Introduction
Angiogenesis is one of the essential causes of tumor
progression (1). The VEGF/VEGF
receptor pathway, in particular, contributes to several processes
in tumor angiogenesis (2). Indeed,
in the clinical setting, anti-angiogenic therapy targeting VEGF has
been demonstrated to be effective for several kinds of cancers
including colorectal cancer.
Bevacizumab, a humanized anti-VEGF monoclonal
antibody, is usually administered in combination with
chemotherapeutic agents. Although these combination therapies
improve overall survival and progression-free survival in
colorectal cancer patients, almost all responders ultimately
develop disease progression (PD) (3). Because endothelial cells are
genetically stable, it has been considered that refractoriness to
combination therapy with chemotherapeutic agents plus bevacizumab
is mostly caused by resistance to the chemotherapeutic agents
rather than to bevacizumab (4,5). In
this context, second-line therapies including bevacizumab
(bevacizumab beyond progression therapy; BBP therapy) were
intensively studied and were found to demonstrate clinical benefits
in patients with metastatic colorectal and breast cancer (6,7).
However, VEGF-independent angiogenesis has been reported as a
mechanism of resistance to anti-VEGF therapy (8). Therefore, resistance to bevacizumab
cannot be excluded as a mechanism of resistance to combination
therapy with chemotherapeutic agents plus bevacizumab. We
hypothesized that BBP therapy is effective even after a tumor
acquires resistance to bevacizumab, and we investigated the
effective mechanism of BBP therapy.
To this end, we used the combination of capecitabine
plus bevacizumab as BBP therapy because this combination was used
for 26% of patients in the metastatic colorectal cancer BBP therapy
trial and 60% of patients in the breast cancer BBP therapy trial
(6,7). Capecitabine is a prodrug of 5-FU and
is designed to generate high levels of 5-FU in tumor cells. It has
been reported that capecitabine and 5-FU inhibited angiogenesis in
colon and gastric cancer models by suppressing the secretion of
angiogenic factors from tumor cells (9,10).
In this study, we used a xenograft model of a human
colon cancer cell line with acquired resistance to bevacizumab
(hereafter, bevacizumab PD model) to investigate the antitumor
effect of the combination of capecitabine plus bevacizumab and its
mechanism of action in terms of angiogenic factors produced by
stromal cells and tumor cells.
Materials and methods
Test agents
Bevacizumab and capecitabine were provided by F.
Hoffmann-La Roche (Basel, Switzerland) as a liquid and fine powder,
respectively. Human immunoglobulin G (HuIgG) was purchased from MP
Biomedicals (Solon, OH, USA). Capecitabine was dissolved in 40 mM
citrate buffer (pH 6.0) containing 5% gum arabic (capecitabine
vehicle).
Animals
Five-week-old male BALB/c-nu (CAnN.Cg-Foxn1
<nu>/CrlCrlj) mice were obtained from Charles River
Laboratories Japan (Yokohama, Japan). All animals were allowed to
acclimatize and recover from shipping-related stress for at least 4
days prior to the study. The health of the mice was monitored by
daily observation. The animals were allowed free access to
chlorinated water and irradiated food, and the animals were kept
under a controlled light-dark cycle (12 h–12 h). All animal
experiments were reviewed and approved by the Institutional Animal
Care and Use Committee at Chugai Pharmaceutical Co., Ltd.
Cell lines and culture conditions
Five human colorectal cancer cell lines (COLO 205,
HCT-8, HCT-116, LS411N, and HT-29), and two mouse cancer cell lines
(B16-F1 and LLC1) were used in this study. All cancer cell lines
were purchased from American Type Culture Collection (Manassas, VA,
USA). COLO 205 and LS411N were maintained in RPMI-1640 supplemented
with 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 4.5 g/l
glucose, 1.5 g/l sodium bicarbonate, and 10% FBS at 37°C under 5%
CO2. HCT-116 and HT-29 were maintained in McCoy's 5A
supplemented with 10% FBS at 37°C under 5% CO2. HCT-8
was maintained in RPMI-1640 supplemented with 1 mM sodium pyruvate
and 10% horse serum at 37°C under 5% CO2. B16-F1 and
LLC1 were maintained in D-MEM supplemented with 10% FBS at 37°C
under 5% CO2.
In vivo tumor growth inhibition by
bevacizumab
Each mouse was inoculated subcutaneously into the
right flank with 5×106 cells/mouse of human colorectal
cancer cell line (either COLO 205, HCT-8, HCT 116, LS411N or
HT-29). Several weeks after tumor inoculation, mice were randomly
allocated to control and treatment groups. Bevacizumab (5 mg/kg)
was administered intraperitoneally (i.p.) once a week for 3 weeks.
To evaluate the antitumor activity of the test agents, tumor volume
(TV) was measured twice a week. The tumor volume was estimated from
the equation TV = ab2/2, where a and
b are tumor length and width, respectively. Tumor volume
ratios were calculated by dividing mean tumor volume on day 8 by
mean tumor volume on day 1 and mean tumor volume on day 22 by mean
tumor volume on day 15 in both the bevacizumab and control
groups.
Combination of bevacizumab plus
chemotherapy in the bevacizumab PD model
Mice inoculated with HT-29 cells that had been
treated with bevacizumab (5 mg/kg) on days 1 and 8 (1st treatment)
were randomly allocated to control IgG plus capecitabine vehicle
(control), bevacizumab, capecitabine, and bevacizumab plus
capecitabine groups on day 29. Bevacizumab (5 mg/kg) was
administered i.p. Once a week and capecitabine (359 mg/kg) was
administered orally (p.o.) every day for 3 weeks (2nd treatment).
To evaluate the antitumor activity and the tolerability of the test
agents, TV and body weight was measured twice a week.
Quantification of microvessel density in
tumor tissues
HT-29 tumor tissues were resected from the
bevacizumab PD model on day 50, and microvessel density (MVD) was
evaluated immunohistochemically by using a monoclonal anti-mouse
CD31 antibody (rat anti-mouse CD31 monoclonal antibody, clone MEC
13.3; BD Biosciences, Franklin Lakes, NJ, USA). Immunohistochemical
staining was performed as described previously (11) using 5-µm-thick sections from
freshly frozen tissues. MVD (%) was calculated from the ratio of
the CD31-positive staining area to the total observation area.
Fields excluding necrotic areas were analyzed. Positive staining
areas were calculated by using imaging analysis software (Definiens
Tissue Studio; Definiens, Munich, Germany).
Flow cytometric analysis of
tumor-infiltrating cells
HT-29 tumors were collected from xenografted mice on
day 26 after inoculation. Single cell suspensions were prepared by
mincing the tumors, followed by treating with a Tumor Dissociation
kit for human tumor tissue (Miltenyi Biotec GmbH, Bergisch
Gladbach, Germany) in a gentleMACS Dissociator (Miltenyi Biotec).
The cells were stained with antibodies to mouse CD11b
(PerCP/Cy5.5), Gr1 (APC), and CD45 (FITC) (BioLegend, San Diego,
CA, USA) at 1 µg/ml and analyzed by using FACSAria
(Becton-Dickinson) and FlowJo software (Tree Star, Ashland, OR,
USA).
Measurement of angiogenesis-related
proteins
HT-29 tumor tissues collected on day 50 from the
bevacizumab PD model were homogenized with cell lysis buffer (Cell
Signaling Technology, Danvers, MA, USA) with protease inhibitor
cocktail (Sigma-Aldrich, St. Louis, MO, USA) and phosphatase
inhibitor cocktail (Nacalai Tesque, Kyoto, Japan). The supernatant
after centrifugation (14,000 × g, 5 min, 4°C) was used for the
assays. The protein concentration of the supernatant was quantified
using a Direct Detect spectrometer (Merck Millipore, Frankfurter,
Germany). The relative expression profiles of human and murine
angiogenesis-related proteins were analyzed by membrane-based
antibody array (Proteome Profiler Angiogenesis Array kit; R&D
Systems, Minneapolis, MN, USA) according to the manufacturer's
protocol. The human angiogenesis array simultaneously detect the
relative levels of 55 angiogenesis-related proteins (Activin A,
ADAMTS-1, Angiogenin, Angiopoietin-1, Angiopoietin-2,
Angiostatin/Plasminogen, Amphiregulin, Artemin, Tissue
Factor/Factor III, CXCL16, DPPIV/CD26, EGF, EG-VEGF,
Endoglin/CD105, Endostatin/Collagen XVIII, Endothelin-1, FGF
acidic, FGF basic, FGF-4, FGF-7/KGF, GDNF, GM-CSF, HB-EGF, HGF,
IGFBP-1, IGFBP-2, IGFBP-3, IL-1β, CXCL8/IL-8, TGF-β1, Leptin,
CCL2/MCP-1, CCL3/MIP-1α, MMP-8, MMP-9, NRG1-β1, Pentraxin 3,
PD-ECGF, PDGF-AA, PDGF-AB/PDGF-BB, Persephin, CXCL4/PF4, PlGF,
Prolactin, Serpin B5/Maspin, Serpin E1/PAI-1, Serpin F1/PEDF,
TIMP-1, TIMP-4, Thrombospondin-1, Thrombospondin-2, uPA, Vasohibin,
VEGF, VEGF-C). The murine angiogenesis array simultaneously detects
the relative levels of 53 angiogenesis-related proteins (ADAMTS1,
Amphiregulin, Angiogenin, Angiopoietin-1, Angiopoietin-3,
CCL2/MCP-1, CCL3/MIP-1α, CXCL1, CXCL10, SDF-1, CXCL16, CXCL4,
IGFBP-10, DLL4, DPPIV, EGF, Endoglin, Endostatin, FGF acidic, FGF
basic, FGF-7/KGF, Fractalkine, GM-CSF, HB-EGF, HGF, IGFBP-1,
IGFBP-2, IGFBP-3, IL-1α, IL-1β, IL-10, Leptin, MMP-3, MMP-8, MMP-9,
IGFBP-9, Osteopontin, PD-ECGF, PDGF-AA, PDGF-AB/BB, Pentraxin-3,
PlGF-2, Prolactin, Proliferin, Serpin E1/PAI-1, Serpin F1/PEDF,
Thrombospondin-2, TIMP-1, TIMP-4, Coagulation Factor III, VEGF and
VEGF-B). The array was hybridized with 280 µg of total
protein. The concentrations of human VEGF, galectin-1, and
galectin-3 were measured by a Quantikine ELISA kit (R&D
Systems).
In vitro cell treatment with 5-FU
HT-29 cells were seeded on 24-well plates at
4×104 cells/well and were incubated overnight at 37°C
under 5% CO2. The cells were then treated with
5-fluorouracil (5-FU) at 0.4, 2, or 20 µM for 24 h.
Statistical analysis
The Wilcoxon test and Dunnett's test were used, with
P<0.05 considered statistically significant. Statistical
analyses were carried out using the SAS preclinical package (SAS
Institute, Cary, NC, USA).
Results
Establishing the bevacizumab PD
model
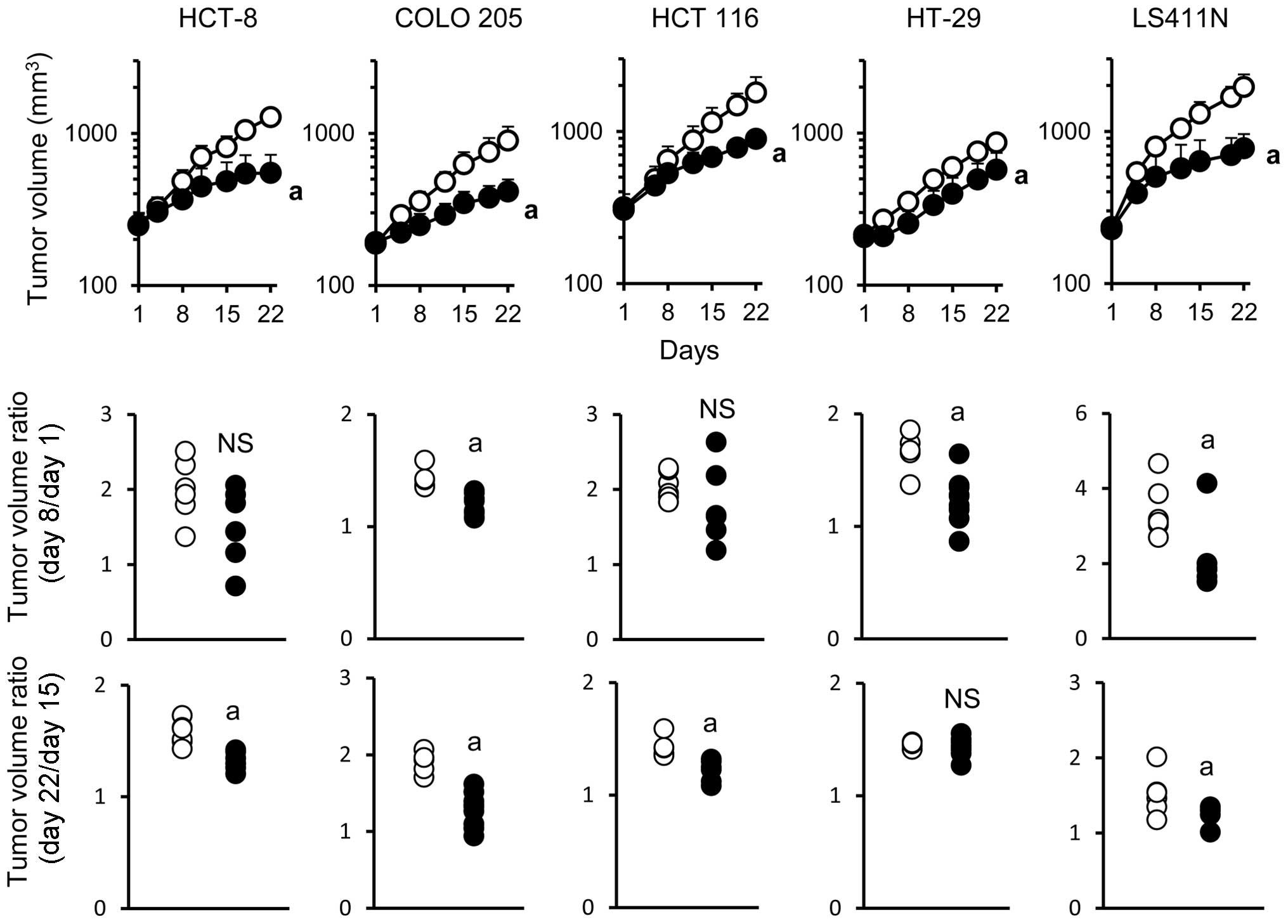
The antitumor activity of bevacizumab monotherapy
was examined in five human colorectal cancer xenograft models in
which tumors produced VEGF (Table
I). Bevacizumab exhibited significant antitumor activity in all
of the five models when evaluated on day 22. To analyze in detail
the mode of antitumor activity of bevacizumab during the treatment,
tumor volume ratio was calculated separately in the early phase
(days 1–8) and in the late phase (days 15–22). Acquisition of
resistance to bevacizumab was evaluated by comparing the
differences in tumor volume ratio in the early and late phases of
treatment. In the COLO 205 and LS411N models, bevacizumab exhibited
a significant antitumor activity in both the early and late phase.
In the HCT-8 and HCT 116 models, bevacizumab exhibited a
significant antitumor activity only in the late phase. Of note, in
the HT-29 model, bevacizumab exhibited significant antitumor
activity in the early phase but not in the late phase (Fig. 1) indicating that the HT-29 model
became non-sensitive to bevacizumab during treatment.
 | Table IVEGF expression levels in tumor tissue
of colorectal cancer xenograft models. |
Table I
VEGF expression levels in tumor tissue
of colorectal cancer xenograft models.
| Tumor type | VEGF (pg per mg
protein) |
|---|
| HCT-8 | 1,669 |
| COLO 205 | 1,778 |
| HCT-116 | 2,539 |
| HT-29 | 3,132 |
| LS411N | 3,540 |
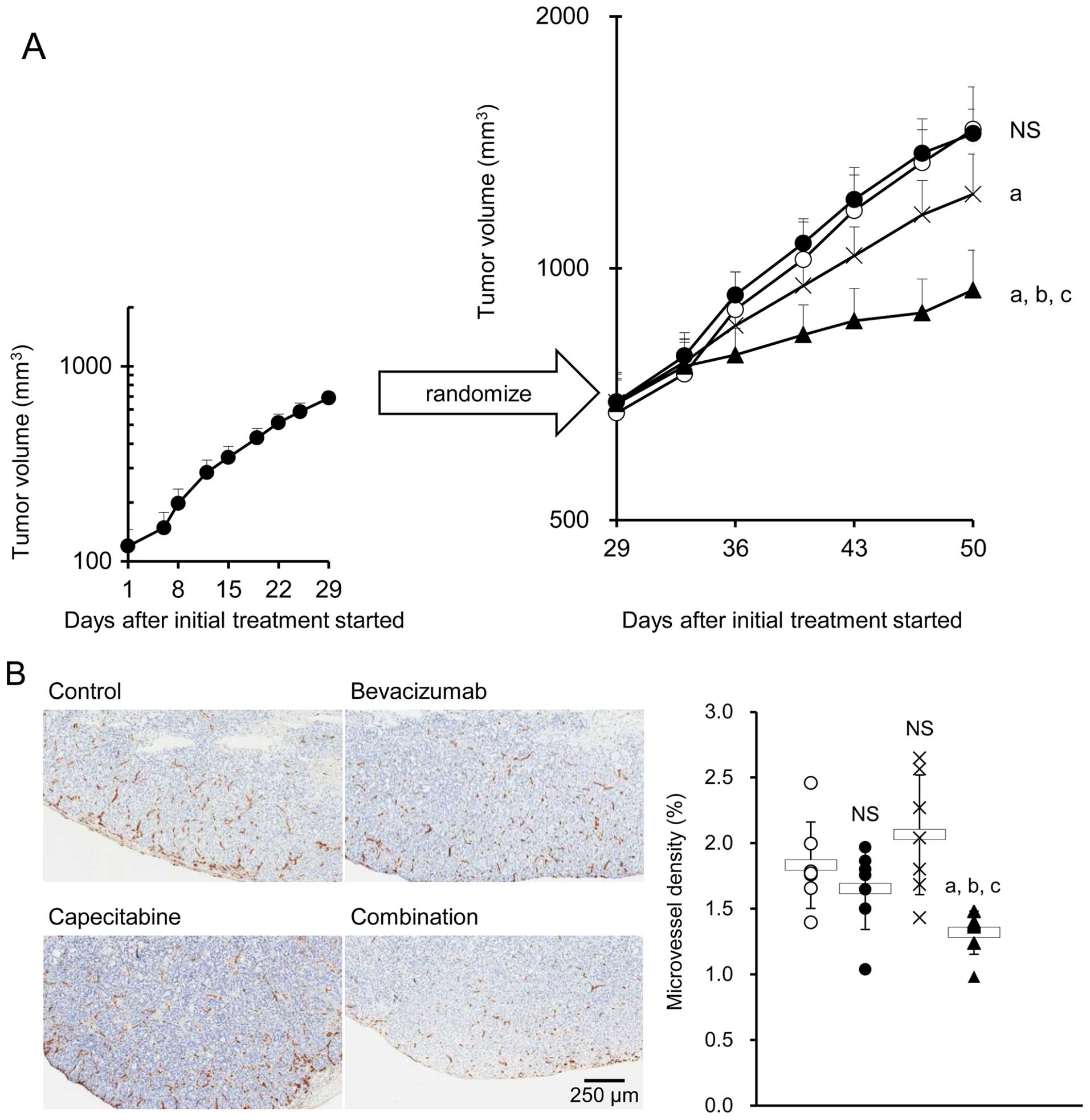
Effect of combination therapy with
bevacizumab plus capecitabine on tumor growth and MVD in the
bevacizumab PD model
The HT-29 xenografted mice treated with bevacizumab
(the bevacizumab PD model mice) were then treated with either
control IgG plus capecitabine vehicle, bevacizumab, capecitabine,
or bevacizumab plus capecitabine. As was expected, bevacizumab
alone did not exhibit any significant antitumor effect, indicating
that the HT-29 tumors pretreated with bevacizumab were refractory
to bevacizumab. Capecitabine alone exhibited a significant
antitumor effect versus control. The combination of bevacizumab
plus capecitabine exhibited a stronger antitumor effect than
capecitabine alone (Fig. 2A). No
difference in MVD was observed in tumor tissues from the control,
bevacizumab, and capecitabine groups. However, a significant
decrease in MVD was observed in the combination group compared with
the groups treated with each drug alone (Fig. 2B).
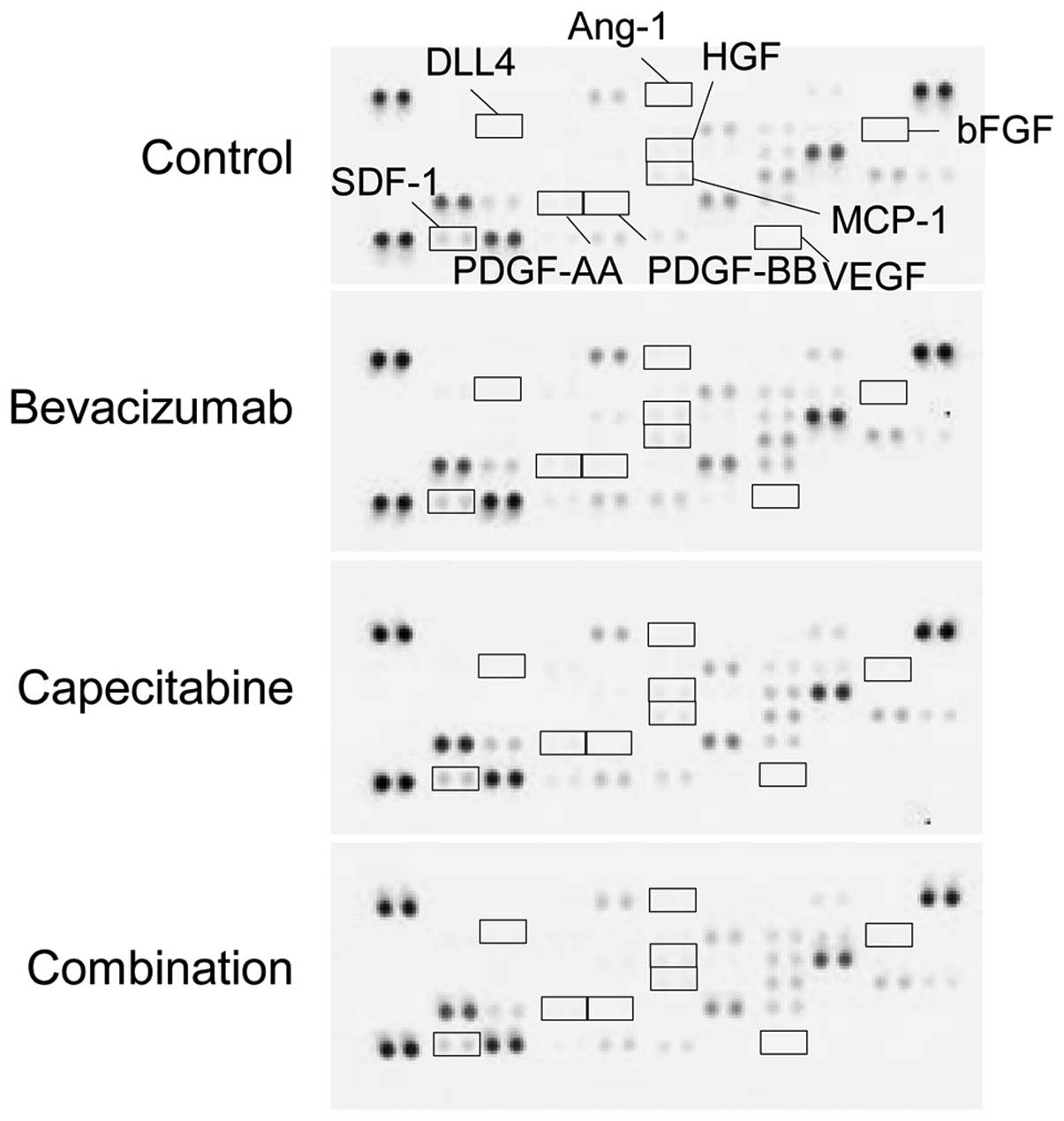
Intratumoral stromal cell-derived
angiogenic factors
To investigate whether stromal cell-derived
angiogenic factors are involved in sensitivity to combination
therapy with bevacizumab, we examined the change in expression of
murine angiogenesis-related factors in tumors from each treatment
group. VEGF, bFGF, HGF, PDGF-AA, PDGF-BB, angiopoietin-1 (Ang-1),
and δ-like ligand 4 (DLL4) were not detected. Although SDF-1 and
MCP-1 were detected, none of the treatment groups showed any
difference in these angiogenic factors (Fig. 3).
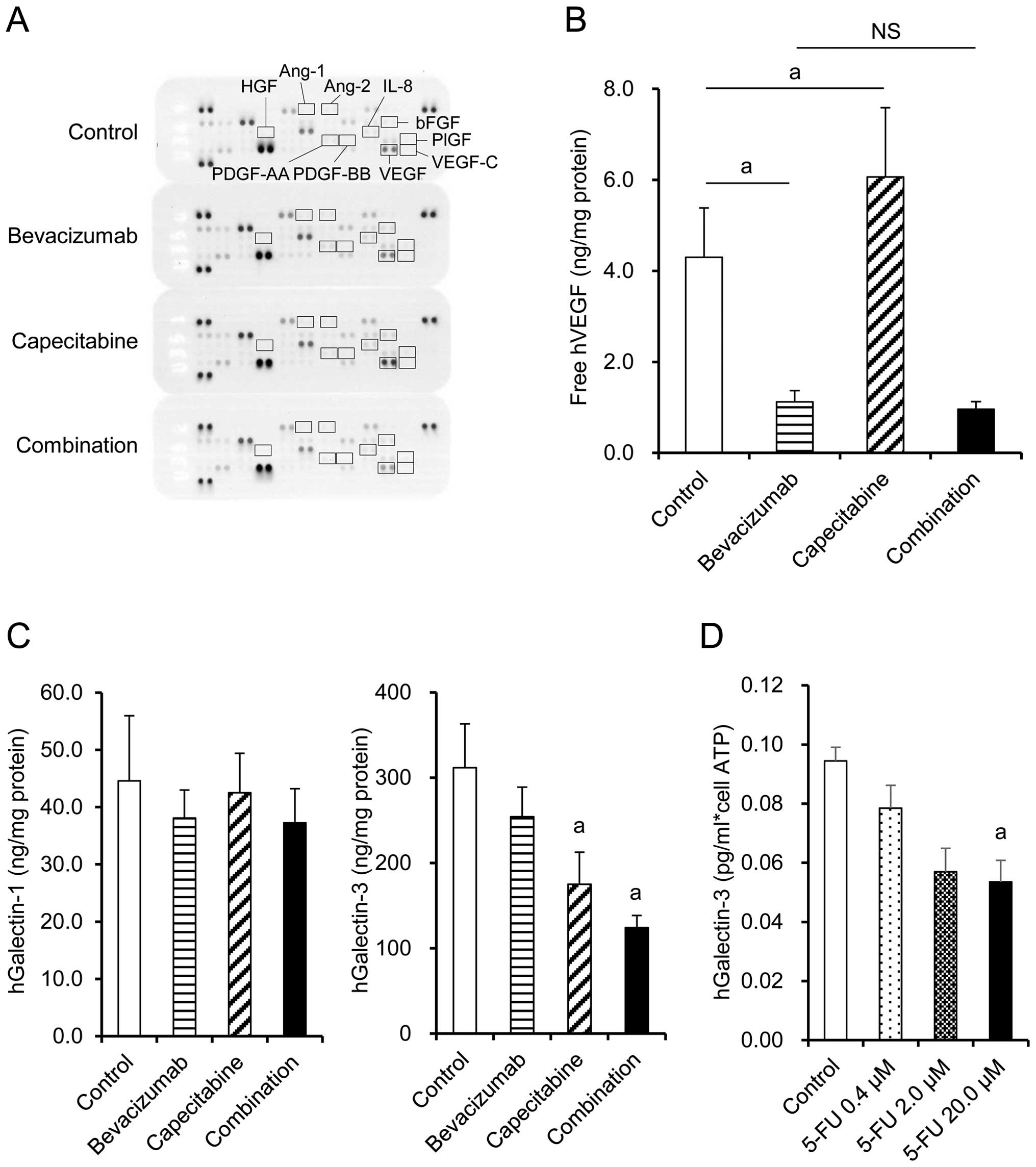
Tumor cell-derived angiogenic
factors
Expression of human angiogenesis-related factors in
tumors from each treatment group was examined by Proteome Profiler
Angiogenesis Array. VEGF, bFGF, and IL-8 were detected, whereas
PDGF-AA, PDGF-BB, VEGF-C, HGF, PlGF, angiopoietin-1 (Ang-1), and
angiopoietin-2 (Ang-2) were not detected regardless of the
treatment (Fig. 4A). bFGF and IL-8
were similar in the capecitabine group, bevacizumab group, and
combination group (Fig. 4A). VEGF
level was significantly increased by capecitabine treatment
compared with the control group, while VEGF was unsurprisingly
neutralized by the bevacizumab-containing treatments when measured
by ELISA (Fig. 4B). Galectin-3
levels were significantly decreased in the capecitabine and
combination groups compared with the control group, whereas no
significant difference was observed in galectin-1 levels (Fig. 4C). When the HT-29 cells were treated
in vitro with 5-FU, an active metabolite of capecitabine,
production of galectin-3 per cell was inhibited in a dose-dependent
manner (Fig. 4D). These data
suggested that capecitabine inhibited galectin-3 production by
HT-29 cells via a mechanism independent of cell growth
inhibition.
Discussion
In colorectal cancer, it was unclear whether
maintenance with a combination of bevacizumab plus a
chemotherapeutic agent would benefit patients who had acquired
resistance to bevacizumab (12). In
this study, it was found that the combination of bevacizumab plus
capecitabine exhibited a strong antitumor effect on xenografted
human colorectal cancer that had acquired resistance to bevacizumab
in vivo. The mechanism of action of the combination of
bevacizumab plus capecitabine was analyzed in terms of tumor
angiogenesis.
We previously reported that the molecular targeted
drugs trastuzumab and erlotinib exhibited antitumor effects when
administered with chemotherapeutic agents after the tumor had
acquired resistance to monotherapy with each if the tumor kept
expressing the target molecules (13,14).
If a tumor continues to express VEGF, combining bevacizumab with
second-line chemotherapy after resistance has been acquired, may be
an appropriate therapy. In order to establish a colorectal cancer
xenograft model that develops resistance to bevacizumab, five human
colorectal cancer xenografts which produced VEGF (Table I) were treated with bevacizumab.
Bevacizumab was given as the sole regimen to avoid development of
resistance to chemotherapeutic agents. As a result, a bevacizumab
PD model was established using the HT-29 xenograft model and was
used in the following experiments.
We investigated the antitumor effect of the
combination of bevacizumab plus capecitabine by using the HT-29 PD
model. Of note, the combination of bevacizumab plus capecitabine
exhibited a significantly stronger antitumor effect than
capecitabine alone (Fig. 2A). These
data suggested that it is important to continue to inhibit VEGF by
bevacizumab in combination with an appropriate chemotherapeutic
agent even after the tumor has acquired resistance to bevacizumab
per se. Indeed, the combination group showed a significant
reduction in MVD, while the groups receiving bevacizumab or
capecitabine alone did not at this stage (Fig. 2B). These results suggested that a
synergistic anti-angiogenic effect was one of the mechanisms
underlying the stronger antitumor effect of the combination
treatment.
To examine the mechanism through which the
combination treatment exerted an anti-angiogenic effect in the
bevacizumab PD model, we analyzed the effect of capecitabine on the
production of several angiogenic factors by both tumor cells and
stromal cells. Since it has been reported that the infiltration of
CD11b+/Gr1+ cells into tumors directly or
indirectly produces other angiogenic factors causing resistance to
anti-VEGF treatment (15), we
analyzed stromal cell-derived angiogenic factors and the number of
CD11b+/Gr1+ cells in the tumor in the HT-29
PD model. However, the number of CD11b+/Gr1+
cells was as few as in the anti-VEGF antibody sensitive tumor
B16-F1 (data not shown). It has also been reported that bFGF,
Ang-1, DLL4, HGF, SDF-1, MCP-1, PDGF-AA, and PDGF-BB from stromal
cells are implicated in the development of resistance to VEGF
inhibition (16–19). MCP-1 and SDF-1 were detected in the
HT-29 PD model, but the levels were similar in the capecitabine,
bevacizumab, and combination groups (Fig. 3). There were no obvious differences
in any of the other murine angiogenic factors detected in the
capecitabine, bevacizumab, or combination groups (Fig. 3). Therefore, the mechanism of action
of the combination of bevacizumab plus capecitabine in this model
could not be explained by the stromal cell-derived angiogenic
factors tested.
Next, we investigated whether tumor cell-derived
angiogenic factors were involved in the anti-angiogenic effect of
the combination therapy. We examined bFGF, IL-8, PDGF-AA, PDGF-BB,
VEGF-C, Ang-1, Ang-2, HGF, and PlGF, which are reported to be
angiogenic factors for tumors (20–22).
PDGF-AA, PDGF-BB, VEGF-C, Ang-1, Ang-2, HGF, and PlGF were not
detected in the HT-29 PD model. bFGF and IL-8 were detected but
their levels were similar in the capecitabine, bevacizumab and
combination groups (Fig. 4A). There
were no obvious differences in any of the other human angiogenic
factors detected in the capecitabine, bevacizumab, or combination
groups (Fig. 4A). Therefore, the
anti-angiogenic effect of the combination therapy in this model
could not be explained by the factors tested.
Recently, galectins, members of a family of
carbohydrate-binding proteins with multiple functions, were
reported to be potent prognostic marker in colorectal cancer and to
promote angiogenesis (23,24). Galectin-1 maintains angiogenesis in
anti-VEGF-refractory tumors (25),
and galectin-3 has been shown as an important mediator of VEGF- as
well as FGF-mediated angiogenic response (26–28).
In light of these findings, we investigated the intratumoral levels
of human galectin-1, galectin-3, and VEGF in each treatment group.
No significant change was observed in galectin-1 level. However,
galectin-3 level was significantly decreased in the capecitabine
and combination groups compared with level in the control group
(Fig. 4C). In addition, 5-FU, an
active metabolite of capecitabine, directly inhibited galectin-3
production by HT-29 cells in vitro (Fig. 4D). Since VEGF- and bFGF-mediated
angiogenesis has been reported to be greatly reduced by galectin-3
inhibitors such as dominant negative galectin-3 or in galectin-3
knockdown cells in vitro and also in Gal3−/− mice
(26), the decrease in intratumor
galectin-3 level by capecitabine in our model was suggested to
contribute, at least in part, to synergistic inhibition of
angiogenesis in combination with anti-VEGF antibody. Despite the
reduction of galectin-3, no difference in MVD was observed between
tumor tissues in the control and capecitabine groups. It has been
reported that conventional 5-FU treatment may promote tumor
angiogenesis by increasing the production of VEGF (9,10). In
contrast with galectin-3 levels, VEGF levels indeed increased in
the capecitabine treatment group in our model (Fig. 4B). These bilateral effects of
capecitabine on the production of angiogenic factors may be a
reason for the apparent absence of anti-angiogenic effect by
capecitabine alone. However, by neutralization of VEGF through
concurrent administration of bevacizumab, a synergistic
anti-angiogenic effect emerged (Fig.
4B). These data raise the possibility that the inhibition of
galectin-3 production by capecitabine and the neutralization of
VEGF by bevacizumab were one of the mechanisms underlying the
effect of combination treatment.
Regarding the development of resistance to
bevacizumab in the HT-29 model, we investigated whether galectin-3
plays a role in acquisition of resistance. However, galectin-3
levels in the tumors did not change before and after developing PD
(data not shown). Galectin-3 interacts with Mgat5-modified
N-glycans on cytokine receptors such as EGFR, IGFR, PDGFR, and
bFGFR, and activates downstream signal transduction pathways
(29). Therefore, alteration in the
expression of any of these receptors may cause galectin-3-induced
resistance to bevacizumab. Thus, further investigations are
required to clarify the mechanisms of resistance to bevacizumab in
this model.
In this study, we indicated that combination therapy
with bevacizumab plus capecitabine could exert strong antitumor
activity, even after resistance to bevacizumab was acquired, by
recovering the anti-angiogenic effect, thus suggesting the clinical
relevance for treatment with bevacizumab in combination therapy
beyond PD.
Acknowledgments
The authors thank Masako Miyazaki and Hiromi
Sawamura from Chugai Pharmaceutical Co., Ltd, for technical
assistance in the experiments, and also thank Naoki Harada,
Kazushige Mori, and Kaori Fujimoto-Ouchi for support and special
advice in this study.
References
|
1
|
Ferrara N and Kerbel RS: Angiogenesis as a
therapeutic target. Nature. 438:967–974. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ellis LM and Hicklin DJ: VEGF-targeted
therapy: Mechanisms of anti-tumour activity. Nat Rev Cancer.
8:579–591. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hurwitz HI, Tebbutt NC, Kabbinavar F,
Giantonio BJ, Guan ZZ, Mitchell L, Waterkamp D and Tabernero J:
Efficacy and safety of bevacizumab in metastatic colorectal cancer:
Pooled analysis from seven randomized controlled trials.
Oncologist. 18:1004–1012. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Klement G, Baruchel S, Rak J, Man S, Clark
K, Hicklin DJ, Bohlen P and Kerbel RS: Continuous low-dose therapy
with vinblastine and VEGF receptor-2 antibody induces sustained
tumor regression without overt toxicity. J Clin Invest.
105:R15–R24. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Klement G, Huang P, Mayer B, Green SK, Man
S, Bohlen P, Hicklin D and Kerbel RS: Differences in therapeutic
indexes of combination metronomic chemotherapy and an anti-VEGFR-2
antibody in multidrug-resistant human breast cancer xenografts.
Clin Cancer Res. 8:221–232. 2002.PubMed/NCBI
|
|
6
|
Bennouna J, Sastre J, Arnold D, Österlund
P, Greil R, Van Cutsem E, von Moos R, Viéitez JM, Bouché O, Borg C,
et al ML18147 Study Investigators: Continuation of bevacizumab
after first progression in metastatic colorectal cancer (ML18147):
A randomised phase 3 trial. Lancet Oncol. 14:29–37. 2013.
View Article : Google Scholar
|
|
7
|
von Minckwitz G, Puglisi F, Cortes J,
Vrdoljak E, Marschner N, Zielinski C, Villanueva C, Romieu G, Lang
I, Ciruelos E, et al: Bevacizumab plus chemotherapy versus
chemotherapy alone as second-line treatment for patients with
HER2-negative locally recurrent or metastatic breast cancer after
first-line treatment with bevacizumab plus chemotherapy (TANIA): An
open-label, randomised phase 3 trial. Lancet Oncol. 15:1269–1278.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jubb AM, Oates AJ, Holden S and Koeppen H:
Predicting benefit from anti-angiogenic agents in malignancy. Nat
Rev Cancer. 6:626–635. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shi H, Jiang J, Ji J, Shi M, Cai Q, Chen
X, Yu Y, Liu B, Zhu Z and Zhang J: Anti-angiogenesis participates
in antitumor effects of metronomic capecitabine on colon cancer.
Cancer Lett. 349:128–135. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yuan F, Shi H, Ji J, Cai Q, Chen X, Yu Y,
Liu B, Zhu Z and Zhang J: Capecitabine metronomic chemotherapy
inhibits the proliferation of gastric cancer cells through
anti-angiogenesis. Oncol Rep. 33:1753–1762. 2015.PubMed/NCBI
|
|
11
|
Yanagisawa M, Yorozu K, Kurasawa M, Nakano
K, Furugaki K, Yamashita Y, Mori K and Fujimoto-Ouchi K:
Bevacizumab improves the delivery and efficacy of paclitaxel.
Anticancer Drugs. 21:687–694. 2010.PubMed/NCBI
|
|
12
|
Kopetz S, Hoff PM, Morris JS, Wolff RA,
Eng C, Glover KY, Adinin R, Overman MJ, Valero V, Wen S, et al:
Phase II trial of infusional fluorouracil, irinotecan, and
bevacizumab for metastatic colorectal cancer: Efficacy and
circulating angiogenic biomarkers associated with therapeutic
resistance. J Clin Oncol. 28:453–459. 2010. View Article : Google Scholar :
|
|
13
|
Fujimoto-Ouchi K, Sekiguchi F, Yamamoto K,
Shirane M, Yamashita Y and Mori K: Preclinical study of prolonged
administration of trastuzumab as combination therapy after disease
progression during trastuzumab monotherapy. Cancer Chemother
Pharmacol. 66:269–276. 2010. View Article : Google Scholar
|
|
14
|
Iwai T, Moriya Y, Shirane M,
Fujimoto-Ouchi K and Mori K: Continuous inhibition of epidermal
growth factor receptor phosphorylation by erlotinib enhances
antitumor activity of chemotherapy in erlotinib-resistant tumor
xenografts. Oncol Rep. 27:923–928. 2012.PubMed/NCBI
|
|
15
|
Maniati E and Hagemann T: IL-17 mediates
resistance to anti-VEGF therapy. Nat Med. 19:1092–1094. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Casanovas O, Hicklin DJ, Bergers G and
Hanahan D: Drug resistance by evasion of antiangiogenic targeting
of VEGF signaling in late-stage pancreatic islet tumors. Cancer
Cell. 8:299–309. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Erber R, Thurnher A, Katsen AD, Groth G,
Kerger H, Hammes HP, Menger MD, Ullrich A and Vajkoczy P: Combined
inhibition of VEGF and PDGF signaling enforces tumor vessel
regression by interfering with pericyte-mediated endothelial cell
survival mechanisms. FASEB J. 18:338–340. 2004.
|
|
18
|
Crawford Y, Kasman I, Yu L, Zhong C, Wu X,
Modrusan Z, Kaminker J and Ferrara N: PDGF-C mediates the
angiogenic and tumorigenic properties of fibroblasts associated
with tumors refractory to anti-VEGF treatment. Cancer Cell.
15:21–34. 2009. View Article : Google Scholar
|
|
19
|
Bergers G and Hanahan D: Modes of
resistance to anti-angiogenic therapy. Nat Rev Cancer. 8:592–603.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yamashita-Kashima Y, Fujimoto-Ouchi K,
Yorozu K, Kurasawa M, Yanagisawa M, Yasuno H and Mori K: Biomarkers
for antitumor activity of bevacizumab in gastric cancer models. BMC
Cancer. 12:372012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Waugh DJ and Wilson C: The interleukin-8
pathway in cancer. Clin Cancer Res. 14:6735–6741. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li D, Xie K, Ding G, Li J, Chen K, Li H,
Qian J, Jiang C and Fang J: Tumor resistance to anti-VEGF therapy
through up-regulation of VEGF-C expression. Cancer Lett. 346:45–52.
2014. View Article : Google Scholar
|
|
23
|
Griffioen AW and Thijssen VL: Galectins in
tumor angiogenesis. Ann Transl Med. 2:902014.PubMed/NCBI
|
|
24
|
Endo K, Kohnoe S, Tsujita E, Watanabe A,
Nakashima H, Baba H and Maehara Y: Galectin-3 expression is a
potent prognostic marker in colorectal cancer. Anticancer Res.
25:3117–3121. 2005.PubMed/NCBI
|
|
25
|
Croci DO, Cerliani JP, Dalotto-Moreno T,
Méndez-Huergo SP, Mascanfroni ID, Dergan-Dylon S, Toscano MA,
Caramelo JJ, García-Vallejo JJ, Ouyang J, et al:
Glycosylation-dependent lectin-receptor interactions preserve
angiogenesis in anti-VEGF refractory tumors. Cell. 156:744–758.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Markowska AI, Liu FT and Panjwani N:
Galectin-3 is an important mediator of VEGF- and bFGF-mediated
angiogenic response. J Exp Med. 207:1981–1993. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Markowska AI, Jefferies KC and Panjwani N:
Galectin-3 protein modulates cell surface expression and activation
of vascular endothelial growth factor receptor 2 in human
endothelial cells. J Biol Chem. 286:29913–29921. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
D'Haene N, Sauvage S, Maris C, Adanja I,
Le Mercier M, Decaestecker C, Baum L and Salmon I: VEGFR1 and
VEGFR2 involvement in extracellular galectin-1- and
galectin-3-induced angiogenesis. PLoS One. 8:e670292013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Partridge EA, Le Roy C, Di Guglielmo GM,
Pawling J, Cheung P, Granovsky M, Nabi IR, Wrana JL and Dennis JW:
Regulation of cytokine receptors by Golgi N-glycan processing and
endocytosis. Science. 306:120–124. 2004. View Article : Google Scholar : PubMed/NCBI
|