Introduction
It is known that many anticancer drugs directly
induce cancer cell death through apoptosis or necrosis, and it is
now increasingly accepted that activating apoptosis is a major part
of the efficacy of some of the most effective anticancer drugs
(1). Anticancer drugs have been
reported to induce apoptosis in various cancers; these include
various chemotherapeutics such as cisplatin (2), doxorubicin (3), paclitaxel (4), and etoposide (5). Besides chemotherapies, various
therapeutics whether they are immunotherapy, radiation therapy or
gene therapy, also trigger apoptosis or programmed cell death in
their target cancer cells.
Apoptosis is characterized by condensation and
fragmentation of nuclear chromatin, compaction of cytoplasmic
organelles, dilatation of the endoplasmic reticulum, a decrease in
cell volume and alterations to the plasma membrane (6). The remaining cell components are then
engulfed by neighboring cells or macrophages. In normal cell
biology, apoptosis also provides a means of purging unwanted cells
and that may include defective cells. Apoptotic signaling can
proceed in two pathways: originating the extrinsic death pathway,
via death receptors expressed on the plasma membranes of cells or
initiated intrinsic mitochondria-mediated pathway, which contain
several proteins that regulate apoptosis. Among the six different
death receptors, Fas (CD95), tumor necrosis factor receptor 1
(TNFR1), death receptor (DR)3, DR4, DR5, and DR6, the Fas system is
a major signaling pathway involved in regulation of apoptosis
according to several studies (7).
The interaction of Fas with FasL induces apoptotic cell death and
altered expression of Fas/FasL has also been implicated in
pathogenesis of diseases associated with immune regulation
(8). Ligation of the Fas receptor
on the cell surface results in its trimerization and recruitment of
the adaptor molecule FADD and procaspase-8 and form the
death-inducing signaling complex (DISC). Procaspase-8 is activated
at the DISC by an autocatalytic cleavage. Cells expressing and
recruiting sufficient amounts of procaspase-8 at the DISC undergo
apoptosis through direct activation of caspase-3 by active
caspase-8. In contrast, cells with limited amounts of active
caspase-8 at the DISC cannot proceed through this direct pathway
and depend on the release of cytochrome c, which leads to
caspase-9 activation and initiation of a proteolytic caspase
cascade (9).
Reactive oxidative species (ROS) have been
implicated in the induction or enhancement of apoptosis (10) through stress stimuli such as
exposure to UV, other forms of irradiation, or cytotoxic drugs
(11). However, the role of ROS in
Fas-induced cell death remains uncertain (12). UV, cycloheximide, c-Jun N-terminal
kinase (JNK), and cytotoxic drugs are also reported to trigger
FADD-dependent apoptosis in cancer cells (12–14).
The mechanism of how FADD-caspase-8 cascade is activated under
these stress stimuli is not completely understood. An interesting
possibility is that there is clustering and thus activation of Fas
receptor and its interaction with FADD induced by the stress
stimuli, as suggested by several studies (12–14).
It has been reported that the activation of the Fas
receptor, which belongs to the TNF receptor family, results in a
signal transduction pathway that most likely uses the production of
ROS (15). As for the intracellular
source of ROS generated with Fas-mediated signaling, both NADPH
oxidase (16) and the mitochondrial
electron transport chain have been implicated in different cell
types (17). Counteracting the
generated ROS, various antioxidants, such as
N-acetyl-cysteine (NAC), superoxide dismutase (SOD)
overexpression or catalase, block Fas-induced apoptosis (12).
Resveratrol, trans-3,5,4′-trihydroxystilbene,
has demonstrated various potential activities including anticancer
effects, anti-oxidant effects, and cardioprotective effects
(18–20). Resveratrol is found in grapes,
berries and peanuts (21), and many
resveratrol derivatives have been reported to possess diverse
biological effects, including anti-inflammatory (22), antitumor (23) and antiarrhythmic effects (24). In particular, the various analogues
of resveratrol include: a novel resveratrol-salicylate hybrid
analog with anti-inflammatory and antioxidant properties (25); astringinin, displaying a potent
anti-melanoma effect (26);
3,4,5-trihydroxy-trans-stilbene, with an apoptotic activity
(27); the synthetic
triacetoxystilbene (TAS), with anti-inflammatory activity (28); HS-1793, a synthetic resveratrol
analogue, with a modulatory effect on tumor-derived T cells
(29); and 8-ADEQ, also a synthetic
resveratrol analogue that displays cell cycle arrest activity
(23).
Isoquinolines, in particular, have attracted
research interest as potential anticancer agents as they can
interfere with cellular initiation, promotion and progression in
the multiple stages of carcinogenesis (30). For instance, berberine, an
isoquinoline alkaloid, inhibits the metastatic potential of breast
cancer cells via modulation of the Akt pathway (31). In this study, we examined the
molecular mechanisms underlying the proapoptotic activities of
8-ADEQ in HL-60 human promyelocytic leukemia cells. From our study,
8-ADEQ potentially represents an apoptosis-inducing agent with
applications in leukemia therapy.
Materials and methods
Chemical and reagents
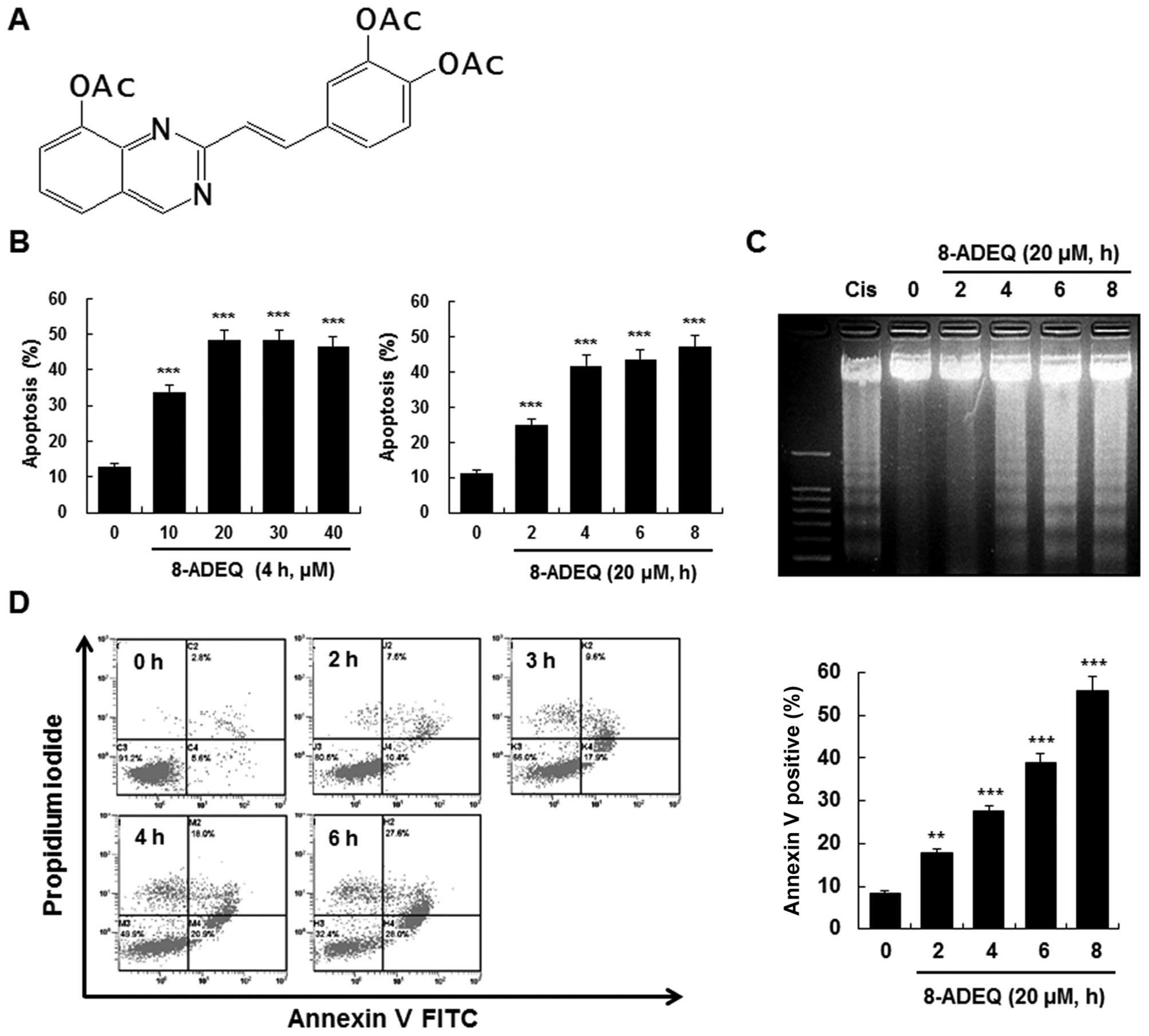
8-ADEQ (Fig. 1A) was
prepared as previously described (22). RPMI-1640 medium, DMEM, FBS,
penicillin, and streptomycin were obtained from Life Technologies
Inc. MTT, anti β-actin antibody, protein A, CCCP (carbonyl cyanide
m-chlorophenylhydrazone), DAPI (4′,6-diamidino-2-phenylindole),
PMSF (phenylmethylsulfonyl fluoride), dithiothreitol, NAC
(N-acetylcysteine), BSO (buthionine sulfoximine), DCFH-DA
(dichloro-dihydro-fluorescein diacetate), acrylamide,
bisacrylamide, sodium dodecyl sulfate, and all other chemicals were
purchased from Sigma Chemical Co. and anti-caspase-8, −9, FADD
antibodies were obtained from Pharmingen. Antibodies for caspase-3,
−6, PARP, Fas, FasL and the peroxidase conjugated secondary
antibody were purchased from Santa Cruz Biotechnology, Inc.
DiOC6 (3,3′-dihexyloxacarbocyanine iodide) was perchased
from Molecular Probes. Proteinase K, ribonuclease A, and TEMED were
perchased from Bio-Rad Laboratories. Caspase inhibitors
(Ac-DEVD-CHO, z-DEVD-fmk, z-IETD-fmk, z-LEHD-fmk, z-VAD-fmk) were
purchased from Enzyme System Products, Inc. SiRNA and transfection
reagents for FADD were purchased from Quiagen, Inc.
Cell culture
HL-60 human promyelocytic leukemia, U-937 human
histocytic lymphoma, Jurkat human acute T cell leukemia, HeLa human
negroid cervix epitheloid carcinoma, A172 human glioblastoma, A549
human lung adenocarcinoma cells were obtained from the Korean Cell
Line Bank and cultured in RPMI-1640 or DMEM media supplemented with
10% heat-inactivated FBS, penicillin (100 U/ml), and streptomycin
sulfate (100 µg/ml). Cells were maintained at 37°C in an atmosphere
of 5% CO2 in air.
MTT assay
The cells were seeded in each well containing 100 µl
of the medium supplemented with 10% FBS in a 96-well plate. After
24 h, various concentrations of 8-ADEQ were added. After 48 h, 50
µl of MTT (5 mg/ml stock solution, in PBS) was added, and the
plates were incubated for an additional 4 h. After centrifugation,
the medium was discarded and the formazan blue, which formed in the
cells, was dissolved with 100 µl DMSO. The optical density was
measured at 540 nm.
TUNEL assay
After treatment of cells with 8-ADEQ (20 µM) for
indicated times, cells were harvested and washed twice with
ice-cold PBS. Then the cells were fixed and smeared with 4%
paraformaldehyde on the slide glass. The fixed cells were incubated
in permeabilization solution for 2 min on ice. TUNEL was performed
using a kit (In situ Cell Death Detection kit) according to the
standard protocol provided by the manufacturer. The 50-µl TUNEL
reaction mixture was added on each sample, and the cells were
incubated for 1 h at 37°C in a humidified atmosphere in the dark.
After washed twice, the cells were incubated with converter-AP for
additional 30 min. Substrate solution was added, and the slides
were incubated for 10 min at room temperature in the dark, followed
by mounting coverslip and then examined under a light
microscope.
DAPI assay
Cells were lysed in a solution containing 5 mM
Tris-HCl (pH 7.4), 1 mM EDTA, and 0.5% (w/v) Triton X-100 for 20
min on ice. The lysate and supernatant after centrifugatation at
25,000 g for 20 min were sonicated for 10 sec, and the level of DNA
in each fraction was measured by a fluorometric method using DAPI.
The amount of the fragmented DNA was calculated as the ratio of the
amount of DNA in the supernatant to that in the lysate.
DNA laddering
Cells under different treatments were collected,
washed with ice-cold PBS twice and then lysed in 1 ml of lysis
buffer (50 mM Tris, 20 mM EDTA, 0.1% NP-40, pH 7.4) for 60 min at
room temperature. The lysates were subsequently incubated with 5 µl
of RNase A (1 mg/ml) for 60 min at 37°C. Proteinase K (10 mg/ml, 10
µl) was then added to the lysates and incubated at 50°C overnight.
DNA was extracted with an equal volume of phenol: chloroform:
isoamyl alcohol (25:24:1). Purified DNA was resuspended in TE
buffer (10 mM Tris, 1 mM EDTA, pH 8.0). Electrophoresis was
performed in a 2% (w/v) agarose gel in 40 mM Tris-acetate buffer
(pH 7.4) at 50 V for 1 h. The fragmented DNA was visualized by
staining with ethidium bromide after electrophoresis (32).
Annexin V and PI double staining by
flow cytometry
During apoptosis, exposure of phosphatidylserine on
the exterior surface of the plasma membrane can be detected by the
binding of fluorescenated Annexin V (Annexin V-FITC). This assay is
combined with analysis of the exclusion of the plasma membrane
integrity probe PI (33). For
Annexin V and PI double staining, cells were suspended with 100 µl
of binding buffer (10 mM HEPES/NaOH, 140 mM NaCl, 2.5 mM
CaCl2, pH 7.4) and stained with 5 µl of FITC-conjugated
Annexin V and 5 µl of PI (50 µg/ml). The mixture was incubated for
15 min at room temperature in the dark and analyzed by the
fluorescence-activated cell sorting (FACS) cater-plus flow
cytometry (Becton-Dickinson Co., Heidelberg, Germany).
Luminescent caspase activity
assay
The activity of caspases was measured using the
specific substrates; caspase-3 (Ac-DEVD-pNA), caspase-8 substrate
(Ac-IETD-pNA), and caspase-9 substrate (Ac-LEHD-pNA). The cells
were incubated with caspase-8 inhibitor (z-IETD-fmk) or not, for 1
h at 37°C, followed by the addition of 8-ADEQ (20 µM). After 4 h of
incubation in the absence of caspase-8 inhibitor, cells were
harvested, and then washed twice with cold PBS. The
Caspase-Glo® 3/7, 8, 9 assay kits were used for
detecting the specific caspase activities. Each Caspase-Glo reagent
was prepared by adding the specific buffer to the specific
luminogenic substrate and mixing. The equal volume of each
Caspase-Glo reagent was added to the sample in the assay well. The
luminescence of each sample was measured in a plate-reading
luminometer.
Western blot analysis
Cells were harvested and washed twice with cold PBS.
Cell pellets were then lysed in ice-cold cell extraction buffer (50
mM HEPES, pH 7.0, 250 mM NaCl, 5 mM EDTA, 0.1% NP-40, 0.1 mM PMSF,
0.5 mM dithiothreitol, 5 mM NaF, 0.5 mM Na orthovanadate)
containing 5 µg/ml each of leupeptin and aprotinin and incubated
for 30 min at 4°C. Cell debris was removed by microcentrifugation
(10,000 g, 5 min), followed by quick freezing of the supernatants.
Cellular proteins (50 µg) were electroblotted onto nitrocellulose
membrane following separation on a 10–15% SDS-polyacrylamide gel
electrophoresis. The immunoblot was incubated for 1 h with blocking
solution (5% skim milk in TTBS), and then incubated for 4 h with a
1:1,000 dilution of primary antibody. Blots were washed three times
with TTBS, and then incubated with a 1:1,000 dilution of
horseradish peroxidase-conjugated secondary antibody for 1 h at
room temperature, washed again three times with TTBS, and then
developed by enhanced chemiluminescence.
Analysis of mitochondrial membrane
potential (MMP, ΔΨm)
Changes in mitochondrial transmembrane potential
were monitored by flow cytometric analysis. Cells were incubated
with 50 nM DiOC6 for 30 min, washed twice with PBS, and
analyzed by flow cytometric analysis (Becton-Dickinson Co.) with
excitation and emission settings of 484 and 500 nm, respectively.
To ensure that DiOC6 uptake was specific for ∆Ψm,
we also treated cells with 100 µM CCCP. CCCP was used as a
reference depolarizing agent of mitochondrial permeability
transition.
Preparation of cytosolic proteins
Cells were collected by centrifugation at 200 g for
10 min at 4°C. The cells were then washed twice with ice-cold PBS,
and centrifuged at 200 g for 5 min. The cell pellet obtained was
then resuspended in ice-cold cell extraction buffer (20 mM
HEPES-KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1
mM EGTA, 1 mM dithiothreitol, 100 µM PMSF, and protease inhibitor
cocktail) for 30 min on ice. The cells were then homogenized with a
glass dounce and a B-type pestle (80 strokes), homogenates were
spun at 15,000 g for 15 min at 4°C, and the supernatant (cytosolic
fraction) was removed whilst taking care to avoid the pellet. The
resulting supernatant was fractionated in SDS-polyacrylamide gels
and transferred to nitrocellulose membranes fot immunoblot analysis
using the indicated primary antibodies. Immunopositive bands were
visualized by ECL kit (Amersham, Buckinghamshire, UK).
Immunoprecipitation
Cells treated with 8-ADEQ were harvested and washed
twice with cold PBS. The cell pellets were resuspended and lysed in
EBC buffer (50 mM Tris, pH 8.0, 120 mM NaCl, 0.5% NP-40, 5 µg/ml
leupeptin, 10 µg/ml aprotinin, 50 µg/ml PMSF, 0.2 mM sodium
ortho-vanadate, 100 mM NaF) for 30 min at 4°C. After centrifugation
(10,000 g, 5 min), protein concentrations were determined. Equal
amount of protein (100 µg) was incubated with anti-Fas and
anti-FADD polyclonal antibodies for 12 h at 4°C, followed by
incubation with 20 µl protein A-Sepharose beads for 4 h. The
protein complex was washed 4 times with EBC buffer and released
from the beads by boiling in 6X sample buffer (350 mM Tris, pH 6.8,
10% SDS, 30% β-mercaptoethanol, 6% glycerol, 0.12% bromophenol
blue) for 5 min. The reaction mixture was then resolved by a 10–15%
SDS-PAGE gel, transferred to nitrocellulose membrane and probed
with anti-caspase-8, anti-Fas, anti-FasL monoclonal antibody. The
blot was developed by enhanced chemiluminescence.
RNA interference
RNA interference of FADD was performed using a
21-base pair (bp) (including a 2-deoxynucleotide overhang) siRNA
duplexes purchased from Qiagen. The sense strand nucleotide strand
was UCACAGACUUUGGACAAAGdTdT. For transfection, siRNA duplexes (200
nM) were introduced into the cells (2×105 cells/ml)
using RNAiFect transfection reagent (Qiagen) according to the
manufacturer's instructions.
ROS detection (DCFH-DA assay)
DCFH-DA was used to measure levels of ROS. The cells
were harvested and suspended in media. DCFH-DA (20 µM) was then
added and incubated for 30 min at 37°C. DCFH-DA was taken up by
cells and, on deacetylation, formed a nonfluorescent DCFH. On
oxidation, this became DCF. The fluorescent intensity was measured
by flow cytometry. The histograms were analyzed by indicating the
MFI.
Statistical analysis
Data presented are the means ± SD of results from
three independent experiments. Statistically significant values
were compared using ANOVA and Dunnett's post hoc test, and P-values
of <0.05 were considered statistically significant.
Results
8-ADEQ inhibits cell growth in various
cancer cell lines
We first examined the cytotoxicity of 8-ADEQ using
MTT assay in various cancer cell lines. 8-ADEQ showed different
degrees of cytotoxicity on these cells as judged by IC50
and its value ranges from 7.59 to 143.88 µM (Table I). Interestingly, 8-ADEQ was found
to have significant cytotoxic effects on hematological cancer cell
lines, such as HL-60, U937, and Jurkat. Since HL-60 cells were
found to be the most sensitive to 8-ADEQ, further experiments were
performed to evaluate the effects of 8-ADEQ on apoptosis and to
identify the molecular mechanism involved in HL-60 cells.
 | Table I.Cytotoxic activity of 8-ADEQ on
various cancer cell lines. |
Table I.
Cytotoxic activity of 8-ADEQ on
various cancer cell lines.
|
| Cell lines | IC50
(µM) |
|---|
| HL-60 | Human promyelocytic
leukemia |
7.59±1.54 |
| U-937 | Human histocytic
lymphoma |
9.43±2.85 |
| Jurkat | Human acute T cell
leukemia |
12.84±1.73 |
| HeLa | Human cervical
epitheloid carcinoma |
73.62±6.62 |
| A172 | Human
glioblastoma |
25.75±2.63 |
| A549 | Human lung
adenocarcinoma | 143.88±13.35 |
8-ADEQ induces apoptosis in HL-60
cells
To determine whether the cytotoxic effect of 8-ADEQ
is associated with the induction of apoptosis, HL-60 cells were
treated with 8-ADEQ and the DNA fragmentation in the cells was
evaluated. The DAPI assay revealed that 8-ADEQ induced DNA
fragmentation in a dose- and time-dependent manner (Fig. 1B). To further characterize the end
stage of apoptosis induced by 8-ADEQ, we examined whether 8-ADEQ
induces a typical ladder pattern of internucleosomal DNA
fragmentation, a hallmark of apoptosis. The laddering pattern of
internucleosomal DNA fragmentation was found to occur in a
time-dependent manner after treating HL-60 cells with 8-ADEQ
(Fig. 1C). Furthermore, we assessed
the translocation of phosphatidylserine (PS) using Annexin V and PI
double staining. Treatment of HL-60 cells with 8-ADEQ resulted in a
significant time-dependent enhancement in both the early and late
stages of apoptosis (Fig. 1D).
8-ADEQ-induced apoptosis involves
caspase, the loss of ∆Ψm, and the release of cytochrome c
The caspase family of cysteine proteases plays a
central role in apoptosis, a controlled demolition of the cellular
architecture in response to diverse stimuli (34). To determine whether 8-ADEQ-induced
apoptosis is involved in the activation of the caspase cascade,
HL-60 cells were treated with 20 µM 8-ADEQ for various time-points,
and activation of caspases was measured by western blotting. In
consequence, time-dependent cleavage of procaspase-8, −9, −3 and −6
was observed by 20 µM 8-ADEQ treatment. The activations of caspases
were accompanied by the degradation of PARP, a target for caspase-3
during the apoptotic response (35). Cleavage of PARP was evident at 4 h
post 8-ADEQ treatment and coincided with the appearance of
activated caspase-3 (Fig. 2A). The
increased enzymatic activity of caspase-3, −8, −9 was detected
after the 8-ADEQ treatment, coincident with the observed cleaved
forms of the caspases (Fig. 2B).
The activation of caspase-8 was detected earlier than that of the
other monitored caspases.
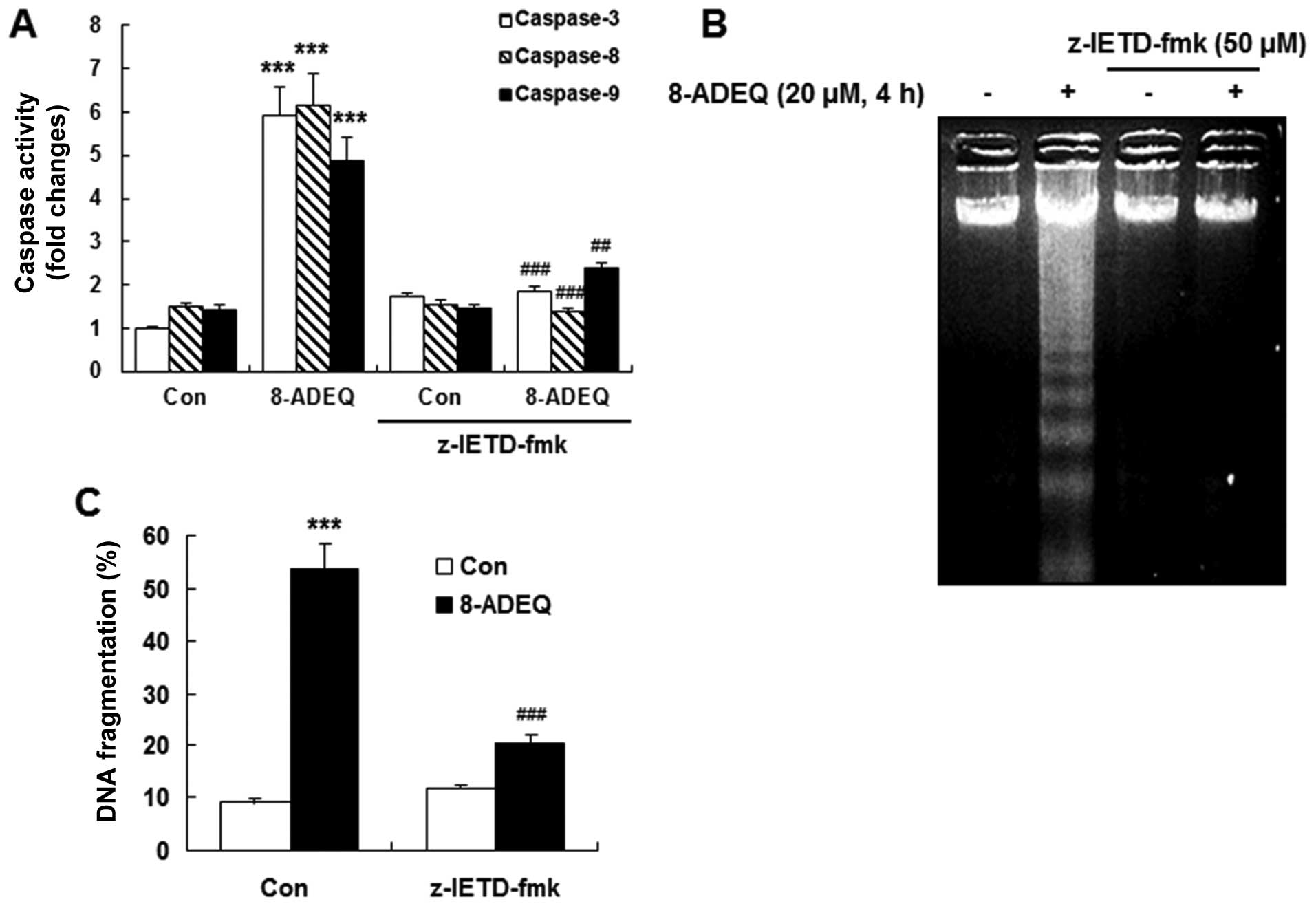
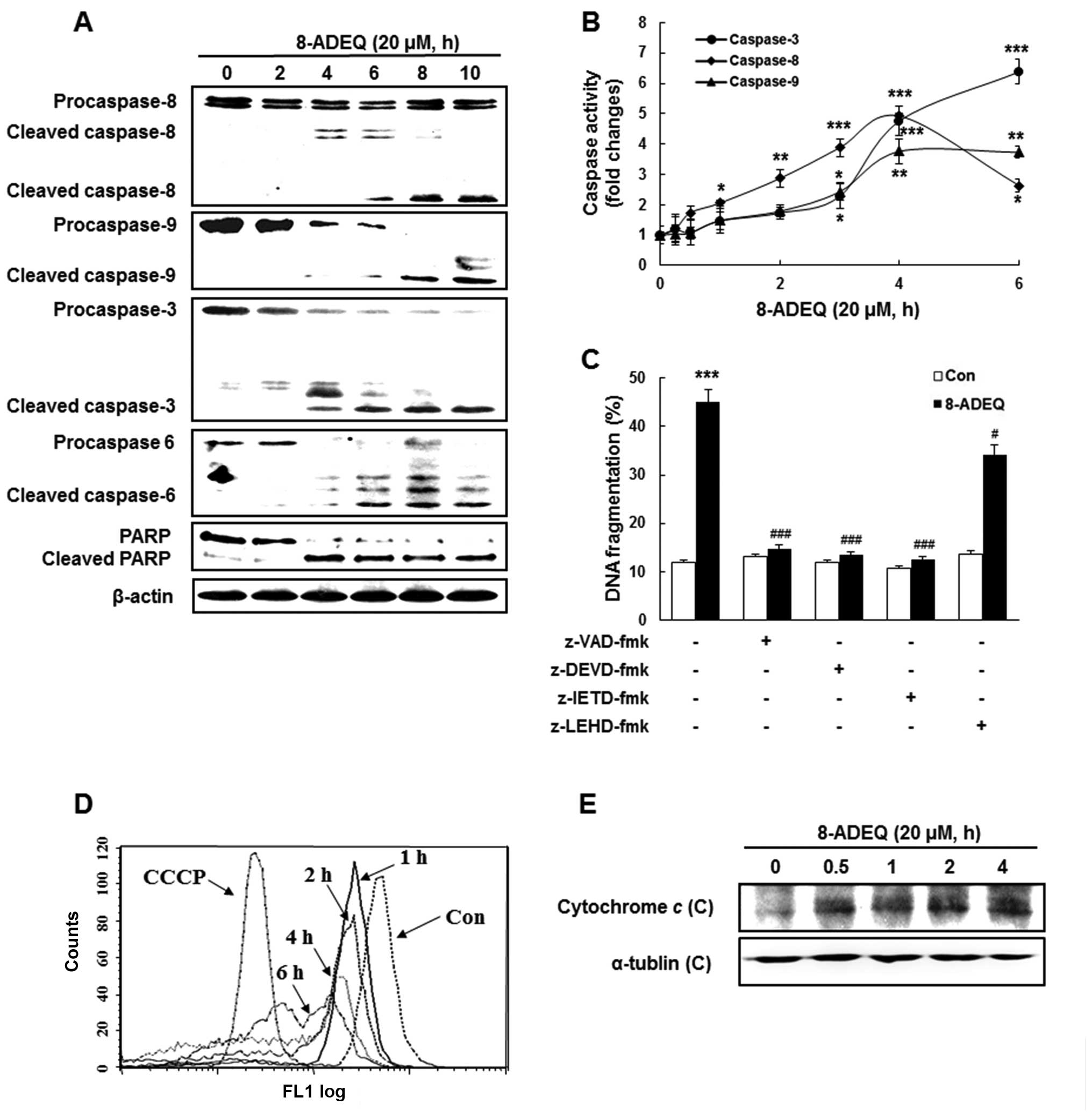 | Figure 2.8-ADEQ-treatment leads to caspase
activation, loss of mitochondrial membrane potential, and cytosolic
release of cytochrome c in HL-60 cells. (A) HL-60 cells were
treated with 20 µM 8-ADEQ for the indicated times. Each protein
fraction was detected by western blotting using specific
antibodies. The amount of β-actin was used as an internal control.
(B) 8-ADEQ induced caspase-3, −8, −9 activation as measured by a
luminometric assay. HL-60 cells were treated with 20 µM 8-ADEQ for
the indicated times. (C) HL-60 cells were pretreated with or
without 50 µM z-VAD-fmk, z-DEVD-fmk, z-IETD-fmk, z-LEHD-fmk for 1
h, and then treated with 20 µM 8-ADEQ for 4 h. The quantification
of DNA fragmentation was with the DAPI assay. (D) Cells were
treated with 20 µM 8-ADEQ for the indicated times, stained with
DiOC6, and analyzed by flow cytometry. CCCP (100 µM) was
used as a positive control. (E) HL-60 cells were treated with 20 µM
8-ADEQ for the indicated times, and the cytosolic (C) fractions
were prepared as described in Materials and methods. Cytochrome
c levels were analyzed by western blotting. α-tubulin was
used as the internal control. Data presented are the means ± SD of
results from three independent experiments. *P<0.05,
**P<0.01, ***P<0.001 vs. control group,
#P<0.05, ###P<0.001 vs. 8-ADEQ-treated
group. |
To determine whether the activation of caspases was
required for the induction of apoptosis by 8-ADEQ, we pretreated
HL-60 cells with a number of caspase inhibitors. z-VAD-fmk (a broad
caspase inhibitor), z-DEVD-fmk (a caspase-3 inhibitor), and
z-IETD-fmk (a caspase-8 inhibitor), apparently inhibited
8-ADEQ-induced DNA fragmentation, whereas z-LEHD-fmk, an inhibitor
of caspase-9 activity, had little effect on reducing DNA
fragmentation compared to the other inhibitors (Fig. 2C). The results indicated that
procaspase-3 and procaspase-8 were involved in the 8-ADEQ-induced
apoptosis in HL-60 cells. These observations indicated that the
caspase cascade may be involved in 8-ADEQ-induced apoptosis. Thus
caspase-8 and −3 dependent pathways are the main stream in
8-ADEQ-induced apoptosis. Because 8-ADEQ also induced caspase-9
cleavage, an initial caspase in the mitochondrial apoptotic
pathway, we investigated whether 8-ADEQ was capable of inducing
∆Ψm depolarization using DiOC6, a
mitochondria-specific voltage-dependent dye. Treatment of cells
with 8-ADEQ at 20 µM caused the dissipation of ∆Ψm in a
time-dependent manner, as did CCCP, a positive control (Fig. 2D). Furthermore, the levels of
cytosolic cytochrome c were elevated by 8-ADEQ in HL-60
cells, suggesting the involvement of the mitochondrial apoptotic
pathway (Fig. 2E).
8-ADEQ induces apoptosis via the
caspase-8-dependent pathway
Because 8-ADEQ induced caspase-8 activation at an
early time-point, we further examined the mechanism of caspase-8
activation during 8-ADEQ-induced apoptosis using the caspase-8
inhibitor, z-IETD-fmk. Treatment with z-IETD-fmk completely
inhibited 8-ADEQ-induced caspase-3, −8, and −9 activation using the
caspase activity assay (Fig. 3A).
In addition, 8-ADEQ-induced internucleosomal DNA fragmentation was
significantly abolished in the presence of z-IETD-fmk (Fig. 3B and C). These results indicated
that the procaspase-8 activation is essential in the 8-ADEQ-induced
apoptosis.
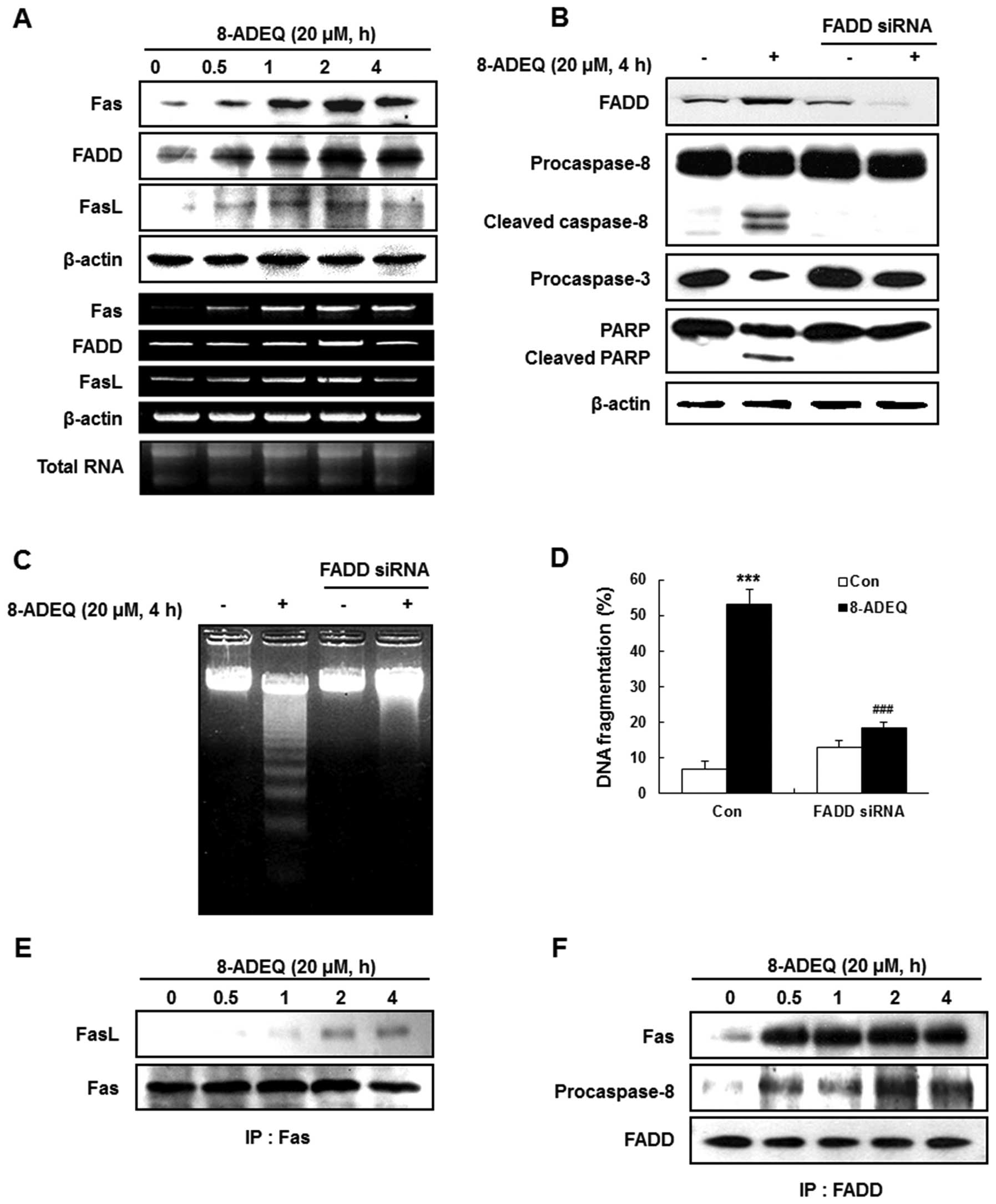
8-ADEQ upregulates Fas and its ligand
and increases the formation of DISC
We demonstrated that procaspase-8 was activated, and
the activated caspase-8 was required in the 8-ADEQ-induced
apoptosis. Caspase-8 acts as the initiator caspase in apoptosis
that results from ligand-driven engagement of various death
receptors such as TNFR1 and Fas (36), or from stimuli that include
oxidative stress and DNA damage (12). To enhance the caspase-8 initiation,
an adaptor protein, FADD is also required to be recruited to the
receptor called DISC (37). To
determine whether 8-ADEQ-induced caspase-8 activation involves the
death receptor pathway, HL-60 cells were treated with 20 µM 8-ADEQ
for various time-points, and the Fas, FasL and FADD protein and
mRNA level changes were monitored by western blotting and RT-PCR,
respectively. The expression levels of the death receptor related
proteins and their mRNA (Fas, FADD and FasL) were apparently
increased by 8-ADEQ in a time-dependent manner (Fig. 4A).
It has been known that FADD plays an important role
to induce an apoptosis signal (37). To further confirm the requirement of
FADD in 8-ADEQ-induced apoptosis, we transiently transfected HL-60
cells with FADD siRNA, leading to a complete reduction of the
protein levels of FADD in the cells (Fig. 4B). The FADD siRNA-transfected cells
were then treated with 8-ADEQ (20 µM) for 4 h. The knockdown of
FADD significantly prevented the 8-ADEQ-induced cleavage of
procaspase-8, procaspase-3, and PARP (Fig. 4B). We also observed that
8-ADEQ-induced DNA fragmentation was completely prevented in the
FADD siRNA-transfected cells, as seen by either the 2% agarose gel
electrophoresis or DAPI staining (Fig.
4C and D). Therefore, FADD-caspase-8-mediated apoptosis plays a
pivotal role in the observed apoptotic induction by 8-ADEQ in HL-60
cells.
Fas and FasL are cell surface membrane proteins that
belong to the TNF-α superfamily proteins and FasL trimer binds
three Fas molecules (36). Because
the death domains present in each receptor chain have a propensity
to associate with one another, Fas ligation leads to clustering of
the death domains. Then, an adaptor protein, FADD also binds its
own death domain to the clustered receptor death domains. FADD also
contains a death effector domain that binds to an analogous domain
repeated in procaspase-8, as part of forming the DISC. Upon
recruitment by FADD, procaspase-8 oligomerization drives its
activation through self-cleavage (38). To identify whether the DISC
formation results in activation of procaspase-8 in 8-ADEQ-induced
apoptosis, we immunoprecipitated Fas and detected bound FasL; FADD
and detected bound Fas and procaspase-8 by western blotting. We
observed that Fas is associated with FADD, FasL, and procaspase-8
and all these proteins were detected in the Fas
immunoprecipitations and at a time-point earlier than that required
for first detection of typical apoptotic features (Fig. 4E and F). These data suggested that
8-ADEQ treatment leads to formation of DISC and that it precedes
the activation of procaspase-8.
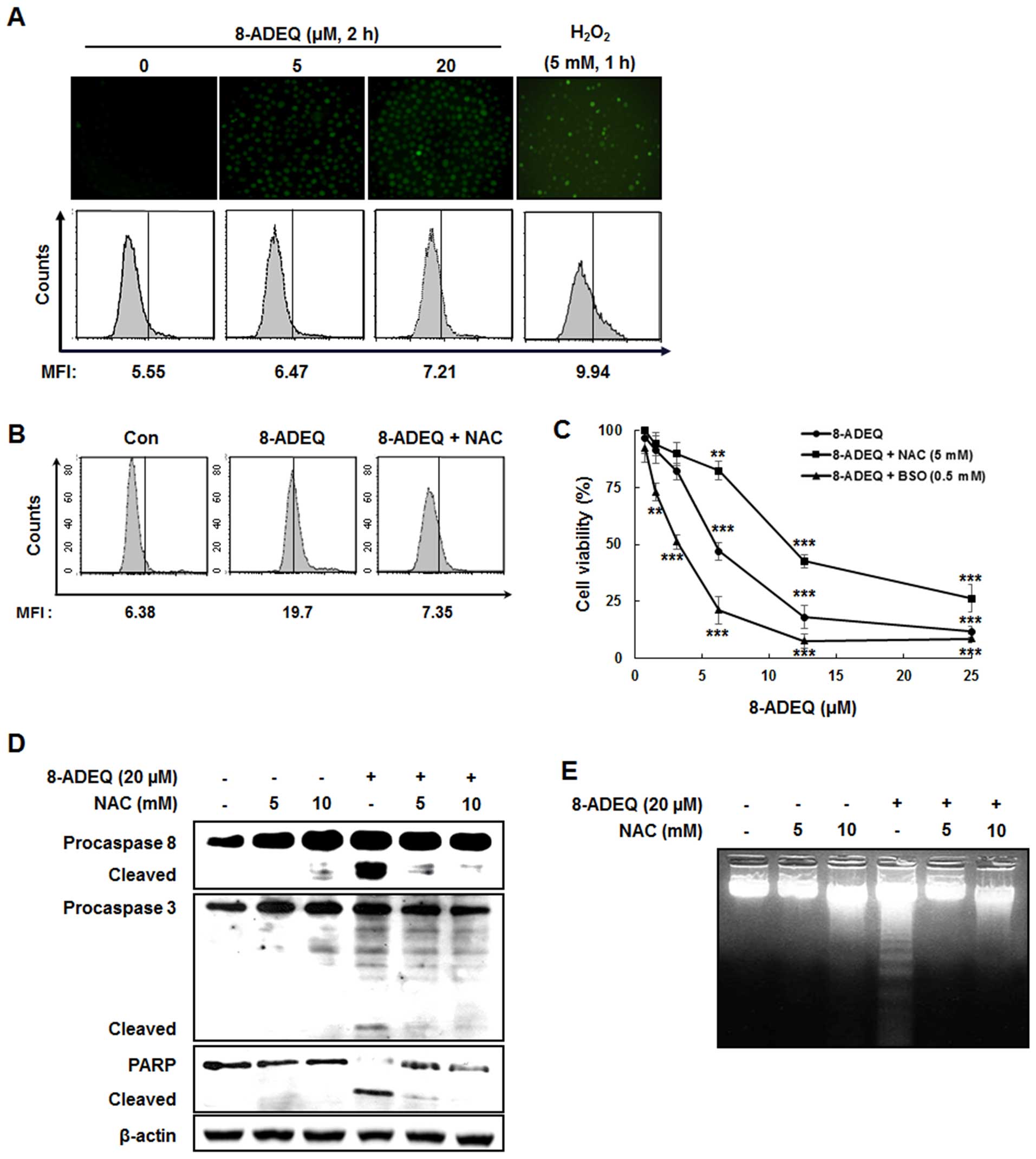
8-ADEQ induces ROS-mediated
apoptosis
It is known that formation of ROS is an early signal
that mediates apoptosis (39), and
a recent study indicated that activation of Fas was linked to
generation of ROS in some cell lines (40). To test the possibility that ROS are
required for 8-ADEQ-induced apoptosis, we first measured the levels
of ROS using a flow cytometric assay. After treatment with 8-ADEQ
for 2 h, generation of ROS appeared in a concentration-dependent
manner (Fig. 5A) and this ROS
generation was blocked by pretreatment of NAC, antioxidant
(Fig. 5B). To determinate whether
the generation of ROS is a crucial step in 8-ADEQ-induced cell
death, we examined the cell viability of 8-ADEQ-treated HL-60 cells
in the presence of NAC, or BSO, an inhibitor of GSH synthesis.
Pretreatment of cells with NAC completely inhibited the
8-ADEQ-induced cell death, whereas BSO pretreatment, resulting in
GSH depletion, and enhanced cell death by 8-ADEQ (Fig. 5C). Furthermore, NAC potently
inhibited the 8-ADEQ-induced activation of caspase-8, and −3 and
DNA fragmentation (Fig. 5D and E),
and this indicated that ROS was required for 8-ADEQ-induced
apoptosis.
Discussion
As resveratrol has been found to have a variety of
desirable effects with respect to various cell types (41), a number of resveratrol analogues
were also evaluated for their biological activities (21,26,27).
With respect to various lead compounds, a series of
styrylquinazoline derivatives were synthesized with the variation
of the benzene ring to a quinazoline ring. There are reported
pharmacological activities of these styrylquinazoline derivatives;
these include HIV integrase inhibitors and PGE2
production inhibitors (22,42).
(E)-8-acetoxy-2-[2-(3,4-diacetoxyphenyl)ethenyl]-quinazoline
(8-ADEQ) was also found to have anti-proliferative effects on human
cervical cancer HeLa cells by DNA damage-mediated G2/M
cell cycle arrest via activation of both Chk1/2-Cdc25 and
p53-p21CIP1/WAF1 through the ATM/ATR pathways (23). However, the cytotoxic and
apoptosis-inducing activities of 8-ADEQ on leukemia had not been
studied in detail. In this study, we first determined that
8-ADEQ-treated HL-60 cells went into apoptotic cell death,
characterized by several morphological changes, membrane blebbing,
chromatin condensation, DNA cleavages, and PS exposure.
Apoptosis is a fundamental cellular activity and
provides protection against cancer development by eliminating
genetically altered cells and hyper-proliferative cells. It has
been demonstrated that defects in apoptosis signaling pathways
contribute to carcinogenesis and chemoresistance (43). As mentioned above, there are two
major apoptotic pathways: the extrinsic death receptor-mediated
pathway and the intrinsic mitochondria-mediated pathway. Both of
these pathways are regulated by caspases, which are responsible,
either directly or indirectly, for cleavage of cellular proteins, a
characteristic of apoptosis (34).
In this study, 8-ADEQ increased the levels of caspase-8, −9, −3,
and −6 and cleaved PARP in a time-dependent manner. By monitoring
the kinetic activities of caspase-8, −3 and −9 by using
luminometric assay with specific substrates, caspase-8 was
initially activated, followed by the other caspases including
caspase-3 and −9. Pretreatment with various caspase inhibitors
markedly prevented 8-ADEQ-induced DNA fragmentation (Fig. 2). These findings indicated that
caspase-3 and −8 play fundamental roles in 8-ADEQ-induced apoptosis
in HL-60 cells. It is noteworthy that specific inhibition of
caspase-9 by z-LEHD-fmk had only partial inhibitory effect on
8-ADEQ-stimulated apoptosis. In this regard, it appears that
activation of the intrinsic mitochondria-mediated pathway alone is
not sufficient to explain the apoptotic effects of 8-ADEQ in HL-60
leukemia cells. The next interesting result emerged after the use
of a specific caspase-8 inhibitor. In general, the activation of
procaspase-8 precedes those of procaspase-3, and caspase-3 is
activated prior to caspase-6, and may be responsible for activation
of caspase-6 (34). Our data
clearly showed that the inhibition of caspase-8 activity blocked
the 8-ADEQ-induced activation of caspase-8, −3 and −9, as seen by
using luminogenic substrates. In addition, it was found that a
caspase-8 inhibitor significantly inhibited 8-ADEQ-induced
apoptosis as determined by DNA fragmentation. Therefore, we suggest
that apoptosis induced by 8-ADEQ was mediated via a
caspase-8-dependent mechanism.
Caspase-8 acts as the initiator caspase in apoptosis
resulting from ligand-driven engagement of death receptors such as
FasL, TRAIL, and TNFR1 (36), or
from the stimuli, including oxidative stress and DNA damage
(12). In this study, Fas
expression was markedly increased in HL-60 cells treated with
8-ADEQ, while that for FasL was only mildly increased. We also
showed that 8-ADEQ changed not only the expression of Fas-mediated
proteins but also their recruitment to Fas. 8-ADEQ resulted in
oligomerization of receptors at the cell membrane and formation of
DISC that included the adapter protein FADD. This led to activation
of caspase-8, and is consistent with the notion presented by many
reports (37,44). Our results conclusively suggest that
activation of caspase-8 is the initial step in 8-ADEQ-induced
apoptosis through DISC formation with FasL, Fas, FADD, and
procaspase-8. In support of this, cells transfected with FADD siRNA
showed a clear diminution of 8-ADEQ-induced cleavage of caspase-8,
caspase-3 and PARP and DNA fragmentation. Therefore, 8-ADEQ-induced
caspase-8 activation is probably due to the stimulation of the
death receptors.
Many apoptosis-inducing agents are accompanied by
loss of MMP, representing a decisive point of irreversible process
in the induction of apoptosis (45). From loss of MMP, cytochrome c
and other pro-apoptotic proteins are released from the
mitochondrial intermembrane space into the cytosol where a caspase
cascade is initiated (46).
Cytochrome c release into cytosol by 8-ADEQ indicated that
MMP was reduced during 8-ADEQ-induced apoptosis. Bid, a BH3-only
proapoptotic member of the Bcl-2 family, undergoes proteolysis by
caspase-8, previously activated by cell surface death receptors
such as Fas and TNF. Truncated Bid (tBid) translocates to
mitochondria and binds to anti-apoptotic proteins such as Bcl-2 and
Bcl-xL, and that leads to a conformational change in Bax,
mitochondrial depolarization, and cytochrome c release from
mitochondria (47).
Conformationally altered Bax also stimulates the cytochrome
c-releasing activity of Bid by cell surface death receptors
such as Fas and TNF and is antagonized by Bcl-2 (45). It is not known whether
apoptosis-inducing effects of 8-ADEQ through MMP depolarization and
cytosolic cytochrome c release are caused by a death
receptor-caspase-8-dependent pathway or are a direct effect on
mitochondria, and this remains to be determined.
Several reports have suggested that oxidative stress
is a common mediator of several systems of apoptosis (40), although the role of ROS in apoptosis
induction remains controversial. It has been reported that ROS act
in the oxidation of the mitochondrial pores, leading to the release
of cytochrome c from the intracellular membrane (10). On the other hand, it is shown that
ROS promote the upstream elements of Fas-mediated signaling, as the
cell death induced by Fas receptor has been suggested to be
redox-regulated, or dependent on ROS (40). The mechanism by which 8-ADEQ
promotes DISC formation appears to be dependent on ROS generation
since the 8-ADEQ-induced ROS generation and activation of caspase-8
and caspase-3 and DNA fragmentation were prevented by antioxidant,
NAC, treatment. Therefore, the effects of ROS may be on caspase-8
or other initiator death receptors. It is not yet known whether
8-ADEQ directly generates ROS, which then affects Fas expression
and the association of Fas-related protein, or 8-ADEQ-induced Fas
receptor activation induces ROS and then alters the redox status.
Here we showed that 8-ADEQ induced oxidative stress, and induced
DISC assembly, followed by caspase-8 activation and apoptosis.
These data support other reports on modifying cellular oxidative
status or levels of antioxidant defenses can alter the sensitivity
to Fas-induced apoptosis (48).
In conclusion, our results demonstrated that 8-ADEQ
inhibited the viability of HL-60 cells and these effects were
mediated through induction of apoptosis. 8-ADEQ induced the
activation of caspase-3, −6, −8, and −9, and accumulation of ROS.
In addition, a caspase-8 inhibitor completely abolished caspase-3
and caspase-9 activation, and subsequent DNA fragmentation by
8-ADEQ. Therefore, caspase-8 plays a key role in 8-ADEQ-stimulated
apoptosis through a potent induction of Fas-mediated signaling.
Moreover, the antioxidant NAC significantly attenuated the 8-ADEQ
effects on caspase-8, and caspase-3 activities and DNA
fragmentation. Based on these findings, we suggest that 8-ADEQ, via
its potent apoptotic activity, has the potential as a therapeutic
and chemopreventive agent for leukemia.
Acknowledgements
This study was supported by the research grant from
the Korea Food Research Institute (E0145202).
References
|
1
|
Debatin KM: Apoptosis pathways in cancer
and cancer therapy. Cancer Immunol Immunother. 53:153–159. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Previati M, Lanzoni I, Corbacella E,
Magosso S, Guaran V, Martini A and Capitani S: Cisplatin-induced
apoptosis in human promyelocytic leukemia cells. Int J Mol Med.
18:511–516. 2006.PubMed/NCBI
|
|
3
|
Fulda S, Strauss G, Meyer E and Debatin
KM: Functional CD95 ligand and CD95 death-inducing signaling
complex in activation-induced cell death and doxorubicin-induced
apoptosis in leukemic T cells. Blood. 95:301–308. 2000.PubMed/NCBI
|
|
4
|
Odoux C and Albers A: Additive effects of
TRAIL and paclitaxel on cancer cells: Implications for advances in
cancer therapy. Vitam Horm. 67:385–407. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Karpinich NO, Tafani M, Rothman RJ, Russo
MA and Farber JL: The course of etoposide-induced apoptosis from
damage to DNA and p53 activation to mitochondrial release of
cytochrome c. J Biol Chem. 277:16547–16552. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Clarke PG: Apoptosis: From morphological
types of cell death to interacting pathways. Trends Pharmacol Sci.
23:308–309; author reply 310. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wajant H: The Fas signaling pathway: More
than a paradigm. Science. 296:1635–1636. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Debatin KM and Krammer PH: Death receptors
in chemotherapy and cancer. Oncogene. 23:2950–2966. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kaufmann SH and Earnshaw WC: Induction of
apoptosis by cancer chemotherapy. Exp Cell Res. 256:42–49. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Giles GI: The redox regulation of thiol
dependent signaling pathways in cancer. Curr Pharm Des.
12:4427–4443. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Roos WP and Kaina B: DNA damage-induced
cell death by apoptosis. Trends Mol Med. 12:440–450. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Devadas S, Hinshaw JA, Zaritskaya L and
Williams MS: Fas-stimulated generation of reactive oxygen species
or exogenous oxidative stress sensitize cells to Fas-mediated
apoptosis. Free Radic Biol Med. 35:648–661. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Micheau O, Solary E, Hammann A and
Dimanche-Boitrel MT: Fas ligand-independent, FADD-mediated
activation of the Fas death pathway by anticancer drugs. J Biol
Chem. 274:7987–7992. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Santiago B, Galindo M, Palao G and Pablos
JL: Intracellular regulation of Fas-induced apoptosis in human
fibroblasts by extracellular factors and cycloheximide. J Immunol.
172:560–566. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shen HM and Pervaiz S: TNF receptor
superfamily-induced cell death: Redox-dependent execution. FASEB J.
20:1589–1598. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reinehr R, Becker S, Eberle A,
Grether-Beck S and Häussinger D: Involvement of NADPH oxidase
isoforms and Src family kinases in CD95-dependent hepatocyte
apoptosis. J Biol Chem. 280:27179–27194. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bishayee K, Mondal J, Sikdar S and
Khuda-Bukhsh AR: Condurango (Gonolobus condurango) extract
activates fas receptor and depolarizes mitochondrial membrane
potential to induce ROS-dependent apoptosis in cancer cells in
vitro: CE-treatment on HeLa: a ROS-dependent mechanism. J
Pharmacopuncture. 18:32–41. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Frémont L: Biological effects of
resveratrol. Life Sci. 66:663–673. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bhat KP and Pezzuto JM: Cancer
chemopreventive activity of resveratrol. Ann NY Acad Sci.
957:210–229. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Savouret JF and Quesne M: Resveratrol and
cancer: A review. Biomed Pharmacother. 56:84–87. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bavaresco L, Fregoni C, Cantù E and
Trevisan M: Stilbene compounds: From the grapevine to wine. Drugs
Exp Clin Res. 25:57–63. 1999.PubMed/NCBI
|
|
22
|
Park JH, Min HY, Kim SS, Lee JY, Lee SK
and Lee YS: Styrylquinazolines: A new class of inhibitors on
prostaglandin E2 production in lipopolysaccharide-activated
macrophage cells. Arch Pharm (Weinheim). 337:20–24. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim JY, Choi HE, Lee HH, Shin JS, Shin DH,
Choi JH, Lee YS and Lee KT: Resveratrol analogue
(E)-8-acetoxy-2-[2-(3,4-diacetoxyphenyl)ethenyl]-quinazoline
induces G2/M cell cycle arrest through the activation of
ATM/ATR in human cervical carcinoma HeLa cells. Oncol Rep.
33:2639–2647. 2015.PubMed/NCBI
|
|
24
|
Hung LM, Chen JK, Lee RS, Liang HC and Su
MJ: Beneficial effects of astringinin, a resveratrol analogue, on
the ischemia and reperfusion damage in rat heart. Free Radic Biol
Med. 30:877–883. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Aldawsari FS, Aguiar RP, Wiirzler LA,
Aguayo-Ortiz R, Aljuhani N, Cuman RK, Medina-Franco JL, Siraki AG
and Velázquez-Martínez CA: Anti-inflammatory and antioxidant
properties of a novel resveratrol-salicylate hybrid analog. Bioorg
Med Chem Lett. 26:1411–1415. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Larrosa M, Tomás-Barberán FA and Espín JC:
The grape and wine polyphenol piceatannol is a potent inducer of
apoptosis in human SK-Mel-28 melanoma cells. Eur J Nutr.
43:275–284. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang Y, Wang B, Cheng J, Yang L, Liu ZL,
Balan K, Pantazis P, Wyche JH and Han Z: FADD-dependent apoptosis
induction in Jurkat leukemia T-cells by the resveratrol analogue,
3,4,5-trihydroxy-trans-stilbene. Biochem Pharmacol. 69:249–254.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Richard N, Porath D, Radspieler A and
Schwager J: Effects of resveratrol, piceatannol,
tri-acetoxystilbene, and genistein on the inflammatory response of
human peripheral blood leukocytes. Mol Nutr Food Res. 49:431–442.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Choi YJ, Yang KM, Kim SD, Yoo YH, Lee SW,
Seo SY, Suh H, Yee ST, Jeong MH and Jo WS: Resveratrol analogue
HS-1793 induces the modulation of tumor-derived T cells. Exp Ther
Med. 3:592–598. 2012.PubMed/NCBI
|
|
30
|
Ortín I, González JF, Cuesta EL,
Manguan-García C, Perona R and Avendaño C: Cytotoxicity mechanisms
of pyrazino[1,2-b]isoquinoline-4-ones and SAR studies. Bioorg Med
Chem. 17:8040–8047. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kuo HP, Chuang TC, Tsai SC, Tseng HH, Hsu
SC, Chen YC, Kuo CL, Kuo YH, Liu JY and Kao MC: Berberine, an
isoquinoline alkaloid, inhibits the metastatic potential of breast
cancer cells via Akt pathway modulation. J Agric Food Chem.
60:9649–9658. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Khodarev NN, Sokolova IA and Vaughan AT:
Mechanisms of induction of apoptotic DNA fragmentation. Int J
Radiat Biol. 73:455–467. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pozarowski P, Grabarek J and Darzynkiewicz
Z: Flow cytometry of apoptosis. Curr Protoc Cell Biol. Chapter 18:
Unit 18.8. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fan TJ, Han LH, Cong RS and Liang J:
Caspase family proteases and apoptosis. Acta Biochim Biophys Sin
(Shanghai). 37:719–727. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kaufmann SH, Desnoyers S, Ottaviano Y,
Davidson NE and Poirier GG: Specific proteolytic cleavage of
poly(ADP-ribose) polymerase: An early marker of
chemotherapy-induced apoptosis. Cancer Res. 53:3976–3985.
1993.PubMed/NCBI
|
|
36
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: A
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Imtiyaz HZ, Rosenberg S, Zhang Y, Rahman
ZS, Hou YJ, Manser T and Zhang J: The Fas-associated death domain
protein is required in apoptosis and TLR-induced proliferative
responses in B cells. J Immunol. 176:6852–6861. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Muzio M: Signalling by proteolysis: Death
receptors induce apoptosis. Int J Clin Lab Res. 28:141–147. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nordberg J and Arnér ES: Reactive oxygen
species, antioxidants, and the mammalian thioredoxin system. Free
Radic Biol Med. 31:1287–1312. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Circu ML and Aw TY: Reactive oxygen
species, cellular redox systems, and apoptosis. Free Radic Biol
Med. 48:749–762. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bhat KPL, Kosmeder JW II and Pezzuto JM:
Biological effects of resveratrol. Antioxid Redox Signal.
3:1041–1064. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee JY, Park JH, Lee SJ, Park H and Lee
YS: Styrylquinazoline derivatives as HIV-1 integrase inhibitors.
Arch Pharm (Weinheim). 335:277–282. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hickman JA: Apoptosis induced by
anticancer drugs. Cancer Metastasis Rev. 11:121–139. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jin Z and El-Deiry WS: Distinct sgnaling
pathways in TRAIL- versus tumor necrosis factor-induced apoptosis.
Mol Cell Biol. 26:8136–8148. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tait SW and Green DR: Mitochondria and
cell death: Outer membrane permeabilization and beyond. Nat Rev Mol
Cell Biol. 11:621–632. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang X, Zhang S, Zhu S, Chen S, Han J,
Gao K, Zeng JZ and Yan X: Identification of mitochondria-targeting
anticancer compounds by an in vitro strategy. Anal Chem.
86:5232–5237. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Desagher S, Osen-Sand A, Nichols A, Eskes
R, Montessuit S, Lauper S, Maundrell K, Antonsson B and Martinou
JC: Bid-induced conformational change of Bax is responsible for
mitochondrial cytochrome c release during apoptosis. J Cell Biol.
144:891–901. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Trachootham D, Lu W, Ogasawara MA, Nilsa
RD and Huang P: Redox regulation of cell survival. Antioxid Redox
Signal. 10:1343–1374. 2008. View Article : Google Scholar : PubMed/NCBI
|