Introduction
Renal cell carcinoma (RCC) is the most lethal
urologic cancer, representing 2–3% of all cancers (1). Although surgery is considered the
primary curative therapy for patients with RCC, the prognosis for
patients with metastatic disease is poor, with a 5-year survival
rate of <10% (2).
Clear cell renal cell carcinoma (ccRCC), which
accounts for ~80% of all RCCs, is characterized by inactivation of
the von Hippel-Lindau (VHL) tumor suppressor gene (3). Drugs targeting the HIF axis (including
mTOR inhibitors) have been approved for advanced RCC (4). However, the efficacy is thought to be
limited, and treatment response is not long lasting; therefore, the
overall survival of ccRCC remains poor (5,6). Since
the pathogenesis of ccRCC is quite complicated, it is unrealistic
to expect that any single mechanism will uncover the full process.
Additional tumorigenic events are supposed to contribute to the
genesis and development of ccRCCs (7,8).
The Notch signaling controls a variety of cellular
processes (9). Notch signaling is
initiated through the interactions between the plasma-embedded
Notch receptors (Notch1-4) and cell surface ligands (Jagged1 and
Jagged2, and δ-like 1, 2 and 4) present on adjacent cells. The
intracellular domain of Notch (ICN), is cleaved from the plasma
membrane and translocates into the nucleus, leading to
transcription of its downstream targets such as Hes and Hey
(10).
In RCC, Notch seems to play an ‛oncogenic' role; it
was previously reported that the Notch signaling cascade was
constitutively active in cell lines (8), and high expression of Notch was
associated with increased risk of metastasis (11). Our previous study revealed the
elevated level of Notch1 and Jagged1 in ccRCC tissues compared with
in normal kidney tissues (12). We
also reported that Jagged1 overexpression may predict poor outcome
in RCC patients (13). However, how
Notch plays the oncogenic function and whether Notch pathway is a
target in RCC remains unclear.
Since Notch1 was detected overexpressed in ccRCC
(13), in the present study, we
exclusively suppressed Notch1, explored how the inhibition would
influence the survival of ccRCC cells, and detected the potential
mechanism. The present study provides the information on the
mechanism of tumorigenesis of RCC and highlights the potential use
of Notch inhibition as a novel treatment for RCC.
Materials and methods
Reagents and antibody
The antibody used against Notch1 (polyclonal rabbit
anti-human Notch1; ab27526) was purchased from Abcam (Cambridge,
UK). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was purchased
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Tissue
culture media and fetal bovine serum (FBS) were purchased from
Gibco (Fullerton, CA, USA). Cell cycle analysis was performed using
the Coulter DNA Prep™ reagents kit (Beckman Coulter, Fullerton, CA,
USA). The Annexin V-fluorescein isothiocyanate (FITC) apoptosis
detection kit was purchased from Beckman Coulter. Human renal
carcinoma cell lines, 786-0 and Caki-1, were obtained from the
Shanghai Institute of Cell Biology, Chinese Academy of Sciences
(Shanghai, China). Other materials will be introduced in the
following context.
Cell culture
Human renal carcinoma cell lines, 786-0, and Caki-1,
were cultured in Dulbecco's modified Eagle's medium (Invitrogen
Life Technologies, Carlsbad, CA, USA) supplemented with 10% FBS, 2
mM L-glutamine, 100 U/ml penicillin and 0.1 mg/ml streptomycin in a
humidified atmosphere with 5% CO2 incubator at 37°C.
Western blotting
The cells were solubilized in a lysis buffer on ice.
All lysates were centrifuged at 4°C at 10,000 × g for 10 min. The
protein concentration was determined using the bicinchoninic acid
protein assay kit (Pierce Biotechnology, Rockford, IL, USA). An
amount of 100 mg protein content was electrophoresed in 8% SDS-PAGE
and blotted on a nitrocellulose membrane. The membrane was blocked
with 5% bovine serum albumin in 1X Tris-buffered saline (TBS)
buffer at room temperature for 2 h and incubated Notch1 (1:200) at
4°C overnight. After three washes for 15 min in TBS, the membrane
was incubated with the peroxidase-conjugated mouse anti-goat IgG
antibody for 2 h at room temperature. Immunoreactive proteins were
visualized by an enhanced chemiluminescence system (Immobilon,
Millipore, Billerica, MA, USA) and GAPDH was used as the control
for protein loading.
Reverse transcriptase-polymerase chain
reaction
Total RNA from the cells was isolated with RNAiso
Plus (Takara, Japan). The RNA was reverse transcribed with a
PrimeScript RT reagent kit (Takara). All the procedures were
conducted in accordance with the manufacturer's instructions. The
resulting cDNA was quantified by RT-PCR using SYBR Premix Ex Taq
(Takara). GAPDH was used as a housekeeping gene. The primer
sequences were: GAGGCGTGGCAGACTATGC (forward) and
CTTGTACTCCGTCAGCGTGA (reverse).
RNA interference
siRNA duplexes were produced by Shanghai GenePharma
Co., Inc. (Shanghai, China) against human Notch1. Scrambled control
siRNA, which was used as a negative control (NC), was also designed
and obtained from GenePharma Co., Inc. (Table I).
 | Table IPrimers used in the present study. |
Table I
Primers used in the present study.
| Gene name | Primer sequence |
|---|
| Si-Notch1 |
GCACGCGGAUUAAUUUGCAdTdT |
|
UGCAAAUUAAUCCGCGUGCdTdT |
| Negative control |
UUCUCCGAACGUGUCACGUTT |
|
ACGUGACACGUUCGGAGAATT |
Growing cells were seeded at 2×105
cells/well into a 6-well tissue culture dish. The transfection was
performed using Lipofectamine 2000 reagent (Invitrogen, Carlsbad,
CA, USA) following the manufacturer's instructions. In the
preliminary test, different concentrations of siRNA 0, 25, 50 and
100 nM were tested. We found 50 nM Si-Notch1 achieved an efficient
inhibition of Notch1. When we increased the concentration of
Si-Notch1, the efficacy did not improve significantly. The final
concentration of siRNA added to the cells was determined to be 50
nM. The cells were cultured in the presence of transfection mixture
for 24 h; the transfection mixture was replaced by fresh RPMI-1640
medium.
Cell viability assay
Cell viability assay was performed as previously
described (12). Briefly, cells
were seeded into 96-well plates at a density of 1.0×104
cells/well. Following overnight incubation, the cells were treated
with siRNA (50 nM) or NC for 48 h. Thereafter, the medium was
removed and 20 µl MTT [5 mg/ml in phosphate-buffered saline
(PBS)] was added to each well. Following incubation for 4 h at
37°C, the supernatant was removed and the formazan crystals were
solubilized by adding 150 µl dimethyl sulfoxide (DMSO).
Viable cells were detected by measuring absorbance at 490 nm using
MRX II absorbance reader (Dynex Technologies, Chantill, VA,
USA).
Colony formation assay
Cells were counted and plated at 500 cells/well in a
6-well plate. After 2 weeks, the cells were washed with PBS, fixed
with methanol for 15 min at room temperature, and stained with
crystal violet for 30 min and after that the number of colonies was
counted.
Apoptosis
Detection of apoptotic cells by flow cytometry.
Cells were plated in 6-well plates (2 ml/well) at a density of
5×105 cells/ml and incubated overnight. After siRNA
treatment for 48 h, the cells were collected and washed with PBS,
followed by resuspension in binding buffer at a concentration of
1×106 cells/ml. A total of 100 ml (1×105
cells) of the solution was removed and mixed with Annexin V-FITC
and propidium iodide (PI) according to the manufacturer's
instructions. The mixed solution was incubated in the dark at room
temperature for 15 min, 400 µl dilution buffer was then
added to each tube and cell apoptosis analysis was performed using
the FC500 flow cytometry system (Beckman Coulter) within 1 h.
Analysis of cell cycle distribution
After exposed to Si-Notch1 or NC (50 nM) for 48 h at
37°C, cells were harvested, washed with cold PBS, fixed with 70%
ethanol and stored at 4°C for subsequent cell cycle analysis. For
detecting DNA content, cells were incubated in the dark at room
temperature with 0.5 ml RNase A for 20 min and with 1 ml PI for 20
min. The DNA content of the cells was measured using the Beckman
Coulter FC500 flow cytometry system. The percentage of cells in G1,
S and G2/M phases was calculated.
Cell migration and invasion assays
For the in vitro migration and invasion
assays, 5×104 cells were resuspended in serum-free
medium and placed in the upper Transwell chamber (8.0-µm PC;
Corning Inc., Corning, NY, USA). The lower chamber contained medium
with 10% FBS. After 24 h of incubation, the migrating cells in the
lower chamber or invading cells on the bottom of each well were
stained with 4′,6-diamidino-2-phenylindole (1 mg/ml; Beijing
Solarbio Science and Technology Co., Ltd., Beijing, China) followed
by fixation in methyl alcohol for 30 min. Then, the number of cells
in six randomly selected microscopic fields (magnification, ×100)
was counted with a BX51 fluorescence microscope (Olympus
Corporation, Tokyo, Japan).
Statistical analysis
Statistical analyses were performed using a
statistical software package (SPSS, version 16.0; SPSS, Inc.,
Chicago, IL, USA). For MTT results, cell cycle analysis,
colony-formation as well as migration and invasion assays, data
from three independent experiments are presented as the mean values
with standard deviations. The differences were evaluated using
two-tailed Student's t-tests. P<0.05 was considered to indicate
a statistically significant difference and all P-values were
two-sided.
Results
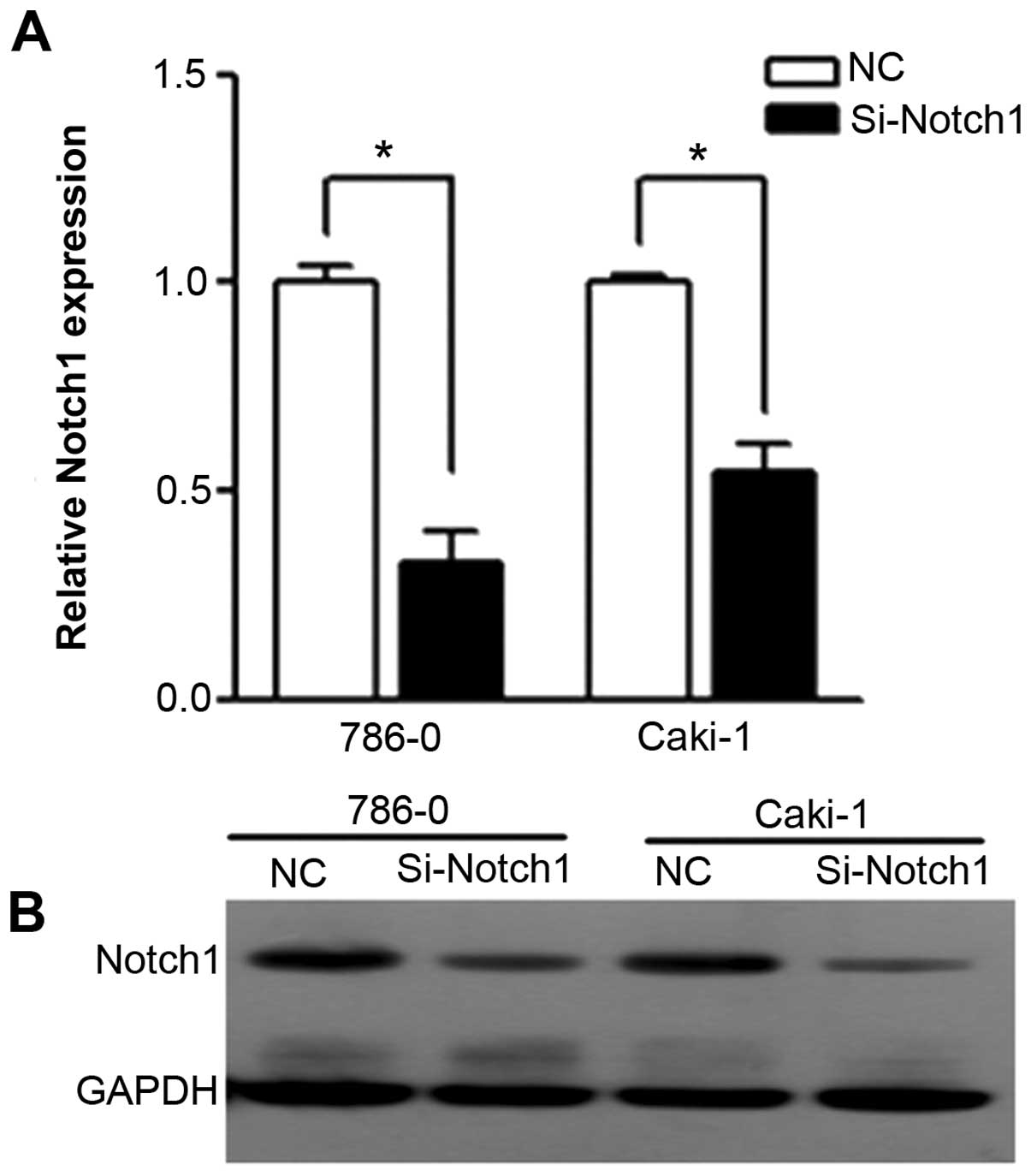
Notch1 expression is markedly decreased
by siRNA
We used RT-PCR and western blotting to confirm the
efficiency of siRNA on Notch1 expression. After transfection,
expression of Notch1 in Si-Notch1 group was reduced as compared
with in the NC group (Fig. 1). For
PCR, we detected a 67.3 and 45.2% reduction of Notch1 in 786-0 and
Caki-1 cell lines, respectively. For western blotting, the protein
of Notch1 was expressed with specific bands at 80 kDa. Notch1
protein was found to be downregulated in Si-Notch1-treated cell
lines compared with the NC group for both cell lines.
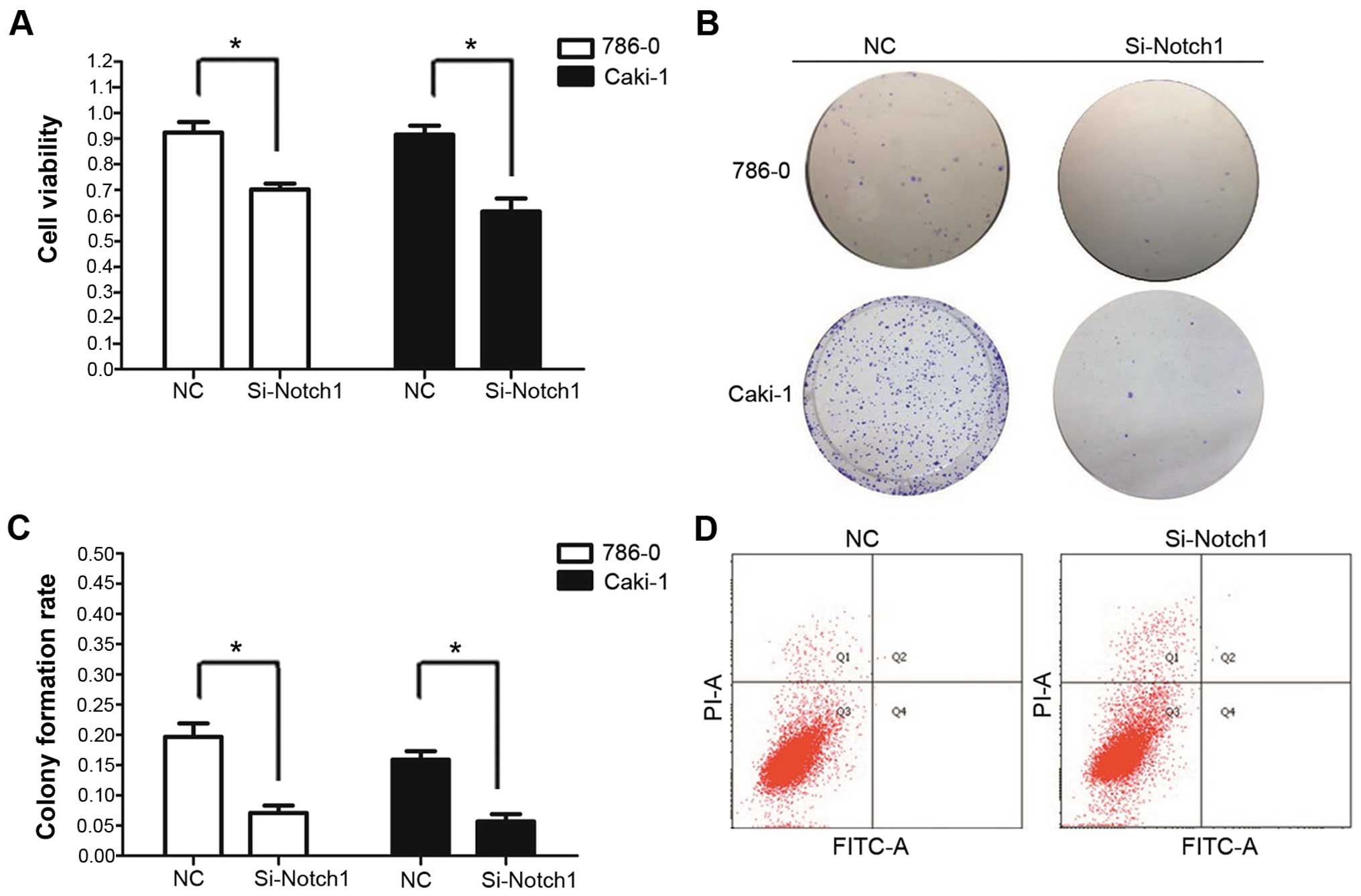
Notch1 inhibition by RNA interference
inhibits growth of tumor cells
To determine whether Notch1 signaling was
responsible for cell proliferation in ccRCC, we interrupted the
signaling pathway by RNA interference of Notch1. An MTT assay was
carried out to measure the proliferation status of the cells. As
shown in Fig. 2, inhibition of cell
proliferation was detected in Si-Notch1-treated cell lines. After
48 h, in NC group, percentage of cell viability was 94.4±4.1 and
91.6±3.5% in 786-0 and Caki-1 cell lines, while in Si-Notch1 group
it was 70.2±2.3 and 61.6±5.1% in 786-0 and Caki-1 cell lines.
Compared to the NC group, the cell growth of Si-Notch1 group was
decreased by 24.0 and 32.8% after 48 h of incubation, in 786-0 and
Caki-1 cells, respectively.
We subsequently confirmed the inhibitory effects of
Si-Notch1 through colony formation assays. The assay further
confirmed that Si-Notch1 treatment inhibited cell proliferation.
After 14 days, the colony formation rates of Si-Notch1 trans-fected
cells were 7.1±1.2 and 5.7±1.2%, in 786-0 and Caki-1, respectively.
In comparison, the colony formation rates of NC transfected cells
were 19.7±2.2 and 15.9±1.4%, respectively. The cell colony
formation rate of Si-Notch1 group was clearly suppressed as
compared to the NC group, in both 786-0 and Caki-1 cell lines
(P=0.003 and P=0.001, respectively) (Fig. 2).
To determine whether the Notch1 inhibition would
cause cell apoptosis, flow cytometry was further used to identify
the cell death types. The 786-0 and Caki-1 RCC cells treated with
Si-Notch1 did not show obvious apoptosis.
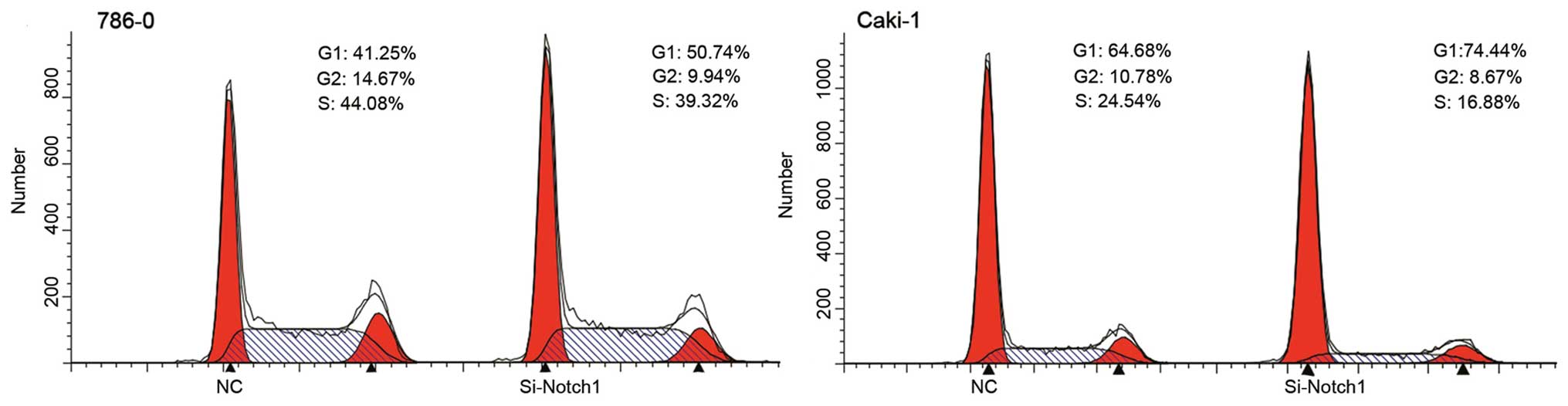
Notch1 inhibition by RNA interference
induces G1/S phase cell cycle arrest
Based on the growth inhibitory response of siRNA
treatment in cells, its effect on cell cycle distribution was next
examined. Both cell types were incubated with Si-Notch1 or NC for
48 h after which cell cycle analysis was performed.
Si-Notch1-treated cells arrested the cell cycle at the G1/S phase
(Table II, Fig. 3), suggesting that the MTT assay
indicated cell cycle arrest.
| Table IINegative control (NC) and Si-Notch1
cells. |
Table II
Negative control (NC) and Si-Notch1
cells.
| Phase | 786-0 (%)
| Phase | Caki-1 (%)
|
|---|
| NC | Si-Notch1 | NC | Si-Notch1 |
|---|
| G1 | 41.29±2.31 | 53.06±2.54 | G1 | 61.38±3.19 | 73.32±4.99 |
| S | 43.62±1.34 | 37.47±4.94 | S | 26.84±1.13 | 15.76±3.12 |
| G2 | 15.09±1.08 | 9.47±3.14 | G2 | 12.11±1.93 | 10.91±2.73 |
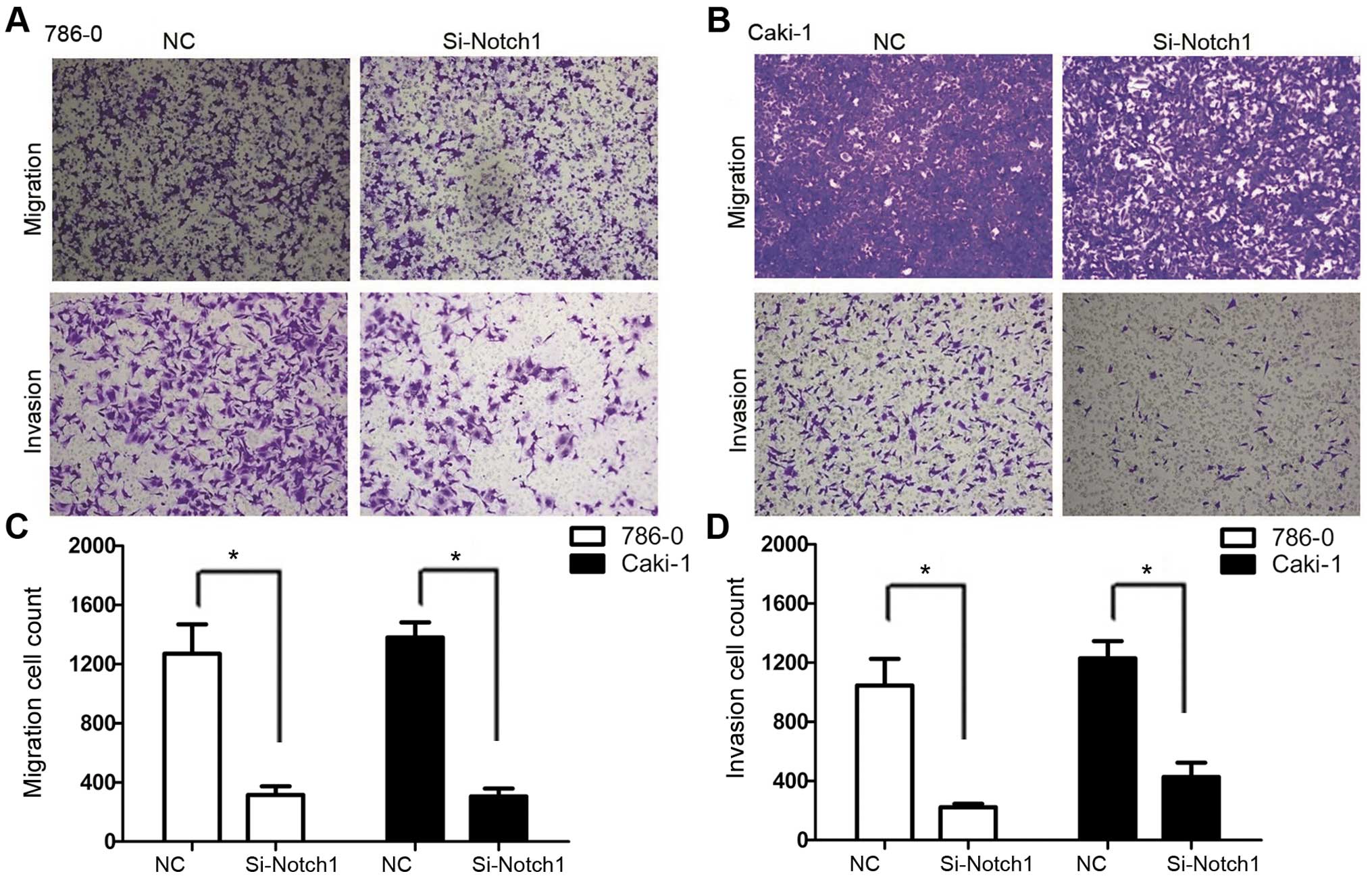
Notch1 inhibition by RNA Interference
inhibits cell migration and invasion
We used Transwell migration and invasion assays to
compare the cell migration and invasive capacity in each group.
Fig. 4 shows results for 786-0 and
Caki-1 cell lines, the Si-Notch1 group had a reduced rate of
migration as well as invasion. For 786-0 cell line, the migration
cell count of Si-Notch1 group was 316±58, while it was 1,271±198 in
NC group (P=0.009). In addition, the invasive cell count of
Si-Notch1-treated cells was 223±22, compared to 1,046±180 with the
NC group (P=0.015). For Caki-1 cell line, the migration cell count
was 306±53 and 1,380±102 in Si-Notch1 and NC groups, respectively
(P<0.001). Moreover, the invasive cell count was 427±97 and
1,229±116 in Si-Notch1 and NC group, respectively (P=0.001).
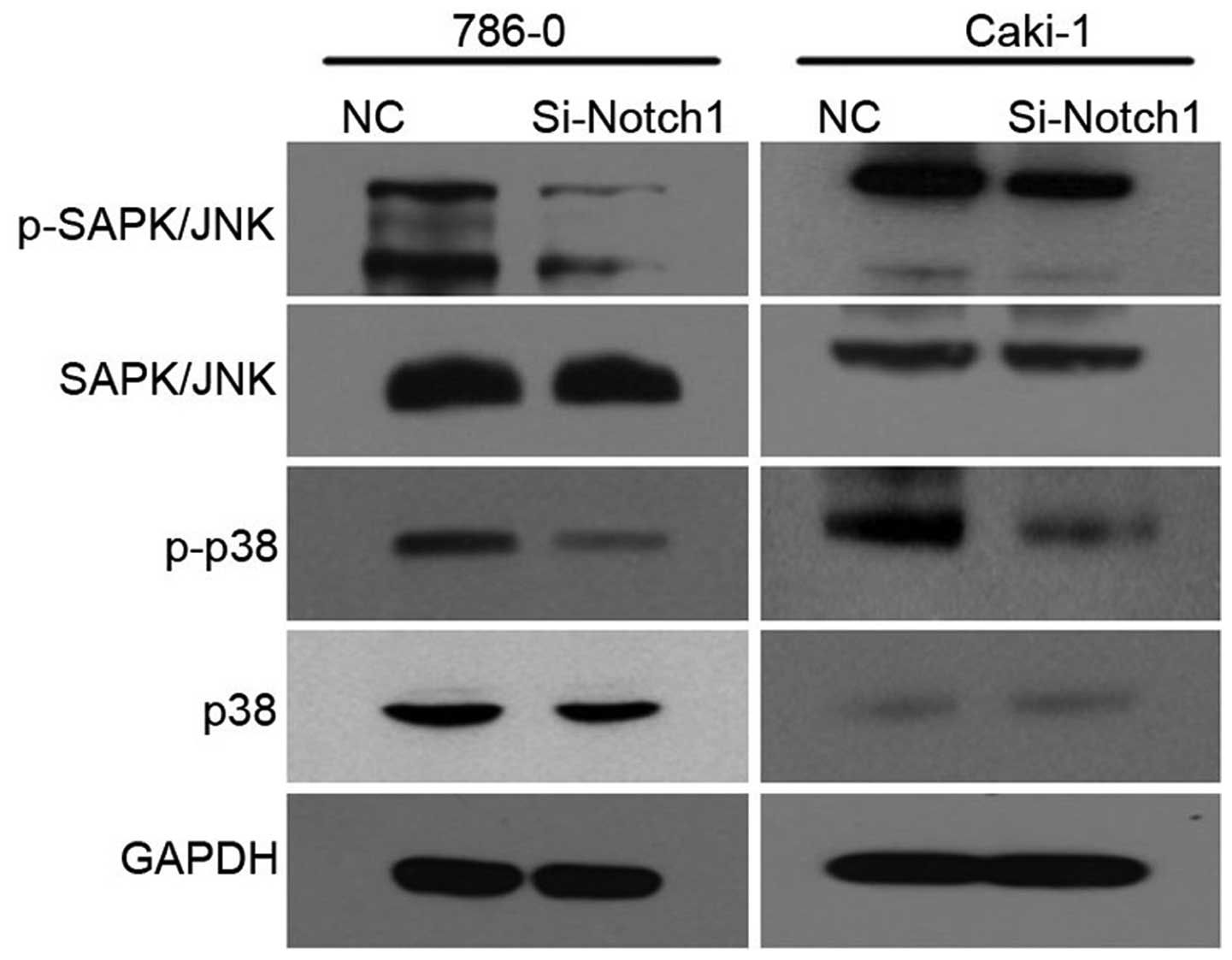
MAPK pathway is regulated by Notch1
inhibition
The cell lines were treated with Si-Notch1 or NC for
48 h, respectively. In addition, the levels of p38 and JNK were
measured by western blotting. As shown in Fig. 5, the levels of phospho-p38 and
phospho-JNK were downregulated at 48 h in Si-Notch1 group as
compared with the NC group, for both 786-0 and Caki-1 cell
lines.
Discussion
RCC is globally the 13th most common cancer
(14,15). Over the past several years, targeted
therapies have increasingly become available and have shown
considerable promise for the treatment of RCC; however, even with
such therapies life expectancy is generally only extended by less
than one year (16). New
therapeutic targets are needed and thus identification of such
targets could lead to the design of new drugs for use in RCC
patients.
The Notch pathway is critical in the determination
of cell fates by regulating cell growth, differentiation and
apoptosis (9). In RCC, Notch
signaling cascade was active in human ccRCC cell lines independent
of the VHL/HIF pathway; Notch1 and the Notch ligand Jagged1 were
expressed at significantly higher levels in ccRCC tumors than in
normal human renal tissue (8). The
expression level of Notch was correlated with the Furman grading,
TNM staging as well as prognosis in RCC cases (13). As a basis for the present study, we
discovered that Notch1 was overexpressed in RCC cell lines as well
as kidney cancer tissues (12,13).
Thus, we took Notch1 as our target to test how the inhibition of
Notch1 exclusively would influence the biological behavior of RCC
cells.
In the present study, inhibition of Notch1 with
siRNA led to a considerable decrease of cell proliferation and
induced cell cycle arrest. Cell colony forming was also inhibited
by Si-Notch1 interference. Moreover, the capacity of invasion and
migration were suppressed by Si-Notch1 treatment. The results
support the therapeutic effect of Si-Notch1 for ccRCC. Though
apoptosis analysis did not show a positive result, we suppose the
inhibitory effect was mainly achieved by cell cycle arrest. The
present study implied that the receptor Notch1, at least played a
significant role in Notch pathway and thus could be considered as a
promising therapeutic target. In fact, Notch pathway has been taken
as a treatment target in various malignancies, and the results
seemed to be encouraging (17,18).
Though non-selective inhibition like Notch γ-secretase inhibitor
has been reported, the side-effects could be serious when it is
applied in experiments in vivo (18). Considering that, a selective
inhibition of Notch seemed to be more reasonable, and given the
limitation of kinase inhibitors, a comprehensive evaluation of
Notch inhibition would be an alternative choice for ccRCC.
The mechanism involved with the oncogenic role of
Notch may be multiple (19,20). The MAPKs comprise a well-studied
family of serine threonine kinases that play important regulatory
roles in the cells (21). Among the
MAPK signaling pathways, JNKs and p38-MAPKs were identified to be
activated in response to a variety of cellular and environmental
stress such as changes in osmolarity, DNA damage, heat shock,
ischemia, cytokines, UV irradiation and oxidative stress (22,23).
Although JNKs are primarily attributed to
proapoptotic cell death or tumor suppression in response to a
variety of stress, emerging evidence suggests that JNKs, play a
role in the malignant transformation of cells and in tumorigenesis
(24). Research suggests that JNK
signaling contributed to cancer development (25,26).
The roles of p38 in invasion and metastasis have also been reported
(27), and activation of p38 has
been shown in various cancers to be the mechanism promoting
expression of MMP-2 (28,29). In the present study, we observed a
reduction in p-SAPK/JNK and p-p38 in Si-Notch1 transfected cells,
suggesting that JNK/p38 may serve downstream of the Notch pathway.
This result may partly illustrate why inhibition of the Notch
pathway leads to considerable inhibition of cell proliferation, and
clear restriction of the invasion and migration capability. We
demonstrated that the oncogenic effect of Notch1 is at least
partially mediated through regulation of the JNK/p38 pathway in two
ccRCC cell lines.
Deficiencies remain in the present study. Firstly,
though we blocked the Notch pathway with a specific receptor, we
consider that knockout of Notch1 would make the results stronger.
Secondly, as a conservative pathway, Notch has a comprehensive
network of its targets. To detect the downstream targets of Notch
pathway, more research needs to be carried out in our following
study.
In conclusion, the present study indicated that
Notch signaling is important in the tumorigenesis of RCC. JNK/p38
may serve as an important molecular mechanism for Notch1-mediated
carcinogenesis of ccRCC. Notch1 inhibition has the potential of
being a novel therapeutic regimen for RCC.
Acknowledgments
The present study was supported by a grant from the
Natural Science Foundation of Ningbo, China (2011A610054), as well
as the Medicine and Hygiene Program of Zhejiang Province, China
(2014KYB231). We thank Dr Wang Xiao for his assistance in study
design and instruction on the experiments.
References
|
1
|
Siegel R, Ma J, Zou Z and Jemal A: Cancer
statistics, 2014. CA Cancer J Clin. 64:9–29. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schrader AJ, Varga Z, Hegele A, Pfoertner
S, Olbert P and Hofmann R: Second-line strategies for metastatic
renal cell carcinoma: Classics and novel approaches. J Cancer Res
Clin Oncol. 132:137–149. 2006. View Article : Google Scholar
|
|
3
|
Vogelzang NJ and Stadler WM: Kidney
cancer. Lancet. 352:1691–1696. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Motzer RJ, Hutson TE, Tomczak P,
Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik
C, Kim ST, et al: Sunitinib versus interferon alfa in metastatic
renal-cell carcinoma. N Engl J Med. 356:115–124. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang L, Yu SY and Hu GY: Pituitary
metastasis from a renal cell carcinoma progressed after sorafenib
treatment. Chin J Cancer. 32:353–356. 2013. View Article : Google Scholar :
|
|
6
|
Zhang ZL, Li YH, Xiong YH, Hou GL, Yao K,
Dong P, Liu ZW, Han H, Qin ZK and Zhou FJ: Oncological outcome of
surgical treatment in 336 patients with renal cell carcinoma. Chin
J Cancer. 29:995–999. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Majid S, Saini S and Dahiya R: Wnt
signaling pathways in urological cancers: Past decades and still
growing. Mol Cancer. 11:72012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sjölund J, Johansson M, Manna S, Norin C,
Pietras A, Beckman S, Nilsson E, Ljungberg B and Axelson H:
Suppression of renal cell carcinoma growth by inhibition of Notch
signaling in vitro and in vivo. J Clin Invest. 118:217–228. 2008.
View Article : Google Scholar
|
|
9
|
Artavanis-Tsakonas S, Rand MD and Lake RJ:
Notch signaling: Cell fate control and signal integration in
development. Science. 284:770–776. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Iso T, Kedes L and Hamamori Y: HES and
HERP families: Multiple effectors of the Notch signaling pathway. J
Cell Physiol. 194:237–255. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ai Q, Ma X, Huang Q, Liu S, Shi T, Zhang
C, Zhu M, Zhang Y, Wang B, Ni D, et al: High-level expression of
Notch1 increased the risk of metastasis in T1 stage clear cell
renal cell carcinoma. PLoS One. 7:e350222012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu K, Zhang L, Lin Y, Yang K and Cheng Y:
Inhibition of γ-secretase induces G2/M arrest and triggers
apoptosis in renal cell carcinoma. Oncol Lett. 8:55–61.
2014.PubMed/NCBI
|
|
13
|
Wu K, Xu L, Zhang L, Lin Z and Hou J: High
Jagged1 expression predicts poor outcome in clear cell renal cell
carcinoma. Jpn J Clin Oncol. 41:411–416. 2011. View Article : Google Scholar
|
|
14
|
Chow WH, Dong LM and Devesa SS:
Epidemiology and risk factors for kidney cancer. Nat Rev Urol.
7:245–257. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ljungberg B, Campbell SC, Choi HY, Jacqmin
D, Lee JE, Weikert S and Kiemeney LA: The epidemiology of renal
cell carcinoma. Eur Urol. 60:615–621. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Escudier B, Eisen T, Stadler WM, Szczylik
C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA,
et al TARGET Study Group: Sorafenib in advanced clear-cell
renal-cell carcinoma. N Engl J Med. 356:125–134. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rasul S, Balasubramanian R, Filipović A,
Slade MJ, Yagüe E and Coombes RC: Inhibition of gamma-secretase
induces G2/M arrest and triggers apoptosis in breast cancer cells.
Br J Cancer. 100:1879–1888. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wei P, Walls M, Qiu M, Ding R, Denlinger
RH, Wong A, Tsaparikos K, Jani JP, Hosea N, Sands M, et al:
Evaluation of selective gamma-secretase inhibitor PF-03084014 for
its antitumor efficacy and gastrointestinal safety to guide optimal
clinical trial design. Mol Cancer Ther. 9:1618–1628. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hellström M, Phng LK, Hofmann JJ, Wallgard
E, Coultas L, Lindblom P, Alva J, Nilsson AK, Karlsson L, Gaiano N,
et al: Dll4 signalling through Notch1 regulates formation of tip
cells during angiogenesis. Nature. 445:776–780. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Qi R, An H, Yu Y, Zhang M, Liu S, Xu H,
Guo Z, Cheng T and Cao X: Notch1 signaling inhibits growth of human
hepatocellular carcinoma through induction of cell cycle arrest and
apoptosis. Cancer Res. 63:8323–8329. 2003.PubMed/NCBI
|
|
21
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chang Q, Zhang Y, Beezhold KJ, Bhatia D,
Zhao H, Chen J, Castranova V, Shi X and Chen F: Sustained JNK1
activation is associated with altered histone H3 methylations in
human liver cancer. J Hepatol. 50:323–333. 2009. View Article : Google Scholar
|
|
23
|
Wada T and Penninger JM: Mitogen-activated
protein kinases in apoptosis regulation. Oncogene. 23:2838–2849.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu J and Lin A: Role of JNK activation in
apoptosis: A double-edged sword. Cell Res. 15:36–42. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Barbarulo A, Iansante V, Chaidos A, Naresh
K, Rahemtulla A, Franzoso G, Karadimitris A, Haskard DO, Papa S and
Bubici C: Poly(ADP-ribose) polymerase family member 14 (PARP14) is
a novel effector of the JNK2-dependent pro-survival signal in
multiple myeloma. Oncogene. 32:4231–4242. 2013. View Article : Google Scholar
|
|
26
|
Hui L, Zatloukal K, Scheuch H, Stepniak E
and Wagner EF: Proliferation of human HCC cells and chemically
induced mouse liver cancers requires JNK1-dependent p21
downregulation. J Clin Invest. 118:3943–3953. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
del Barco Barrantes I and Nebreda AR:
Roles of p38 MAPKs in invasion and metastasis. Biochem Soc Trans.
40:79–84. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim ES, Kim MS and Moon A: TGF-β-induced
upregulation of MMP-2 and MMP-9 depends on p38 MAPK, but not ERK
signaling in MCF10A human breast epithelial cells. Int J Oncol.
25:1375–1382. 2004.PubMed/NCBI
|
|
29
|
Kumar B, Koul S, Petersen J, Khandrika L,
Hwa JS, Meacham RB, Wilson S and Koul HK: p38 mitogen-activated
protein kinase-driven MAPKAPK2 regulates invasion of bladder cancer
by modulation of MMP-2 and MMP-9 activity. Cancer Res. 70:832–841.
2010. View Article : Google Scholar : PubMed/NCBI
|