Introduction
Hepatocellular carcinoma (HCC) is one of the most
common malignant tumors worldwide; this disease ranks fifth in
global morbidity and is the third primary cause of cancer-related
mortality (1). Approximately
700,000 new liver cancer cases and deaths are reported annually
worldwide, half of which occur in China (2). HCC accounts for 70–85% of total liver
cancer cases (3). Most patients
with HCC are already in middle and advanced stages when properly
diagnosed because of lack of effective diagnostic approaches;
although HCC could be treated by excision, therapeutic approaches
for this disease are limited, resulting in poor prognosis. Hence,
novel specific biomarkers for early diagnosis and prognosis
evaluation of HCC must be developed.
Human DEK is a highly abundant chromatin
architectural protein (4–7). This protein was initially found in the
DEK-CAN fusion protein in acute myeloid leukemia (AML) (8,9). Many
studies showed that the DEK protein significantly influences cell
cycle and participates in multiple cell functions, such as
maintaining the integrity of heterochromatin (10), transcriptional regulation (11), mRNA splicing (12), DNA replication (13) and damage repair and susceptibility
(14). The DEK protein is also
related to cell apoptosis (15,16).
The relationship between DEK and tumor has been increasingly
investigated. Studies showed that DEK is overexpressed in multiple
malignant human tumors, such as bladder (17), colorectal (18) and gastric cancer (19). In particular, DEK is upregulated in
primary HCCs compared with that in matched nonmalignant liver
tissues. Thus, DEK is related to poor prognosis and could be used
as an independent tumor biomarker for predicting the prognosis of
tumors. DEK is also correlated with the occurrence and
development of multiple malignant human tumors. However, only few
studies reported the overexpression regulatory mechanism of
DEK in tumors, particularly the transcriptional regulatory
mechanism of DEK in HCC.
DNA methylation has been extensively studied in
epigenetics. Researchers reported the presence of abnormal DNA
methylation during the formation and development of multiple
tumors. DNA methylation occurs when the methyl group
(−CH3) is transferred from S-adenosylmethionine to the
5′ cytosine of the DNA sequence under the catalysis of DNA
methyltransferases (20).
Methylation in the genome of mammals mainly occurs at the CpG site.
The CpG sequence is not uniformly distributed in the genome; the
sequence appears less frequently in most regions than other
dinucleotide sequences but is more frequent in some regions. A CpG
island is a region with high cytosine and guanine contents,
sequence length >200–500 bp, GC content >50% and CpG
proportion >0.6 (21). The CpG
island mainly exists in the gene promoter region and near the first
exon region. Among normal human genomes, the CpG island in genes
frequently expressed in cells, such as housekeeping genes, is under
non-methylated state. Moreover, inactive genes are normally under
high-methylated state, which inhibits their expression. CpG sites
on non-CpG islands are under methylated state, which could be
preserved stably through cell division (22).
CpG island in tumor cells manifests methylation
abnormality. CpG island methylation in promoter regions is related
to transcriptional silencing; conversely, CpG site methylation
beyond the promoter region is called genosome methylation, which is
related to transcriptional activation. Low methylation levels of
the entire genome and high methylation levels of the CpG island in
a local region occur simultaneously during tumor formation
(23). This abnormal methylation is
mainly presented as follows. High methylation levels in the
promoter region results in the silencing of cancer suppressor gene,
whereas low methylation levels of some oncogenes results in the
activation of gene transcription. Given that DEK is a
candidate biomarker for tumor diagnosis, the relationship between
its overexpression and promoter methylation level, as well as
transcriptional regulation of the DEK gene, requires further
study to elucidate the regulatory mechanism of DEK
overexpression in HCC. The present study aims to provide additional
basis for conducting DEK research on HCC treatment.
Materials and methods
Cell culture
Human HCC cell lines and normal hepatic L02 cells
were obtained from the American Type Culture Collection and the
Chinese Academy of Sciences, respectively. The cells were cultured
in Dulbecco's modified Eagle's medium (DMED; Gibco-Invitrogen,
Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS) in an
atmosphere of 5% CO2 at 37°C. Normal hepatic tissues
were supplied by the PLA General Hospital after receiving informed
consent from the patients and obtaining the approval of hospital
authorities. The tissues were stored in liquid nitrogen. The
institutional ethics committee approved this study.
Western blot analysis
Cells were harvested by trypsinization, counted, and
ultrasonically lysed with 200 μl of 1X sodium dodecyl
sulfate (SDS) loading buffer per 1×106 cells. Protein
samples were prepared and loaded at 20 μl per lane for
SDS-PAGE. The samples were electrotransferred onto nitrocellulose
filter membrane, and non-specific binding was blocked in
Tris-buffered saline with Tween buffer containing 5% non-fat dried
milk (Yili). Western blot analysis was performed with DEK and AP-2α
(16448-1-AP and 13019-3-AP; Proteintech) antibodies, followed by
anti-rabbit IgG conjugated with horseradish peroxidase. GAPDH
(TA-08; ZSGB-Bio, Beijing, China) was used as loading control.
Chemiluminescence signals were quantified using SuperSignal West
Femto Maximum Sensitivity Substrate (Pierce).
Real-time quantitative PCR
Total RNA was isolated from cultured cells by using
the SV Total RNA Isolation system of RNA extraction kit (Promega,
Madison, WI, USA) following the manufacturer's instructions. Total
RNA (1 μg) was used to synthesize cDNAs by using GoScript™
Reverse Transcription system (Promega). RT-qPCR reactions using
SuperReal PreMix Plus (SYBR-Green; Tiangen Biotech Co., Beijing,
China) were performed using Roche LightCycler 480 System. The
expression of the gene of interest was normalized to that of
B2M, and relative expression was calculated using
comparative Ct method. PCR primers were as follows: DEK,
forward, 5′-CAGGCACTGTGTCCTCAT-3′ and reverse,
5′-CATTTGGTTCGCTTAGCCT-3′; B2M, forward,
5′-GGCTATCCAGCGTACTCC-3′ and reverse, 5′-ACGGCAGGCATACTCATC-3′.
Reporter gene constructs
Genomic DNA was extracted from eight cell lines and
normal hepatic tissues by using QIAamp DNA Mini kit (Qiagen,
Hilden, Germany) following the manufacturer's instructions.
DEK promoter CpG island region was truncated into four
fragments, namely, CpG1 (−732 bp/−305 bp), CpG2-1 (−310 bp/+35 bp),
CpG2-2 (−167 bp/+35 bp) and CpG3 (−730 bp/+57 bp). Four pairs of
primers for PCR amplification were designed by Primer Premier 5.0
software (Table I). The
amplification products were cloned into the pGL3-Basic luciferase
reporter gene vector (Promega) and named as pGL3-DEK/CpG1,
pGL3-DEK/CpG2-1, pGL3-DEK/CpG2-2 and
pGL3-DEK/CpG3. The linear plasmids digested by
HindIII of the four recombinant plasmids were treated with
SssI methylation enzyme, and their methylation status was
identified using BstUI (methylation-sensitive restriction enzyme).
The completely methylated linear plasmids were digested by
NheI to obtain the full methylation of the four target
fragments, which were cloned into the pGL3-Basic vector. The
resulting vectors were called pGL3-Basic/CpG1 (Me),
pGL3-Basic/CpG2-1 (Me), pGL3-Basic/CpG2-2 (Me) and pGL3-Basic/CpG3
(Me).
 | Table IPrimer sequences for luciferase
reporter gene vector construction. |
Table I
Primer sequences for luciferase
reporter gene vector construction.
| Name | Primer sequences
(5′-3′) | Length (bp) |
|---|
| CpG1 |
F-CTAGCTAGCAAAGCATCTGCATAGATGACCTAG | 428 |
|
R-CCCAAGCTTCCTGGAAAAGATGATGAGCAGTC | |
| CpG2-1 |
F-CTAGCTAGCTCCAGGAAGCGACCGTGGAAACAATAAAC | 345 |
|
R-CCCAAGCTTTTCAAAATGGCGGTTCGGGAAGGAG | |
| CpG2-2 |
F-CTAGCTAGCACTCCAGGCGCAGCCGGGGAGA | 202 |
|
R-CCCAAGCTTTTCAAAATGGCGGTTCGGGAAGGAG | |
| CpG3 |
F-CTAGCTAGCAGCATCTGCATAGATGACCTAGAACTC | 787 |
|
R-CCCAAGCTTGCTCCCCAGAATCAACAAGATTTTC | |
Transfection and luciferase assays
The cells were plated in 96-well or 24-well plates,
grown to 60–80% confluence, and transfected with small-interfering
RNA (siRNA) or plasmids by using Lipofectamine® 2000
reagent (Invitrogen) according to the manufacturer's instructions.
Cells transfected with luciferase constructs were harvested and
lysed in Passive lysis buffer (Promega) after 48 h. The cell
lysates were analyzed for firefly and Renilla luciferase
activity by using the Dual-Luciferase reporter assay system
(Promega) as recommended by the manufacturer. Firefly luciferase
activity was normalized to Renilla luciferase activity as
internal control.
Bisulfite genomic sequencing
The genomic DNA of eight HCC cell lines and normal
hepatic tissue was treated with sodium bisulfite by
EpiTect® Fast Bisulfite Conversion DNA kit (Qiagen). PCR
amplification fragment should not be too long (<500 bp) because
the treated genomic DNA becomes unstable and easy to break. Thus,
we divided the CpG island sequence into two short segments to
amplify 294 and 381 bp in the DEK promoter −595 bp/−302 bp
and −329 bp/+52 bp, respectively. The corresponding segments were
named C1 and C2. We then designed two pairs of primers (Table II) for the treated C1 and C2 for
bisulfite sequencing PCR (BSP). BSP was performed in 40 cycles of
98°C for 10 sec, 55°C for 30 sec, and 72°C for 30 sec. Finally, the
PCR products were cloned into the pMD18-T vector (Takara). At least
10 clones were randomly selected for bisulfite genome sequencing
(BGS) to analyze the methylation status of the DEK promoter
CpG island.
| Table IIPrimer sequences for BSP. |
Table II
Primer sequences for BSP.
| Name | Primer sequences
(5′-3′) | Length (bp) |
|---|
| C1 |
F-TTTTATATTTATAGGGGTGTAAATTTATGT | 294 |
|
R-CTTCCTAAAAAAAATAATAAACAATCCC | |
| C2 |
F-GGGATTGTTTATTATTTTTTTTAGGAAG | 381 |
|
R-CCAAAATCAACAAAATTTTCAAAATAAC | |
Prediction of transcription factor
binding sites of DEK promoter CpG island region
Bioinformatics methods were used to predict the CpG
island location in the DEK promoter region near the sequence
from −1000 to +400 bp range by MethPrimer online software
(http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi).
The transcription factor binding sites of DEK promoter CpG
island region were predicted by TFSEARCH online software
(http://www.cbrc.jp/research/db/TFSEARCH.html).
Point and deletion mutation assays
We analyzed point and deletion mutation of the
binding site 'GCCCGCGGC' of the transcription factor AP-2α in the
CpG2-2 region. Point mutation is the substitution of the underlined
consensus sequence 'GCCCGCGGC' into 'TAACGCGGC'. Deletion mutation
includes the deletion of the underlined part of 'GCCCGCGGC' and the removal of all
'GCCCGCGGC'. The three kinds of mutational CpG2-2 fragments,
namely, pGL3-basic/PM, pGL3-basic/FM and pGL3-basic/AM, were cloned
into the pGL3-Basic vector (Promega). Finally, HepG2 cells were
transfected with the plasmids by using Lipofectamine®
2000 reagent (Invitrogen) according to the manufacturer's
instructions. Blank plasmid pGL3-basic and pGL3-DEK/CpG2-2
were used as negative and positive controls. Luciferase activity
was assayed after 24 h.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assay was
performed with EpiQuik ChIP kit (P-2002-1; EpiGentek, Farmingdale,
NY, USA) following the manufacturer's instructions. A total of
1×106 HepG2 cells were fixed in 1% formaldehyde at room
temperature for 10 min. The cells were then lysed and chromatin was
sheared by sonication. The DNA-protein complex was
immunoprecipitated with anti-RNA Pol II antibody (P-2002-1;
EpiGentek), anti-IgG antibody (P-2002-1; EpiGentek), and anti-AP-2α
antibody (13019-3-AP; Proteintech). After reverse cross-linking and
DNA purification, DNA from input (1:20 diluted) or
immunoprecipitated samples were assayed by PCR. The products were
separated by 3% agarose gel electrophoresis. The primers for the
DEK promoter were (forward) 5′-GCGACCGTGGAAACAATAAACA-3′ and
(reverse) 5′-TGCCTCCGCGGGAAGCTC-3′. Primers for the positive
control GAPDH were (forward) 5′-ACGTAGCTCAGGCCTCAAGA-3′ and
(reverse) 5′-GCGGGCTCAATTTATAGAAAC-3′ (P-2002-1; EpiGentek).
Overexpression and small-interfering RNA
interference assays of transcription factor AP-2α
AP-2α expression plasmid was constructed according
to the CDS coding region of TFAP-2α (NM_001032280.2) in the
National Center for Biotechnology Information website (www.ncbi.nlm.nih.gov/). The modified CDS of TFAP-2α
was inserted with the His tag sequence 'CATCACCATCACCATCAC' behind
the transcription start site 'ATG' and added with two restriction
enzyme cutting sites on both ends, namely, 5′-end EcoRI
'GAATTC' and 3′-end HindIII 'TTCGAA'. The modified CDS was
then cloned into the pcDNA3.1 (−) vector (Invitrogen) and named
pcDNA3.1 (−)-hAP2. The vector was transfected into HepG2 cells, and
the expression levels of AP-2α and DEK were assayed.
In addition, AP-2α siRNA interference sequences and
siRNA-negative control (NC) (Table
III) used in this study were designed and synthesized by
GenePharma. Equimolar amounts of three oligonucleotides specific
for AP-2α were combined before transfecting 7721 cells.
Blank control, NC, and the experiment group were set up for the
transfection experiment. Total RNA was extracted using SV Total RNA
Isolation system of RNA extraction kit (Promega) after 24 h of
transfection. Total protein was extracted after transfection of 48
h. Finally, the mRNA and protein expression levels of AP-2α
and DEK were investigated by RT-PCR and western blot
analyses. The primer sequences for AP-2α and DEK are
shown in Table IV.
| Table IIIAP-2α small-interfering RNA
interference sequences. |
Table III
AP-2α small-interfering RNA
interference sequences.
| Name | Sense (5′-3′) | Antisense
(5′-3′) |
|---|
| siRNA-1 |
CCAGAUCAAACUGUAAUUAdTdT |
UAAUUACAGUUUGAUCUGGdTdT |
| siRNA-2 |
GGAAGAUCUUUAAGAGAAAdTdT |
UUUCUCUUAAAGAUCUUCCdTdT |
| siRNA-3 |
CCUGCUCACAUCACUAGUAdTdT |
UACUAGUGAUGUGAGCAGGdTdT |
| siRNA-NC UU |
CUCCGAACGUGUCACGUdTdT |
ACGUGACACGUUCGGAGAAdTdT |
| Table IVPrimer sequences for reverse
transcription-PCR. |
Table IV
Primer sequences for reverse
transcription-PCR.
| Name | Primer sequences
(5′-3′) | Length (bp) |
|---|
| DEK |
F-CAGGCACTGTGTCCTCAT | 316 |
|
R-CATTTGGTTCGCTTAGCCT | |
| AP-2α |
F-CGTCTCCGCCATCCCTATTA | 364 |
|
R-GGTTTCGCACACGTACCCAA | |
| B2M |
F-GGCTATCCAGCGTACTCC | 247 |
|
R-ACGGCAGGCATACTCATC | |
Statistical analysis
Statistical analysis was performed using SPSS
software. Results were analyzed by Student's t-test, and
differences were considered statistically significant at P<0.05.
All in vitro experiments were performed in triplicate, and
the average result is reported. Error bars depict standard
error.
Results
DEK is overexpressed in HCC
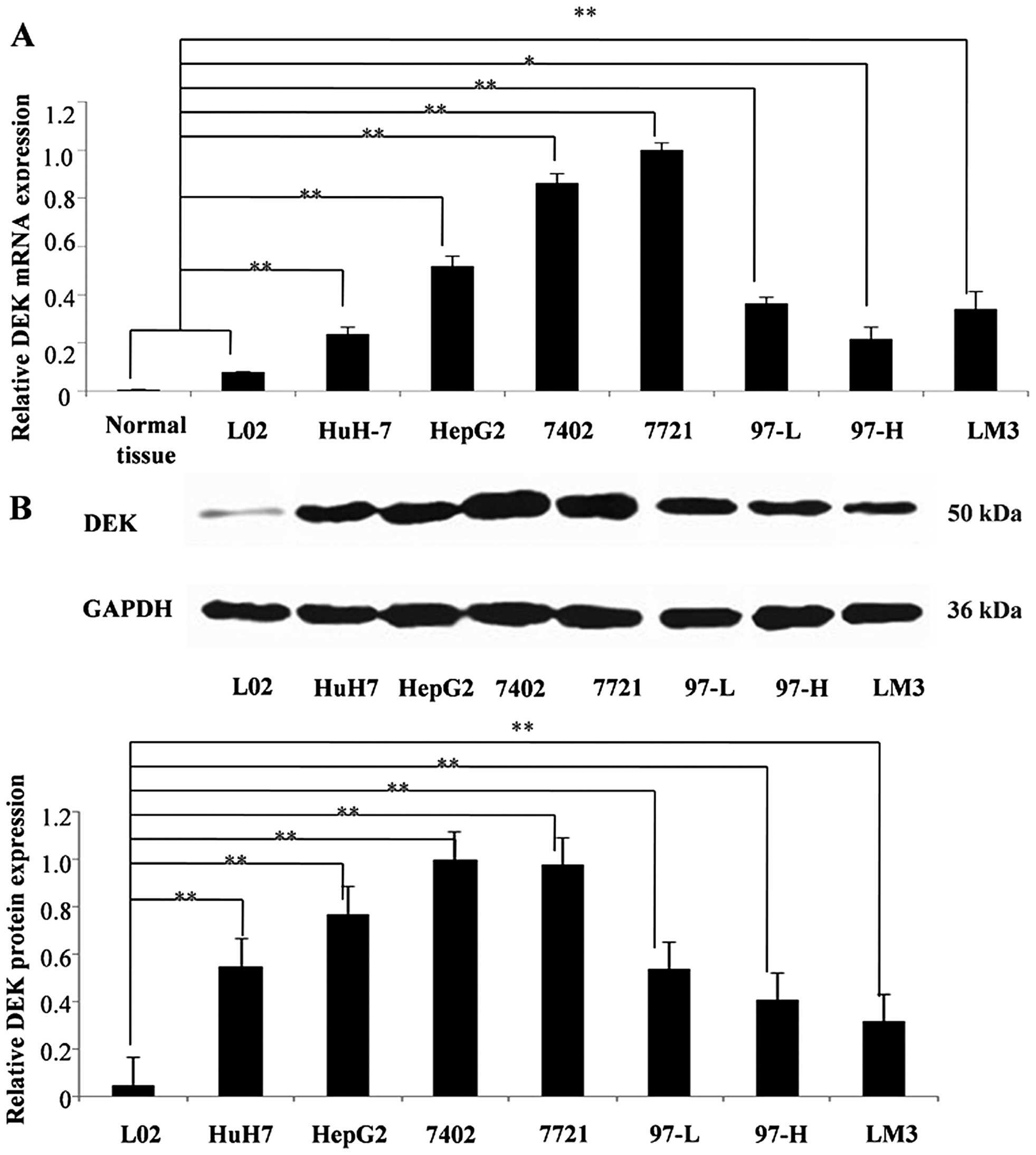
Fluorescent quantitative PCR and western blot
analyses were performed to determine the mRNA and protein
expression levels of DEK in HCC cell lines, respectively. The
results showed that DEK mRNA and protein were overexpressed
in all HCC cell lines compared with those in normal hepatic cell
line L02 and tissues (Fig. 1).
Gel-Pro Analyzer was used to quantify the western blotting results,
and two-tailed Student's t-test indicated that DEK mRNA and
protein expression levels in the seven HCC cell lines (P<0.05 or
P<0.01) significantly differed from those in L02 and normal
hepatic tissues. Among the cell lines, 7402 and 7721 HCC cells
exhibited the highest DEK expression level. DEK was found to be
overexpressed in HCC.
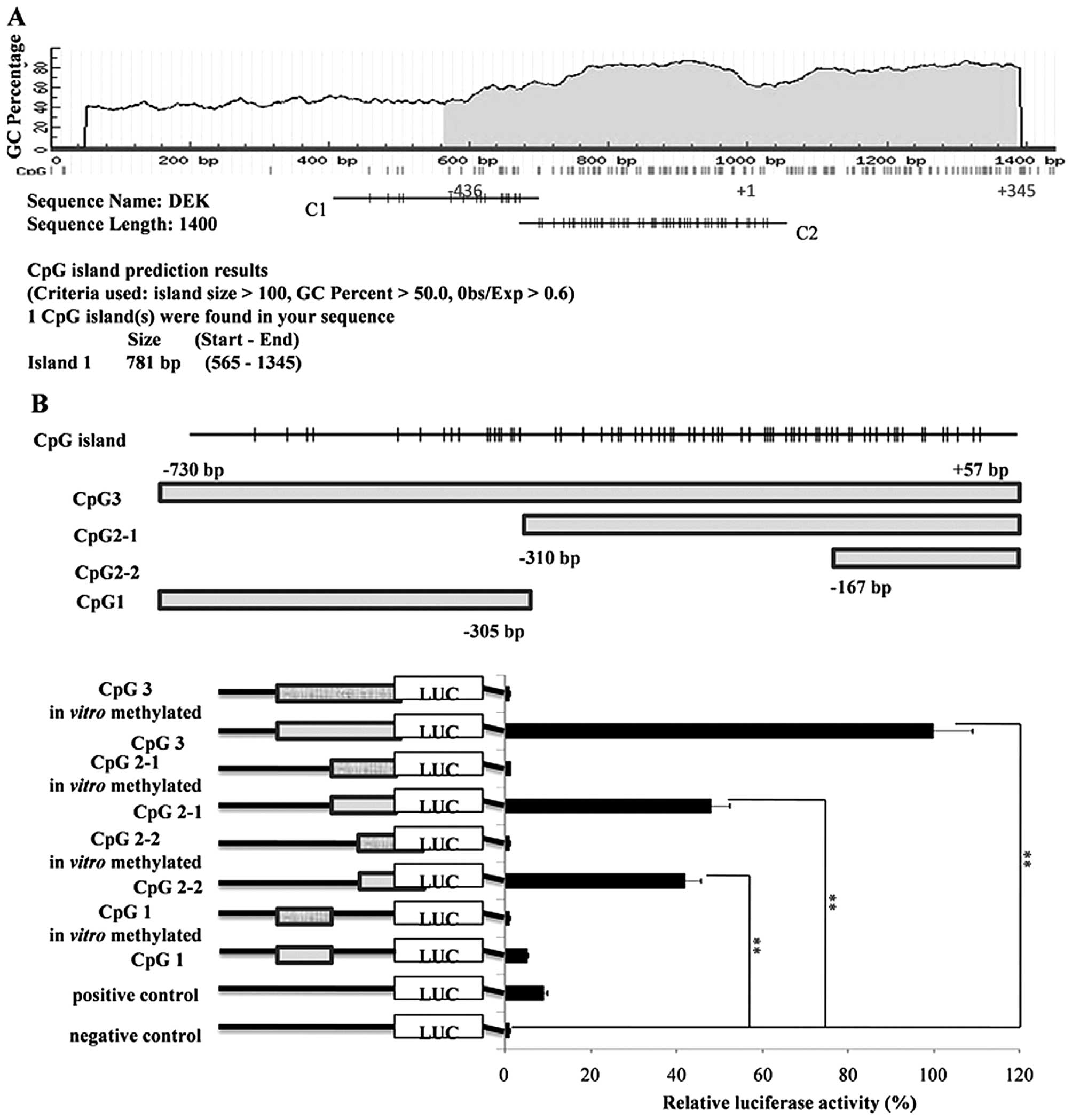
CpG2-2 (−167 bp/+35 bp) is the core
promoter region of DEK
Sequences near the DEK promoter region within
−1000 bp/+400 bp were analyzed through bioinformatics methods with
the MethPrimer online software. Potential CpG island was located
within −436 bp/+345 bp (Fig. 2A).
The extension of the DEK core promoter was then identified.
A series of reporter gene constructs was obtained by
progressive-type truncation strategy, transfected into HepG2 cells,
and then analyzed with the Dual-Luciferase reporter assay system.
The results demonstrated that among the constructs without
methylation in vitro, the activities of
pGL3-DEK/CpG3, pGL3-DEK/CpG2-1, and
pGL3-DEK/CpG2-2 were higher than those of
pGL3-DEK/CpG1 compared with the controls (P<0.01).
However, the activities of pGL3-DEK/CpG2-1 and
pGL3-DEK/CpG2-2 were not significantly different from that
of pGL3-DEK/CpG3 (P>0.05). Further analysis showed that
pGL3-DEK/CpG2-2 reserved most of the activity of
pGL3-DEK/CpG2-1 (P>0.05) (Fig. 2B); hence, CpG2-2 (−167 bp/+35 bp) is
the minimum core promoter region of the DEK gene. In
addition, no activity was found in HepG2 cells for constructs
methylated in vitro compared with that in NC. This result
illustrates that hypermethylation of the DEK promoter
inhibited its activity in HCC.
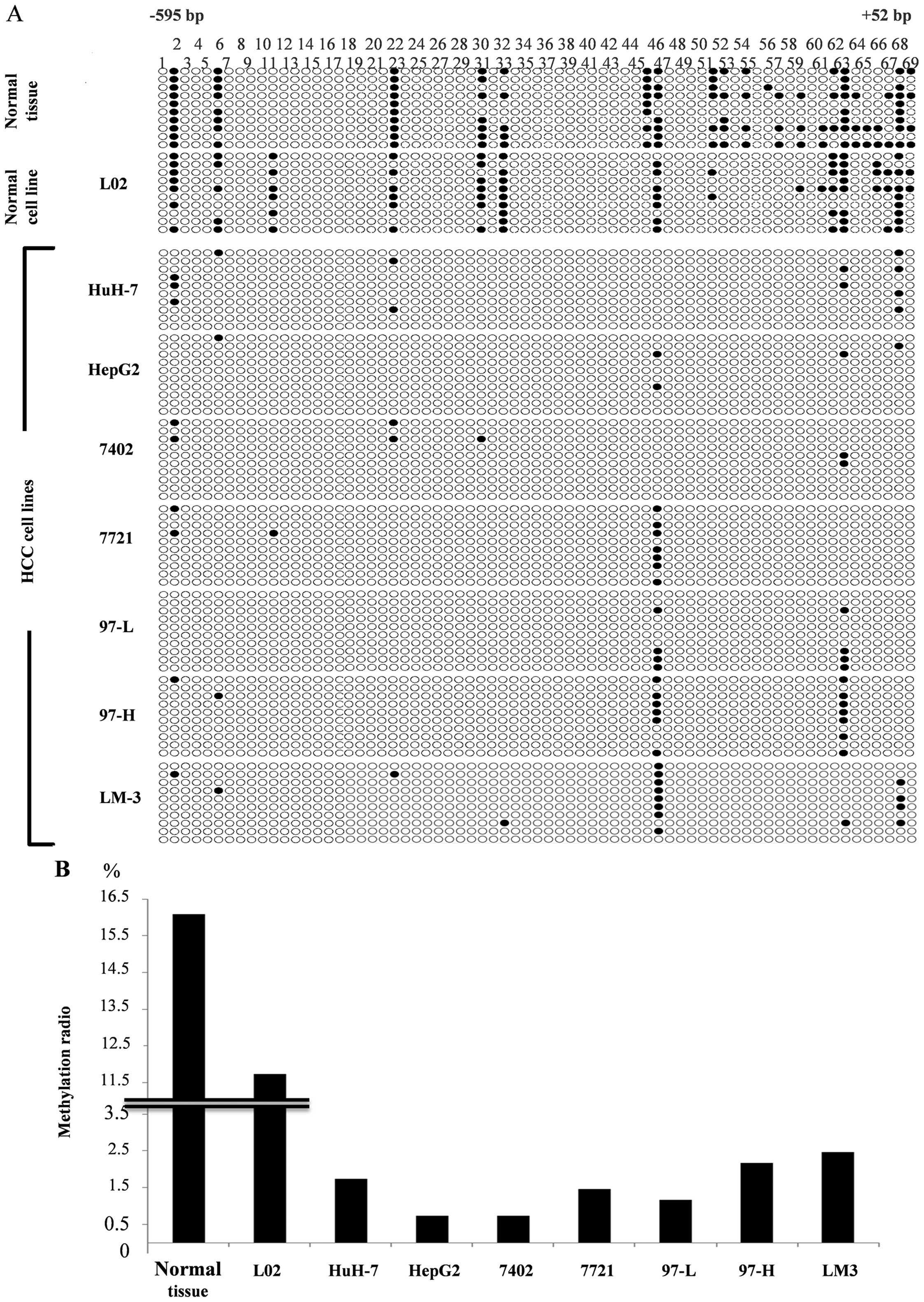
DEK promoter in HCC is commonly under
low-methylation state
BSP and BGS were performed using genomic DNA of
eight HCC cell lines and normal hepatic tissues to investigate
whether the methylation level of the DEK promoter is related
to its overexpression in HCC. We encoded the sequencing results
into QUMA online software for comparative analysis. The atlas shown
in Fig. 3A illustrates the detailed
CpG methylation sites of DEK promoters. The results proved
that the methylation levels of DEK promoters in normal
hepatic tissues and normal hepatic L02 were significantly higher
than those in the HCC cell lines; moreover, methylation sites were
mostly located in the CpG2-2 region. We then calculated the total
ratio of methylation sites and plotted the methylation ratio
histogram (Fig. 3B) to investigate
the methylation level of DEK promoters in all cell lines and
tissues. The result showed that DEK promoters in HCC were commonly
under low-methylation state compared with those in normal cells and
tissues; hence, DEK overexpression in HCC was correlated
with the low-methylation level of the promoters.
Transcription factor binding sites of DEK
core promoter prediction
We speculated that the DEK core promoter
region may contain important transcription factor binding sites
that play pivotal roles in maintaining the basal transcriptional
activity of DEK. Hence, we analyzed the core promoter region
CpG2-2 (−167 bp/+35 bp) by using TFSEARCH online software. The
results showed that DNA binding sites for four transcription
factors, namely, nuclear factor Y (NF-Y), AP-2α, E2F and Ying Yang
1 (YY1), were found in the CpG2-2 region. According to the BGS
results (Fig. 3A), transcription
factor binding sites containing CpG methylation sites in normal
hepatic cells and tissues were screened; these sites included
AP-2α, E2F and YY1 (Fig. 4A). Among
these sites, the binding site forAP-2α presented the most
significant methylation difference compared with that in HCC and
was located in sites no. 45 and 46 CpG (Fig. 3A). The results suggested that
DEK overexpression in HCC was correlated with the
demethylation of the AP-2α transcription factor binding site. In
addition, transcription factors NF-Y, E2F and YY1 were previously
reported to regulate DEK expression (24,25).
We will focus on the regulation effect of the transcription factor
AP-2α for DEK expression in further studies.
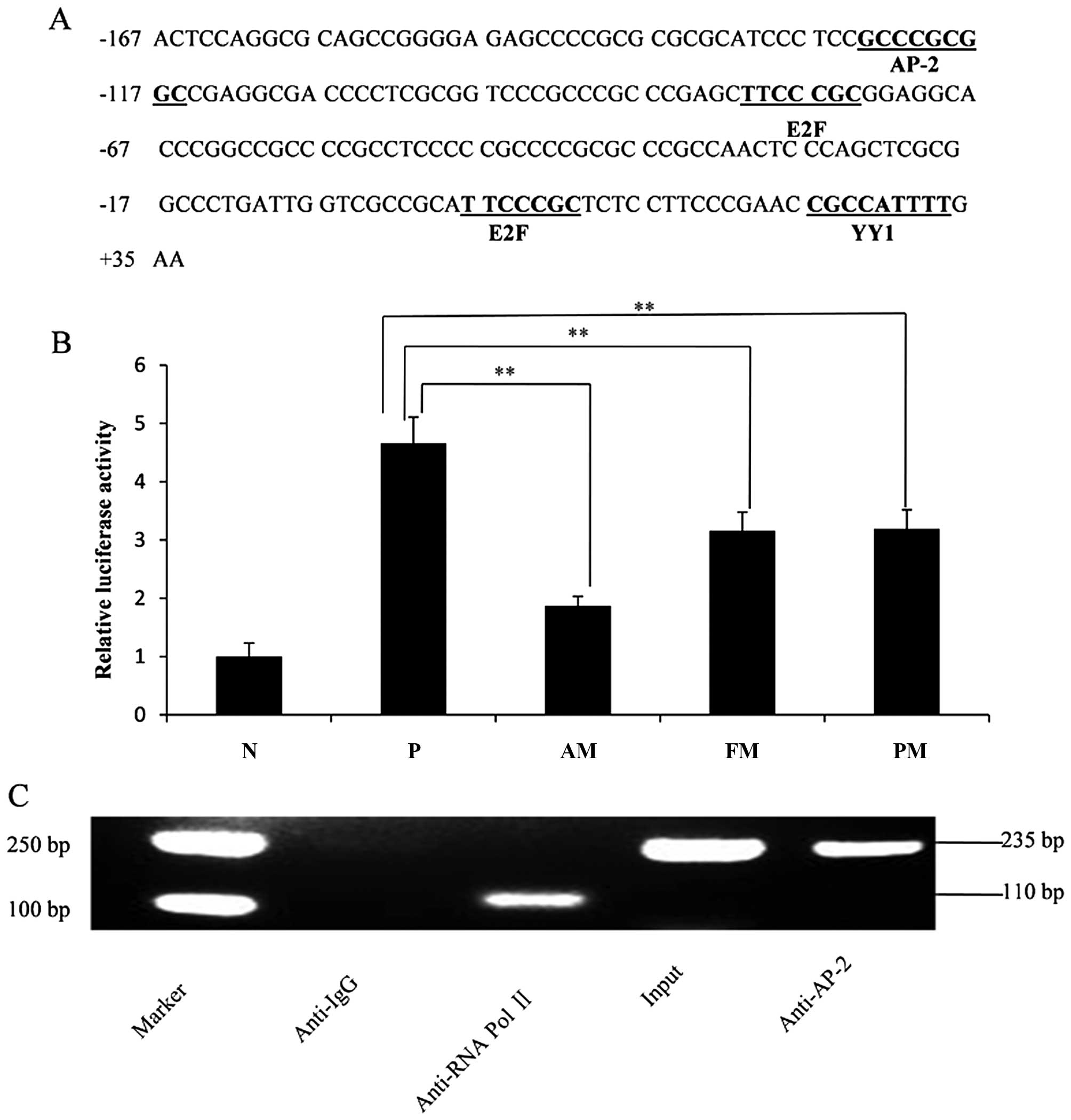 | Figure 4Characterization of transcription
factor binding sites. (A) Nucleotide sequence of the DEK
core promoter was located from −167 bp to +35 bp. Putative
transcription factor binding sites were underlined. (B) Mutation
analysis of putative transcription factor AP-2α binding site. HepG2
cells were transfected with the pGL3-basic vector, the wild plasmid
pGL3-DEK/CpG2-2, three nucleotide substitution mutant
variant (pGL3-basic/PM), six site-deletion mutant variant
(pGL3-basic/FM), and all site-deletion mutant variant
(pGL3-basic/AM). Luciferase activity was measured after 24 h. The
negative control was normalized to 1, and others were compared with
the normalized level. N, negative control; P, positive control; PM,
point mutation; FM, fraction deletion; AM, all deletion.
Transfections were performed in triplicate for each experiment
(**P<0.01). (C) The binding of AP-2α to the DEK core
promoter in vivo using Chromatin immunoprecipitation assay.
The nucleoprotein complex from HepG2 cells was immunoprecipitated
with anti-IgG, anti-RNA Pol II and anti-AP-2α antibodies,
respectively. Then, the precipitated and purified DNA fragments
were subjected to PCR. Anti-IgG antibody (negative control) failed
to precipitate any protein-DNA complex in vivo; anti-RNA Pol
II antibody (positive control) precipitated the RNA Pol II bound to
the GAPDH promoter and the PCR amplified products were 110 bp (RNA
Pol II is considered to be enriched in the GAPDH gene
promoter); anti-AP-2α antibody precipitated AP-2α proteins bound to
the DEK core promoter which contains the predicted AP-2α
binding sites 'GCCCGCGGC' in vivo and its PCR amplified
products were 235 bp. The input served as the internal reference
and measure the efficiency of ChIP. |
AP-2α binding site plays a dominant role
in maintaining DEK core promoter activity
We evaluated the effect of the predicted AP-2α
transcription factor binding site on DEK core promoter activity. We
performed point and deletion mutations in the wild-type plasmid
pGL3-DEK/CpG2-2 and transfected the constructs into HepG2
cells. Luciferase activities were determined after 24 h. The
results showed that DEK core promoter activity was
significantly reduced after point and deletion mutations of the
AP-2α transcription factor binding site (P<0.01). In particular,
all deletion mutation-type pGL3-basic/AM led to ~60% reduction in
DEK core promoter activity compared with that in the wild-type
pGL3-DEK/CpG2-2 (Fig. 4B).
This result indicated that the AP-2α transcription factor binding
site plays a dominant role in maintaining the transcription
activity of the DEK core promoter. We also predict that the
newly found transcription factor AP-2α may significantly regulate
DEK expression.
Transcription factor AP-2α binds to the
DEK core promoter in vivo
AP-2α is a transcription factor that binds to the
promoter regions of its target genes, which contain the binding
sites 'GCCCGCGGC', and mediates the transcription of the target
genes. We performed ChIP assay to validate whether AP-2α interacts
with the DEK core promoter in vivo. The nucleoprotein
complex prepared from HepG2 cells was immunoprecipitated with
anti-RNA Pol II, anti-IgG and anti-AP-2α antibodies. The
precipitated DNA was subjected to PCR with primers flanking the
region containing the AP-2α binding site. The PCR results showed
that anti-AP-2α antibody precipitated proteins were bound in
vivo to the amplified sequence of the DEK promoter; by
contrast, non-specific IgG antibody (NC antibody) failed to
precipitate proteins bound in vivo in this sequence
(Fig. 4C). Thus, the transcription
factor AP-2α could bind to the DEK core promoter region
in vivo.
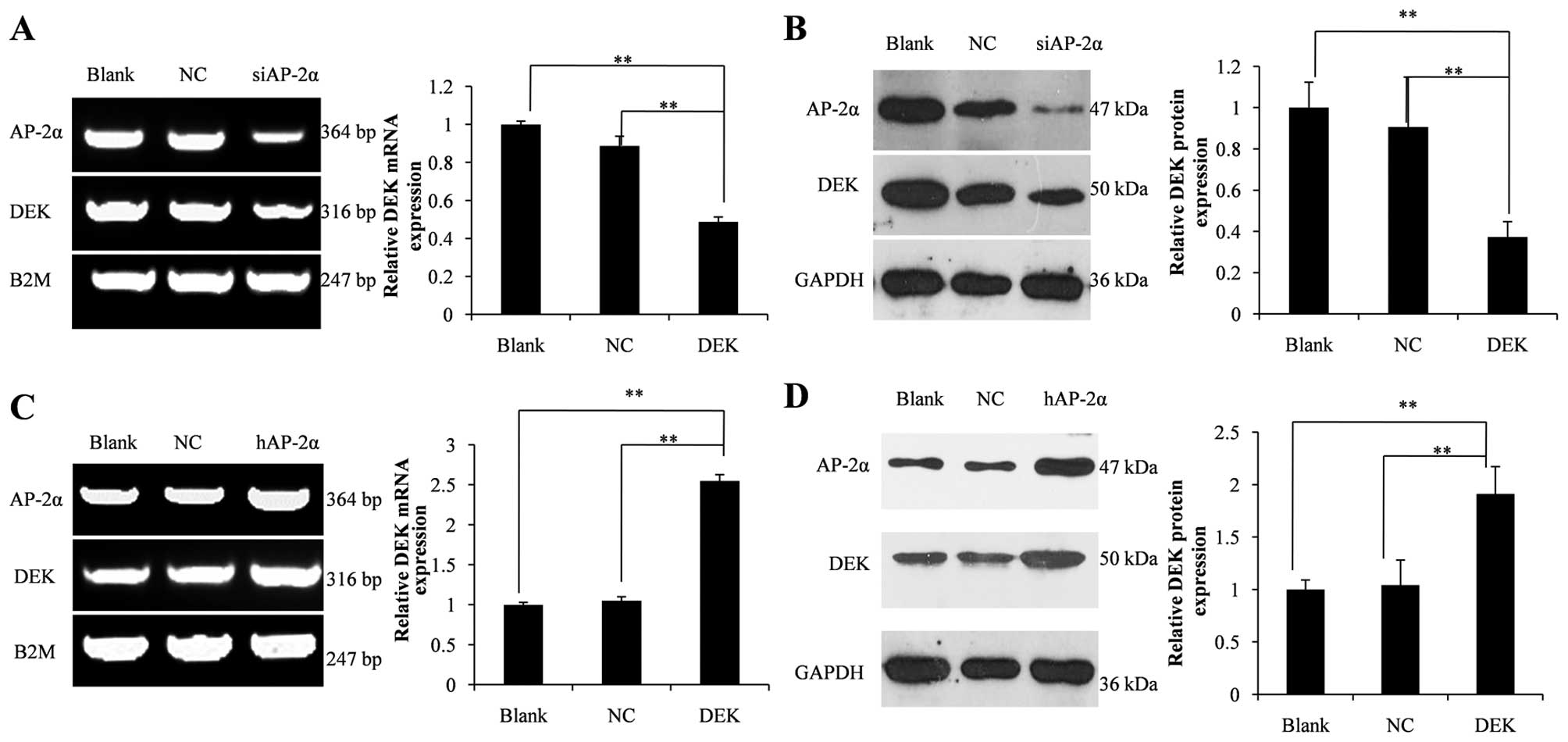
Transcription factor AP-2α upregulates
the expression level of the DEK gene
AP-2α siRNA or AP-2α expression vector
pcDNA3.1 (−)-hAP2 was transfected into 7721 or HepG2 cells to
determine the role of AP-2α in regulating DEK expression.
DEK mRNA and protein levels were detected by RT-PCR and
western blot analyses, respectively. The results illustrated that
siRNA mediated 53% knockdown of AP-2α mRNA and 80% knockdown of the
AP-2α protein, resulting in reduction in DEK mRNA
level by 51% and DEK protein level by 63% (Fig. 5A and B). By contrast, overexpression
of AP-2α mRNA by 1.9-fold and AP-2α protein by 2.0-fold led
to 2.5-fold increase in DEK mRNA level and a substantial
increase of 90% in the DEK protein level compared with those in the
blank control (Fig. 5C and D).
Thus, we conclude that the expression of DEK was mediated by the
transcription factor AP-2α.
Discussion
We performed RT-qPCR and western blot analyses to
verify that DEK was really overexpressed in HCC. The mRNA and
protein expression levels of DEK increased, which were identical
with the results reported in previous studies (26–28).
However, minimal information is available on the regulation of DEK
expression. The overexpression of DEK in HCC was initially reported
to be of S-phase dependency and could exert carcinogenic effect by
inhibiting p53 activity (29).
Previous studies showed that the proportion of DEK mRNA
overexpression in HCC tissues was rather high, which may be related
to the conversion of normal hepatic tissues into HCC (26). In addition, reports suggested that
DEK, as an independent risk factor in HCC, promoted tumor
invasiveness, resulting in the poor prognosis of HCC (27). Hence, the abnormal overexpression of
DEK in tumors would probably become a biomarker for early diagnosis
or vicious transformation. Nevertheless, studies on the
transcriptional regulatory mechanism of the abnormal overexpression
of DEK in tumors were not comprehensive. The DEK promoter
contains transcription factor binding sites bound by transcription
factors E2F, YY1, ERα, NF-Y and c-myc. E2F and YY1 were bound to
the sequences near the DEK transcription start site, which
induced the formation of a transcriptional initiation complex to
activate the relevant passageways and boost tumor formation.
Several studies on the transcriptional regulatory
mechanism of genes in tumors revealed that decreased methylation
levels of oncogenes are common in human tumors. Methylation level
is inversely correlated with gene expression. The low-methylation
level of many genes in tumor tissues occurs in the promoter region.
The low-methylation level of LINE-1 in chronic granulocytic
leukemia could initiate its sense transcription and antisense
transcription of c-MET (30). Numerous activated and amplified
c-fos genes were found in ovarian cancer tissues of mice,
primarily because of the low-methylation level of the c-fos
gene (31). Proto-oncogene
c-myc was also under the low-methylation state in tumor
tissues, which led to significant upregulation of gene expression
and then accelerated cell malignant proliferation, resulting in
tumor occurrence (32). Additional
studies reported that the promoter region of estrogen receptor (ER)
regulatory gene pS2 in breast cancer was lowly methylated,
which is significantly related to the upregulation of ER (33). Moreover, the low-methylation level
of multiple genes often occurs in the same kind of tumor. For
example, genes, such as claudin 4, LCN2, TFF2,
S100A4 and mesothelin, are all lowly methylated in
pancreatic cancer (34).
The present study revealed that the functional
minimal core promoter CpG2-2 is located within the 202 bp region at
position −167 bp/+35 bp relative to the transcription start site by
Dual-Luciferase reporter assay. BGS was performed to evaluate the
effect of methylation level on DEK promoter transcriptional
activity. The results suggested that DEK promoter in HCC
cell lines was commonly under low-methylation state compared with
normal hepatic tissues and normal hepatic cell line L02. Moreover,
methylation sites focused on CpG2-2 region, which indicated that
DEK overexpression in HCC is related to the low-methylation level
of the core promoter. Hypermethylation exerted a remarkable
inhibiting effect on its transcriptional activity. According to the
BGS results, AP-2α transcription factor binding site predicted by
TFSEARCH online software was screened as the most hypermethylated
site in normal hepatic cells and tissues. Furthermore, we verified
whether transcription factor AP-2α regulates DEK expression. We
performed point and deletion mutations of the AP-2α DNA binding
site and ChIP assays. The results demonstrated that the AP-2α
binding site was crucial for the transcription activity of the
DEK core promoter, and AP-2α transcription factor could bind
to the DEK core promoter region in vivo. Knocking
down endogenous AP-2α led to reduction in DEK expression.
Conversely, overexpression of AP-2α upregulated DEK expression. The
transcription factor AP-2α regulates DEK expression by directly
binding to the AP-2α binding site.
In summary, the present study reveals that the
DEK core promoter is located in the −167 bp/+35 bp region.
DEK overexpression in HCC is regulated by the transcription factor
AP-2α and closely correlated with the methylation level of the core
promoter. This study further reveals the transcriptional regulatory
mechanism of DEK overexpression in HCC and provides additional
basis for early diagnosis, prognosis judgment, and genetic therapy
in HCC.
Acknowledgments
We are grateful to the PLA General Hospital for
providing the normal hepatic tissue sample. The present study was
supported by the National Natural Science Foundation of China
(grant no. 81201762).
References
|
1
|
Raza A and Sood GK: Hepatocellular
carcinoma review: Current treatment, and evidence-based medicine.
World J Gastroenterol. 20:4115–4127. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar
|
|
3
|
Perz JF, Armstrong GL, Farrington LA,
Hutin YJ and Bell BP: The contributions of hepatitis B virus and
hepatitis C virus infections to cirrhosis and primary liver cancer
worldwide. J Hepatol. 45:529–538. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hu HG, Scholten I, Gruss C and Knippers R:
The distribution of the DEK protein in mammalian chromatin. Biochem
Biophys Res Commun. 358:1008–1014. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Waldmann T, Baack M, Richter N and Gruss
C: Structure-specific binding of the proto-oncogene protein DEK to
DNA. Nucleic Acids Res. 31:7003–7010. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Waldmann T, Eckerich C, Baack M and Gruss
C: The ubiquitous chromatin protein DEK alters the structure of DNA
by introducing positive supercoils. J Biol Chem. 277:24988–24994.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Waldmann T, Scholten I, Kappes F, Hu HG
and Knippers R: The DEK protein: an abundant and ubiquitous
constituent of mammalian chromatin. Gene. 343:1–9. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
von Lindern M, Fornerod M, van Baal S,
Jaegle M, de Wit T, Buijs A and Grosveld G: The translocation
(6;9), associated with a specific subtype of acute myeloid
leukemia, results in the fusion of two genes, dek and can, and the
expression of a chimeric, leukemia-specific dek-can mRNA. Mol Cell
Biol. 12:1687–1697. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Soekarman D, von Lindern M, Daenen S, de
Jong B, Fonatsch C, Heinze B, Bartram C, Hagemeijer A and Grosveld
G: The translocation (6;9) (p23;q34) shows consistent rearrangement
of two genes and defines a myeloproliferative disorder with
specific clinical features. Blood. 79:2990–2997. 1992.PubMed/NCBI
|
|
10
|
Kappes F, Waldmann T, Mathew V, Yu J,
Zhang L, Khodadoust MS, Chinnaiyan AM, Luger K, Erhardt S,
Schneider R, et al: The DEK oncoprotein is a Su(var) that is
essential to heterochromatin integrity. Genes Dev. 25:673–678.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fu GK, Grosveld G and Markovitz DM: DEK,
an autoantigen involved in a chromosomal translocation in acute
myelogenous leukemia, binds to the HIV-2 enhancer. Proc Natl Acad
Sci USA. 94:1811–1815. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McGarvey T, Rosonina E, McCracken S, Li Q,
Arnaout R, Mientjes E, Nickerson JA, Awrey D, Greenblatt J,
Grosveld G, et al: The acute myeloid leukemia-associated protein,
DEK, forms a splicing-dependent interaction with exon-product
complexes. J Cell Biol. 150:309–320. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Alexiadis V, Waldmann T, Andersen J, Mann
M, Knippers R and Gruss C: The protein encoded by the
proto-oncogene DEK changes the topology of chromatin and reduces
the efficiency of DNA replication in a chromatin-specific manner.
Genes Dev. 14:1308–1312. 2000.PubMed/NCBI
|
|
14
|
Kavanaugh GM, Wise-Draper TM, Morreale RJ,
Morrison MA, Gole B, Schwemberger S, Tichy ED, Lu L, Babcock GF,
Wells JM, et al: The human DEK oncogene regulates DNA damage
response signaling and repair. Nucleic Acids Res. 39:7465–7476.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim DW, Chae JI, Kim JY, Pak JH, Koo DB,
Bahk YY and Seo SB: Proteomic analysis of apoptosis related
proteins regulated by proto-oncogene protein DEK. J Cell Biochem.
106:1048–1059. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee KS, Kim DW, Kim JY, Choo JK, Yu K and
Seo SB: Caspase-dependent apoptosis induction by targeted
expression of DEK in Drosophila involves histone acetylation
inhibition. J Cell Biochem. 103:1283–1293. 2008. View Article : Google Scholar
|
|
17
|
Casas S, Nagy B, Elonen E, Aventín A,
Larramendy ML, Sierra J, Ruutu T and Knuutila S: Aberrant
expression of HOXA9, DEK, CBL and CSF1R in acute myeloid leukemia.
Leuk Lymphoma. 44:1935–1941. 2003. View Article : Google Scholar
|
|
18
|
Lin L, Piao J, Gao W, Piao Y, Jin G, Ma Y,
Li J and Lin Z: DEK over expression as an independent biomarker for
poor prognosis in colorectal cancer. BMC Cancer. 13:3662013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Piao J, Shang Y, Liu S, Piao Y, Cui X, Li
Y and Lin Z: High expression of DEK predicts poor prognosis of
gastric adenocarcinoma. Diagn Pathol. 9:672014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ahmad I and Rao DN: Chemistry and biology
of DNA methyltransferases. Crit Rev Biochem Mol Biol. 31:361–380.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gardiner-Garden M and Frommer M: CpG
islands in vertebrate genomes. J Mol Biol. 196:261–282. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cottrell SE: Molecular diagnostic
applications of DNA methylation technology. Clin Biochem.
37:595–604. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stresemann C, Brueckner B, Musch T,
Stopper H and Lyko F: Functional diversity of DNA methyltransferase
inhibitors in human cancer cell lines. Cancer Res. 66:2794–2800.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sitwala KV, Adams K and Markovitz DM: YY1
and NF-Y binding sites regulate the transcriptional activity of the
dek and dek-can promoter. Oncogene. 21:8862–8870. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Carro MS, Spiga FM, Quarto M, Di Ninni V,
Volorio S, Alcalay M and Müller H: DEK Expression is controlled by
E2F and deregulated in diverse tumor types. Cell Cycle.
5:1202–1207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lü ZL, Luo DZ and Wen JM: Expression and
significance of tumor-related genes in HCC. World J Gastroenterol.
11:3850–3854. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin LJ and Chen LT: The role of DEK
protein in hepatocellular carcinoma for progression and prognosis.
Pak J Med Sci. 29:778–782. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yi HC, Liu YL, You P, Pan JS, Zhou JY, Liu
ZJ and Zhang ZY: Overexpression of DEK gene is correlated with poor
prognosis in hepatocellular carcinoma. Mol Med Rep. 11:1318–1323.
2015.
|
|
29
|
Kondoh N, Wakatsuki T, Ryo A, Hada A,
Aihara T, Horiuchi S, Goseki N, Matsubara O, Takenaka K, Shichita
M, et al: Identification and characterization of genes associated
with human hepatocellular carcinogenesis. Cancer Res. 59:4990–4996.
1999.PubMed/NCBI
|
|
30
|
Roman-Gomez J, Jimenez-Velasco A, Agirre
X, Cervantes F, Sanchez J, Garate L, Barrios M, Castillejo JA,
Navarro G, Colomer D, et al: Promoter hypomethylation of the LINE-1
retrotransposable elements activates sense/antisense transcription
and marks the progression of chronic myeloid leukemia. Oncogene.
24:7213–7223. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li S, Hansman R, Newbold R, Davis B,
McLachlan JA and Barrett JC: Neonatal diethylstilbestrol exposure
induces persistent elevation of c-fos expression and
hypomethylation in its exon-4 in mouse uterus. Mol Carcinog.
38:78–84. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dang CV, O'donnell KA and Juopperi T: The
great MYC escape in tumorigenesis. Cancer Cell. 8:177–178. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rodriguez BAT, Weng YI, Liu TM, Zuo T, Hsu
PY, Lin CH, Cheng AL, Cui H, Yan PS and Huang TH: Estrogen-mediated
epigenetic repression of the imprinted gene cyclin-dependent kinase
inhibitor 1C in breast cancer cells. Carcinogenesis. 32:812–821.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sato N, Maitra A, Fukushima N, van Heek
NT, Matsubayashi H, Iacobuzio-Donahue CA, Rosty C and Goggins M:
Frequent hypomethylation of multiple genes overexpressed in
pancreatic ductal adenocarcinoma. Cancer Res. 63:4158–4166.
2003.PubMed/NCBI
|