Introduction
Lung cancer is one of the leading causes of
cancer-related death in the world (1). Treatments of lung cancer vary from
stage to stage (2). Common
chemotherapy and radiotherapy cause undesirable side-effects.
Conventional therapies at a later stage of cancer development and
poor prognosis limit the survival rate of lung cancer patients
(3,4). Previous research found that crocodile
blood displayed marked effects on cancer cells not only with
anti-23 strains of bacteria, including those resistant to
antibiotics, but also with antioxidant and anti-inflammatory
properties (5,6). Yet the mechanism remains unclear. Our
present study aimed to evaluate the effects of crocodile tissue
extracts on a lung cancer cell line and studied the related
mechanisms of action. An attempt was made to characterize the
active fractions.
Materials and methods
Cell culture
The A549 human lung adenocarcinoma cell line was
obtained from the American Type Culture Collection (ATCC; Manassas,
VA, USA). Culturing was maintained according to their
recommendations. The A549 cell line was cultured in F-12K medium
(Invitrogen Life Technologies, Carlsbad, CA, USA) with 10% fetal
bovine serum (FBS) and 1% penicillin-streptomycin (PS) (both from
Gibco, Grand Island, NY, USA) antibiotic solution. All cell
cultures and subcultures were incubated at 37°C with 5%
CO2.
Whole blood and fraction preparation
Crocodile blood powder was purchased from a local
vendor and dissolved in phosphate-buffered saline (PBS). After
sonification, the mixture was centrifuged at 10,000 × g for 20 min.
The supernatant was collected. After precipitation by ammonium
sulfate overnight at 4°C, the mixture was centrifuged at 10,000 × g
for 20 min. The supernatant and residues were collected and
desalted by Sephadex G25.
Fast protein liquid chromatography (FPLC)
analysis
An ÄKTA FPLC system (Amersham Pharmacia Biotech,
Sweden) and a Sephadex G25 column (50×4.6 mm; GE Healthcare, USA)
were used. The samples were separated at 5 ml/min. The mobile phase
was distilled water with a constant elution component. The column
temperature was approximately 25°C and detected at 280 nm. The
detector with conductance was recruited.
High performance liquid chromatography
(HPLC) analysis
An Agilent 1100 system (Agilent Technologies, Palo
Alto, CA, USA) and a Prevail C18 column (250×4.6 mm I.D., 5
µm; Alltech, USA) were used. The samples were separated by
HPLC at a flow rate of 0.8 ml/min. The mobile phase consisted of
0.1% trifluoroacetic acid in water (A) and acetonitrile (B) with a
gradient elution of 5% B at 0–5 min; 20% B at 6–15 min; 30% B at
16–25 min; 60% B at 26–35 min; 90% B at 36–45 min; 5% B at 46–50
min. The column temperature was set at 25°C with injection volume
of 10 µl. Detection was set at 280 nm.
Assessment of cell viability by MTT
assays
A549 cells were seeded into a 96-well plate at a
density of 1×104 cells/well. Cells were treated with
different concentrations of sample solution for 24, 48 and 72 h,
respectively, and those without treatment as control. A total of 50
µl of 3-(4,5-dimethylthiazol-2-yl)-2,5-di-phenyltetrazolium
bromide (MTT; Sigma-Aldrich, St. Louis, MO, USA) solution was added
to each well and incubated for 1 h. The purple formazan formed was
dissolved in 200 µl of DMSO and quantified by measuring the
absorbance at 540 nm with a microplate reader (Tecan Infinite M200;
Tecan Group, Switzerland). The relative cell viability was compared
to the control. The half maximal inhibitory concentration
(IC50) was determined according to the relative cell
viability curve.
Measurement of cytotoxicity
A549 cells were seeded into a 96-well plate at a
density of 1×104 cells/well. Cells were treated with
different concentrations of sample solution for 24 h. Measurement
was carried out according to the manufacturer's instructions
(CytoTox 96 Non-Radioactive Cytotoxicity Assay kit, G1780; Promega,
Madison, WI, USA). In brief, after centrifugation at 250 × g for 4
min, 50 µl of the supernatant was transferred into a new
96-well plate. A total of 50 µl of substrate was added and
the mixture was incubated for 30 min at room temperature.
Measurement was carried out using a microplate reader at 490
nm.
DNA fragmentation
A549 cells were seeded in 100-mm dishes at a
confluency of 5×106. The cells were treated with 200
µg/ml of S2 for 24, 48 and 72 h, respectively and harvested
with trypsin (Invitrogen Life Technologies) and PBS. The cell
pellets were then re-suspended in 500 µl of DNA digestion
buffer with Proteinase K (BioVision, USA) added to a 0.5 mg/ml
final concentration overnight at 55°C with gentle shaking. A total
of 700 µl of neutralized phenol/chloroform/isoamyl alcohol
(25:24:1) was added and mixed fairly vigorously. The samples were
spinned at 10,000 × g for 5 min and 500 µl of the upper
phase was transferred to a new microfuge tube. DNA was precipitated
with 1 ml 100% ethanol at room temperature and centrifuged at
10,000 × g. The sample was then washed with 70% ethanol twice and
air dried. The DNA precipitate was dissolved in 50 µl of TE
buffer and electrophoresed on a 1.5% agarose gel with GelRed
Nucleic Acid Gel Stain (Biotium, Hayward, CA, USA) and detected
under UV light.
Cell cycle analysis
After A549 cells grew to 80% confluency on 100-mm
culture dishes, different concentrations of S2 were added after 24
h. The cells were harvested with trypsin and washed with cold PBS
twice. After centrifugation at 250 × g for 5 min, the cell pellets
were re-suspended in cold 70% ethanol and fixed at −20°C overnight.
The supernatant was discarded after centrifugation at 250 × g for 5
min. The cell pellets were washed with cold PBS twice and
re-suspended with 0.5 ml propidium iodide (PI) solution. After
incubation at 37°C in the dark, the cells were transferred into a 5
ml polystyrene round-bottom tube and sorted with 10,000
events/sample in triplicate using a flow cytometer (BD FACSCanto;
BD Biosciences, USA). The results were analyzed with ModFit 3.0
software.
Detection of apoptosis
A549 cells were cultured into 100-mm dishes and
treated with different concentrations of S2 for 24 h. After
harvesting with trypsin and washing twice with cold PBS, detection
was carried out according to the manufacturer's instructions (FITC
Annexin V Apoptosis Detection kit; BD Biosciences). In brief, the
cell pellets were re-suspended in 0.5 ml of 1X binding buffer. A
total of 5 µl Annexin V-FITC solution and 5 µl 1X PI
solution were added to 100 µl of the cell suspension. The
mixture was incubated for 15 min in the dark at room temperature.
Before injecting into the flow cytometer, 0.4 ml 1X binding buffer
was added to each sample mixture. Analysis was achieved with 10,000
events/sample in triplicate using a flow cytometer (BD FACSCanto;
BD Biosciences). The results were analyzed with WinMDI 2.9
software.
Mitochondrial membrane permeability
A549 cells were cultured into 6-well plates and
treated with different concentrations of S2 for 3 h. After
harvesting with trypsin and washing twice with cold PBS, the cell
pellets were re-suspended in 0.5 ml of JC-1 solution (MitoScreen
kit; BD Biosciences). After incubation for 15 min at 37°C in the
dark, the cell mixture was centrifuged at 250 × g for 5 min and the
supernatant was discarded. Before re-suspending with 0.5 ml of 1X
assay buffer solution, the cell pellets were washed twice with 1 ml
of 1X binding buffer at room temperature. Analysis was achieved
with 10,000 events/sample in triplicate using a flow cytometer. The
results were analyzed with WinMDI 2.9 software.
Measurement of reactive oxygen species
(ROS)
A549 cells were cultured in 6-well plates with
coverslips inside at a density of 1×105 cells/well and
treated with different concentrations of S2 for 24 h. After washing
with warm Hank's Balanced Salt Solution (HBSS; Gibco, Gaithersburg,
MD, USA), a sufficient amount of 25 µM
carboxy-H2DCFDA (Image-iT™ LIVE Green Reactive Oxygen
Species Detection kit; BD Biosciences) working solution was applied
to the coverslips followed by a 30-min incubation at 37°C in the
dark. To stain the nucleus, a final concentration of 1.0 µM
of Hoechst was added to the cells at the last 5 min of incubation.
The coverslips were washed with warm HBSS (Thermo Fisher
Scientific, USA) prior to reversely cover the slides with mounting
medium. The slides were detected under a fluorescence microscope
under a excitation/emission wavelength of 350/461 nm for Hoechst
and 495/529 nm for carboxy-H2DCFDA.
Activities of caspase-3/7
A549 cells were seeded onto 96-well plates at the
concentration of 1×104 cells/well. The cells were
treated with different concentrations of S2 for 24 h. A total of
100 µl of 1X caspase substrate Z-DEVD solution (Apo-ONE
Homogeneous Caspase-3/7 Assay kit; Promega) was added to each well
(1:1). The mixture was gently mixed using a plate shaker at 250 × g
and incubated at room temperature for 3 h. The fluorescence of each
well was measured at 485/520 nm using a microplate reader.
Detection of nuclei, mitochondria and
cytochrome c
A549 cells were cultured into 6-well plates with
coverslips inside at a density of 1×105cells/well and
treated with different concentrations of S2 for 24 h. An optimal
concentration of mitochondrial dye MitoTracker Red (M7512;
Invitrogen Life Technologies) was added and incubated for 30 min at
37°C. After washing with warm HBSS, 1 ml of 4% paraformaldehyde was
added for 30 min, followed by 0.5% of Triton-X for 15 min. After
washing with PBS, 3% bovine serum albumin (BSA) was added. An
optimal concentration of purified mouse anti-cytochrome c
(BD Sciences) and goat anti-mouse antibody conjugating FITC (Thermo
Fisher Scientific) were added to the coverslips. At the last 5 min
of incubation, the nuclear staining dye Hoechst (33342; Molecular
Probes) was added. All coverslips were mounted to the slides
conversely. Scanning and detection were processed under confocal
laser scanning microscopy (FV1000; Olympus).
Western blotting
A549 cells were seeded in 100-mm dishes followed by
24 h of treatment with different concentrations of S2. After
harvesting and washing, the cell pellets were re-suspended in whole
cell lysis buffer on ice. The cell lysate was collected by
centrifugation at 10,000 × g at 4°C for 30 min after incubation
overnight at −20°C. The protein concentration of each sample
supernatant was quantified by using the DC Protein Assay kit
(Bio-Rad, Hercules, CA, USA). After normalization, 80 µg of
proteins were loaded to 4–12% SDS-PAGE to separate under a voltage
of 80–120 V. After transferring to the polyvinylidene fluoride
(PVDF) membrane (Millipore Corp., USA), the membrane was blocked
with non-fat dry milk (5% w/v) or BSA at room temperature for 1 h.
Dilution of the primary antibodies and secondary antibodies were
adjusted according to the manufacturer's instructions (Cell
Signaling Technologies, Danvers, MA, USA; Santa Cruz
Biotechnology). Blots were developed using the ECL
Chemiluminescence Detection Reagent (GE Healthcare) according to
the manufacturer's instructions. Densitometric analysis was
performed with ImageJ software.
Statistical analysis
Statistical analysis of the raw data was carried out
by one-way analysis of variance (ANOVA) followed by Tukey's post
hoc test. The data are expressed as means ± SD as indicated.
Significance was accepted at p<0.05.
Results
Chromatographic analysis of crocodile
blood extracts and S2 fractions
The fractions collected from the precipitation and
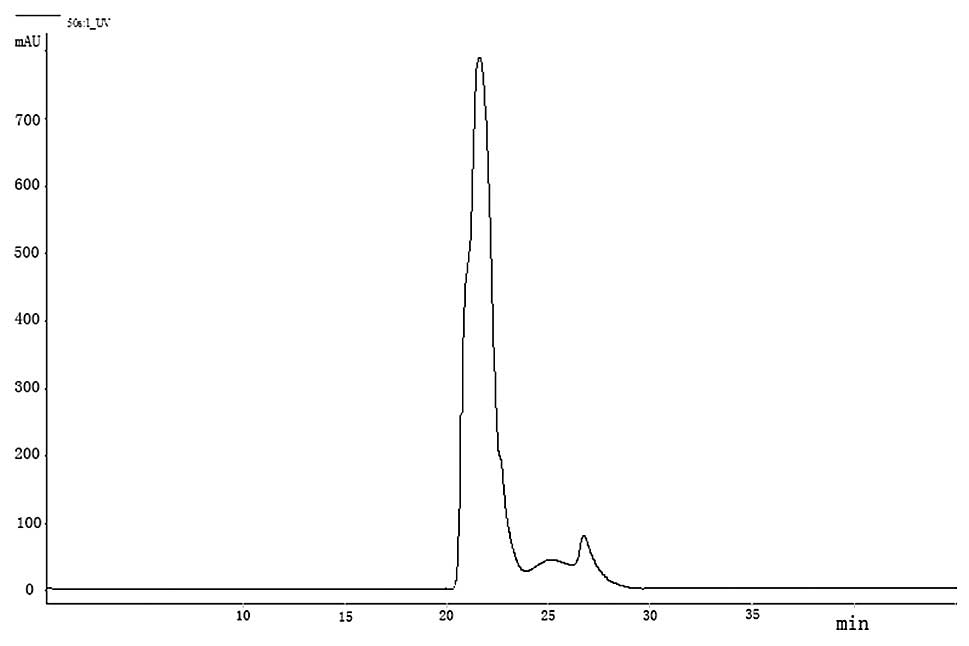
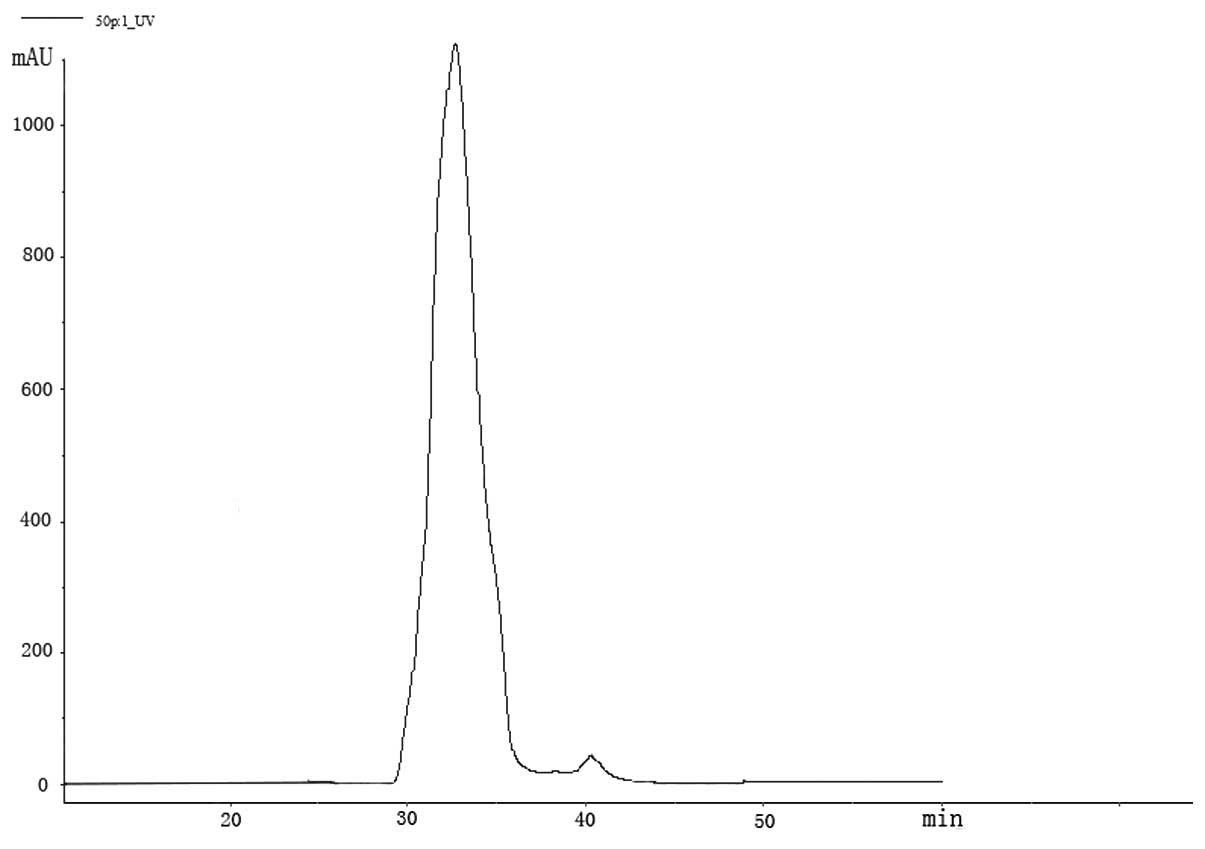
supernatant were separated according to molecular size (Figs. 1 and 2). The chromatogram profile showed a
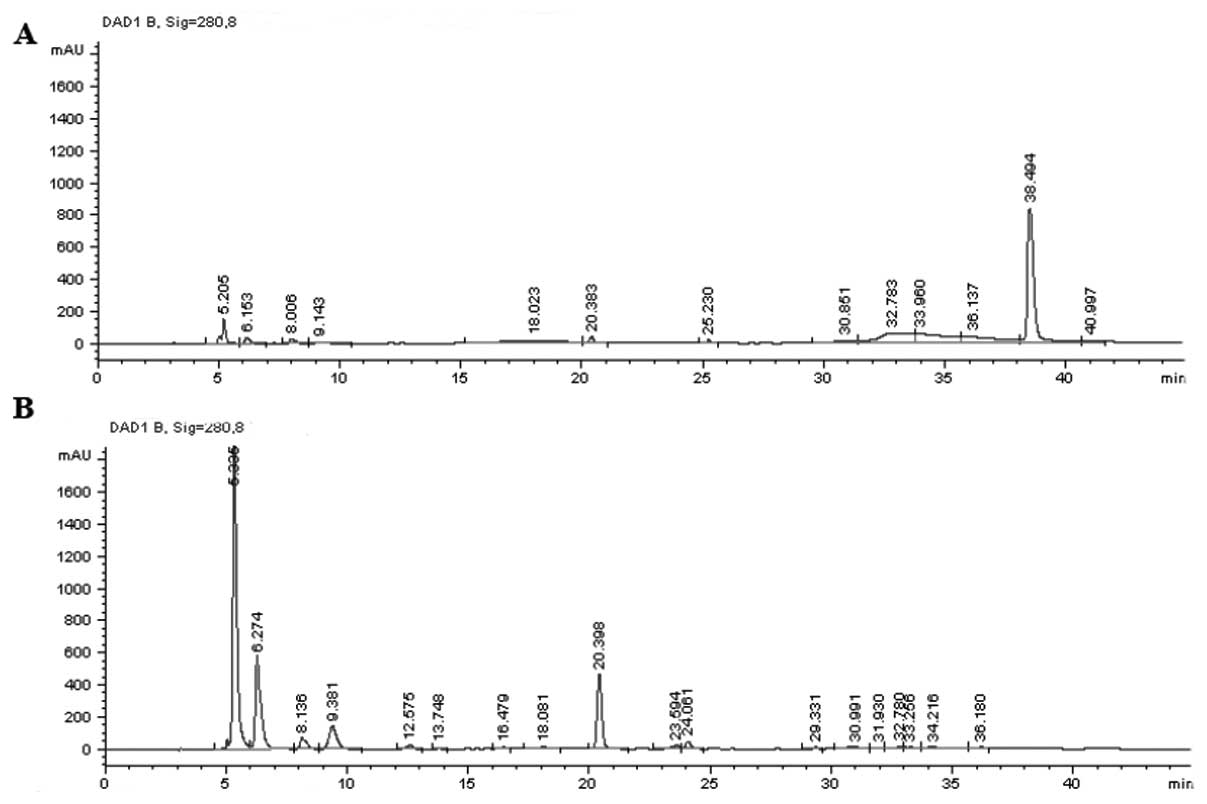
higher concentration of less polarity in whole blood (Fig. 3A). After precipitation, the less
polar parts could not be found but the most polar portion were
eluted (Fig. 3B).
Detection of apoptosis and cell cycle
distribution
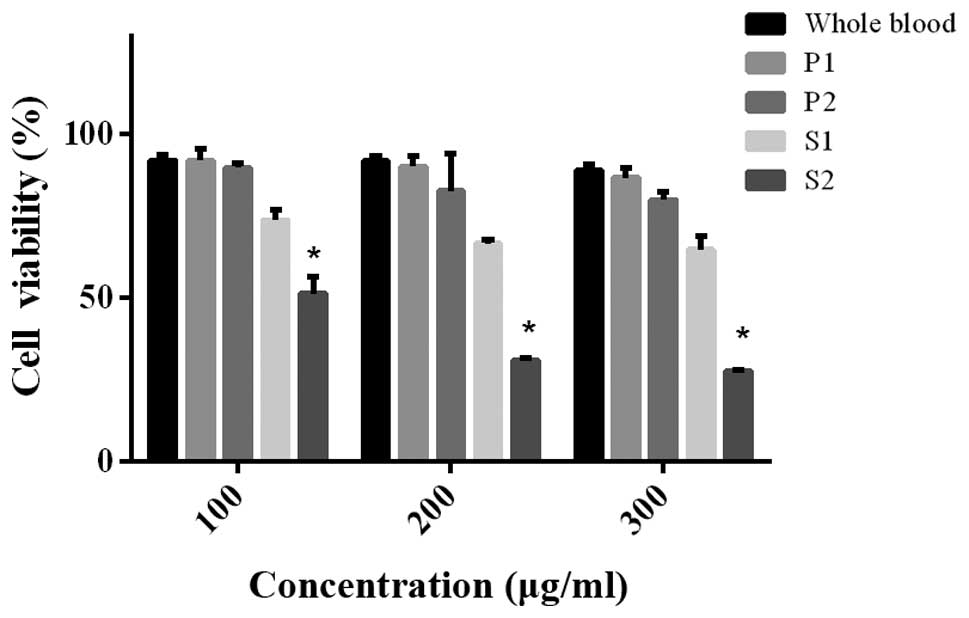
Cell viability showed no significant change after
treatment with residues including P1 and P2, but decreased after
treatment with S1 and S2. S2 displayed more significant efficacy
(Fig. 4). The whole blood extract
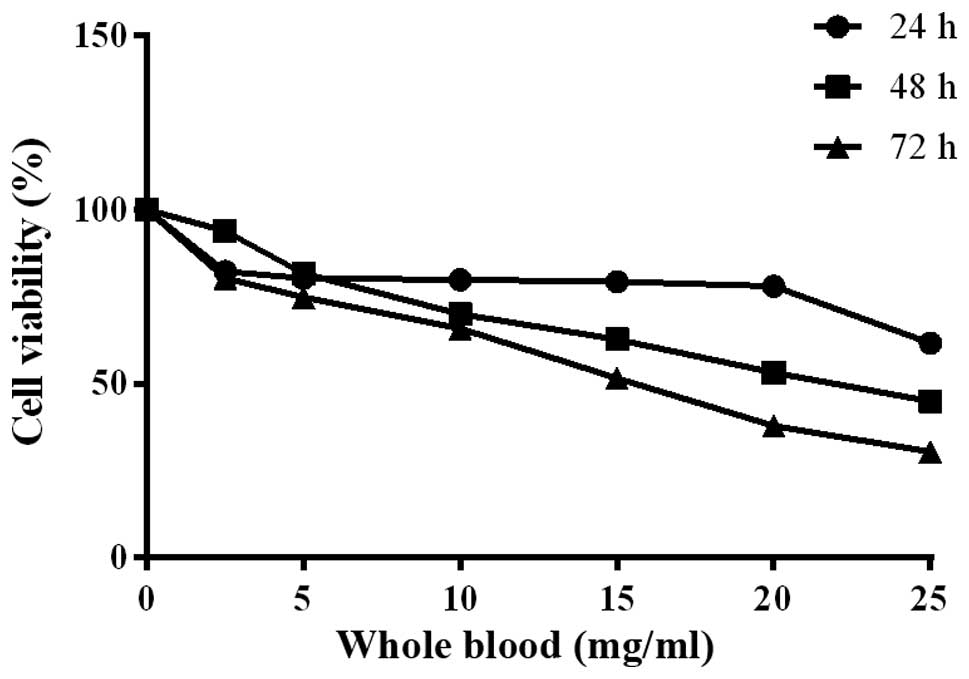
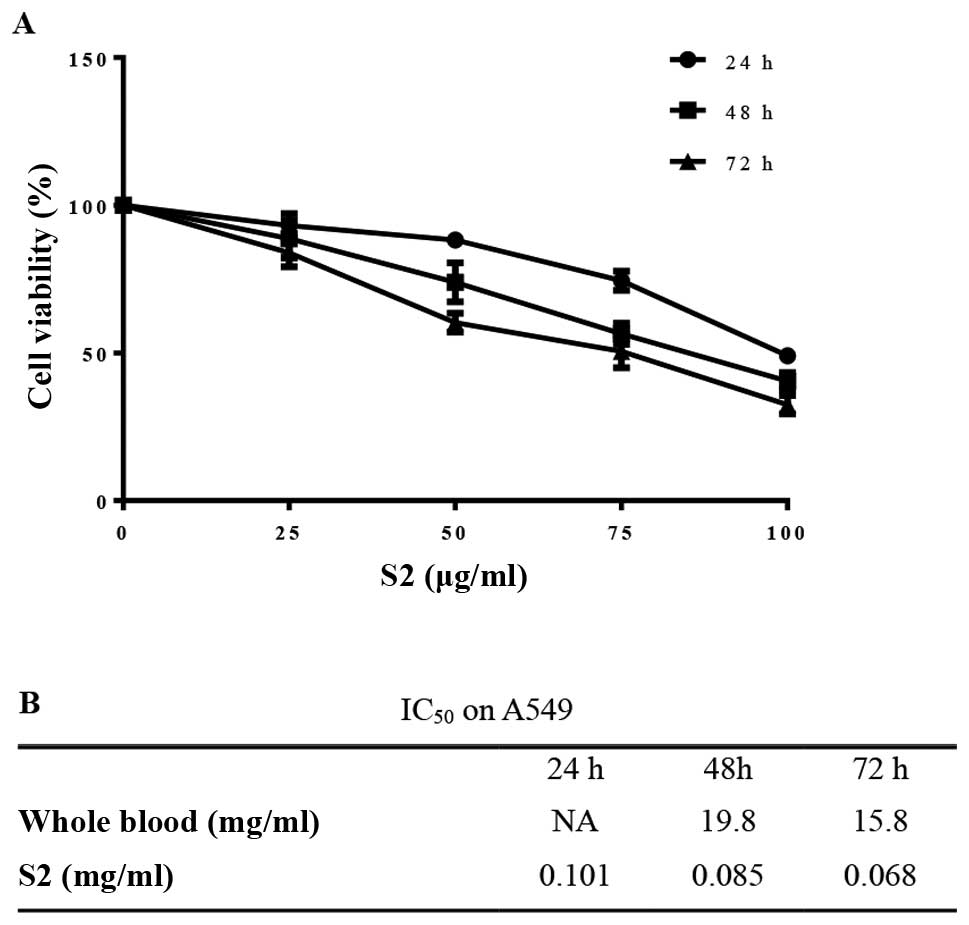
displayed inhibitory effects on cell viability (Fig. 5). Cell growth was prohibited in a
dose- and time-dependent manner after treatment with S2 (Fig. 6A) with a low IC50 value
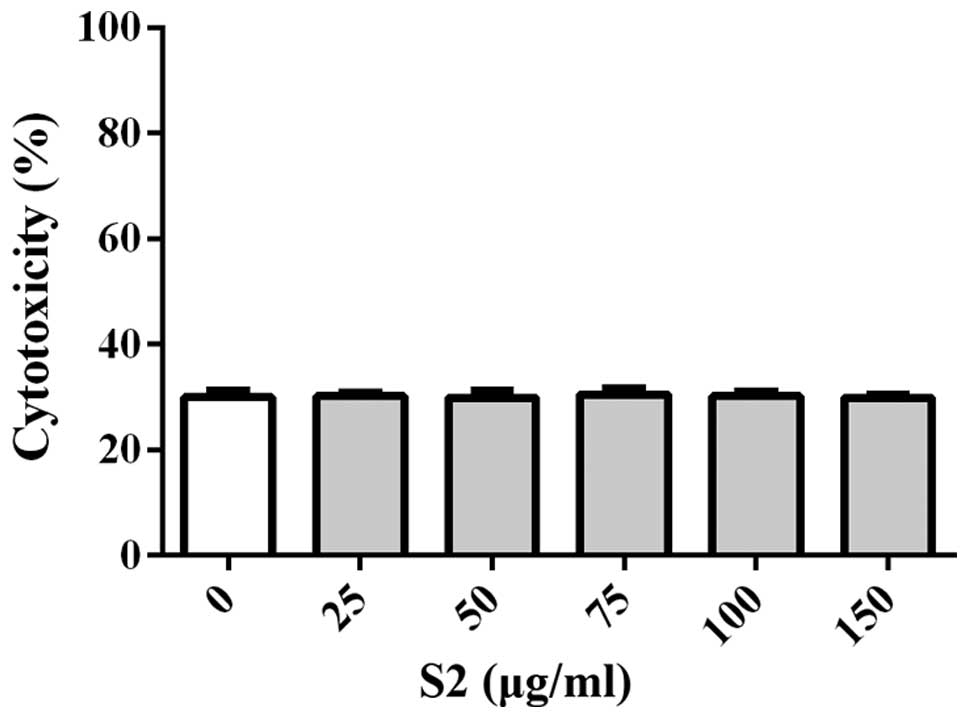
(Fig. 6B). Treatment with S2
revealed no cytotoxicity to cells concurrent with increasing
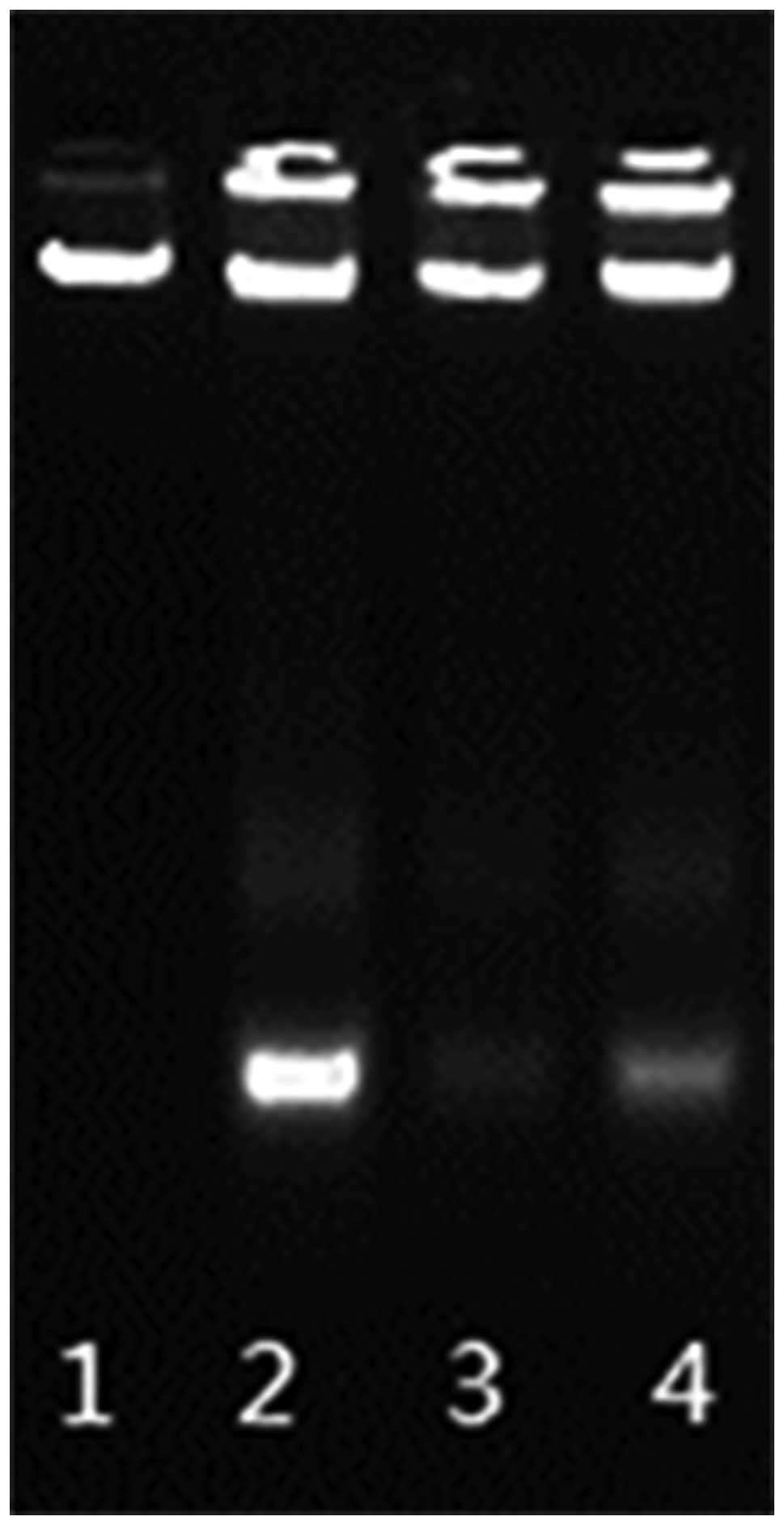
concentrations (Fig. 7). DNA ladder
was found in the early 24 h of treatment while fragments were not
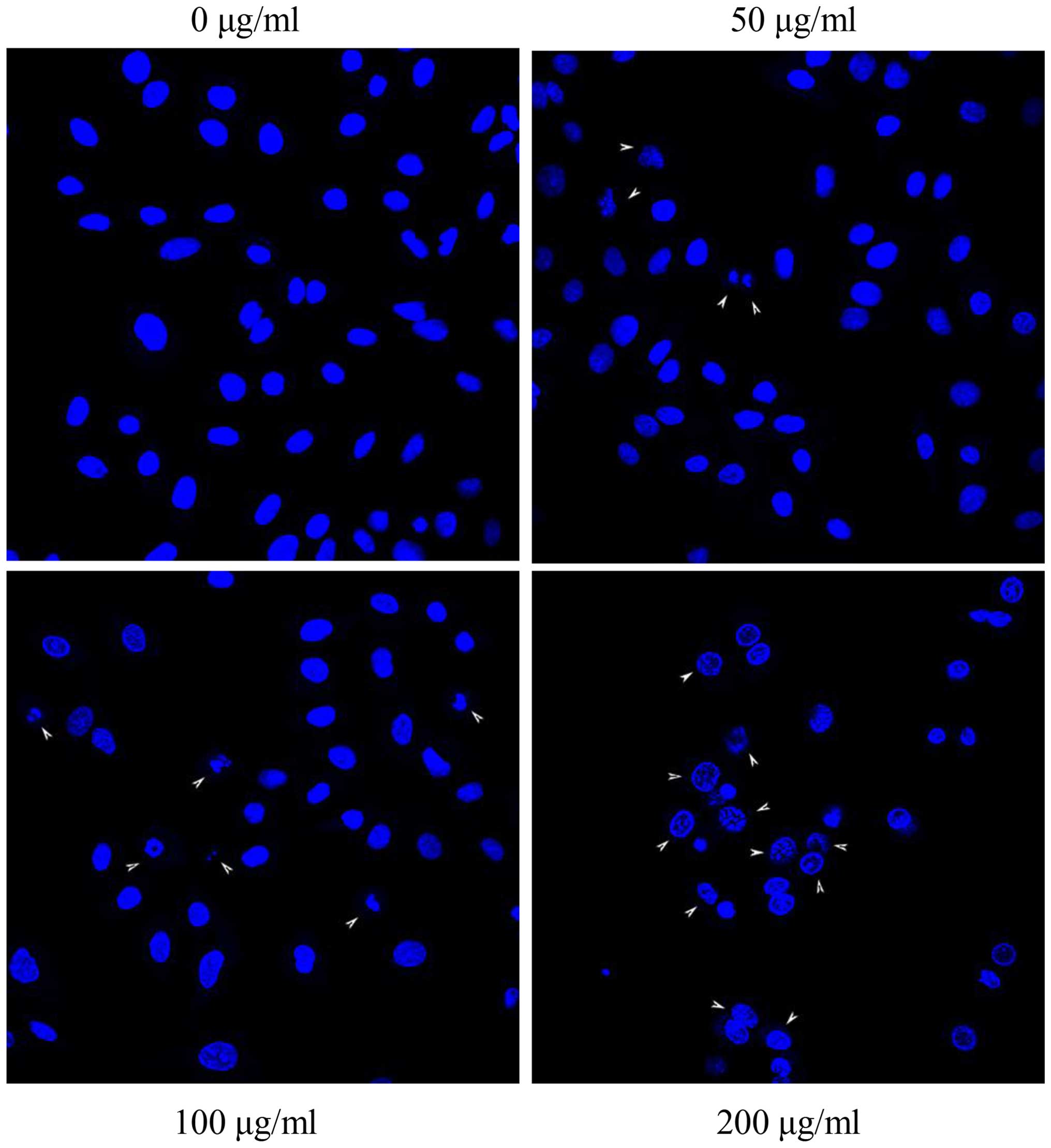
detected in the control under the same exposure time (Fig. 8). DNA condensations were observed
after treatment with S2 after 24 h (Fig. 9). There was a significant increase
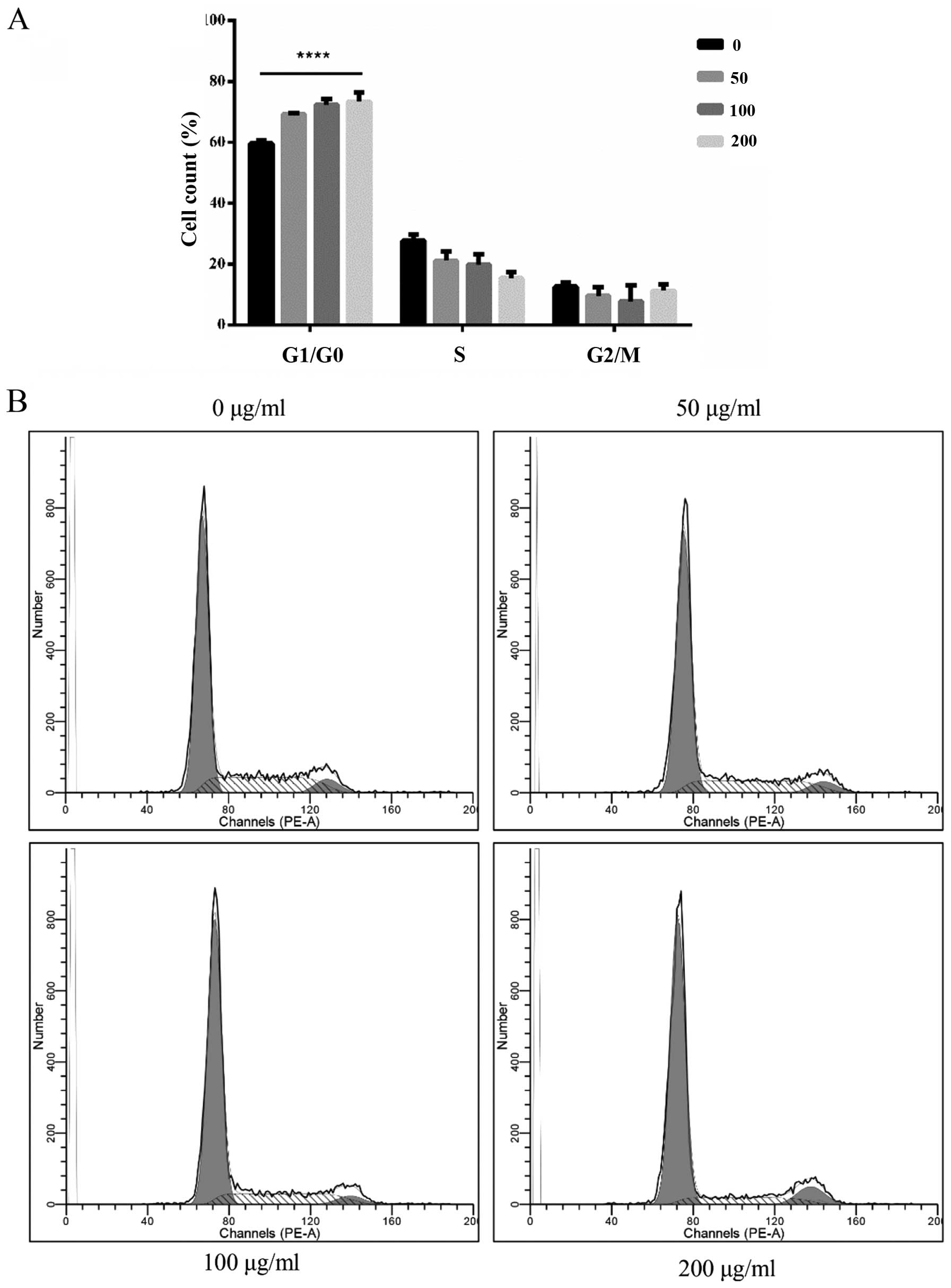
in the percentage of cells in the G0/G1 phase compared to that
noted in the control but a decreasing trend in the S phase, as well
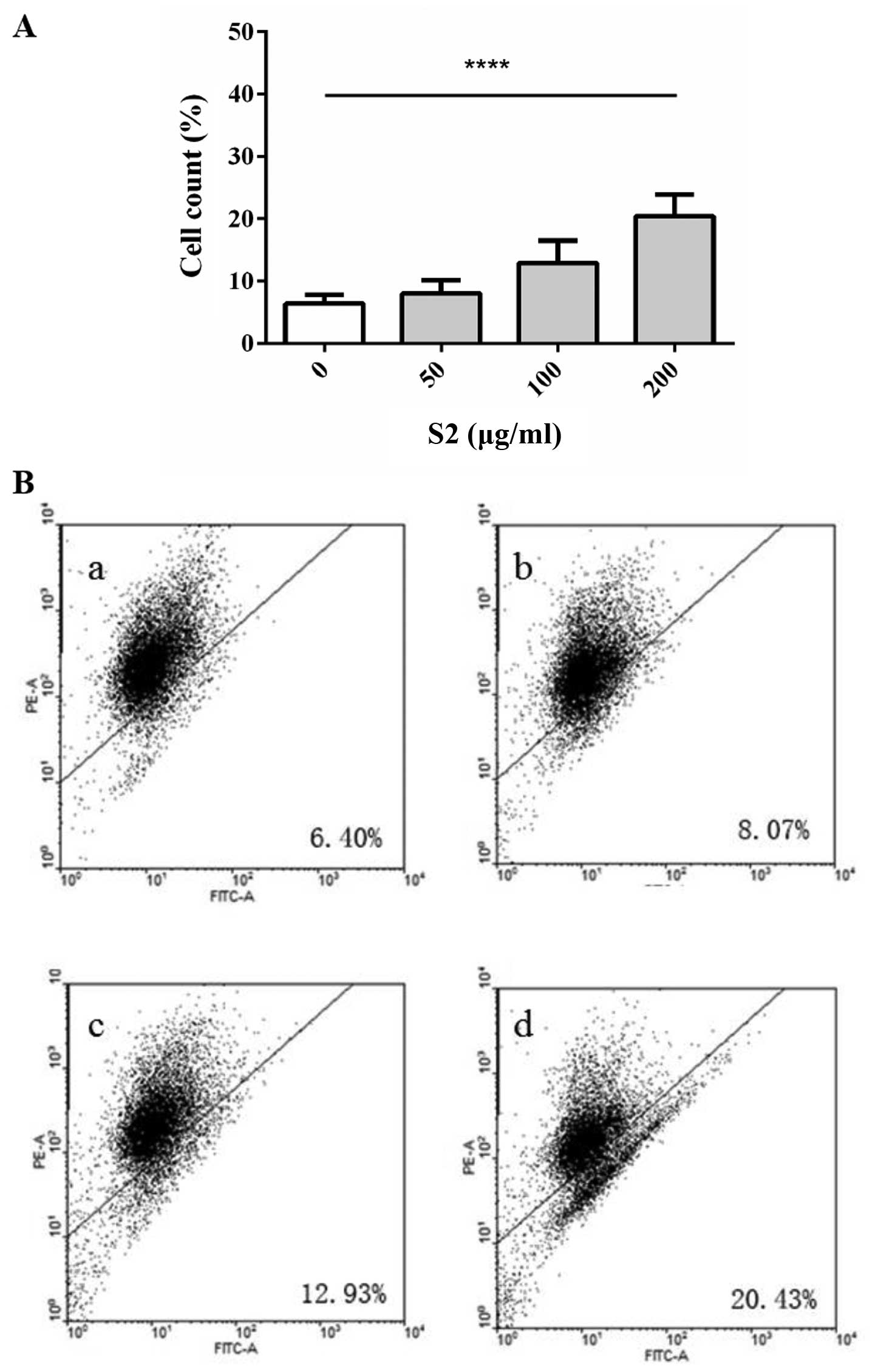
as the slight changes in the G2/M phase were not obvious (Fig. 10). The percentage of FITC-positive
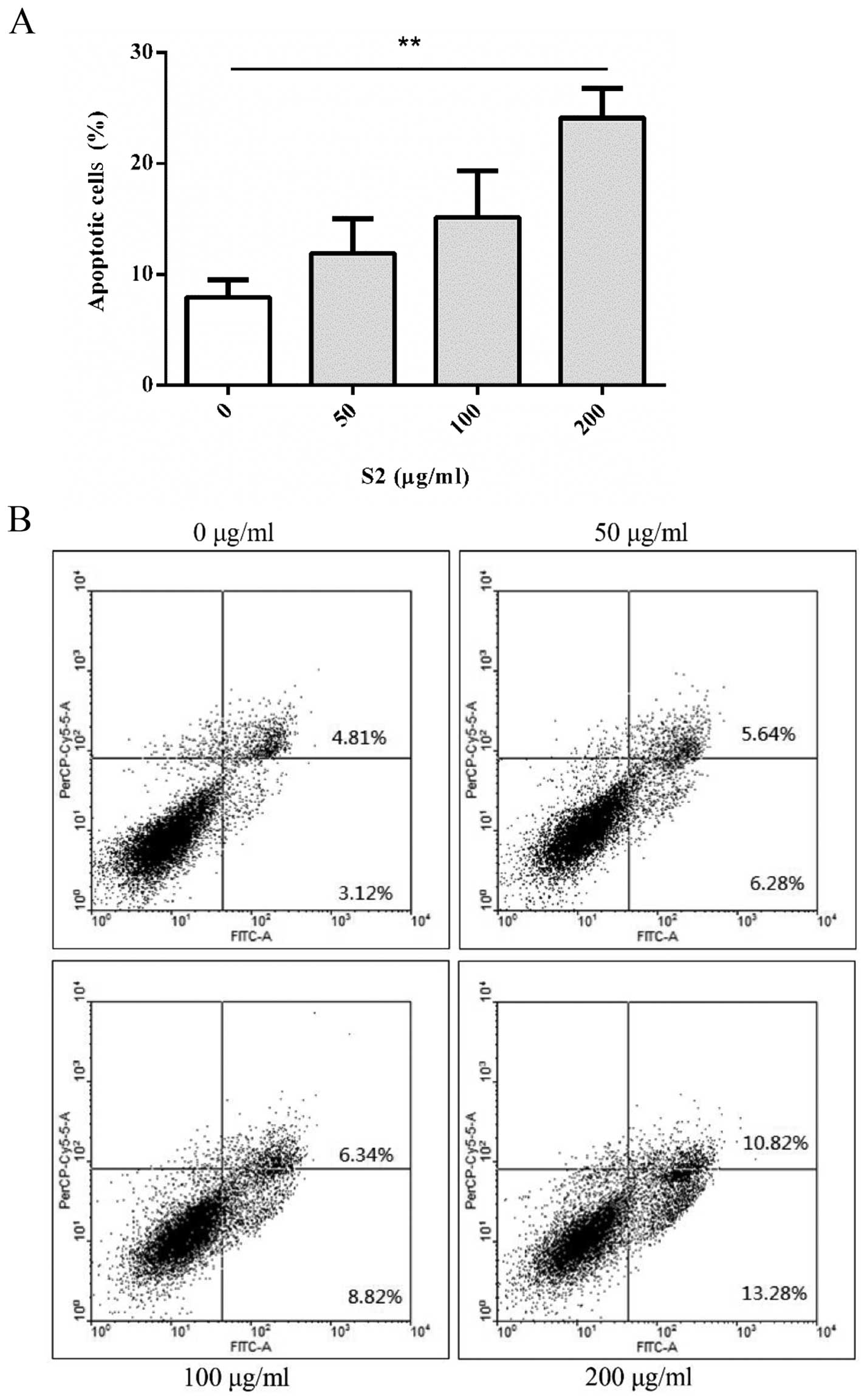
and double-positive cells showed an increasing trend with S2
concentrations (Fig. 11). The
appearance of ROS in the control group was not detected but the
amount and intensities of ROS increased (Fig. 12). After treatment with various
concentrations of S2, the cell numbers decreased with the increase
in S2 concentrations with a signal of higher intensity of FITC and
lower intensity of PE (Fig. 13B).
Moreover, at the higher concentration of S2, a more obvious change
was observed after 24 h (Fig.
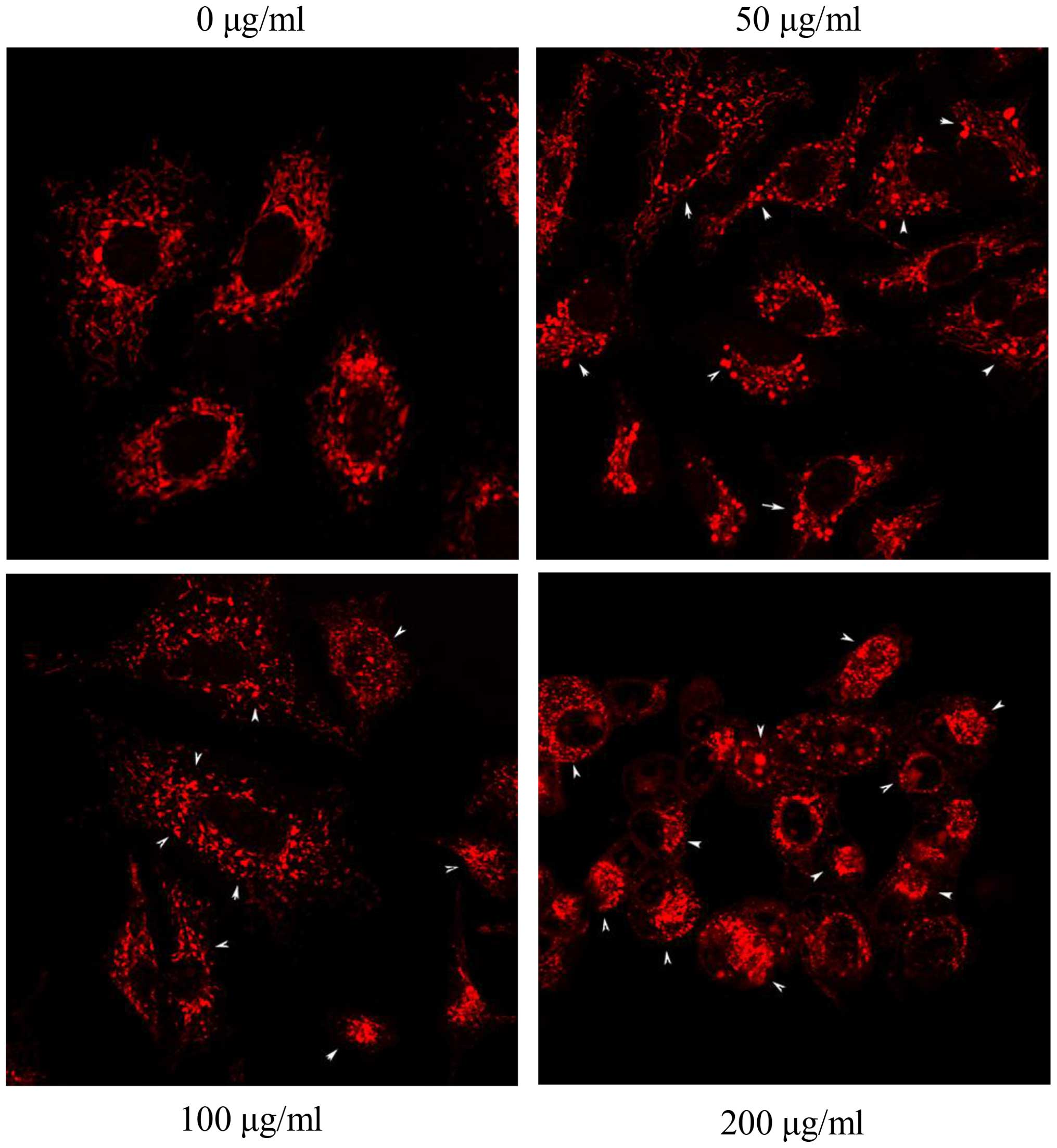
13A). Aggregations were found in mitochondria followed by
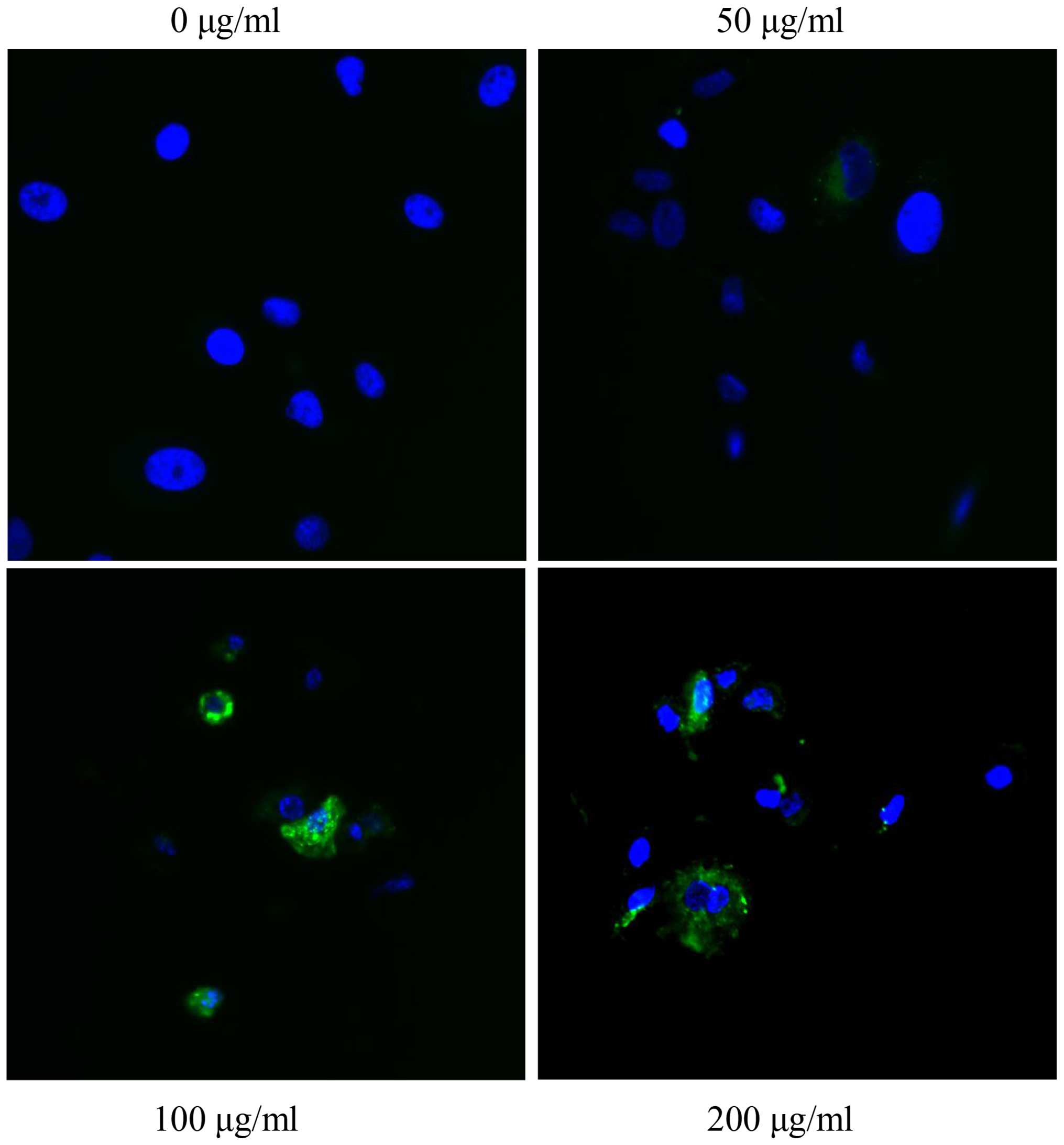
treatment with S2 (Fig. 14). The
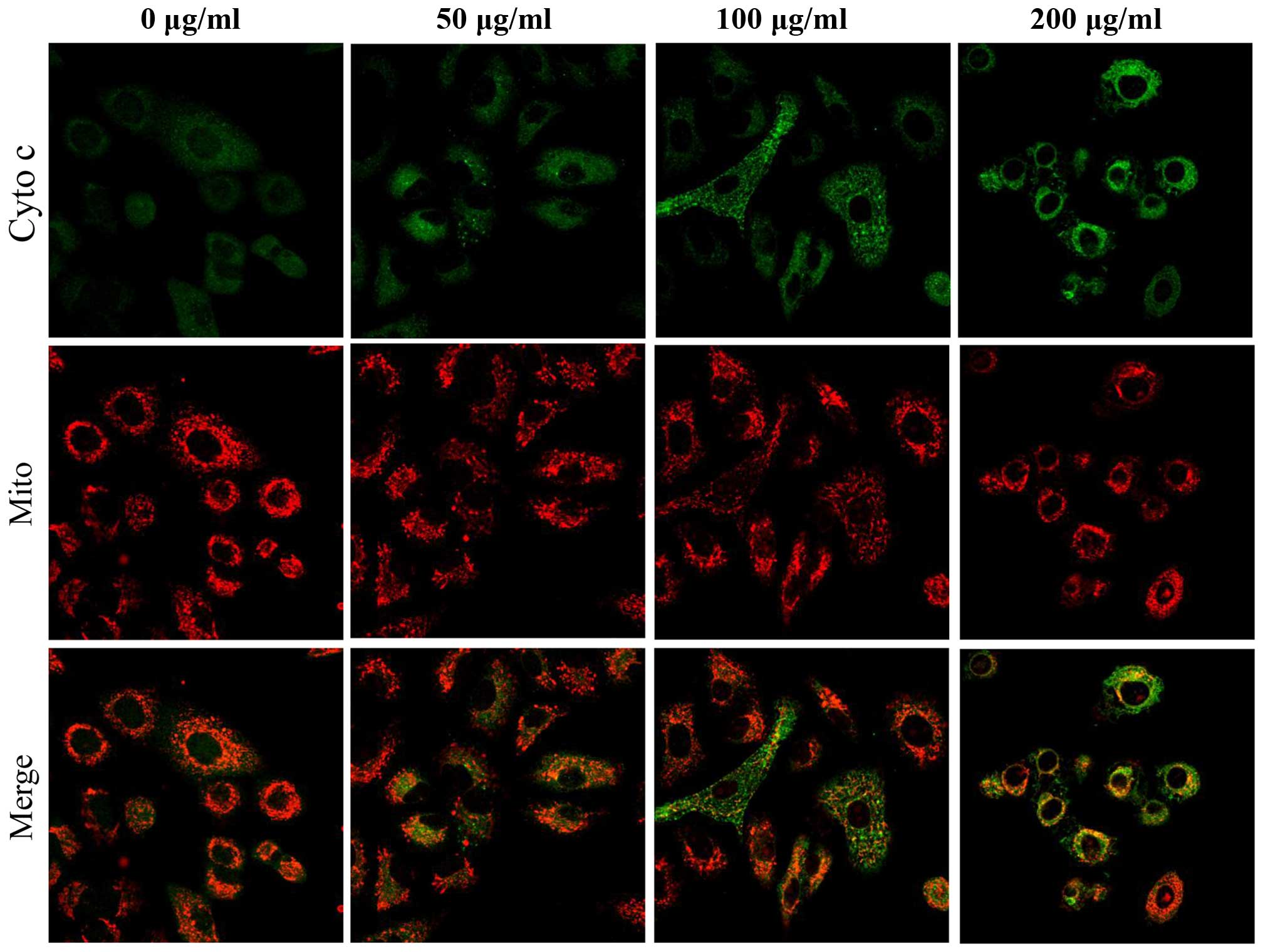
co-localization of green vs. red signal shifted after treatment
with S2 (Fig. 15). The fluorescent
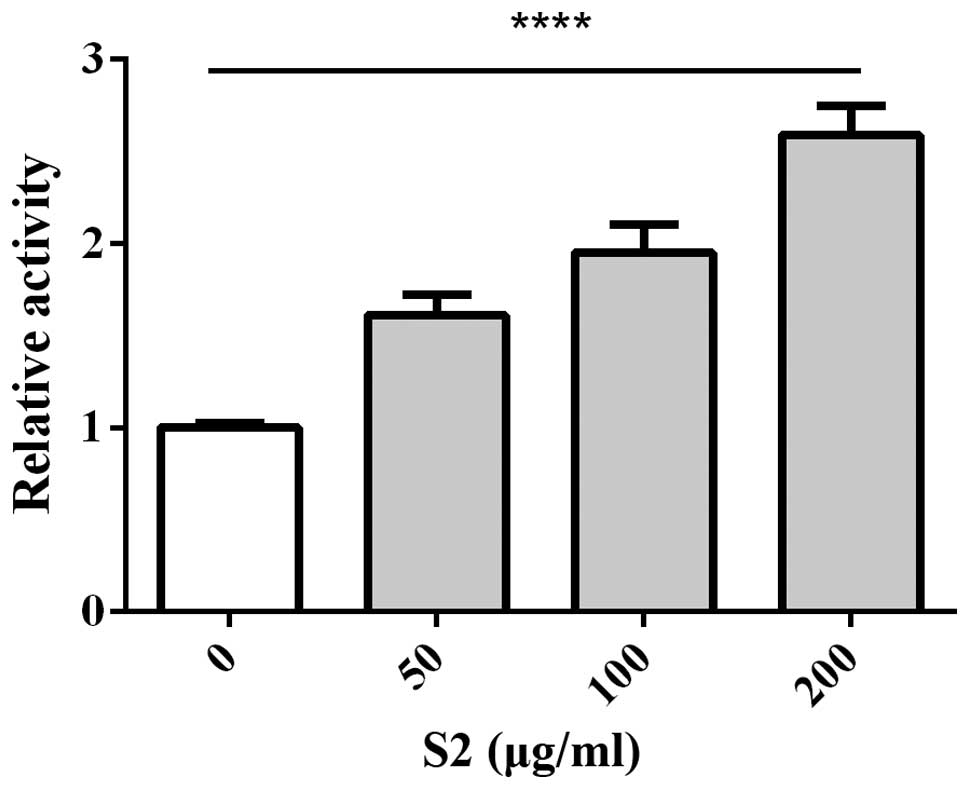
intensity of caspase-3/7 showed a significant increase (Fig. 16).
Expression levels of proteins
Following treatment with various concentrations of
S2, the expression level of Bcl-2 decreased and the trend became
significant when the S2 concentrations increased whereas expression
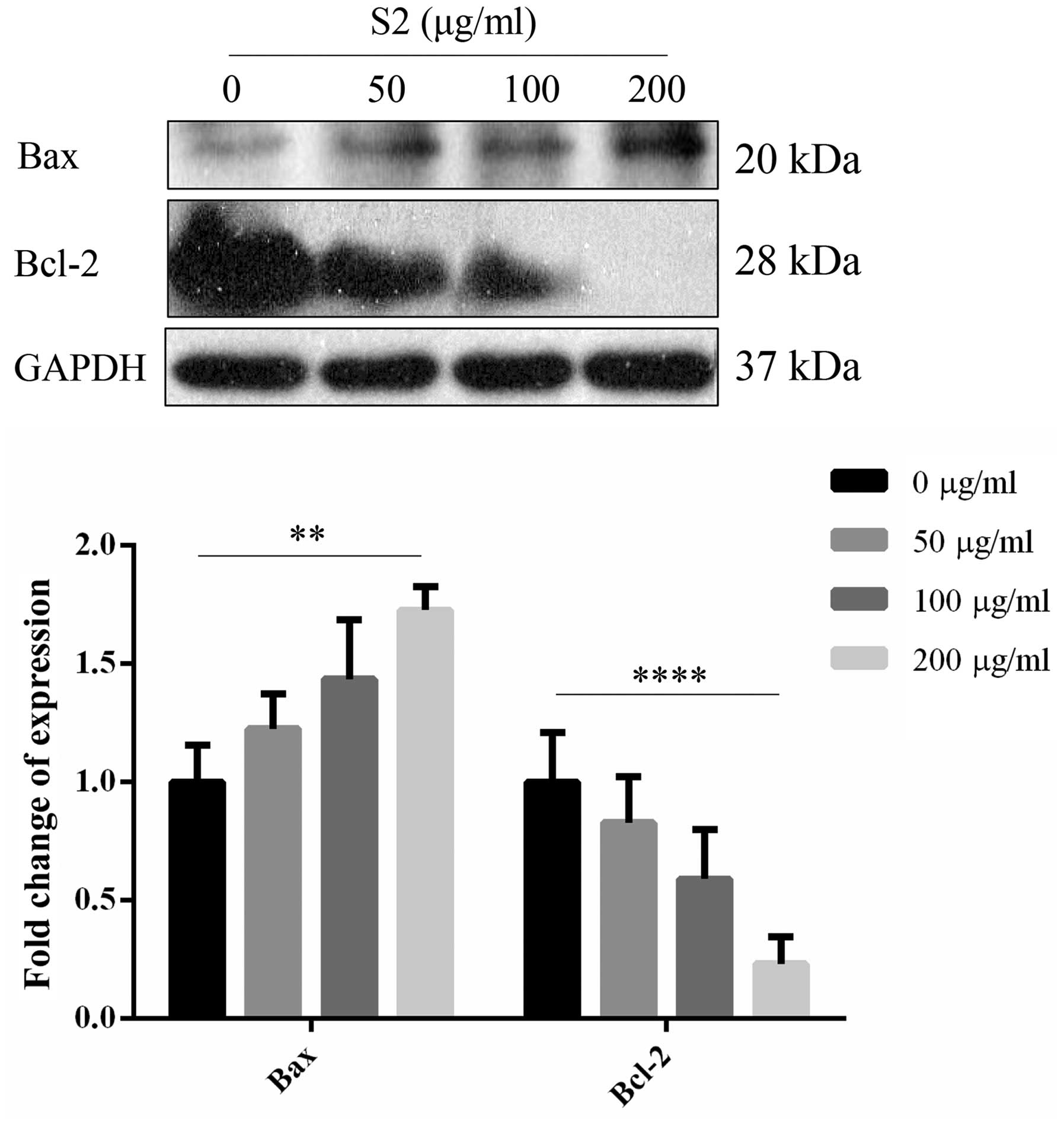
of Bax showed a total opposite trend. With increasing
concentrations, an upregulated expression level of Bax was noted
(Fig. 17). Expression levels of
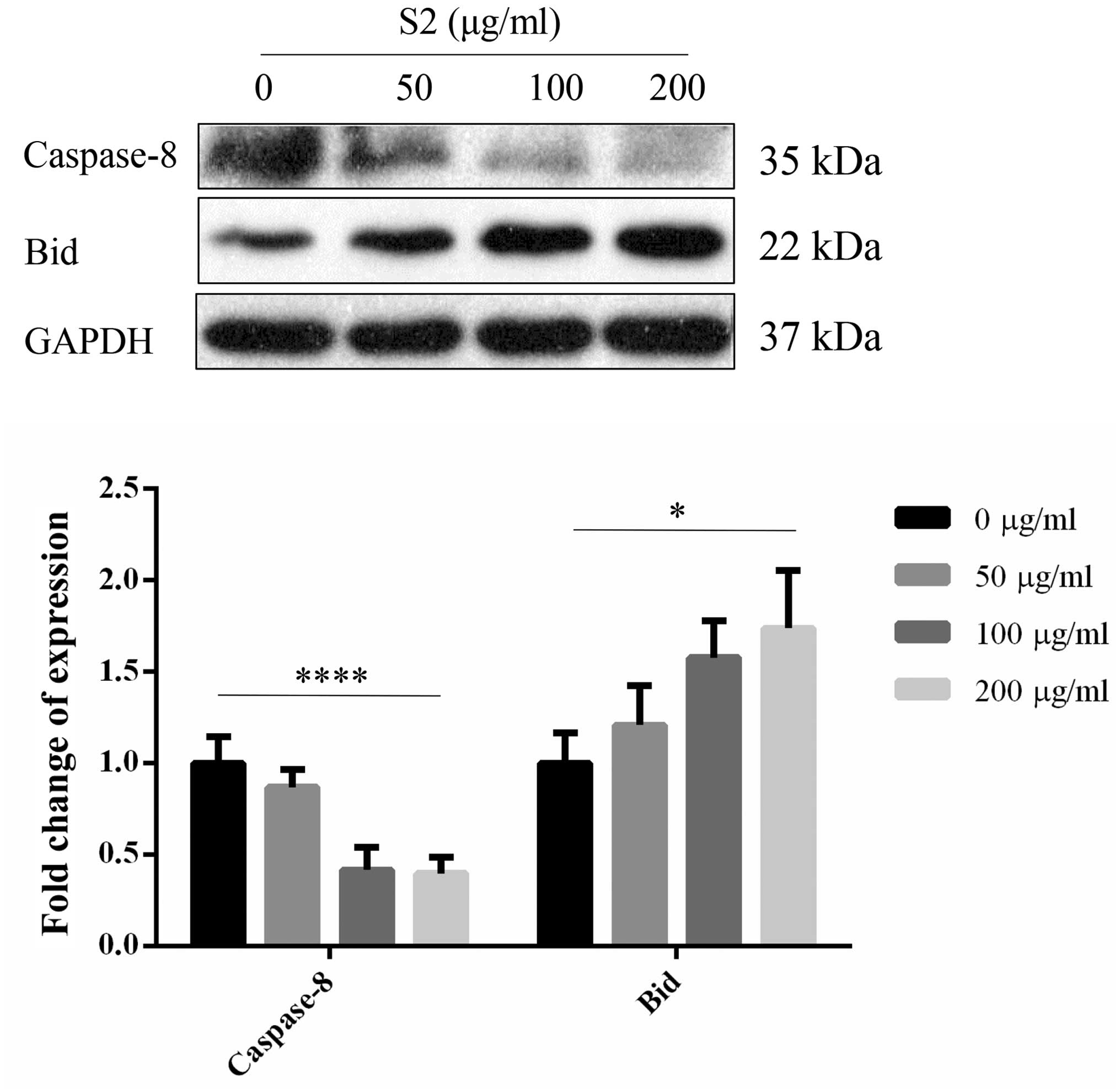
another protein in the Bcl-2 family, Bid, displayed an increasing
trend with a reverse change in caspase-8 (Fig. 18). The expression of caspase family
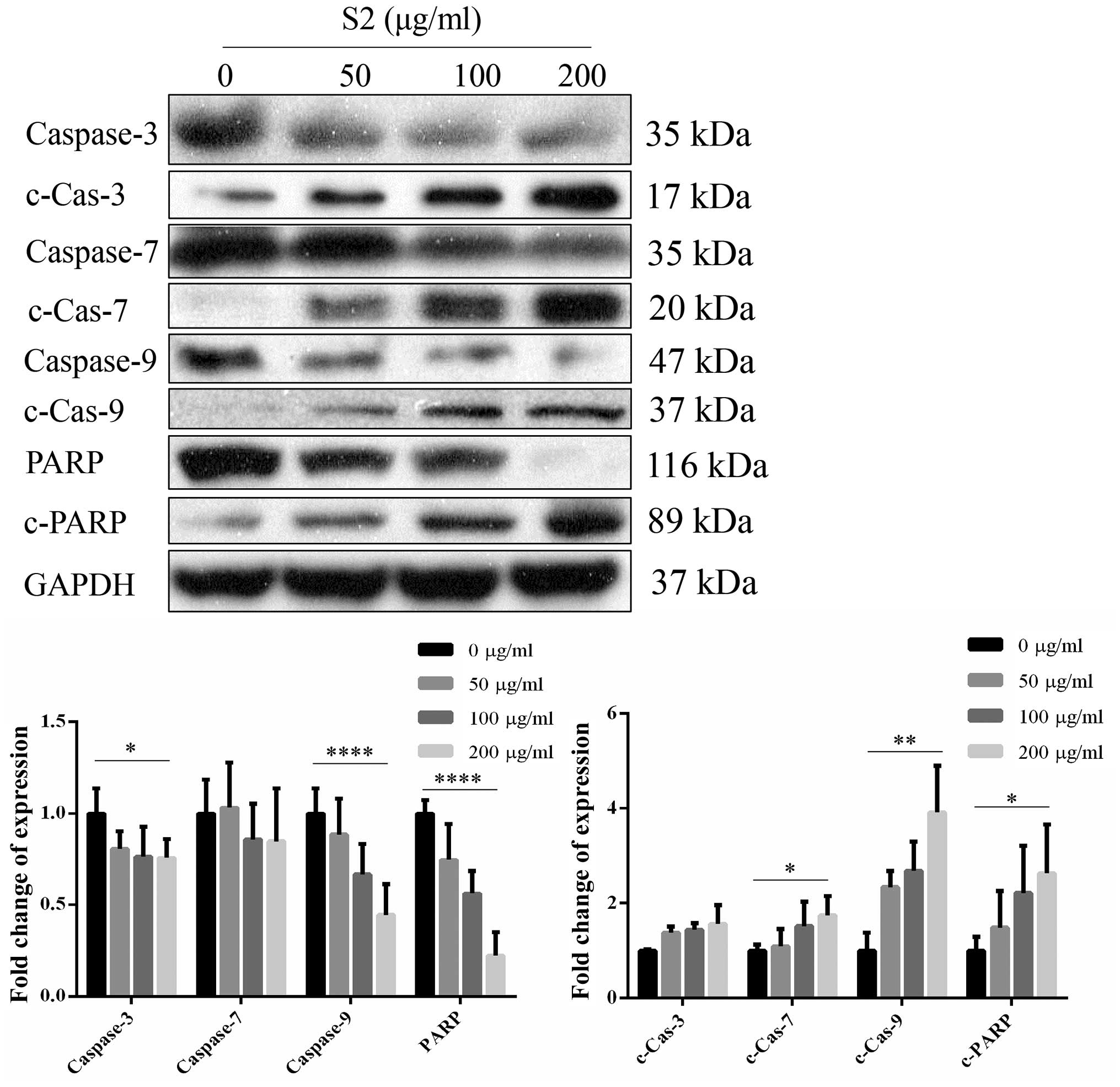
proteins showed similar changes, with downregulation of wild types
and upregulation of the cleaved forms. The expression of PARP
decreased together with the decreasing expression of cleaved PARP
(Fig. 19). The expression level of
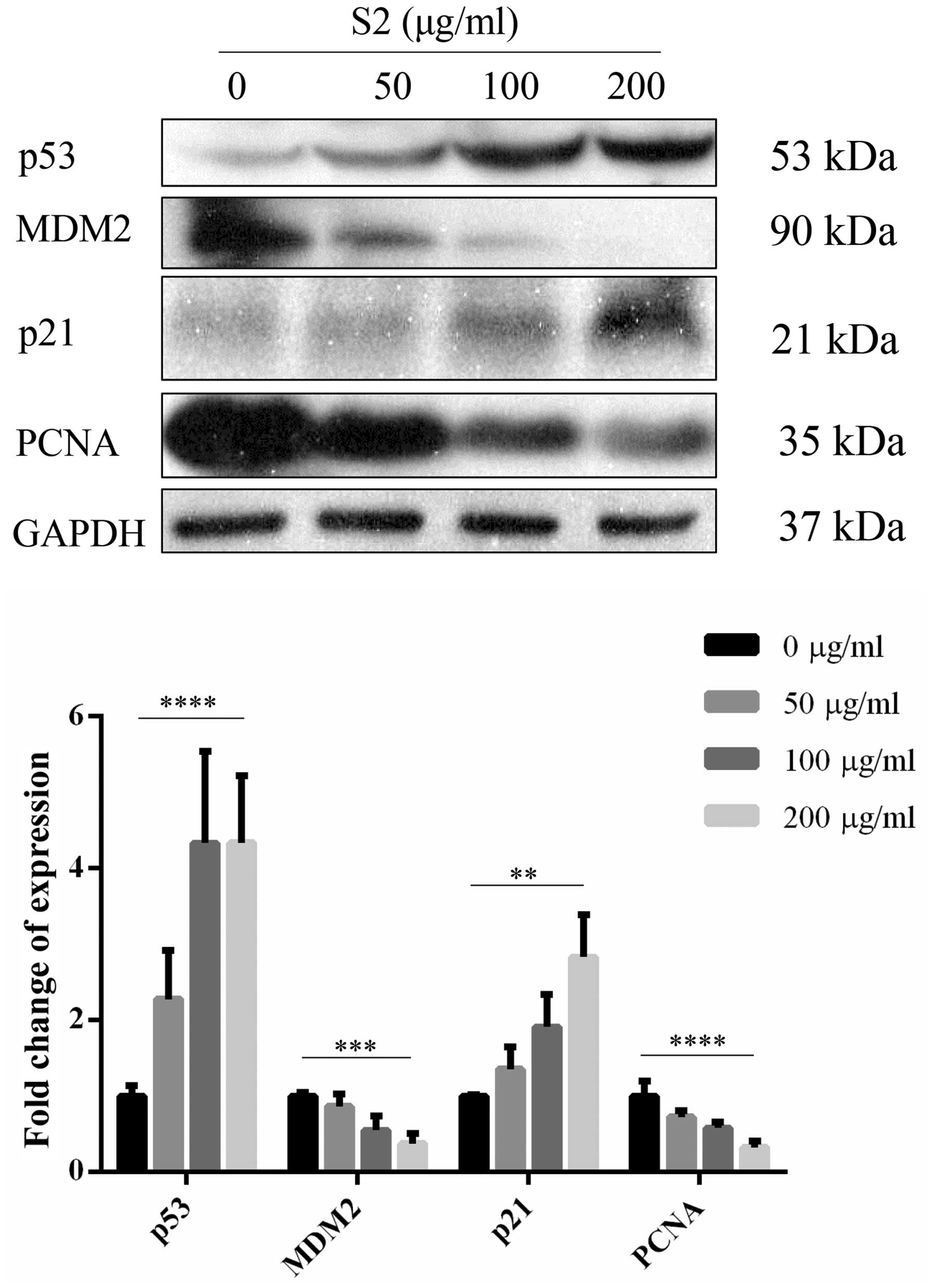
cell cycle-related proteins, MDM2 and PCNA, showed a decrease when
p21 and p53 were upregulated (Fig.
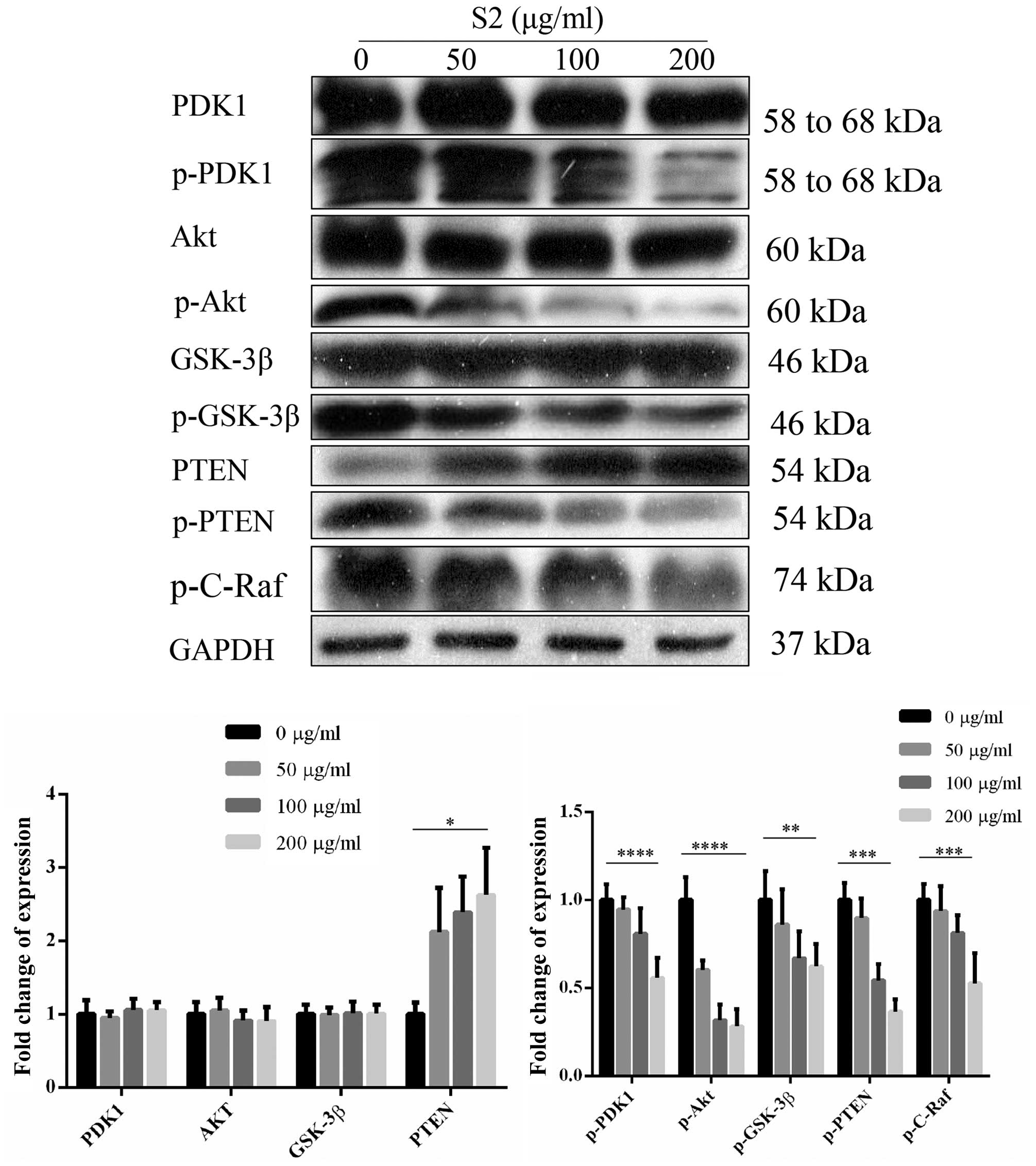
20). The expression of proteins associated with the PI3K/AKT
survival pathway, wild-type Akt, PDK1 and GSK-3β showed no
significant changes compared to the control. In contrast, the
phosphorylated forms of the above proteins and c-Raf were
decreased. Furthermore, wild-type PTEN showed an increasing trend
while phosphorylated PTEN exhibited a significant decrease
(Fig. 21).
Discussion
The active fractions of the alligator blood extract
were applied to study their effects on colorectal cancer cell
lines. The S2 fractions showed the highest effects on cell growth
inhibition which indicated that the S2 fractions contained the
active components. The HPLC profile suggested that the active
components might remain in the polar parts but could not be further
separated. When cells were at the last stage of apoptosis, DNA
condensation and cleavage of certain substrates occurred and led to
DNA fragmentation (7). S2 induced
DNA fragmentation with apoptosis (8). S2 induced cell cycle arrest in the
G0/G1 phase. Several proteins associated with cell cycle arrest
were detected. MDM2 is a negative regulator of p53 protein. Through
binding to p53, MDM2 exerts its function in facilitating the
degradation of p53, which subsequently prohibits cell cycle arrest
(9). Downregulation of MDM2 enables
the implementation of p53 in regulating cell cycle progression and
DNA repair (10). A decrease in
PCNA promotes the expression of p21 (11). A previous study reported that even
when cells were undergoing cell cycle arrest, they stop
duplication, but cell death may not occur (12). In the present study, we observed
that S2 induced not only cell cycle arrest, but apoptosis. S2
induced the generation and increase of ROS, which facilitated the
rupture of cells (13). The
increase in ROS caused disturbance in the mitochondrial membrane
potential (14). At higher
concentrations of S2, the change in mitochondrial membrane
permeability reinforced the changes in the mitochondria. The
mitochondrial respiratory activity and the membrane potential were
found to be modulated while the collapse in the permeability was
observed in early apoptosis (15).
It is believed that mitochondrial aggregation is an up-stream
event. The release of cytochrome c plays a critical role in
mediating the apoptotic intrinsic pathway (16). Cytochrome c was found to be
released from the mitochondria which resulted from changes in the
mitochondria such as aggregation, an increase in ROS and Bcl-2,
along with a decrease in membrane potential and Bax. Cytochrome
c that binds to pro-caspase-9 forms the apoptosome causing a
decreasing expression level of pro-caspase-9 and the increasing
level of its activated form (16).
Downregulation of caspase-8 and upregulation of Bid reveal
activation of the extrinsic pathway. The activation of caspase-9 or
the activation of caspase-8 triggers the caspase cascade. Active
caspase family proteins cleave certain key substrates and the cells
are digested through apoptosis (17). The inactivation of PARP transforms
cell death mechanism from necrosis to apoptosis by protecting the
integrity of NAD+ and ATP formation (18). If the cleavage of PARP is blocked,
necrosis presents as the dominating form of cell death resulting
from the continued activation of PARP (19). To study the linkage between cell
cycle arrest and apoptosis, the PI3K/AKT pathway was examined. PDK1
and AKT showed no obvious changes whereas the phosphorylated forms
showed a decreasing trend indicating that inhibition in the initial
AKT pathway was via reduction of the active phosphorylated form of
the target proteins. The downstream substrate of active AKT,
GSK-3β, inhibits the activities of p21 and cyclin D1; therefore it
enables cells to undergo cell cycle arrest (20). The phosphorylation by AKT resulted
in the inhibitory effect on GSK-3β that led to the dysfunction in
the regulation. Furthermore, the inhibition of phosphorylated
GSK-3β caused the upregulation of p21 to promote cell cycle arrest.
Another substrate of active AKT, MDM2, was downregulated resulting
in the upregulation of p53 and subsequently inhibited the
activities of Bcl-2 (21). The
upregulated expression level of p53 caused cell cycle arrest, but
also led to apoptosis (22). The
increased expression of active PTEN and the decreased expression of
its inactive form, phosphorylated PTEN indicated inhibitory effects
on the PI3K/AKT pathway. Change in PTEN activity affects cell cycle
progression and apoptosis of cancer cells (23). S2 induced cell cycle arrest and
apoptosis through the intrinsic and extrinsic pathways of
apoptosis. By modulating the activity of PTEN, S2 inhibited the
expression of the PI3K/AKT survival pathway resulting in not only
cell cycle arrest but also apoptosis.
Acknowledgments
This study was supported in part by Mr. Terry Wong
of HTL Holdings Ltd.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hirsch FR, Spreafico A, Novello S, Wood
MD, Simms L and Papotti M: The prognostic and predictive role of
histology in advanced non-small cell lung cancer: A literature
review. J Thorac Oncol. 3:1468–1481. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Crinò L, Weder W, van Meerbeeck J and
Felip E; ESMO Guidelines Working Group: Early stage and locally
advanced (non-metastatic) non-small-cell lung cancer: ESMO Clinical
Practice Guidelines for diagnosis, treatment and follow-up. Ann
Oncol. 21(Suppl 5): v103–v115. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pata S, Yaraksa N, Daduang S, Temsiripong
Y, Svasti J, Araki T and Thammasirirak S: Characterization of the
novel antibacterial peptide Leucrocin from crocodile (Crocodylus
siamensis) white blood cell extracts. Dev Comp Immunol. 35:545–553.
2011. View Article : Google Scholar
|
|
5
|
Phosri S, Mahakunakorn P, Lueangsakulthai
J, Jangpromma N, Swatsitang P, Daduang S, Dhiravisit A and
Thammasirirak S: An investigation of antioxidant and
anti-inflammatory activities from blood components of crocodile
(Crocodylus siamensis). Protein J. 33:484–492. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alley MC, Scudiero DA, Monks A, Hursey ML,
Czerwinski MJ, Fine DL, Abbott BJ, Mayo JG, Shoemaker RH and Boyd
MR: Feasibility of drug screening with panels of human tumor cell
lines using a microculture tetrazolium assay. Cancer Res.
48:589–601. 1988.PubMed/NCBI
|
|
7
|
Kressel M and Groscurth P: Distinction of
apoptotic and necrotic cell death by in situ labelling of
fragmented DNA. Cell Tissue Res. 278:549–556. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Abraham RT: Cell cycle checkpoint
signaling through the ATM and ATR kinases. Genes Dev. 15:2177–2196.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Waga S, Hannon GJ, Beach D and Stillman B:
The p21 inhibitor of cyclin-dependent kinases controls DNA
replication by interaction with PCNA. Nature. 369:574–578. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bellamy CO, Clarke AR, Wyllie AH and
Harrison DJ: p53 Deficiency in liver reduces local control of
survival and proliferation, but does not affect apoptosis after DNA
damage. FASEB J. 11:591–599. 1997.PubMed/NCBI
|
|
12
|
Green DR: Apoptotic pathways: Ten minutes
to dead. Cell. 121:671–674. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cui H, Kong Y and Zhang H: Oxidative
stress, mitochondrial dysfunction, and aging. J Signal Transduct.
2012:6463542012. View Article : Google Scholar
|
|
14
|
Aon MA, Cortassa S and O'Rourke B:
Mitochondrial oscillations in physiology and pathophysiology. Adv
Exp Med Biol. 641:98–117. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Haga N, Fujita N and Tsuruo T:
Mitochondrial aggregation precedes cytochrome c release from
mitochondria during apoptosis. Oncogene. 22:5579–5585. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Denecker G, Vercammen D, Declercq W and
Vandenabeele P: Apoptotic and necrotic cell death induced by death
domain receptors. Cell Mol Life Sci. 58:356–370. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fuertes MA, Castilla J, Alonso C and Pérez
JM: Cisplatin biochemical mechanism of action: From cytotoxicity to
induction of cell death through interconnections between apoptotic
and necrotic pathways. Curr Med Chem. 10:257–266. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang W, Zhang Y, Li Y, Wu Z and Zhu D:
Myostatin induces cyclin D1 degradation to cause cell cycle arrest
through a phosphatidylinositol 3-kinase/AKT/GSK-3 β pathway and is
antagonized by insulin-like growth factor 1. J Biol Chem.
282:3799–3808. 2007. View Article : Google Scholar
|
|
20
|
Vousden KH and Ryan KM: p53 and
metabolism. Nat Rev Cancer. 9:691–700. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Finlan LE and Hupp TR: The life cycle of
p53: A key target in drug development. Apoptosis Pathways as
Targets for Novel Therapies in Cancer and Other Diseases. Los M and
Gibson SB: Springer; New York, NY, USA: pp. 157–172. 2005,
View Article : Google Scholar
|
|
22
|
Yan X, Fraser M, Qiu Q and Tsang BK:
Overexpression of PTEN sensitizes human ovarian cancer cells to
cisplatin-induced apoptosis in a p53-dependent manner. Gynecol
Oncol. 102:348–355. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Moghaddam SJ, Li H, Cho SN, Dishop MK,
Wistuba II, Ji L, Kurie JM, Dickey BF and Demayo FJ: Promotion of
lung carcinogenesis by chronic obstructive pulmonary disease-like
airway inflammation in a K-ras-induced mouse model. Am J Respir
Cell Mol Biol. 40:443–453. 2009. View Article : Google Scholar
|