Introduction
Esophageal carcinoma is a common alimentary canal
malignancy with high incidence. There are ~0.3104 million new
malignancies worldwide each year. Despite extensive advancement in
its diagnosis and treatment, recurrence of esophageal carcinoma
remains the main cause of high mortality after treatment for these
patients. Current treatments, including surgical intervention,
radiotherapy (RT) and chemotherapy, are moderately effective in
early-stage cases, but are less effective in more advanced cases
(1). For example, RT is one of the
most important methods for patients at every stage of esophageal
carcinoma, however, patient prognosis remains unsatisfactory and
unpredictable due to profound radioresistance. Thus, identification
of key molecules or pathways specifically expressed in esophageal
carcinoma that are essential for the growth of cancer cells may
provide novel therapeutic targets and ultimately lead to improved
survival. Research over the past years clearly implicates multiple
genetic alterations in the development and progression of
esophageal carcinoma (2), including
those that have important functions in cell adhesion, signal
transduction, cell differentiation, metastasis, DNA damage and
repair (3).
B-cell-specific Moloney murine leukemia virus
integration site-1 (BMI-1), a member of the polycomb group of
transcriptional repressors, has been detected in a variety of human
carcinoma specimens, from pre-cancerous to advanced stages. In
particular, BMI-1 is overexpressed in a number of malignancies
(4,5). Recently, it has been reported that
BMI-1 is also overexpressed in alimentary canal cancers,
particularly in esophageal carcinoma, which suggests that BMI-1 may
confer radioresistance to esophageal carcinoma (6). Gene chip analysis also showed that
BMI-l predicts cancer metastasis (7), promotes cancer cell proliferation and
invasion, causes resistance to apoptosis and enhances transfer
capabilities (8). Moreover, as an
active heterodimer E3 ligase, BMI-1 may induce the ubiquitination
and phosphorylation of H2AX, which is thought to be a critical
sensor that can initiate DNA damage response (DDR) (9–11). Our
research has also shown that H2AX is associated with the
radiosensitization of esophageal cancer cells (12). However, how the function of BMI-1 in
radiosensitivity is regulated remains a fundamental question that
needs to be answered to elucidate the essential mechanisms
controlling DDR.
To identify the regulatory function of BMI-1 in DDR,
we conducted proteomic analysis to systematically identify BMI-1
interacting proteins and the related mechanisms. However, to the
best of our knowledge, the mechanisms by which BMI-1 promotes the
proliferation of esophageal carcinoma cells after RT have not been
reported, to date. Given the important role of BMI-1 in ionizing
radiation-induced DDR, we hypothesized that silencing of BMI-1 may
cause DNA damage pathway defects and thus increase
radiosensitivity. In the present study, we verificated this
hypothesis in vitro through two different cell lines and
explored the mechanisms through which BMI-1 induces tumor
progression.
Materials and methods
Cell lines and cell culture
The human esophageal squamous cell carcinoma cell
lines ECA109 and TE13 were cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum (FBS) at 37°C in 5%
CO2. The shRNA plasmid of BMI-1 was purchased from the
GeneChem Co. ECA109 and TE13 cells were plated in 12-well plates at
a density of 300 cells/well in triplicates, and plasmid
multiplicity of infection (MOI) was 10. The cells were cultured in
RPMI-1640 + 10% FBS with 5 µg/ml Polybrene for 12 h. Cells
were collected 12 h after transduction with the plasmid and were
selected in puromycin for stable clones. Subsequently, the cells
were used for irradiation as indicated.
X-ray irradiation
Irradiation was performed at room temperature with a
6-MV Siemens linear accelerator (Siemens, Concord, CA, USA) at a
dose rate of 2 Gy/min. After irradiation, the cells were recovered
in an incubator for the indicated time until harvesting.
shRNA transfection
For the shRNA analyses, human BMI-1 small
interfering RNA (shRNA) with the nucleotide sequence:
5′-GCUUAUCCAUUGAAUUCUUUGACCA-3′ (sense) and
5′-UGGUCAAAGAAUUCAAUGGAUAAGC-3′ (antisense), corresponding to part
of the BMI-1 mRNA; and the negative control (NC) scrambled shRNA
(NC-shRNA sense, 5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense,
5′-ACGUGACAGGUUCGGAGAATT-3′) were designed and purchased from
Invitrogen Corp. (Carlsbad, CA, USA). All of the shRNA sequences
were subjected to the Basic Local Alignment Search Tool (BLAST) to
confirm the absence of homology to any additional known coding
sequences in the human genome. Cells were transfected using
Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's
protocol. Briefly, one day prior to transfection, ECA109 and TE13
cells (5×105/well) were cultured in 6-well tissue
culture plates until they reached 50% confluency, and then the
cells were transiently transfected with either BMI-1-shRNA or
NC-shRNA (100 nM).
Quantitative real-time reverse
transcription-polymerase chain reaction (qRT-PCR)
Total RNAs were extracted using TRIzol reagent.
Real-time PCR was then performed using Platinum SYBR-Green qPCR
SuperMix-UDG (Invitrogen) according to the manufacturer's protocol.
Briefly, after the reverse transcription reaction at 42°C for 60
min and 70°C for 5 min, cDNAs were synthesized using the ReverAid
First Strand cDNA Synthesis kit (Thermo, Waltham, MA, USA), and
then initially denatured at 94°C for 30 sec, followed by 40 cycles
of the repeated procedure as follows: denaturation at 94°C for 5
sec, annealing at 56°C for 15 sec and extension at 72°C for 10 sec.
As a control, the levels of glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) expression were also analyzed. Incorporation
of the SYBR-Green dye into PCR products was monitored in real-time
with LightCycler real-time PCR detection system (Roche Applied
Science, Indianapolis, IN, USA), thereby allowing determination of
the threshold cycle (Ct) at which exponential amplification of
products begins. Independent experiments were repeated 3 times for
each sample, and the relative expression levels of genes were
analyzed using the 2−ΔΔCt method (13).
Western blot analysis
The cellular total protein was solubilized in RIPA
lysis buffer (1% Triton X-100, 150 mM NaCl, 10 mM Tris-HCl, pH 7.4,
1 mM EDTA, 1 mM EGTA, pH 8.0, 0.2 mM Na3VO4,
0.2 mM phenylmethylsulfonyl fluoride and 0.5% NP-40). The protein
amount was evaluated using BCA assays and separated on 10% SDS-PAGE
gel, and electrotransferred to polyvinylidene fluoride (PVDF)
membranes (Pierce, Rockford, IL, USA). The membranes were incubated
overnight at 4°C with the indicated antibodies. The specific
antibodies were rabbit BMI-1 (1:10,000), anti-p16 (1:50,00),
anti-rH2AX (1:10,000), rabbit bcl-2 (1:1,000), anti-bax (1:500) and
rabbit β-actin antibodies (1:10,000) (all from Abcam, Cambridge,
MA, USA). After washing for 3×5 min with TBS-T, the membranes were
incubated with secondary anti-rabbit IgG for 1 h at room
temperature away from light. The membranes were scanned for the
relative value of protein expression in gray scale by Image-Pro
Plus software 6.0 (Media Cybernetics, Sliver Spring, MD, USA). The
levels of the protein were calculated as the ratio of the intensity
of protein to that of β-actin. Experiments were carried out in
triplicate wells/time period and repeated 3 times.
Cell proliferation assay
The cells (2×103/well) were cultured in
96-well tissue culture plates until they reached 50% confluency,
and then transfected with a final concentration of 100 nM. After
transfection for 24 h, viability of the cells was determined at 24,
48 and 72 h after irradiation using the CellTiter 96 AQueous One
Solution cell proliferation assay (MTS) (Dojindo Molecular
Technologies, Gaithersburg, MD, USA). Briefly, the cells were
collected and incubated in medium containing 5 mg/ml MTS reagent
(Promega Corporation, Madison, WI, USA) at 37°C for 2 h. The
absorbance at 492 nm was measured by an enzyme-linked immunosorbent
assay (ELISA) plate reader. This experiment was repeated 3
times.
Transwell assay
Cell invasion was estimated using 24-well Transwell
chambers with polycarbonate filters of 8-µm pore size
(Costar), and the chambers were precoated with extracellular matrix
gel obtained from Corning (Corning, NY, USA). Twenty-four hours
after transfection and 1 h after irradiation, an aliquot of
5×104 cells was placed in the upper chamber with 0.2 ml
serum-free medium, whereas the lower chamber (24-well plate) was
loaded with 0.5 ml of medium containing 10% FBS. The chambers were
incubated in a humid atmosphere of 5% CO2 at 37°C for
4.5 h in the invasion assay. After incubation, the cells on the
upper surface of each filter were wiped off with a cotton swab. The
cells on the lower surface of the filter were fixed with 4%
formaldehyde, stained with crystal violet for 8 min, and then
washed with water. For each filter, the number of migrated or
invaded cells in five fields (magnification, ×200) was observed and
counted.
Co-immunoprecipitation (Co-IP) assay
Cell lysates were first pre-cleared with 25 ml of
protein A-agarose (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
The supernatants were immunoprecipitated with 2 mg of anti-rH2AX
(1:100), anti-H2AK119ub (1:100), anti-BMI-1 (1:100) (all from
Abcam) antibody for 2 h at 4°C, followed by incubation with protein
A-agarose overnight at 4°C. Protein A-agarose antigen-antibody
complexes were pelleted by centrifugation at 12,000 × g for 60 sec
at 4°C. The pellets were washed five times with 1 ml IPH buffer,
for 20 min each time at 4°C. Bound proteins were resolved by
SDS-PAGE, followed by western blotting with antibodies against
rH2AX, H2AK119ub and BMI-1.
Analysis of cell cycle distribution and
apoptosis
Both cell cycle distribution and spontaneous
apoptosis events were detected using a FACsCalibur II sorter and
CellQuest FACS system (BD Biosciences, San Jose, CA, USA). To
analyze cell cycle distribution, the cells were transfected using
shRNA for 24 h and stimulated with irradiation for 24 h before
being harvested. Cells were fixed with 70% ethanol at 4°C
overnight, washed twice with phosphate-buffered saline (PBS) and
resuspended in staining solution (50 µg/ml propidium iodide,
1 mg/ml RNase A, 0.1% Triton X-100 in PBS) for 30 min at 37°C in
the dark. To detect the extent of apoptosis under stress
conditions, cells were transfected using shRNA for 24 h before
irradiation and stained with fluorescein isothiocyanate
(FITC)-conjugated Annexin V and 7-amino-actinomycin D (7-AAD) using
the Annexin V-FITC apoptosis detection kit (BD Pharmingen)
according to the manufacturer's protocol.
Colony formation assay
Cell survival curves were generated by a standard
colony formation assay with minor modifications (14). Precooled tumor cells were irradiated
by graded single doses (0–8 Gy) in control, NC and BMI-1 shRNA
groups, seeded in Petri dishes, and cultivated in RPMI-1640
supplemented with 10% FBS. The experiments were repeated at least
twice. Two weeks later, the cells were fixed and stained with
crystal violet (0.6%). Colonies exceeding 50 cells were scored as
survivors.
Statistical analysis
Statistical analysis was conducted with the SPSS
software package version 13.0 (SPSS, Inc., Chicago, IL, USA). All
data are presented as the mean ± standard deviation (SD), and
analyzed using ANOVA with SPSS 13.0. For all tests, a p-value of
<0.05 and 0.01 was considered to be statistically significant
and is indicated by asterisks in the figures. All p-values given
are the results of two-sided tests. Data were obtained from at
least three independent experiments with a similar pattern.
Results
shRNAs targeting the BMI-1 gene and
protein downregulate BMI-1 expression in the ECA109 and TE13 cells
following transfection
To address the functional importance of the BMI-1
gene, we employed shRNA to deplete its expression in ECA109 and
TE13 cells, both of which were treated with NC-shRNA or shRNA
targeting the BMI-1 gene. After transfection for 24 h, the
expression of BMI-1 in cells was examined by real-time quantitative
reverse transcription-polymerase chain reaction (qRT-PCR) and
western blot analysis. The qRT-PCR analysis confirmed that the
levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were
unaffected by transfection of BMI-1-shRNA or NC-shRNA. As shown in
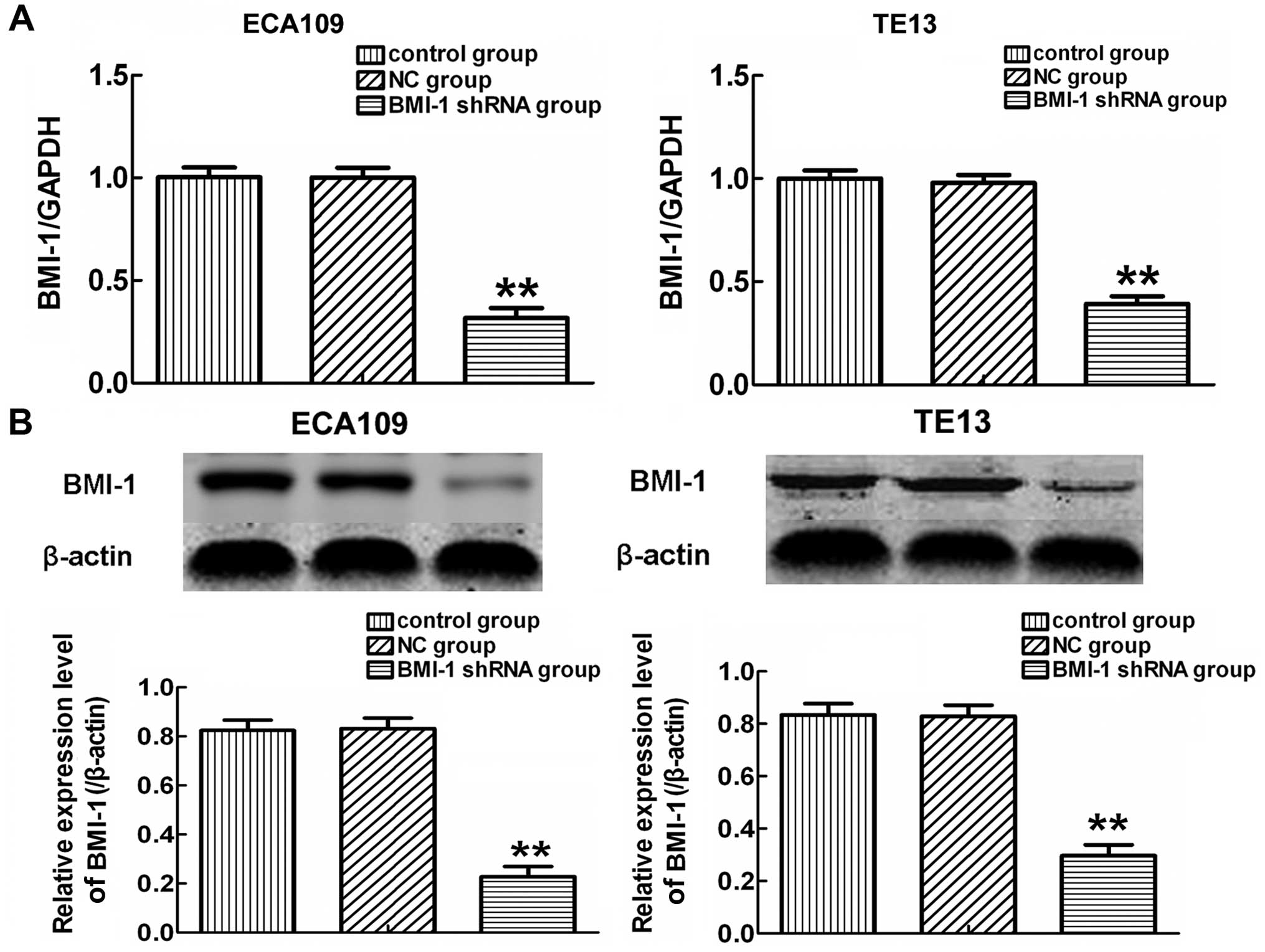
Fig. 1A, qRT-PCR showed that the
expression of BMI-1 in the BMI-1-shRNA group was significantly
lower than the levels in the control and NC-shRNA group after
transfection for 24 h (p<0.01). Similar results were observed in
the western blot analysis (Fig.
1B). These data indicated that BMI-1-specific shRNA effectively
and obviously suppressed the expression of BMI-1 in the ECA109 and
TE13 cells.
Specific knockdown of BMI-1 expression by
shRNA inhibits the growth and improves the radiosensitivity of
ECA109 and TE13 cells after IR in vitro
To evaluate the effect of BMI-1 on ECA109 and TE13
cell proliferation, cells in the 3 groups were harvested at 24 h
after transfection with shRNA of BMI-1 and at 24, 48 and 72 h after
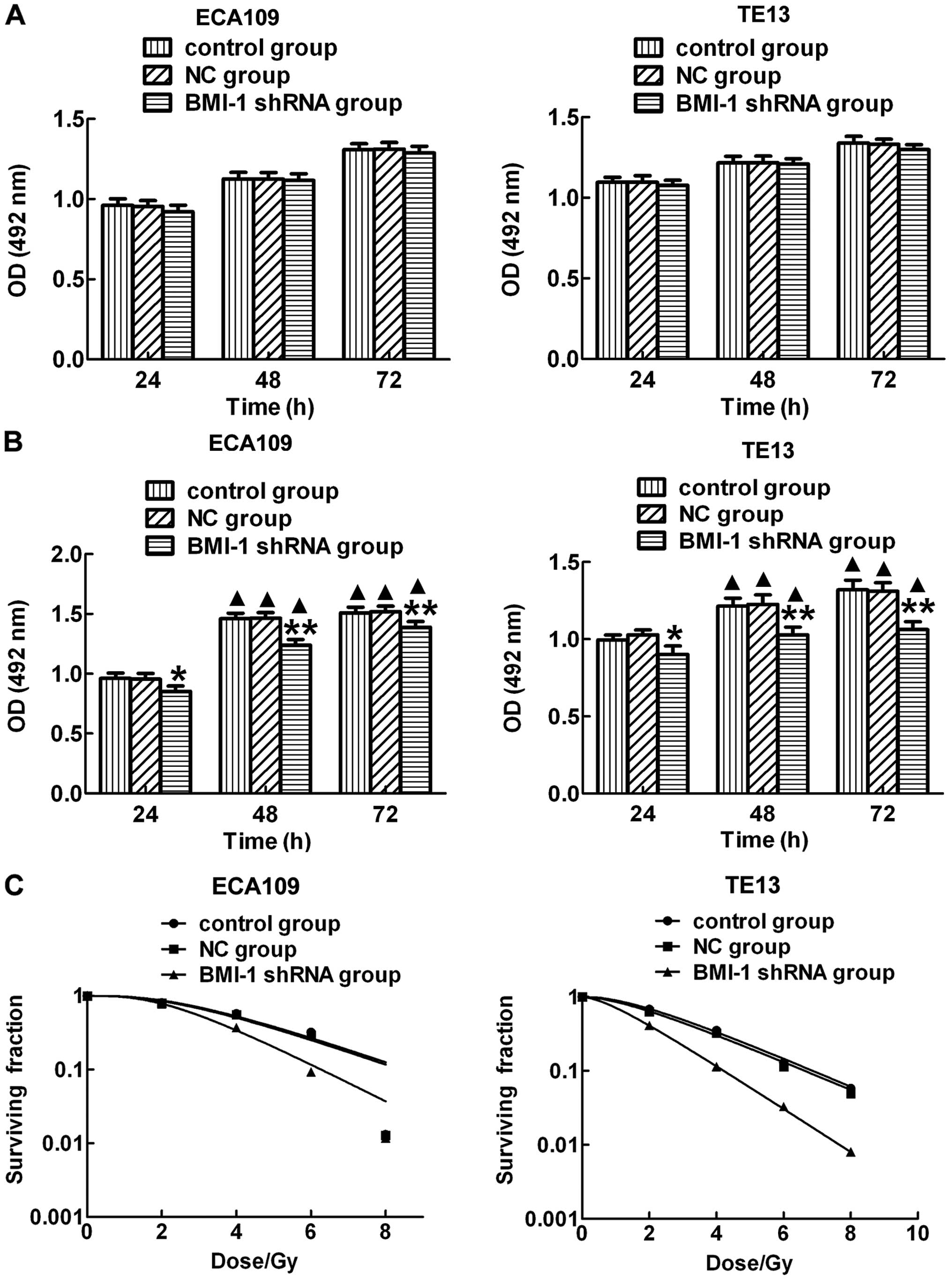
irradiation. MTS assay results showed that the proliferation rates
of the ECA109 and TE13 cells were not significantly different in
the 3 groups at each time point without irradiation (Fig. 2A). However, the proliferation levels
of the ECA109 and TE13 cells were significantly lower in the BMI-1
shRNA group when compared with the proliferation levels noted in
the control and NC-shRNA groups after IR (Fig. 2B). Moreover, the proliferation
levels in each group were obviously higher at 48 and 72 h after IR
than these levels in the corresponding unirradiated groups. In
addition, we also detected the radiation sensitivity in the
different groups after exposure to IR and found that there was no
significant difference between the control and NC groups. However,
cells in the BMI-1 shRNA group had higher sensitivity than that
noted in the other groups (Fig.
2C). These results suggest that downregulation of BMI-1 after
irradiation significantly inhibited the proliferation of the ECA109
and TE13 cells and increased radiation sensitivity.
BMI-1 shRNA inhibits the invasive ability
of the ECA109 and TE13 cells
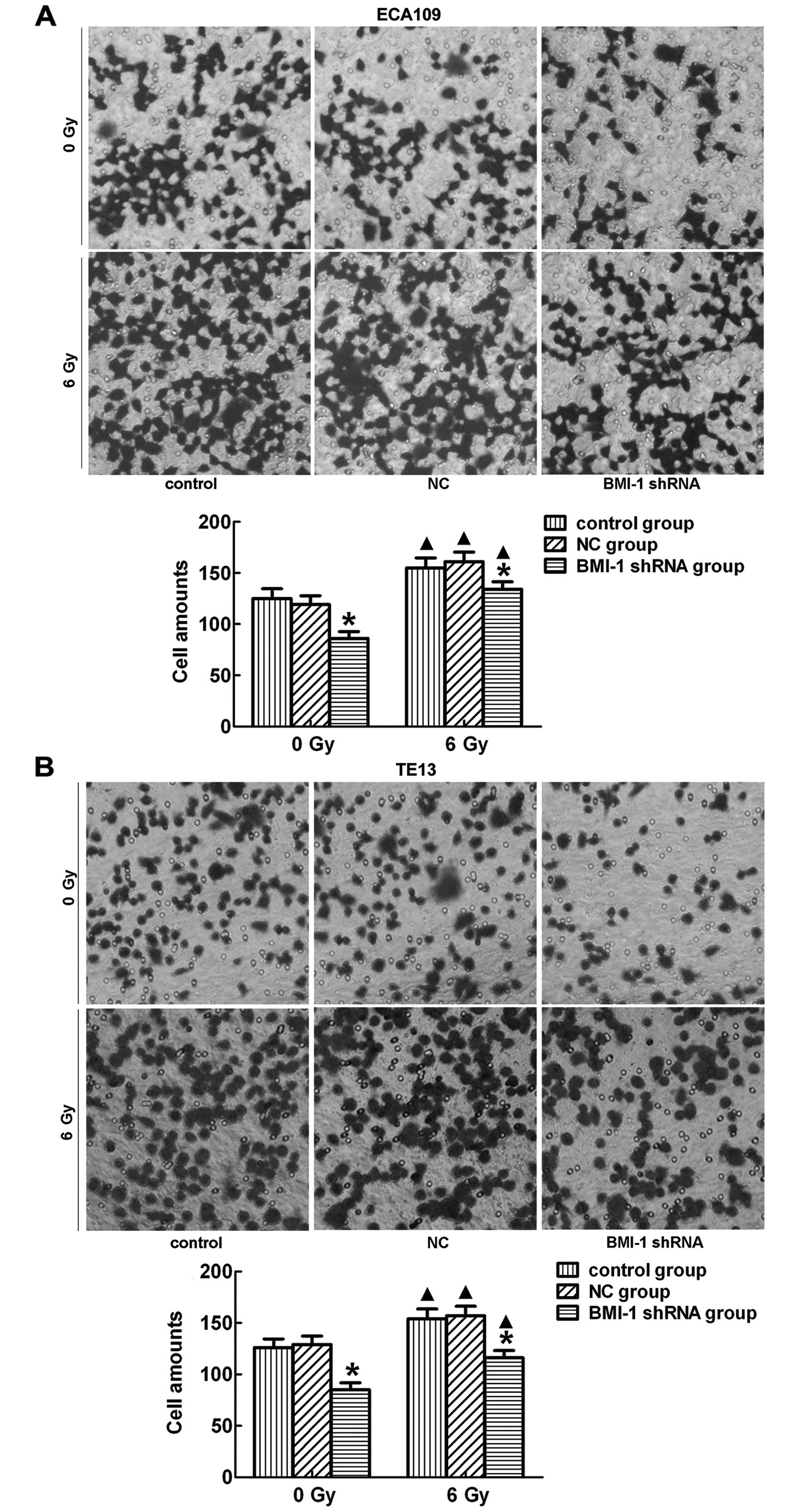
Transwell assay revealed that the ECA109 and TE13
cells transfected with BMI-1 shRNA had a much lower invasive
activity than that noted in the control and NC group cells
(p<0.05) before and after irradiation (Fig. 3A, ECA109; and B, TE13). This suggests that suppression of
BMI-1 had an inhibitory effect on the invasion of cells. There was
no significant difference between the control and NC group.
BMI-1 interacts with the ubiquitination
and phosphorylation of H2AX in ECA109 and TE13 cells in a DNA
damage-induced manner
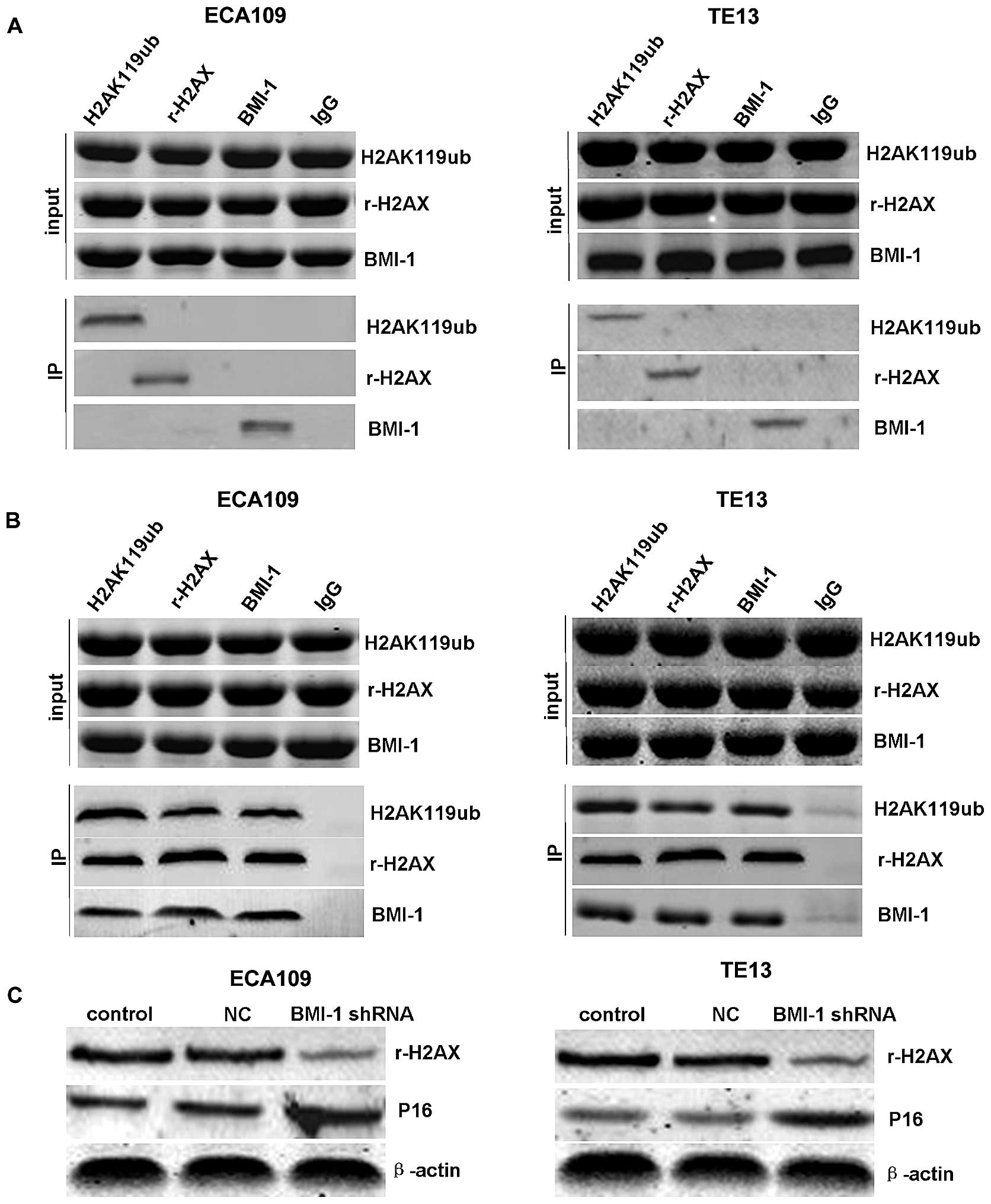
The specificity of this interaction was confirmed by
Co-IP of H2AK119ub, r-H2AX and BMI-1. Notably, the interactions
between BMI-1 and H2AK119ub or r-H2AX were obviously enhanced in
the ECA109 and TE13 cells after IR (Fig. 4B), while there was no significant
interaction without irradiation (Fig.
4A). This modification was induced by DNA damage as we observed
that the ubiquitination and phosphorylation of H2AX were increased
upon IR, indicating that BMI-1 may play an important role in DNA
damage repair through inducing the ubiquitination and
phosphorylation of H2AX. In addition, the downregulation of r-H2AX
and upregulation of P16 were detected in the BMI-1 shRNA group
after knockdown of BMI-1 expression (Fig. 4C).
Mechanisms of radiosensitization of the
silencing of BMI-1
In order to further explore the mechanisms involved
in the promotion of radiosensitization of esophageal cancer cells
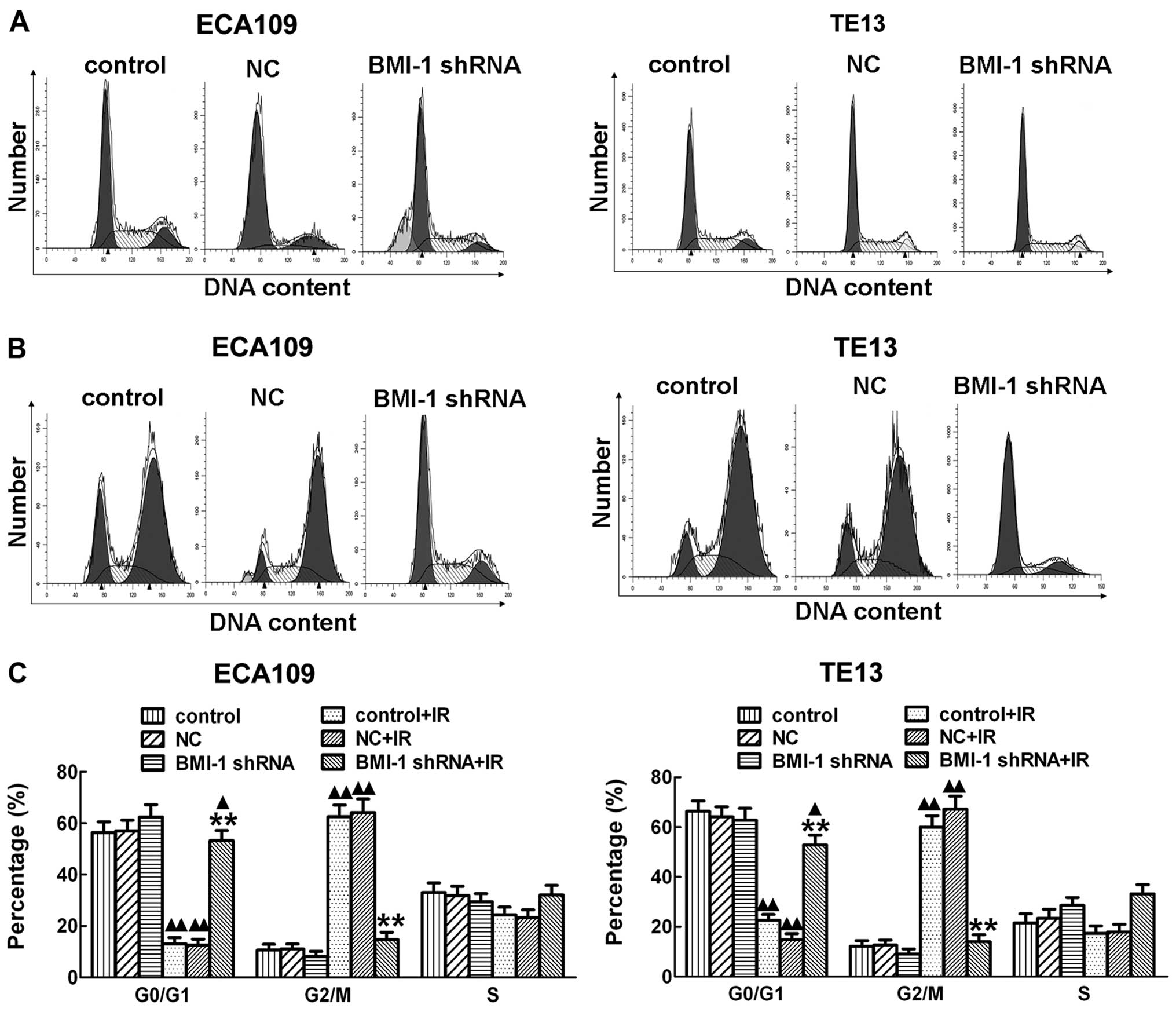
by silencing of BMI-1, flow cytometry was performed and
demonstrated that irradiation obviously induced cell cycle arrest
at the G2/M stage after irradiation for 24 h both in the ECA109 and
TE13 cells in vitro. However, silencing of BMI-1 by shRNA
obviously decreased the cell cycle arrest after irradiation
(Fig. 5B), which allowed much time
for repair of damaged tumor cells. Cancer cell repair may decrease
the killing effect of RT, which induces radioresistance. With no
irradiation, there was no significant difference among the BMI-1
shRNA, control and NC groups (Fig.
5A). Our results showed that knockdown of BMI-1 expression in
esophageal cancer ECA109 and TE13 cells by RNA interference
technology after exposure to irradiation decreased the G2/M phase
arrest in cells to a certain degree, reducing the opportunity of
tumor cells to repair thereby enhancing radiosensitivity.
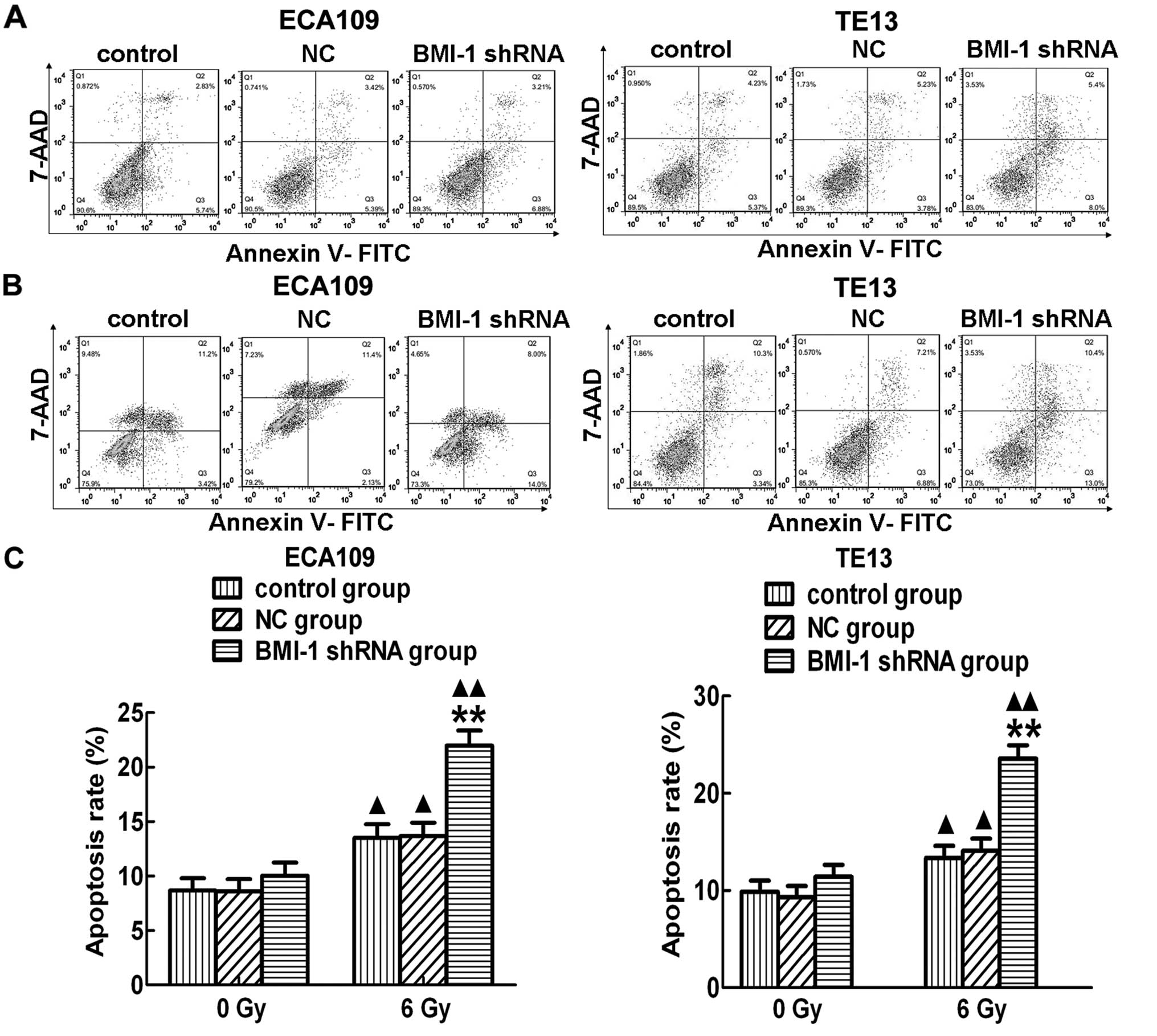
In addition, the apoptosis rate was not obviously
different among the control, NC and BMI-1 shRNA groups, in
vitro without irradiation (Fig.
6A). However, the apoptosis rate in the BMI-1 shRNA group was
significantly higher than that of the other two groups after RT
both in the ECA109 and TE13 cells (Fig.
6B and C). In addition, the apoptosis levels in the different
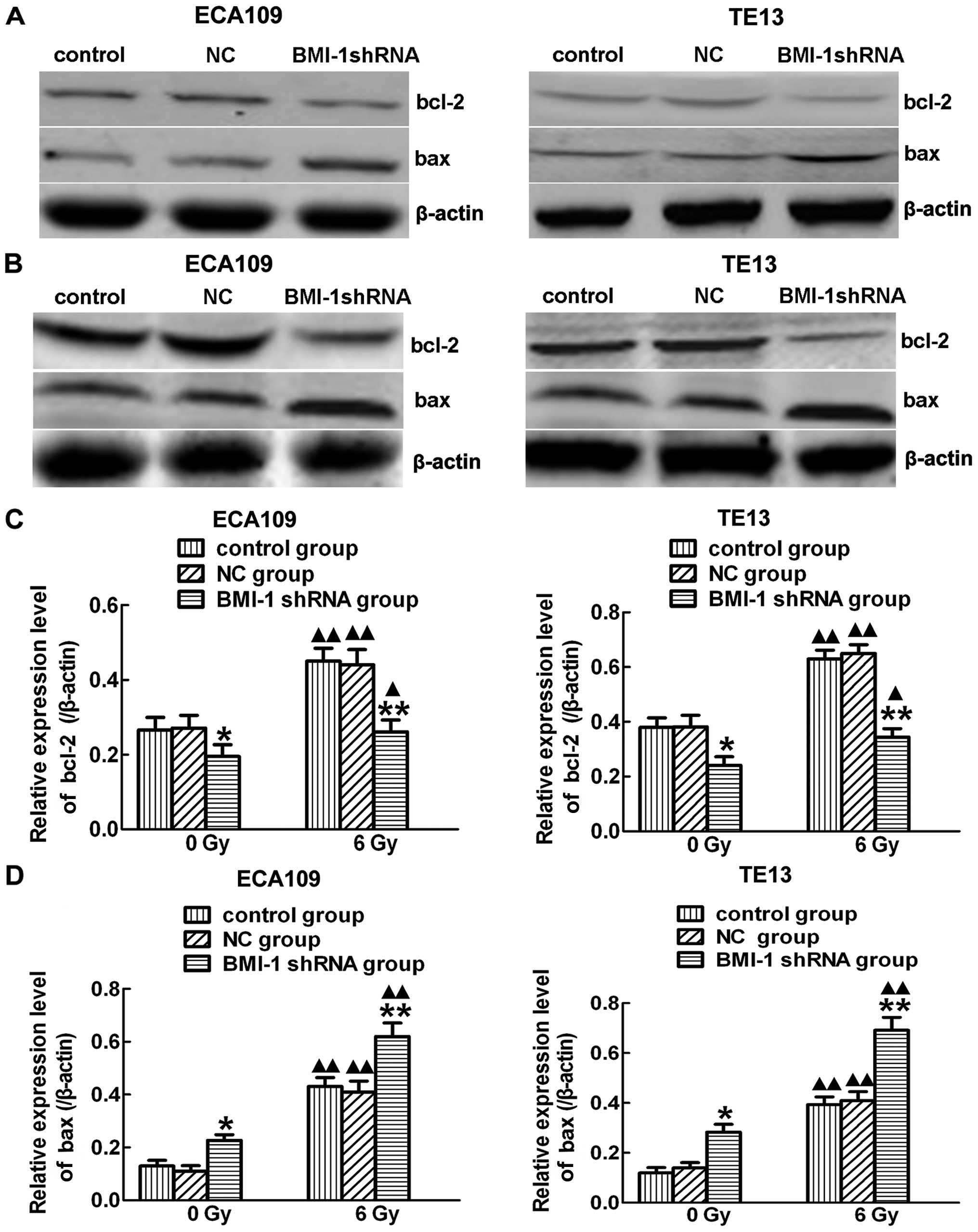
groups were increased after irradiation. In order to further
explore the mechanism, we observed the expression levels of bcl-2
and bax after (Fig. 7B) and before
irradiation (Fig. 7A). Significant
downregulation of bcl-2 (Fig. 7C)
and upregulation of bax (Fig. 7D)
were observed in the BMI-1-shRNA group after treatment with
irradiation. Notably, there were no significant changes among the 3
groups without irradiation. Thus, BMI-1 has the ability to regulate
the cell cycle, induce apoptosis and thus promote radiosensitivity
after IR.
Discussion
Esophageal carcinoma is one of the most frequent
malignant tumors, and is associated with the highest morbidity and
mortality worldwide. For example, the incidence of esophageal
carcinoma is as high as 26.3/100,000 in the US, which is higher
than that in Asia. Radiotherapy (RT) is one of the most important
treatment methods for esophageal carcinoma, but the 5-year survival
rate is only 10 to 15%, accompanied by a high recurrence rate at
~60 to 80%. Despite improvement in the local control rate after
extensive application of new techniques in RT, local failure is
still the main cause of death. Moreover, the molecular mechanisms
behind the development and progression of esophageal carcinoma
after RT are unknown.
BMI-1, one of the core components in polycomb
repressive complex 1, may play an important role in the
immortalization of normal epithelial cells and early malignant
transformation, as well as in the maintenance of the self-renewal
of stem cells and carcinogenesis in esophageal carcinoma. In
addition to their role in development, as one of the polycomb group
proteins, BMI-1 has been reported to be overexpressed in a variety
of human cancers, such as malignant lymphomas and various solid
tumors (15). In the present study,
we designed and constructed an shRNA recombinant expression vector
targeting BMI-1 and detected its expression at both the protein and
mRNA levels after transfection for 24 h. The findings demonstrated
that BMI-1 expression at both the protein and gene levels was
obviously lower in the BMI-1-shRNA group than those noted in the
control and the NC groups, while there was no significant
difference between the control and the NC group, indicating that
the shRNA targeting of BMI-1 decreased the BMI-1 expression, and
may be a potential therapeutic strategy for the gene therapy of
esophageal carcinoma.
In the present study, we also investigated the
effects on proliferation, invasion and radiosensitivity of
esophageal carcinoma cells by BMI-1 shRNA following irradiation.
The suppression of BMI-1 expression in the ECA109 and TE13 cells
significantly inhibited cell proliferation and improved
radiosensitivity after the cells were treated with shRNA and
irradiation at different times. The invasive ability of the cancer
cells in the BMI-1 shRNA group was also obviously suppressed after
irradiation or not. Previous studies have reported that the
downregulation of BMI-1 by adenovirus-mediated delivery of BMI-1
shRNA reduced the proliferation and invasion of cancer cells and
decreased the radioresistance (16,17).
Notably, there were no obvious differences among the control group,
NC and BMI-1-shRNA group without irradiation either in
proliferation or in radiosensitivity, indicating that RT combined
with shRNA effectively inhibits the proliferation of tumor cells
and enhances radiosensitization.
It was reported that BMI-1 is related to the
ubiquitination and phosphorylation of H2AX (18). Recent studies identified that BMI-1
possesses E3 ligase activity for H2AX monoubiquitination and
interacts with H2AX in a DNA damage-induced manner (19). In accordance with previous research,
our results found that interaction between BMI-1 and ub-H2AX or
r-H2AX was obviously enhanced in esophageal carcinoma after
exposure to IR, while there was no significant interaction without
irradiation, indicating a vital role of BMI-1 in DDR and its
potential mechanism. Our pre-trial confirmed that it could increase
radiation sensitivity through knockdown of H2AX in esophageal
cancer and cell cycle arrest at the G0/G1 phase (12). In the present study, we found that
silencing of the BMI-1 gene by shRNA significantly decreased the
phosphorylation of H2AX, followed by upregulation of p16, which is
able to inactivate CDK by directly binding to CDK4 and CDK6,
leading to cell cycle arrest (20).
These data indicated that BMI-1 may play an important role in
regulating the cell cycle through interaction with H2AX.
Cellular responses to DNA damage include cell cycle
arrest and apoptosis. The highest expression of BMI-1 is observed
in the G2/M phase (21,22). Our data revealed that inhibition of
the expression of BMI-1 by shRNA in ECA109 and TE13 cells
significantly induced spontaneous cell apoptosis and eliminated
cell cycle arrest at the G2/M phase of the cell cycle after RT.
However, given no ionizing radiation, there was no statistically
significant difference among the control, NC and BMI-1 shRNA group.
These results are in accordance with previous studies on esophageal
carcinoma (23) and cervical
carcinoma cells (24). Accordingly,
this method of suppressing BMI-1 expression which leads to cell
growth inhibition and the induction of apoptosis, in turn reduces
the proliferation of esophageal carcinoma cells, indicating that it
may have a significant therapeutic effect on esophageal carcinoma
(25,26). In the present study, our data showed
that the apoptosis rate of cells was higher in the BMI-1 shRNA
group than that noted in the control and NC group after exposure to
irradiation, while there was no statistically significant
difference among the 3 groups without RT. Meanwhile, our results
further indicated that the promotive effect of BMI-1 shRNA on the
apoptosis in ECA109 and TE13 cells after treatment with irradiation
was accompanied by upregulated expression of bcl-2 and
downregulation of bax, which are associated with cell apoptosis
after DNA damage. These results are in accordance with previous
studies, which showed that BMI-1 increased cell apoptosis through
regulation of apoptosis-related proteins (27–29),
and further enhanced radiosensitivity.
In summary, our data explored the important role of
BMI-1 in the growth and invasion of esophageal carcinoma by shRNA
silencing of BMI-1 expression after IR or not. In addition,
downregulation of BMI-1 decreased bcl-2 expression, enhanced bax
expression and eliminated IR-induced G2/M arrest by inhibition of
BMI-1 expression. To the best of our knowledge for the first time,
we demonstrated the suppression of BMI-1 expression by shRNA after
treatment with irradiation, indicating that BMI-1 may be an
important gene for the gene targeted therapy of human esophageal
carcinoma.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (no. 81372416) and the Medical
Sciences Key Topics of Hebei Province (nos. 20160183 and
20150305).
References
|
1
|
Sun Y, Liu M, Yang B, Li B and Lu J: Role
of siRNA silencing of MMP-2 gene on invasion and growth of
laryngeal squamous cell carcinoma. Eur Arch Otorhinolaryngol.
265:1385–1391. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhao JX and Xie XL: Regulation of gene
expression in laryngeal carcinama by microRNAs. Int J Pathol Clin
Med. 32:222–225. 2012.
|
|
3
|
Nacerddine K, Beaudry JB, Ginjala V,
Westerman B, Mattiroli F, Song JY, van der Poel H, Ponz OB,
Pritchard C, Cornelissen-Steijger P, et al: Akt-mediated
phosphorylation of Bmi1 modulates its oncogenic potential, E3
ligase activity, and DNA damage repair activity in mouse prostate
cancer. J Clin Invest. 122:1920–1932. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rouhigharabaei L, Ferreiro JF, Put N,
Michaux L, Tousseyn T, Lefebvre C, Gardiner A, De Kelver W,
Demuynck H, Verschuere J, et al: BMI1, the polycomb-group gene, is
recurrently targeted by genomic rearrangements in progressive
B-cell leukemia/lymphoma. Genes Chromosomes Cancer. 52:928–944.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guo BH, Feng Y, Zhang R, Xu LH, Li MZ,
Kung HF, Song LB and Zeng MS: Bmi-1 promotes invasion and
metastasis, and its elevated expression is correlated with an
advanced stage of breast cancer. Mol Cancer. 10:102011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dong Q, Sharma S, Liu H, Chen L, Gu B, Sun
X and Wang G: HDAC inhibitors reverse acquired radio resistance of
KYSE-150R esophageal carcinoma cells by modulating Bmi-1
expression. Toxicol Lett. 224:121–129. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen Y, Lian G, Zhang Q, Zeng L, Qian C,
Chen S and Huang K: Overexpression of Bmi-1 induces the malignant
transformation of gastric epithelial cells in vitro. Oncol Res.
21:33–41. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hoenerhoff MJ, Chu I, Barkan D, Liu ZY,
Datta S, Dimri GP and Green JE: BMI1 cooperates with H-RAS to
induce an aggressive breast cancer phenotype with brain metastases.
Oncogene. 28:3022–3032. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang H, Wang L, Erdjument-Bromage H, Vidal
M, Tempst P, Jones RS and Zhang Y: Role of histone H2A
ubiquitination in Polycomb silencing. Nature. 431:873–878. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Buchwald G, van der Stoop P, Weichenrieder
O, Perrakis A, van Lohuizen M and Sixma TK: Structure and E3-ligase
activity of the Ring-Ring complex of polycomb proteins Bmi1 and
Ring1b. EMBO J. 25:2465–2474. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cao R, Tsukada Y and Zhang Y: Role of
Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol
Cell. 20:845–854. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shi HY and Zhu SC: Radiosensitization of
esophageal cancer cells ECA109 by knockdown of H2AX. Thorac Cancer.
4:1759–7706. 2013. View Article : Google Scholar
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
14
|
Bartek J and Lukas J: Chk1 and Chk2
kinases in checkpoint control and cancer. Cancer Cell. 3:421–429.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Raaphorst FM: Deregulated expression of
Polycomb-group oncogenes in human malignant lymphomas and
epithelial tumors. Hum Mol Genet. 14(Suppl 1): R93–R100. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Song W, Tao K, Li H, Jin C, Song Z, Li J,
Shi H, Li X, Dang Z and Dou K: Bmi-1 is related to proliferation,
survival and poor prognosis in pancreatic cancer. Cancer Sci.
101:1754–1760. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liang W, Zhu D, Cui X, Su J, Liu H, Han J,
Zhao F and Xie W: Knockdown BMI1 expression inhibits proliferation
and invasion in human bladder cancer T24 cells. Mol Cell Biochem.
382:283–291. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ginjala V, Nacerddine K, Kulkarni A, Oza
J, Hill SJ, Yao M, Citterio E, van Lohuizen M and Ganesan S: BMI1
is recruited to DNA breaks and contributes to DNA damage-induced
H2A ubiquitination and repair. Mol Cell Biol. 31:1972–1982. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pan MR, Peng G, Hung WC and Lin SY:
Monoubiquitination of H2AX protein regulates DNA damage response
signaling. J Biol Chem. 286:28599–28607. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Suh HN and Han HJ: Collagen I regulates
the self-renewal of mouse embryonic stem cells through α2β1
integrin- and DDR1-dependent Bmi-1. J Cell Physiol. 226:3422–3432.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu Z, Liu H, Lv X, Liu Y, Li S and Li H:
Knockdown of the Bmi-1 oncogene inhibits cell proliferation and
induces cell apoptosis and is involved in the decrease of Akt
phosphorylation in the human breast carcinoma cell line MCF-7.
Oncol Rep. 25:409–418. 2011.
|
|
22
|
He X, Dong Y, Wu CW, Zhao Z, Ng SS, Chan
FK, Sung JJ and Yu J: MicroRNA-218 inhibits cell cycle progression
and promotes apoptosis in colon cancer by downregulating BMI1
polycomb ring finger oncogene. Mol Med. 18:1491–1498. 2013.
|
|
23
|
Liu WL, Guo XZ, Zhang LJ, Wang JY, Zhang
G, Guan S, Chen YM, Kong QL, Xu LH, Li MZ, et al: Prognostic
relevance of Bmi-1 expression and autoantibodies in esophageal
squamous cell carcinoma. BMC Cancer. 10:4672010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Min L, Shen D-X, Guo X-T, Guan T and Chen
X-D: Clinicopathological and prognostic significance of Bmi-1
expression in human cervical cancer. Acta Obstet Gynecol Scand.
90:737–745. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yao XB, Wang XX, Liu H, Zhang SQ and Zhu
HL: Silencing Bmi-1 expression by RNA interference suppresses the
growth of laryngeal carcinoma cells. Int J Mol Med. 31:1262–1272.
2013.PubMed/NCBI
|
|
26
|
Rizo A, Olthof S, Han L, Vellenga E, de
Haan G and Schuringa JJ: Repression of BMI1 in normal and leukemic
human CD34+ cells impairs self-renewal and induces
apoptosis. Blood. 114:1498–1505. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Crea F, Duhagon Serrat MA, Hurt EM, Thomas
SB, Danesi R and Farrar WL: BMI1 silencing enhances docetaxel
activity and impairs antioxidant response in prostate cancer. Int J
Cancer. 128:1946–1954. 2011. View Article : Google Scholar
|
|
28
|
Siddique HR, Parray A, Tarapore RS, Wang
L, Mukhtar H, Karnes RJ, Deng Y, Konety BR and Saleem M: BMI1
polycomb group protein acts as a master switch for growth and death
of tumor cells: Regulates TCF4-transcriptional factor-induced BCL2
signaling. PLoS One. 8:e606642013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhu D, Wan X, Huang H, Chen X, Liang W,
Zhao F, Lin T, Han J and Xie W: Knockdown of Bmi1 inhibits the
stemness properties and tumorigenicity of human bladder cancer stem
cell-like side population cells. Oncol Rep. 31:727–736. 2014.
|