiveness and
promote metastasis (63). When
cancer cells enter the circulation system, most of the cancer cells
will not survival due to the loss of integrin-dependent adhesion to
ECM components causing anoikis, damage incurred by hemodynamic
shear forces and the predation by cells by the innate immune
system, specifically NK cells (149) (Fig.
3D). The circulating tumor cells can be detected in the
bloodstream of patients using microchip technology, immunomagnetic
nanoscreening and 2-NBDG fluorescence imaging (182–184). In order to survive in the
circulation, tumor cells recruit platelets which, in turn, form a
coat to protect them from the innate immune system. Even if tumor
cells are NK susceptible and cytotoxic NK cells threaten their life
in the blood, platelets are capable of protecting them from
cytolysis by forming a physical shield around cancer cells, thereby
promoting metastasis (185,186).
As platelets become activated, they can release growth factors,
chemokines and protease, which can perpetuate the cohesion of
heteroaggregates containing tumor cells. Platelets can also support
the attachment to the endothelium and thereby contribute to
metastasis (181) (Fig. 3D). During circulation, these
invasive tumor cells may arrest at any distant organ site. When the
new site is ready for metastatic tumor growth, the primary tumors
are able to secrete factors to induce cancer cell extravasation.
For example, the secreted angiopoietin-like-4 (ANGPTL4), as well as
EREG, COX-2, MMP-1, and MMP-2, are able to disrupt pulmonary
vascular endothelial cell-cell junctions to facilitate cancer cell
extravasation (59,187). When cancer cells arrive at a
secondary site, the microenvironment is phenotypically and
functionally distinct from the primary tumor, which may cause some
physical barriers. In order to overcome physical barriers at the
secondary site, primary tumors can secrete factors that perturb the
distant microenvironment (Fig. 3E).
For example, the pre-metastatic niche referred to as interactions
between metastatic tumor cells and their stromal cells (188), have been defined as a new concept,
which describes the tumor microenvironment playing important roles
in the tumor cell survival in the circulation and growth at a
secondary site. Primary tumors can secrete growth factors priming
certain tissues in the metastatic site for tumor cell engraftment
and growth (189). The primary
tumor cells secrete pro-inflammatory factors such as VEGF-A, TGFβ
and TNFα inducing the expression of chemoattractants S100A8 and
S100A9 in lung VE-cadherin+ endothelial cells and
Mac1+ myeloid cells (190,191). But the exact mechanism by which
these chemoattractants elicit cell accumulation is not known.
Hiratsuka et al showed that serum amyloid A (SAA)3 induced
in pre-metastatic lungs by S100A8 and S100A9 has an important role
in the accumulation of myeloid cells and acts as a
positive-feedback regulator for chemoattractant secretion.
Meanwhile, SAA3 can stimulate NF-κB signaling in a TLR4-dependent
manner and facilitate metastasis (192). More interestingly, Kaplan et
al found that bone marrow-derived hematopoietic progenitor
cells that express vascular endothelial growth factor receptor 1
home to tumor-specific pre-metastatic sites and form cellular
clusters before the arrival of tumor cells (193). They first demonstrated that a
non-neoplastic cell population can portend a future metastatic
site. After extravasation, tumor cells utilize the microenvironment
in the metastatic site and form a new tumor microenvironment
supporting metastatic tumor growth (Fig. 3F).
Cancer cells require an enormous variety of genetic
changes to elicit tumorigenesis. The clinical therapy for many
types of human cancers has mainly focused on the malignant cancer
cell itself, and have made great achievements, yet cancer therapy
still remain a great challenge. Currently, the most commonly used
radiotherapy and chemotherapy strategies have serious side effects,
such as destruction of the patient immune system, and patients
rapidly develop therapeutic resistance. As described above, the
tumor microenvironment commonly participates in tumor initiation as
well as progression in many tumor types, providing us with the hope
that therapeutic targeting of these events could be efficient for
cancer therapy. Recent publications provide strong evidence that
tumor stromal cells forming the tumor microenvironment contribute
to chemoresistance. For example, CCR2 null host mice responded
better than a control when treated with doxorubicin. This effect
was induced because myeloid cells can be recruited to
doxorubicin-treated tumors through a stromal CCL2/CCR2
chemokine/chemokine receptor axis leading to chemoresistance
(194). Similarly, during
treatment with platinum analogs, endogenous mesenchymal stem cells
(MSCs) are activated and release polyunsaturated fatty acids which
protect cancer cells against a range of chemotherapeutics (195). These outstanding findings show
that the tumor microenvironment is a potent administrator of
resistance to traditional cytotoxic therapies, mainly chemotherapy
and radiotherapy, and reveal potential available targets to enhance
chemotherapy efficacy in patients. Multitargeted approaches, in
which tumor cells and the tumor microenvironment are simultaneously
inhibited, have been developed in recent years. These multitargeted
approaches have many advantages compared with traditional
therapies. On the one hand, stromal cells in the tumor
microenvironment are presumably genetically stable, while tumor
cells are known to be genetically unstable. Therefore, it is less
likely for cancer patients to accumulate adaptive mutations as well
as rapidly acquire resistance to chemotherapy as well as
radiotherapy. On the other hand, the tumor microenvironment has a
certain similarity in diverse cancer types, thus one therapeutic
target of the tumor microenvironment can be implicated in more than
one type of cancer. Based on research of the tumor
microenvironment, multiple technologies have been developed for the
research of cancer, such as liquid biopsy (196) and in silico molecular
biology (197). The liquid biopsy
is used to analyze tumor DNA in urine for non-muscle invasive
bladder cancer patients and provides a non-invasive approach for
bladder cancer detection (198).
The in silico biomarker profiling technology is used to
identify GLUT4-specific inhibitors for cancer therapy (199). These results made valuable
contributions for the personalized/precision medicine and hold
great potential for personalized detection.
Due to these advantages, targeting the tumor
microenvironment holds great potential for cancer therapy. There
are many tumor-promoting factors in the tumor microenvironment,
suggesting that inhibition of these tumor-promoting factors or
destroying these signaling pathways can prevent the development of
cancer. For example, tumors in a stroma xenograft model treated
with the TGF-β inhibitor exhibited a reduction in blood vessels
(200). As enhancement of GSH
synthesis in CLL cells is possibly a crucial mechanism by which
stromal cells facilitate leukemia cell survival and drug resistance
through providing cysteine (127),
a strategy to destroy stromal protection of CLL cells by inhibiting
the transporter to impact the uptake of cystine by stromal cells as
well as act in concert with traditional drugs may be an efficient
pathway to cure CLL. In addition, the remodeling of ECM can be
regulated by many families of matrix-degrading enzymes such as
heparanase, chymases, MMPs and tryptases (201). Inhibitors of these enzymes such as
PI-88 [an inhibitor for heparanase (202)] may prevent the multistep pathway
in the tumor microenvironment in order to treat cancer. However,
some fragments of basement membrane collagen generated by MMP have
endogenous effects as integrin-mediated suppressors of pathologic
angiogenesis as well as tumor growth (203). This fact indicates that the
delicate balance between the tumor-inhibitory and tumor-promotion
functions of stromal cells should be considered. In addition, the
normal function of stromal cells should not be destroyed following
therapy. More research is needed to develop more efficient
approaches to combat cancer.
This review highlights the evidence for the crucial
role of the tumor microenvironment in tumor progression and
metastasis. As noted, tumor initiation as well as progression are
complex and multistep processes in which the tumor microenvironment
may contribute to its success. In this dynamic progression, the
tumor microenvironment can affect cancer cell proliferation as well
as tumor metastasis. For this reason, research on cancer must
combine the tumor cell-intrinsic pathway with the extrinsic pathway
in the tumor microenvironment. The mysterious role of the tumor
microenvironment is being deciphered in primary and metastatic
tumors, particularly using various new fields such as secreted
miRNAs, metabolism, and pre-metastasis. Targeting the tumor
microenvironment combined with current clinical approaches holds
great potential for developing new efficient therapies. Cancer
medicine must move toward a new era of personalized diagnostics and
therapeutics that aggressively embraces integrative approaches.
This study was supported by grants from the National
Natural Science Foundations of China (nos. 81321002, 81502351),
ISTCPC (2012DFA31370).
|
1
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hanahan D and Coussens LM: Accessories to
the crime: Functions of cells recruited to the tumor
microenvironment. Cancer Cell. 21:309–322. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cirri P and Chiarugi P: Cancer associated
fibroblasts: The dark side of the coin. Am J Cancer Res. 1:482–497.
2011.PubMed/NCBI
|
|
5
|
Gabrilovich DI, Ostrand-Rosenberg S and
Bronte V: Coordinated regulation of myeloid cells by Tumours. Nat
Rev Immunol. 12:253–268. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cheng L, Huang Z, Zhou W, Wu Q, Donnola S,
Liu JK, Fang X, Sloan AE, Mao Y, Lathia JD, et al: Glioblastoma
stem cells generate vascular pericytes to support vessel function
and tumor growth. Cell. 153:139–152. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cornil I, Theodorescu D, Man S, Herlyn M,
Jambrosic J and Kerbel RS: Fibroblast cell interactions with human
melanoma cells affect tumor cell growth as a function of tumor
progression. Proc Natl Acad Sci USA. 88:6028–6032. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Räsänen K and Vaheri A: Activation of
fibroblasts in cancer stroma. Exp Cell Res. 316:2713–2722. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu C, Vickers MF and Kerbel RS:
Interleukin 6: A fibroblast-derived growth inhibitor of human
melanoma cells from early but not advanced stages of tumor
progression. Proc Natl Acad Sci USA. 89:9215–9219. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Leonardi GC, Candido S, Cervello M,
Nicolosi D, Raiti F, Travali S, Spandidos DA and Libra M: The tumor
microenvironment in hepatocellular carcinoma (review). Int J Oncol.
40:1733–1747. 2012.PubMed/NCBI
|
|
11
|
Rønnov-Jessen L and Petersen OW: Induction
of alpha-smooth muscle actin by transforming growth factor-beta 1
in quiescent human breast gland fibroblasts. Implications for
myofibroblast generation in breast neoplasia. Lab Invest.
68:696–707. 1993.PubMed/NCBI
|
|
12
|
Postlethwaite AE, Keski-Oja J, Moses HL
and Kang AH: Stimulation of the chemotactic migration of human
fibroblasts by transforming growth factor beta. J Exp Med.
165:251–256. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mayo LD, Dixon JE, Durden DL, Tonks NK and
Donner DB: PTEN protects p53 from Mdm2 and sensitizes cancer cells
to chemotherapy. J Biol Chem. 277:5484–5489. 2002. View Article : Google Scholar
|
|
14
|
Erez N, Truitt M, Olson P, Arron ST and
Hanahan D: Cancer-associated fibroblasts are activated in incipient
neoplasia to orchestrate tumor-promoting inflammation in an
NF-kappaB-dependent manner. Cancer Cell. 17:135–147. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
De Wever O, Demetter P, Mareel M and
Bracke M: Stromal myofibroblasts are drivers of invasive cancer
growth. Int J Cancer. 123:2229–2238. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schmitt-Gräff A, Desmoulière A and
Gabbiani G: Heterogeneity of myofibroblast phenotypic features: An
example of fibroblastic cell plasticity. Virchows Arch. 425:3–24.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tlsty TD and Hein PW: Know thy neighbor:
Stromal cells can contribute oncogenic signals. Curr Opin Genet
Dev. 11:54–59. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Skalli O, Pelte MF, Peclet MC, Gabbiani G,
Gugliotta P, Bussolati G, Ravazzola M and Orci L: Alpha-smooth
muscle actin, a differentiation marker of smooth muscle cells, is
present in microfilamentous bundles of pericytes. J Histochem
Cytochem. 37:315–321. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
De Wever O and Mareel M: Role of tissue
stroma in cancer cell invasion. J Pathol. 200:429–447. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pittenger MF, Mackay AM, Beck SC, Jaiswal
RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S and
Marshak DR: Multilineage potential of adult human mesenchymal stem
cells. Science. 284:143–147. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rattigan Y, Hsu JM, Mishra PJ, Glod J and
Banerjee D: Interleukin 6 mediated recruitment of mesenchymal stem
cells to the hypoxic tumor milieu. Exp Cell Res. 316:3417–3424.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Karnoub AE, Dash AB, Vo AP, Sullivan A,
Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R and Weinberg
RA: Mesenchymal stem cells within tumour stroma promote breast
cancer metastasis. Nature. 449:557–563. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zeisberg EM, Tarnavski O, Zeisberg M,
Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT,
Roberts AB, et al: Endothelial-to-mesenchymal transition
contributes to cardiac fibrosis. Nat Med. 13:952–961. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zeisberg EM, Potenta S, Xie L, Zeisberg M
and Kalluri R: Discovery of endothelial to mesenchymal transition
as a source for carcinoma-associated fibroblasts. Cancer Res.
67:10123–10128. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Petersen OW, Lind Nielsen H, Gudjonsson T,
Villadsen R, Rønnov-Jessen L and Bissell MJ: The plasticity of
human breast carcinoma cells is more than epithelial to mesenchymal
conversion. Breast Cancer Res. 3:213–217. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Petersen OW, Nielsen HL, Gudjonsson T,
Villadsen R, Rank F, Niebuhr E, Bissell MJ and Rønnov-Jessen L:
Epithelial to mesenchymal transition in human breast cancer can
provide a nonmalignant stroma. Am J Pathol. 162:391–402. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kurose K, Gilley K, Matsumoto S, Watson
PH, Zhou XP and Eng C: Frequent somatic mutations in PTEN and TP53
are mutually exclusive in the stroma of breast carcinomas. Nat
Genet. 32:355–357. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Orimo A and Weinberg RA: Stromal
fibroblasts in cancer: A novel tumor-promoting cell type. Cell
Cycle. 5:1597–1601. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bronisz A, Godlewski J, Wallace JA,
Merchant AS, Nowicki MO, Mathsyaraja H, Srinivasan R, Trimboli AJ,
Martin CK, Li F, et al: Reprogramming of the tumour
microenvironment by stromal PTEN-regulated miR-320. Nat Cell Biol.
14:159–167. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sun X, Mao Y, Wang J, Zu L, Hao M, Cheng
G, Qu Q, Cui D, Keller ET, Chen X, et al: IL-6 secreted by
cancer-associated fibroblasts induces tamoxifen resistance in
luminal breast cancer. Oncogene. Jun 9–2014.Epub ahead of print.
View Article : Google Scholar
|
|
31
|
Sica A: Role of tumour-associated
macrophages in cancer-related inflammation. Exp Oncol. 32:153–158.
2010.
|
|
32
|
Sica A, Larghi P, Mancino A, Rubino L,
Porta C, Totaro MG, Rimoldi M, Biswas SK, Allavena P and Mantovani
A: Macrophage polarization in tumour progression. Semin Cancer
Biol. 18:349–355. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin EY, Nguyen AV, Russell RG and Pollard
JW: Colony-stimulating factor 1 promotes progression of mammary
tumors to malignancy. J Exp Med. 193:727–740. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nesbit M, Schaider H, Miller TH and Herlyn
M: Low-level monocyte chemoattractant protein-1 stimulation of
monocytes leads to tumor formation in nontumorigenic melanoma
cells. J Immunol. 166:6483–6490. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schmid MC, Avraamides CJ, Dippold HC,
Franco I, Foubert P, Ellies LG, Acevedo LM, Manglicmot JR, Song X,
Wrasidlo W, et al: Receptor tyrosine kinases and TLR/IL1Rs
unexpectedly activate myeloid cell PI3kγ, a single convergent point
promoting tumor inflammation and progression. Cancer Cell.
19:715–727. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Balkwill F: Cancer and the chemokine
network. Nat Rev Cancer. 4:540–550. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Qian BZ, Li J, Zhang H, Kitamura T, Zhang
J, Campion LR and Kaiser EA: CCL2 recruits inflammatory monocytes
to facilitate breast-tumour metastasis. Nature. 475:222–225. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mizutani K, Sud S, McGregor NA,
Martinovski G, Rice BT, Craig MJ, Varsos ZS, Roca H and Pienta KJ:
The chemokine CCL2 increases prostate tumor growth and bone
metastasis through macrophage and osteoclast recruitment.
Neoplasia. 11:1235–1242. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Acharyya S, Oskarsson T, Vanharanta S,
Malladi S, Kim J, Morris PG, Manova-Todorova K, Leversha M, Hogg N,
Seshan VE, et al: A CXCL1 paracrine network links cancer
chemoresistance and metastasis. Cell. 150:165–178. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shields JD, Kourtis IC, Tomei AA, Roberts
JM and Swartz MA: Induction of lymphoidlike stroma and immune
escape by tumors that express the chemokine CCL21. Science.
328:749–752. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cremer I, Fridman WH and Sautès-Fridman C:
Tumor microenvironment in NSCLC suppresses NK cells function.
Oncoimmunology. 1:244–246. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhao X, Rong L, Zhao X, Li X, Liu X, Deng
J, Wu H, Xu X, Erben U, Wu P, et al: TNF signaling drives
myeloid-derived suppressor cell accumulation. J Clin Invest.
122:4094–4104. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bayne LJ, Beatty GL, Jhala N, Clark CE,
Rhim AD, Stanger BZ and Vonderheide RH: Tumor-derived
granulocyte-macrophage colony-stimulating factor regulates myeloid
inflammation and T cell immunity in pancreatic cancer. Cancer Cell.
21:822–835. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Würth R, Bajetto A, Harrison JK, Barbieri
F and Florio T: CXCL12 modulation of CXCR4 and CXCR7 activity in
human glioblastoma stem-like cells and regulation of the tumor
micro-environment. Front Cell Neurosci. 8:1442014.
|
|
45
|
Bergers G and Benjamin LE: Tumorigenesis
and the angiogenic switch. Nat Rev Cancer. 3:401–410. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bertolini F, Shaked Y, Mancuso P and
Kerbel RS: The multifaceted circulating endothelial cell in cancer:
Towards marker and target identification. Nat Rev Cancer.
6:835–845. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Volm M, Koomägi R and Mattern J:
Prognostic value of vascular endothelial growth factor and its
receptor FLT-1 in squamous cell lung cancer. Int J Cancer.
74:64–68. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yoshiji H, Gomez DE, Shibuya M and
Thorgeirsson UP: Expression of vascular endothelial growth factor,
its receptor, and other angiogenic factors in human breast cancer.
Cancer Res. 56:2013–2016. 1996.PubMed/NCBI
|
|
49
|
Olson TA, Mohanraj D, Carson LF and
Ramakrishnan S: Vascular permeability factor gene expression in
normal and neoplastic human ovaries. Cancer Res. 54:276–280.
1994.PubMed/NCBI
|
|
50
|
Brown LF, Berse B, Jackman RW, Tognazzi K,
Manseau EJ, Dvorak HF and Senger DR: Increased expression of
vascular permeability factor (vascular endothelial growth factor)
and its receptors in kidney and bladder carcinomas. Am J Pathol.
143:1255–1262. 1993.PubMed/NCBI
|
|
51
|
Asahara T, Takahashi T, Masuda H, Kalka C,
Chen D, Iwaguro H, Inai Y, Silver M and Isner JM: VEGF contributes
to postnatal neovascularization by mobilizing bone marrow-derived
endothelial progenitor cells. EMBO J. 18:3964–3972. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Png KJ, Halberg N, Yoshida M and Tavazoie
SF: A microRNA regulon that mediates endothelial recruitment and
metastasis by cancer cells. Nature. 481:190–194. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zeng Z, Li YC, Jiao ZH, Yao J and Xue Y:
The cross talk between cGMP signal pathway and PKC in pulmonary
endothelial cell angiogenesis. Int J Mol Sci. 15:10185–10198. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Karar J and Maity A: PI3K/AKT/mTOR pathway
in angiogenesis. Front Mol Neurosci. 4:512011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang Z, Martin D, Molinolo AA, Patel V,
Iglesias-Bartolome R, Degese MS, Vitale-Cross L, Chen Q and Gutkind
JS: mTOR co-targeting in cetuximab resistance in head and neck
cancers harboring PIK3CA and RAS mutations. J Natl Cancer Inst.
106:dju2152014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Patel V, Marsh CA, Dorsam RT, Mikelis CM,
Masedunskas A, Amornphimoltham P, Nathan CA, Singh B, Weigert R,
Molinolo AA, et al: Decreased lymphangiogenesis and lymph node
metastasis by mTOR inhibition in head and neck cancer. Cancer Res.
71:7103–7112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Oft M, Heider KH and Beug H: TGFbeta
signaling is necessary for carcinoma cell invasiveness and
metastasis. Curr Biol. 8:1243–1252. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Biswas S, Guix M, Rinehart C, Dugger TC,
Chytil A, Moses HL, Freeman ML and Arteaga CL: Inhibition of
TGF-beta with neutralizing antibodies prevents radiation-induced
acceleration of metastatic cancer progression. J Clin Invest.
117:1305–1313. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Padua D, Zhang XH, Wang Q, Nadal C, Gerald
WL, Gomis RR and Massagué J: TGFbeta primes breast tumors for lung
metastasis seeding through angiopoietin-like 4. Cell. 133:66–77.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Siegel PM, Shu W, Cardiff RD, Muller WJ
and Massagué J: Transforming growth factor beta signaling impairs
Neu-induced mammary tumorigenesis while promoting pulmonary
metastasis. Proc Natl Acad Sci USA. 100:8430–8435. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yin JJ, Selander K, Chirgwin JM, Dallas M,
Grubbs BG, Wieser R, Massagué J, Mundy GR and Guise TA: TGF-beta
signaling blockade inhibits PTHrP secretion by breast cancer cells
and bone metastases development. J Clin Invest. 103:197–206. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Kang Y, HE W, Tulley S, Gupta GP,
Serganova I, Chen CR, Manova-Todorova K, Blasberg R, Gerald WL and
Massagué J: Breast cancer bone metastasis mediated by the Smad
tumor suppressor pathway. Proc Natl Acad Sci USA. 102:13909–13914.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Labelle M, Begum S and Hynes RO: Direct
signaling between platelets and cancer cells induces an
epithelial-mesenchymal-like transition and promotes metastasis.
Cancer Cell. 20:576–590. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Nakamura T, Nishizawa T, Hagiya M, Seki T,
Shimonishi M, Sugimura A, Tashiro K and Shimizu S: Molecular
cloning and expression of human hepatocyte growth factor. Nature.
342:440–443. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Birchmeier C, Birchmeier W, Gherardi E and
Vande Woude GF: Met, metastasis, motility and more. Nat Rev Mol
Cell Biol. 4:915–925. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Matsumoto K and Nakamura T: Hepatocyte
growth factor and the met system as a mediator of tumor-stromal
interactions. Int J Cancer. 119:477–483. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wilson TR, Fridlyand J, Yan Y, Penuel E,
Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, et al:
Widespread potential for growth-factor-driven resistance to
anticancer kinase inhibitors. Nature. 487:505–509. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Straussman R, Morikawa T, Shee K,
Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J,
Frederick DT, et al: Tumour micro-environment elicits innate
resistance to RAF inhibitors through HGF secretion. Nature.
487:500–504. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhou P, Li H, Wheeler S, Grandis JR,
Stabile LA and Egloff AM: Generation of head and neck cancer
patient-derived xenografts with in vivo acquired cetuximab
resistance. Clin Cancer Res. 21(Suppl 4): B392015. View Article : Google Scholar
|
|
70
|
Rossi D and Zlotnik A: The biology of
chemokines and their receptors. Annu Rev Immunol. 18:217–242. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Murphy PM, Baggiolini M, Charo IF, Hébert
CA, Horuk R, Matsushima K, Miller LH, Oppenheim JJ and Power CA:
International union of pharmacology. XXII Nomenclature for
chemokine receptors. Pharmacol Rev. 52:145–176. 2000.PubMed/NCBI
|
|
72
|
Zlotnik A and Yoshie O: Chemokines: A new
classification system and their role in immunity. Immunity.
12:121–127. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Dhawan P and Richmond A: Role of CXCL1 in
tumorigenesis of melanoma. J Leukoc Biol. 72:9–18. 2002.PubMed/NCBI
|
|
74
|
Wang D, Wang H, Brown J, Daikoku T, Ning
W, Shi Q, Richmond A, Strieter R, Dey SK and DuBois RN: CXCL1
induced by prostaglandin E2 promotes angiogenesis in colorectal
cancer. J Exp Med. 203:941–951. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Karunagaran D, Tzahar E, Beerli RR, Chen
X, Graus-Porta D, Ratzkin BJ, Seger R, Hynes NE and Yarden Y:
ErbB-2 is a common auxiliary subunit of NDF and EGF receptors:
Implications for breast cancer. EMBO J. 15:254–264. 1996.PubMed/NCBI
|
|
76
|
Conti I and Rollins BJ: CCL2 (monocyte
chemoattractant protein-1) and cancer. Semin Cancer Biol.
14:149–154. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Craig MJ and Loberg RD: CCL2 (monocyte
chemoattractant protein-1) in cancer bone metastases. Cancer
Metastasis Rev. 25:611–619. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Popivanova BK, Kostadinova FI, Furuichi K,
Shamekh MM, Kondo T, Wada T, Egashira K and Mukaida N: Blockade of
a chemokine, CCL2, reduces chronic colitis-associated
carcinogenesis in mice. Cancer Res. 69:7884–7892. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Rottiers V and Näär AM: MicroRNAs in
metabolism and metabolic disorders. Nat Rev Mol Cell Biol.
13:239–250. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Krützfeldt J and Stoffel M: MicroRNAs: A
new class of regulatory genes affecting metabolism. Cell Metab.
4:9–12. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Kosaka N, Iguchi H, Yoshioka Y, Takeshita
F, Matsuki Y and Ochiya T: Secretory mechanisms and intercellular
transfer of microRNAs in living cells. J Biol Chem.
285:17442–17452. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zhang J, Zhao H, Gao Y and Zhang W:
Secretory miRNAs as novel cancer biomarkers. Biochim Biophys Acta.
1826:32–43. 2012.PubMed/NCBI
|
|
84
|
Xu D and Tahara H: The role of exosomes
and microRNAs in senescence and aging. Adv Drug Deliv Rev.
65:368–375. 2013. View Article : Google Scholar
|
|
85
|
Kosaka N, Iguchi H, Yoshioka Y, Hagiwara
K, Takeshita F and Ochiya T: Competitive interactions of cancer
cells and normal cells via secretory microRNAs. J Biol Chem.
287:1397–1405. 2012. View Article : Google Scholar :
|
|
86
|
Yang M, Chen J, Su F, Yu B, Su F, Lin L,
Liu Y, Huang JD and Song E: Microvesicles secreted by macrophages
shuttle invasion-potentiating microRNAs into breast cancer cells.
Mol Cancer. 10:1172011. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Fabbri M, Paone A, Calore F, Galli R,
Gaudio E, Santhanam R, Lovat F, Fadda P, Mao C, Nuovo GJ, et al:
MicroRNAs bind to Toll-like receptors to induce prometastatic
inflammatory response. Proc Natl Acad Sci USA. 109:E2110–E2116.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Roth C, Rack B, Müller V, Janni W, Pantel
K and Schwarzenbach H: Circulating microRNAs as blood-based markers
for patients with primary and metastatic breast cancer. Breast
Cancer Res. 12:R902010. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Mitchell PS, Parkin RK, Kroh EM, Fritz BR,
Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O'Briant
KC, Allen A, et al: Circulating microRNAs as stable blood-based
markers for cancer detection. Proc Natl Acad Sci USA.
105:10513–10518. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Cairns RA, Harris IS and Mak TW:
Regulation of cancer cell metabolism. Nat Rev Cancer. 11:85–95.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Lunt SY and Vander Heiden MG: Aerobic
glycolysis: Meeting the metabolic requirements of cell
proliferation. Annu Rev Cell Dev Biol. 27:441–464. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Riemann A, Schneider B, Ihling A, Nowak M,
Sauvant C, Thews O and Gekle M: Acidic environment leads to
ROS-induced MAPK signaling in cancer cells. PLoS One. 6:e224452011.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Fischer K, Hoffmann P, Voelkl S,
Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G,
Hoves S, et al: Inhibitory effect of tumor cell-derived lactic acid
on human t cells. Blood. 109:3812–3819. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Walenta S and Mueller-Klieser WF: Lactate:
Mirror and motor of tumor malignancy. Semin Radiat Oncol.
14:267–274. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Rattigan YI, Patel BB, Ackerstaff E,
Sukenick G, Koutcher JA, Glod JW and Banerjee D: Lactate is a
mediator of metabolic cooperation between stromal carcinoma
associated fibroblasts and glycolytic tumor cells in the tumor
microenvironment. Exp Cell Res. 318:326–335. 2012. View Article : Google Scholar :
|
|
97
|
Végran F, Boidot R, Michiels C, Sonveaux P
and Feron O: Lactate influx through the endothelial cell
monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway
that drives tumor angiogenesis. Cancer Res. 71:2550–2560. 2011.
View Article : Google Scholar
|
|
98
|
Semenza GL: Tumor metabolism: Cancer cells
give and take lactate. J Clin Invest. 118:3835–3837.
2008.PubMed/NCBI
|
|
99
|
Feron O: Pyruvate into lactate and back:
From the Warburg effect to symbiotic energy fuel exchange in cancer
cells. Radiother Oncol. 92:329–333. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Sonveaux P, Végran F, Schroeder T, Wergin
MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C,
Jordan BF, et al: Targeting lactate-fueled respiration selectively
kills hypoxic tumor cells in mice. J Clin Invest. 118:3930–3942.
2008.PubMed/NCBI
|
|
101
|
Koukourakis MI, Giatromanolaki A, Harris
AL and Sivridis E: Comparison of metabolic pathways between cancer
cells and stromal cells in colorectal carcinomas: A metabolic
survival role for tumor-associated stroma. Cancer Res. 66:632–637.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Nieman KM, Kenny HA, Penicka CV, Ladanyi
A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB,
Hotamisligil GS, et al: Adipocytes promote ovarian cancer
metastasis and provide energy for rapid tumor growth. Nat Med.
17:1498–1503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Marie SK and Shinjo SM: Metabolism and
brain cancer. Clinics (Sao Paulo). 66(Suppl 1): 33–43. 2011.
View Article : Google Scholar
|
|
104
|
Hirayama A, Kami K, Sugimoto M, Sugawara
M, Toki N, Onozuka H, Kinoshita T, Saito N, Ochiai A, Tomita M, et
al: Quantitative metabolome profiling of colon and stomach cancer
microenvironment by capillary electrophoresis time-of-flight mass
spectrometry. Cancer Res. 69:4918–4925. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Ko YH, Lin Z, Flomenberg N, Pestell RG,
Howell A, Sotgia F, Lisanti MP and Martinez-Outschoorn UE:
Glutamine fuels a vicious cycle of autophagy in the tumor stroma
and oxidative mitochondrial metabolism in epithelial cancer cells:
Implications for preventing chemotherapy resistance. Cancer Biol
Ther. 12:1085–1097. 2011. View Article : Google Scholar
|
|
106
|
Filipp FV, Ratnikov B, De Ingeniis J,
Smith JW, Osterman AL and Scott DA: Glutamine-fueled mitochondrial
metabolism is decoupled from glycolysis in melanoma. Pigment Cell
Melanoma Res. 25:732–739. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Jain M, Nilsson R, Sharma S, Madhusudhan
N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB and Mootha
VK: Metabolite profiling identifies a key role for glycine in rapid
cancer cell proliferation. Science. 336:1040–1044. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Okamoto K, Toyokuni S, Kim WJ, Ogawa O,
Kakehi Y, Arao S, Hiai H and Yoshida O: Overexpression of human
mutT homologue gene messenger RNA in renal-cell carcinoma: evidence
of persistent oxidative stress in cancer. Int J Cancer. 65:437–441.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Gackowski D, Banaszkiewicz Z, Rozalski R,
Jawien A and Olinski R: Persistent oxidative stress in colorectal
carcinoma patients. Int J Cancer. 101:395–397. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Toyokuni S, Okamoto K, Yodoi J and Hiai H:
Persistent oxidative stress in cancer. FEBS Lett. 358:1–3. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Zhou Y, Hileman EO, Plunkett W, Keating MJ
and Huang P: Free radical stress in chronic lymphocytic leukemia
cells and its role in cellular sensitivity to ROS-generating
anticancer agents. Blood. 101:4098–4104. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Szatrowski TP and Nathan CF: Production of
large amounts of hydrogen peroxide by human tumor cells. Cancer
Res. 51:794–798. 1991.PubMed/NCBI
|
|
113
|
Grivennikov SI, Greten FR and Karin M:
Immunity, inflammation, and cancer. Cell. 140:883–899. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Ishikawa K, Takenaga K, Akimoto M,
Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y and Hayashi
J: ROS-generating mitochondrial DNA mutations can regulate tumor
cell metastasis. Science. 320:661–664. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Trachootham D, Alexandre J and Huang P:
Targeting cancer cells by ROS-mediated mechanisms: A radical
therapeutic approach? Nat Rev Drug Discov. 8:579–591. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Martinez-Outschoorn UE, Lin Z, Trimmer C,
Flomenberg N, Wang C, Pavlides S, Pestell RG, Howell A, Sotgia F
and Lisanti MP: Cancer cells metabolically 'fertilize' the tumor
microenvironment with hydrogen peroxide, driving the Warburg
effect: Implications for Pet imaging of human tumors. Cell Cycle.
10:2504–2520. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Baehrecke EH: Autophagy: Dual roles in
life and death? Nat Rev Mol Cell Biol. 6:505–510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Eisenberg-Lerner A and Kimchi A: The
paradox of autophagy and its implication in cancer etiology and
therapy. Apoptosis. 14:376–391. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Pavlides S, Tsirigos A, Migneco G,
Whitaker-Menezes D, Chiavarina B, Flomenberg N, Frank PG, Casimiro
MC, Wang C, Pestell RG, et al: The autophagic tumor stroma model of
cancer: Role of oxidative stress and ketone production in fueling
tumor cell metabolism. Cell Cycle. 9:3485–3505. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Martinez-Outschoorn UE, Balliet RM,
Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, Whitaker-Menezes
D, Daumer KM, Lin Z, Witkiewicz AK, et al: Oxidative stress in
cancer associated fibroblasts drives tumor-stroma co-evolution: A
new paradigm for understanding tumor metabolism, the field effect
and genomic instability in cancer cells. Cell Cycle. 9:3256–3276.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Bonuccelli G, Tsirigos A, Whitaker-Menezes
D, Pavlides S, Pestell RG, Chiavarina B, Frank PG, Flomenberg N,
Howell A, Martinez-Outschoorn UE, et al: Ketones and lactate 'fuel'
tumor growth and metastasis: Evidence that epithelial cancer cells
use oxidative mitochondrial metabolism. Cell Cycle. 9:3506–3514.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Martinez-Outschoorn UE, Trimmer C, Lin Z,
Whitaker-Menezes D, Chiavarina B, Zhou J, Wang C, Pavlides S,
Martinez-Cantarin MP, Capozza F, et al: Autophagy in cancer
associated fibroblasts promotes tumor cell survival: Role of
hypoxia, HIF1 induction and NFκB activation in the tumor stromal
microenvironment. Cell Cycle. 9:3515–3533. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Chiavarina B, Whitaker-Menezes D, Migneco
G, Martinez-Outschoorn UE, Pavlides S, Howell A, Tanowitz HB,
Casimiro MC, Wang C, Pestell RG, et al: HIF1-alpha functions as a
tumor promoter in cancer associated fibroblasts, and as a tumor
suppressor in breast cancer cells: Autophagy drives
compartment-specific oncogenesis. Cell Cycle. 9:3534–3551. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Martinez-Outschoorn UE, Whitaker-Menezes
D, Pavlides S, Chiavarina B, Bonuccelli G, Casey T, Tsirigos A,
Migneco G, Witkiewicz A, Balliet R, et al: The autophagic tumor
stroma model of cancer or 'battery-operated tumor growth': A simple
solution to the autophagy paradox. Cell Cycle. 9:4297–4306. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Salem AF, Whitaker-Menezes D, Lin Z,
Martinez-Outschoorn UE, Tanowitz HB, Al-Zoubi MS, Howell A, Pestell
RG, Sotgia F and Lisanti MP: Two-compartment tumor metabolism:
Autophagy in the tumor microenvironment and oxidative mitochondrial
metabolism (OXPHOS) in cancer cells. Cell Cycle. 11:2545–2556.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Zhang W, Trachootham D, Liu J, Chen G,
Pelicano H, Garcia-Prieto C, Lu W, Burger JA, Croce CM, Plunkett W,
et al: Stromal control of cystine metabolism promotes cancer cell
survival in chronic lymphocytic leukaemia. Nat Cell Biol.
14:276–286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Carmeliet P and Jain RK: Molecular
mechanisms and clinical applications of angiogenesis. Nature.
473:298–307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Risau W: Mechanisms of angiogenesis.
Nature. 386:671–674. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Carmeliet P and Jain RK: Angiogenesis in
cancer and other diseases. Nature. 407:249–257. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Leek RD, Lewis CE, Whitehouse R, Greenall
M, Clarke J and Harris AL: Association of macrophage infiltration
with angiogenesis and prognosis in invasive breast carcinoma.
Cancer Res. 56:4625–4629. 1996.PubMed/NCBI
|
|
132
|
Polverini PJ and Leibovich SJ: Induction
of neovascularization in vivo and endothelial proliferation in
vitro by tumor-associated macrophages. Lab Invest. 51:635–642.
1984.PubMed/NCBI
|
|
133
|
O'Sullivan C, Lewis CE, Harris AL and
McGee JO: Secretion of epidermal growth factor by macrophages
associated with breast carcinoma. Lancet. 342:148–149. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Lewis CE, Leek R, Harris A and McGee JO:
Cytokine regulation of angiogenesis in breast cancer: The role of
tumor-associated macrophages. J Leukoc Biol. 57:747–751.
1995.PubMed/NCBI
|
|
135
|
Harmey JH, Dimitriadis E, Kay E, Redmond
HP and Bouchier-Hayes D: Regulation of macrophage production of
vascular endothelial growth factor (VEGF) by hypoxia and
transforming growth factor beta-1. Ann Surg Oncol. 5:271–278. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Stockmann C, Doedens A, Weidemann A, Zhang
N, Takeda N, Greenberg JI, Cheresh DA and Johnson RS: Deletion of
vascular endothelial growth factor in myeloid cells accelerates
tumori-genesis. Nature. 456:814–818. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Bingle L, Lewis CE, Corke KP, Reed MW and
Brown NJ: Macrophages promote angiogenesis in human breast tumour
spheroids in vivo. Br J Cancer. 94:101–107. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Voronov E, Shouval DS, Krelin Y, Cagnano
E, Benharroch D, Iwakura Y, Dinarello CA and Apte RN: IL-1 is
required for tumor invasiveness and angiogenesis. Proc Natl Acad
Sci USA. 100:2645–2650. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Chen P, Huang Y, Bong R, Ding Y, Song N,
Wang X, Song X and Luo Y: Tumor-associated macrophages promote
angiogenesis and melanoma growth via adrenomedullin in a paracrine
and autocrine manner. Clin Cancer Res. 17:7230–7239. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Mazzieri R, Pucci F, Moi D, Zonari E,
Ranghetti A, Berti A, Politi LS, Gentner B, Brown JL, Naldini L, et
al: Targeting the ANG2/TIE2 axis inhibits tumor growth and
metastasis by impairing angiogenesis and disabling rebounds of
proangiogenic myeloid cells. Cancer Cell. 19:512–526. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Hughes R, Fang HY, Muthana M and Lewis CE:
Role of tumour-associated macrophages in the regulation of
angiogenesis. Tumour-Associated Macrophages. 17–29. 2011.
View Article : Google Scholar
|
|
142
|
Giraudo E, Inoue M and Hanahan D: An
amino-bisphosphonate targets MMP-9-expressing macrophages and
angiogenesis to impair cervical carcinogenesis. J Clin Invest.
114:623–633. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Li ZR, Li YP, Lin ML, Su WR, Zhang WX,
Zhang Y, Yao L and Liang D: Activated macrophages induce
neovascularization through upregulation of MMP-9 and VEGF in rat
corneas. Cornea. 31:1028–1035. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Houghton AM, Grisolano JL, Baumann ML,
Kobayashi DK, Hautamaki RD, Nehring LC, Cornelius LA and Shapiro
SD: Macrophage elastase (matrix metalloproteinase-12) suppresses
growth of lung metastases. Cancer Res. 66:6149–6155. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Guo X, Oshima H, Kitmura T, Taketo MM and
Oshima M: Stromal fibroblasts activated by tumor cells promote
angiogenesis in mouse gastric cancer. J Biol Chem. 283:19864–19871.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Zhang Y, Tang H, Cai J, Zhang T, Guo J,
Feng D and Wang Z: Ovarian cancer-associated fibroblasts contribute
to epithelial ovarian carcinoma metastasis by promoting
angiogenesis, lymphangiogenesis and tumor cell invasion. Cancer
Lett. 303:47–55. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Friedl P and Wolf K: Tumour-cell invasion
and migration: Diversity and escape mechanisms. Nat Rev Cancer.
3:362–374. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Steeg PS: Tumor metastasis: Mechanistic
insights and clinical challenges. Nat Med. 12:895–904. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Valastyan S and Weinberg RA: Tumor
metastasis: Molecular insights and evolving paradigms. Cell.
147:275–292. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Liotta LA, Kleinerman J, Catanzaro P and
Rynbrandt D: Degradation of basement membrane by murine tumor
cells. J Natl Cancer Inst. 58:1427–1431. 1977.PubMed/NCBI
|
|
151
|
Liotta LA: Tumor invasion and metastases -
role of the extracellular matrix: Rhoads memorial Award lecture.
Cancer Res. 46:1–7. 1986.PubMed/NCBI
|
|
152
|
Fingleton B, Vargo-Gogola T, Crawford HC
and Matrisian LM: Matrilysin [MMP-7] expression selects for cells
with reduced sensitivity to apoptosis. Neoplasia. 3:459–468. 2001.
View Article : Google Scholar
|
|
153
|
Noë V, Fingleton B, Jacobs K, Crawford HC,
Vermeulen S, Steelant W, Bruyneel E, Matrisian LM and Mareel M:
Release of an invasion promoter E-cadherin fragment by matrilysin
and stromelysin-1. J Cell Sci. 114:111–118. 2001.
|
|
154
|
Sameni M, Dosescu J, Moin K and Sloane BF:
Functional imaging of proteolysis: Stromal and inflammatory cells
increase tumor proteolysis. Mol Imaging. 2:159–175. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Grimshaw MJ, Hagemann T, Ayhan A, Gillett
CE, Binder C and Balkwill FR: A role for endothelin-2 and its
receptors in breast tumor cell invasion. Cancer Res. 64:2461–2468.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Sahai E: Mechanisms of cancer cell
invasion. Curr Opin Genet Dev. 15:87–96. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Gaggioli C, Hooper S, Hidalgo-Carcedo C,
Grosse R, Marshall JF, Harrington K and Sahai E: Fibroblast-led
collective invasion of carcinoma cells with differing roles for
RhoGTPases in leading and following cells. Nat Cell Biol.
9:1392–1400. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Provenzano PP, Eliceiri KW, Campbell JM,
Inman DR, White JG and Keely PJ: Collagen reorganization at the
tumor-stromal interface facilitates local invasion. BMC Med.
4:382006. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Provenzano PP, Inman DR, Eliceiri KW,
Trier SM and Keely PJ: Contact guidance mediated three-dimensional
cell migration is regulated by rho/rock-dependent matrix
reorganization. Biophys J. 95:5374–5384. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Goetz JG, Minguet S, Navarro-Lérida I,
Lazcano JJ, Samaniego R, Calvo E, Tello M, Osteso-Ibáñez T,
Pellinen T, Echarri A, et al: Biomechanical remodeling of the
microenvironment by stromal caveolin-1 favors tumor invasion and
metastasis. Cell. 146:148–163. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Chen J, Yao Y, Gong C, Yu F, Su S, Chen J,
Liu B, Deng H, Wang F, Lin L, et al: CCL18 from tumor-associated
macrophages promotes breast cancer metastasis via PItPnm3. Cancer
Cell. 19:541–555. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Yamaguchi H, Pixley F and Condeelis J:
Invadopodia and podosomes in tumor invasion. Eur J Cell Biol.
85:213–218. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Yamaguchi H, Lorenz M, Kempiak S,
Sarmiento C, Coniglio S, Symons M, Segall J, Eddy R, Miki H,
Takenawa T, et al: Molecular mechanisms of invadopodium formation:
The role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell
Biol. 168:441–452. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Buccione R, Orth JD and McNiven MA: Foot
and mouth: Podosomes, invadopodia and circular dorsal ruffles. Nat
Rev Mol Cell Biol. 5:647–657. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Calle Y, Chou HC, Thrasher AJ and Jones
GE: Wiskott-Aldrich syndrome protein and the cytoskeletal dynamics
of dendritic cells. J Pathol. 204:460–469. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Linder S and Aepfelbacher M: Podosomes:
Adhesion hot-spots of invasive cells. Trends Cell Biol. 13:376–385.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Mizutani K, Miki H, He H, Maruta H and
Takenawa T: Essential role of neural Wiskott-Aldrich syndrome
protein in podosome formation and degradation of extracellular
matrix in src-transformed fibroblasts. Cancer Res. 62:669–674.
2002.PubMed/NCBI
|
|
168
|
Moreno-Bueno G, Portillo F and Cano A:
Transcriptional regulation of cell polarity in EMT and cancer.
Oncogene. 27:6958–6969. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Liu R, Li J, Xie K, Zhang T, Lei Y, Chen
Y, Zhang L, Huang K, Wang K, Wu H, et al: FGFR4 promotes
stroma-induced epithelial-to-mesenchymal transition in colorectal
cancer. Cancer Res. 73:5926–5935. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Bergers G, Song S, Meyer-Morse N,
Bergsland E and Hanahan D: Benefits of targeting both pericytes and
endothelial cells in the tumor vasculature with kinase inhibitors.
J Clin Invest. 111:1287–1295. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Raza A, Franklin MJ and Dudek AZ:
Pericytes and vessel maturation during tumor angiogenesis and
metastasis. Am J Hematol. 85:593–598. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Cooke VG, LeBleu VS, Keskin D, Khan Z,
O'Connell JT, Teng Y, Duncan MB, Xie L, Maeda G, Vong S, et al:
Pericyte depletion results in hypoxia-associated
epithelial-to-mesenchymal transition and metastasis mediated by met
signaling pathway. Cancer Cell. 21:66–81. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Cano A, Pérez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, Del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Batlle E, Sancho E, Francí C, Domínguez D,
Monfar M, Baulida J and García De Herreros A: The transcription
factor snail is a repressor of E-cadherin gene expression in
epithelial tumour cells. Nat Cell Biol. 2:84–89. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Zhou BP, Deng J, Xia W, Xu J, Li YM,
Gunduz M and Hung MC: Dual regulation of Snail by
GSK-3beta-mediated phosphorylation in control of
epithelial-mesenchymal transition. Nat Cell Biol. 6:931–940. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM
and Zhou BP: Stabilization of snail by NF-kappaB is required for
inflammation-induced cell migration and invasion. Cancer Cell.
15:416–428. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Quigley JP and Armstrong PB: Tumor cell
intravasation alucidated: The chick embryo opens the window. Cell.
94:281–284. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Wyckoff JB, Jones JG, Condeelis JS and
Segall JE: A critical step in metastasis: In vivo analysis of
intravasation at the primary tumor. Cancer Res. 60:2504–2511.
2000.PubMed/NCBI
|
|
180
|
Wolf MJ, Hoos A, Bauer J, Boettcher S,
Knust M, Weber A, Simonavicius N, Schneider C, Lang M, Stürzl M, et
al: Endothelial CCR2 signaling induced by colon carcinoma cells
enables extravasation via the JAK2-Stat5 and p38MAPk pathway.
Cancer Cell. 22:91–105. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Gay LJ and Felding-Habermann B:
Contribution of platelets to tumour metastasis. Nat Rev Cancer.
11:123–134. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Nagrath S, Sequist LV, Maheswaran S, Bell
DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky
A, et al: Isolation of rare circulating tumour cells in cancer
patients by microchip technology. Nature. 450:1235–1239. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Huang YY, Hoshino K, Chen P, Wu CH, Lane
N, Huebschman M, Liu H, Sokolov K, Uhr JW, Frenkel EP, et al:
Immunomagnetic nanoscreening of circulating tumor cells with a
motion controlled microfluidic system. Biomed Microdevices.
15:673–681. 2013. View Article : Google Scholar :
|
|
184
|
Cai H and Peng F: 2-NBDG fluorescence
imaging of hyper-metabolic circulating tumor cells in mouse
xenograft model of breast cancer. J Fluoresc. 23:213–220. 2013.
View Article : Google Scholar
|
|
185
|
Nieswandt B, Hafner M, Echtenacher B and
Männel DN: Lysis of tumor cells by natural killer cells in mice is
impeded by platelets. Cancer Res. 59:1295–1300. 1999.PubMed/NCBI
|
|
186
|
Mbeunkui F and Johann DJ Jr: Cancer and
the tumor micro-environment: A review of an essential relationship.
Cancer Chemother Pharmacol. 63:571–582. 2009. View Article : Google Scholar
|
|
187
|
Gupta GP, Nguyen DX, Chiang AC, Bos PD,
Kim JY, Nadal C, Gomis RR, Manova-Todorova K and Massagué J:
Mediators of vascular remodelling co-opted for sequential steps in
lung metastasis. Nature. 446:765–770. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Psaila B, Kaplan RN, Port ER and Lyden D:
Priming the 'soil' for breast cancer metastasis: The pre-metastatic
niche. Breast Dis. 26:65–74. 2007.PubMed/NCBI
|
|
189
|
Psaila B and Lyden D: The metastatic
niche: Adapting the foreign soil. Nat Rev Cancer. 9:285–293. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Hiratsuka S, Watanabe A, Aburatani H and
Maru Y: Tumour-mediated upregulation of chemoattractants and
recruitment of myeloid cells predetermines lung metastasis. Nat
Cell Biol. 8:1369–1375. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Rafii S and Lyden D: S100 chemokines
mediate bookmarking of premetastatic niches. Nat Cell Biol.
8:1321–1323. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Hiratsuka S, Watanabe A, Sakurai Y,
Akashi-Takamura S, Ishibashi S, Miyake K, Shibuya M, Akira S,
Aburatani H and Maru Y: The S100A8-serum amyloid A3-TLR4 paracrine
cascade establishes a pre-metastatic phase. Nat Cell Biol.
10:1349–1355. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Kaplan RN, Riba RD, Zacharoulis S, Bramley
AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, et
al: VEGFR1-positive haematopoietic bone marrow progenitors initiate
the pre-metastatic niche. Nature. 438:820–827. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Nakasone ES, Askautrud HA, Kees T, Park
JH, Plaks V, Ewald AJ, Fein M, Rasch MG, Tan YX, Qiu J, et al:
Imaging tumor-stroma interactions during chemotherapy reveals
contributions of the microenvironment to resistance. Cancer Cell.
21:488–503. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Roodhart JM, Daenen LG, Stigter EC, Prins
HJ, Gerrits J, Houthuijzen JM, Gerritsen MG, Schipper HS, Backer
MJ, van Amersfoort M, et al: Mesenchymal stem cells induce
resistance to chemotherapy through the release of platinum-induced
fatty acids. Cancer Cell. 20:370–383. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Joosse SA and Pantel K: Tumor-educated
platelets as liquid biopsy in cancer patients. Cancer Cell.
28:552–554. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Wong HK, Fatimy RE, Onodera C, Wei Z, Yi
M, Mohan A, Gowrisankaran S, Karmali P, Marcusson E, Wakimoto H, et
al: The Cancer Genome Atlas Analysis predicts microRNA for
targeting cancer growth and vascularization in glioblastoma. Mol
Ther. 23:1234–1247. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Resnick MJ, Lopatin M, Shore ND, Lam PN,
Helfand B, Abramson RD, Crager M, Bonham M, Tezcan H, Clark-Langone
KM, et al: Analysis of tumor DNA in urine as a highly sensitive
liquid biopsy for patients with non-muscle invasive bladder cancer
(NMIBC). Clin Cancer Res. 22:142016.
|
|
199
|
Mishra RK, Wei C, Hresko RC, Bajpai R,
Heitmeier M, Matulis SM, Nooka AK, Rosen ST, Hruz PW, Schiltz GE,
et al: In Silico modeling-based identification of glucose
transporter 4 (GLUT4)-selective inhibitors for cancer therapy. J
Biol Chem. 290:14441–14453. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Tuxhorn JA, McAlhany SJ, Yang F, Dang TD
and Rowley DR: Inhibition of transforming growth factor-beta
activity decreases angiogenesis in a human prostate cancer-reactive
stroma xenograft model. Cancer Res. 62:6021–6025. 2002.PubMed/NCBI
|
|
201
|
Carmeliet P: Angiogenesis in health and
disease. Nat Med. 9:653–660. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Joyce JA, Freeman C, Meyer-Morse N, Parish
CR and Hanahan D: A functional heparan sulfate mimetic implicates
both heparanase and heparan sulfate in tumor angiogenesis and
invasion in a mouse model of multistage cancer. Oncogene.
24:4037–4051. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Hamano Y, Zeisberg M, Sugimoto H, Lively
JC, Maeshima Y, Yang C, Hynes RO, Werb Z, Sudhakar A and Kalluri R:
Physiological levels of tumstatin, a fragment of collagen IV alpha3
chain, are generated by MMP-9 proteolysis and suppress angiogenesis
via alphav beta3 integrin. Cancer Cell. 3:589–601. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Rudland PS, Platt-Higgins A, El-Tanani M,
De Silva Rudland S, Barraclough R, Winstanley JH, Howitt R and West
CR: Prognostic significance of the metastasis-associated protein
osteopontin in human breast cancer. Cancer Res. 62:3417–3427.
2002.PubMed/NCBI
|
|
205
|
El-Tanani MK, Campbell FC, Kurisetty V,
Jin D, Mccann M and Rudland PS: The regulation and role of
osteopontin in malignant transformation and cancer. Cytokine Growth
Factor Rev. 17:463–474. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
206
|
Tuck AB and Chambers AF: The role of
osteopontin in breast cancer: Clinical and experimental studies. J
Mammary Gland Biol Neoplasia. 6:419–429. 2001. View Article : Google Scholar
|
|
207
|
Weber GF: The metastasis gene osteopontin:
A candidate target for cancer therapy. Biochim Biophys Acta.
1552:61–85. 2001.
|
|
208
|
Matarrese P, Fusco O, Tinari N, Natoli C,
Liu FT, Semeraro ML, Malorni W and Iacobelli S: Galectin-3
overexpression protects from apoptosis by improving cell adhesion
properties. Int J Cancer. 85:545–554. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Inohara H, Honjo Y, Yoshii T, Akahani S,
Yoshida J, Hattori K, Okamoto S, Sawada T, Raz A and Kubo T:
Expression of galectin-3 in fine-needle aspirates as a diagnostic
marker differentiating benign from malignant thyroid neoplasms.
Cancer. 85:2475–2484. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Pinedo HM, Verheul HM, D'Amato RJ and
Folkman J: Involvement of platelets in tumour angiogenesis? Lancet.
352:1775–1777. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Pai R, Soreghan B, Szabo IL, Pavelka M,
Baatar D and Tarnawski AS: Prostaglandin E2 transactivates EGF
receptor: A novel mechanism for promoting colon cancer growth and
gastrointestinal hypertrophy. Nat Med. 8:289–293. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Ikushima H and Miyazono K: TGFbeta
signalling: A complex web in cancer progression. Nat Rev Cancer.
10:415–424. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
213
|
Sierko E and Wojtukiewicz MZ: Platelets
and angiogenesis in malignancy. Semin Thromb Hemost. 30:95–108.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Medina VA and Rivera ES: Histamine
receptors and cancer pharmacology. Br J Pharmacol. 161:755–767.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Moghaddam A, Zhang HT, Fan TP, HU DE, Lees
VC, Turley H, Fox SB, Gatter KC, Harris AL and Bicknell R:
Thymidine phosphorylase is angiogenic and promotes tumor growth.
Proc Natl Acad Sci USA. 92:998–1002. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
216
|
Bronckaers A, Gago F, Balzarini J and
Liekens S: The dual role of thymidine phosphorylase in cancer
development and chemotherapy. Med Res Rev. 29:903–953. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Geiger TR and Peeper DS: Critical role for
TrkB kinase function in anoikis suppression, tumorigenesis, and
metastasis. Cancer Res. 67:6221–6229. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Kim YJ, Borsig L, Varki NM and Varki A:
P-selectin deficiency attenuates tumor growth and metastasis. Proc
Natl Acad Sci USA. 95:9325–9330. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
David M, Wannecq E, Descotes F, Jansen S,
Deux B, Ribeiro J, Serre CM, Grès S, Bendriss-Vermare N, Bollen M,
et al: Cancer cell expression of autotaxin controls bone metastasis
formation in mouse through lysophosphatidic acid-dependent
activation of osteoclasts. PLoS One. 5:e97412010. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Visentin B, Vekich JA, Sibbald BJ, Cavalli
AL, Moreno KM, Matteo RG, Garland WA, Lu Y, Yu S, Hall HS, et al:
Validation of an anti-sphingosine-1-phosphate antibody as a
potential therapeutic in reducing growth, invasion, and
angiogenesis in multiple tumor lineages. Cancer Cell. 9:225–238.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Ruf W and Mueller BM: Thrombin generation
and the pathogenesis of cancer. Semin Thromb Hemost. 32(Suppl 1):
61–68. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Bajou K, Noël A, Gerard RD, Masson V,
Brunner N, Holst-Hansen C, Skobe M, Fusenig NE, Carmeliet P, Collen
D, et al: Absence of host plasminogen activator inhibitor 1
prevents cancer invasion and vascularization. Nat Med. 4:923–928.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
223
|
Falcón BL, Hashizume H, Koumoutsakos P,
Chou J, Bready JV, Coxon A, Oliner JD and McDonald DM: Contrasting
actions of selective inhibitors of angiopoietin-1 and
angiopoietin-2 on the normalization of tumor blood vessels. Am J
Pathol. 175:2159–2170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Payne AS and Cornelius LA: The role of
chemokines in melanoma tumor growth and metastasis. J Invest
Dermatol. 118:915–922. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Wu Y, Li YY, Matsushima K, Baba T and
Mukaida N: CCL3-CCR5 axis regulates intratumoral accumulation of
leukocytes and fibroblasts and promotes angiogenesis in murine lung
metastasis process. J Immunol. 181:6384–6393. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Läubli H, Spanaus KS and Borsig L:
Selectin-mediated activation of endothelial cells induces
expression of CCL5 and promotes metastasis through recruitment of
monocytes. Blood. 114:4583–4591. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Yi F, Jaffe R and Prochownik EV: The CCL6
chemokine is differentially regulated by c-Myc and L-Myc, and
promotes tumorigenesis and metastasis. Cancer Res. 63:2923–2932.
2003.PubMed/NCBI
|
|
228
|
Keeley EC, Mehrad B and Strieter RM: CXC
chemokines in cancer angiogenesis and metastases. Adv Cancer Res.
106:91–111. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Nakamura ES, Koizumi K, Kobayashi M,
Saitoh Y, Arita Y, Nakayama T, Sakurai H, Yoshie O and Saiki I:
RANKL-induced CCL22/macrophage-derived chemokine produced from
osteoclasts potentially promotes the bone metastasis of lung cancer
expressing its receptor CCR4. Clin Exp Metastasis. 23:9–18. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
230
|
Romagnani P, Lasagni L, Annunziato F,
Serio M and Romagnani S: CXC chemokines: The regulatory link
between inflammation and angiogenesis. Trends Immunol. 25:201–209.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Strieter RM, Burdick MD, Gomperts BN,
Belperio JA and Keane MP: CXC chemokines in angiogenesis. Cytokine
Growth Factor Rev. 16:593–609. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
232
|
Feliciano P: CXCL1 and CXCL2 link
metastasis and chemoresistance. Nat Genet. 44:8402012.
|
|
233
|
Keane MP, Belperio JA, Xue YY, Burdick MD
and Strieter RM: Depletion of CXCR2 inhibits tumor growth and
angiogenesis in a murine model of lung cancer. J Immunol.
172:2853–2860. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
234
|
White ES, Flaherty KR, Carskadon S, Brant
A, Iannettoni MD, Yee J, Orringer MB and Arenberg DA: Macrophage
migration inhibitory factor and CXC chemokine expression in
non-small cell lung cancer: Role in angiogenesis and prognosis.
Clin Cancer Res. 9:853–860. 2003.PubMed/NCBI
|
|
235
|
Strieter RM, Burdick MD, Mestas J,
Gomperts B, Keane MP and Belperio JA: Cancer CXC chemokine networks
and tumour angiogenesis. Eur J Cancer. 42:768–778. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
236
|
Miyazaki H, Patel V, Wanzg H, Edmunzs RK,
Gutkind JS and Yeudall WA: Down-regulation of CXCL5 inhibits
squamous carcinogenesis. Cancer Res. 66:4279–4284. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
237
|
Gijsbers K, Gouwy M, Struyf S, Wuyts A,
Proost P, Opdenakker G, Penninckx F, Ectors N, Geboes K and van
Damme J: GCP-2/CXCL6 synergizes with other endothelial cell-derived
chemokines in neutrophil mobilization and is associated with
angiogenesis in gastrointestinal tumors. Exp Cell Res. 303:331–342.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
238
|
Verbeke H, Struyf S, Berghmans N, Van
Coillie E, Opdenakker G, Uyttenhove C, Van Snick J and Van Damme J:
Isotypic neutralizing antibodies against mouse GCP-2/CXCL6 inhibit
melanoma growth and metastasis. Cancer Lett. 302:54–62. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
239
|
Tang Z, Yu M, Miller F, Berk RS, Tromp G
and Kosir MA: Increased invasion through basement membrane by
CXCL7-transfected breast cells. Am J Surg. 196:690–696. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
240
|
Bachelder RE, Wendt MA and Mercurio AM:
Vascular endothelial growth factor promotes breast carcinoma
invasion in an autocrine manner by regulating the chemokine
receptor CXCR4. Cancer Res. 62:7203–7206. 2002.PubMed/NCBI
|
|
241
|
Belperio JA, Phillips RJ, Burdick MD, Lutz
M, Keane M and Strieter R: The SDF-1/CXCL 12/CXCR4 biological axis
in non-small cell lung cancer metastases. Chest. 125(Suppl 5):
156S2004. View Article : Google Scholar : PubMed/NCBI
|
|
242
|
Pan J, Mestas J, Burdick MD, Phillips RJ,
Thomas GV, Reckamp K, Belperio JA and Strieter RM: Stromal derived
factor-1 (SDF-1/CXCL12) and cXcr4 in renal cell carcinoma
metastasis. Mol Cancer. 5:562006. View Article : Google Scholar : PubMed/NCBI
|