Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the
fourth leading cause of cancer deaths worldwide with a 5-year
survival rate of less than 7%, a mortality rate that is nearly
identical to the incidence rate (1,2). The
main reason for the poor survival rate is that the current
diagnostic methods lack the sensitivity and the specificity to
identify markers of PDAC in the early stages, resulting in delayed
diagnosis until the later stages of the disease. In 2015, fewer
than 20% of newly diagnosed patients with pancreatic cancer were
classified as having resectable cancer (3). Therefore, it is necessary to
characterize PDACs precursor lesions and the pathological
mechanisms regulating the genetic progression from normal cells to
PDAC (4).
Pancreatic intraepithelial neoplasia (PanIN) is the
most common pancreatic precursor lesion. Based on increasing
degrees of architectural and nuclear atypia, PanIN are grouped into
three histological stages: PanIN-1 (PanIN-1A and PanIN-1B), PanIN-2
and PanIN-3 (5). Activating
mutations in the KRAS oncogene are found in all three PanIN
stages and over 90% of invasive PDAC (6). A genetically engineered mouse model,
Pdx-cre; KrasG12D, which uses a Pdx1 promoter to drive
the expression of mutant Kras, is able to recapitulate the
progression of human PanIN to PDAC successfully and serves as a
convenient tool to explore the precursor lesions and the
pathological mechanisms of the pancreatic cancer (7).
AP-1 component members, c-Fos and c-Jun, participate
in the regulation of many processes, including proliferation, cell
cycle progression, migration, differentiation, apoptosis and
angiogenesis (8,9). In addition to the specific role of
c-Fos as a differentiation transcription factor for osteoclasts and
bone resorbing cells, c-Fos also has an important role in the tumor
formation and suppression. It has been reported that c-Fos
downregulation in MCF-7/ADR cells results in enhanced apoptosis and
altered expression of apoptosis-associated proteins, including Bax,
Bcl-2, p53 and PUMA (10). The
ERK/c-Fos/MMP-7 pathway plays an important role in modulating the
invasion of colon cancer cells and inhibition of this pathway holds
promise as a treatment for the colon cancer metastasis (11).
In the present study, we investigated the role of
AP-1 in initiating the PanIN-PDAC progression in vivo using
Pdx-cre; KrasG12D mouse model. Our results indicate that
the c-Fos expression, but not the c-Jun expression, increased
during the development from precursor lesions to tumor. We
demonstrated that ERK activates c-Fos to promote the PanIN-PDAC
progression through initiation of proliferation, inflammation and
apoptosis. These results will help to characterize the early
prognosis of the pancreatic cancer.
Materials and methods
Cell culture and transfection
Cell lines were obtained from the Cell Resource
Center in Peking Union Medical College (PUMC; Beijing, China).
Panc-1 cells were cultured in Dulbeccos modified Eagles medium
(DMEM; HyClone Laboratories; Thermo Fisher Scientific) with 10%
fetal bovine serum (FBS) and PCT-3 cells were cultured in RPMI-1640
(HyClone Laboratories; Thermo Fisher Scientific) with 10% FBS in 5%
CO2 at 37°C. Adherent cells were passaged every 2–3 days
with 0.5 mg/ml trypsin (1:250) and 0.53 mM
ethylenediaminetetraacetic acid (EDTA). siRNA oligonucleotides were
designed against c-Fos as follows: GGGGCAAGGTGGA ACAGTTAT. Cells
were transfected using Lipofectamine™ 2000 (Invitrogen, Carlsbad,
CA, USA).
Western blot analysis
Proteins were extracted with SDS lysis buffer [50 mM
Tris-HCl (pH 6.8), 10% glycerol and 2% SDS] and quantified using
the BCA protein assay reagent (Thermo Fisher Scientific). Extracts
were separated on a 12% SDS-PAGE gel and electrophoretically
transferred to PVDF membrane (GE Healthcare). The membrane was
blocked in 5% skim milk for 1 h at room temperature and then
incubated overnight with the indicated antibodies at 4°C.
Antibodies against c-Fos (1:1,000) and GAPDH (1:3,000) were
purchased from Cell Signaling Technology (Danvers, MA, USA). The
membrane was incubated with an anti-rabbit or an anti-mouse IgG-HRP
(Santa Cruz Biotechnology, Santa Cruz, CA, USA) for 1 h at room
temperature. Chemiluminescence was detected using an ECL blot
detection system (Santa Cruz Biotechnology).
Cell proliferation assay
Cell proliferation was assessed by the CCK-8 assay
(Dojindo Molecular Technologies). Cells were seeded at a density of
2,000 cells/well in 96-well plates. A total of 10 µl CCK-8 solution
was added to each well containing 100 µl of culture medium and
incubated for 2 h at 37°C. Absorbance was measured at 450 nm using
a multiwell spectrophotometer (BioTek Instruments, Inc., Winooski,
VT, USA).
Cell cycle assay
Cells were transfected in 6-well plates
(5×105 cells/well). After 48 h, cells were harvested,
washed with cold phosphate-buffered saline (PBS) and fixed in 70%
ethanol overnight at 4°C. After centrifugation at 800–1,000 rpm
three times, cells were re-suspended with 500 µl PBS. Cells were
stained with a solution containing 10 mg/ml RNase A, 0.1% Triton
X-100 and 1 mg/ml propidium iodide. Cell cycle analysis was
performed by fluorescence-activated cell sorting.
Wound-healing assay
Cells were transfected in 6-well plates
(5×105 cells/well). After 24 h, a monolayer of cells was
scratched by a 200 ml tip in serum-free medium. Cell migration was
quantified by the area of migrated cells to the scratched cell-free
zone after 24 h, and measured by ImageJ software.
Mouse model and in vivo treatment
The genetically engineered mouse stains Pdx1-cre and
LSL-KrasG12D used in the present study were purchased
from the National Cancer Institute (NCI; Rockville, MD, USA).
Pdx-cre; KrasG12D mice were generated by crossing
LSL-KrasG12D mice with Pdx1-cre mice. The mutant mice
were genotyped by PCR using primers as followed: lox sense
primer: AGCTAGCCACCATGGCTT GAGTAAGTCTGCA and lox antisense
primer: CCTTTACA AGCGCACGCAGACTGTAGA; cre sense primer:
CTGGA CTACATCTTGAGTTGC and cre antisense primer: GGTG
TACGGTCAGTAAATTTG. Animals were housed in a clean vivarium and fed
standard mouse chow. To accelerate carcinogenesis in Pdx-cre;
KrasG12D mice, an intraperitoneal injection of caerulein
(50 µg/kg/day) was administered. ERK/c-Fos signaling blockade was
accomplished using the inhibitor U0126 (500 µg/kg) every two
days.
Animals were housed in the Experimental Animal
Center of Peking Union Medical College Hospital. They were
maintained in a 12-h light/dark cycle and temperature-controlled
environment with ad libitum access to water and food.
Animals were cared for and studies were performed in accordance
with the principles of the 3Rs (replacement, reduction and
refinement) and guidelines of the National and Beijing Experimental
Animal Welfare Ethics.
Immunohistochemical analyses
Freshly isolated biopsies of the pancreas were fixed
in 10% neutral-buffered formulin (Sigma-Aldrich) for 16 h and
embedded in paraffin. Next, 5-µm-thick sections were
deparaffinized, rehydrated and treated in boiling sodium citrate
buffer (10 mM pH 6.0) for 10 min to unmask antigens. Endogenous
peroxidase activity was quenched by treating the slides with 3%
hydrogen peroxide for 10 min. Sections were blocked in PBS
containing 10% goat serum, 1% BSA and 0.1% Triton X-100 for 1 h at
room temperature prior to being incubated overnight at 4°C with
antibodies: JNK (ab179461, Abcam, 1:100), p38 (ab170099, Abcam,
1:50), caspase-3 (ab13847, Abcam, 1:500), CK19 (10712-1-AP,
Proteintech, 1:100), MMP9 (ab38898, Abcam, 1:100), ERK1/2 (#4695,
Cell Signaling Technology, 1:100), amylase (12540-1-AP,
Proteintech, 1:100), CD45 (ab10558, Abcam, 1:100). After incubation
with secondary biotinylated antibodies (1:200; KPL), the presence
of the antigens was revealed using diaminobenzadine tetrachloride
(DAB; Dako) and counter stained with nuclear red or with
heamatoxylin (blue). The relative quantities of IHC reaction were
accessed by Image-Pro Plus 6.0.
Statistical analyses
Statistical analyzes were performed using the
Students t-test (two-tailed) in Microsoft Excel software
(Microsoft, Redmond, WA, USA). The results are presented as the
mean ± standard deviation of triplicates of each experiment. All
experiments were performed in triplicate unless stated otherwise.
P<0.05 were considered statistically significant.
Results
C-Fos promotes proliferation, cell
cycle and migration in human pancreatic cancer cells
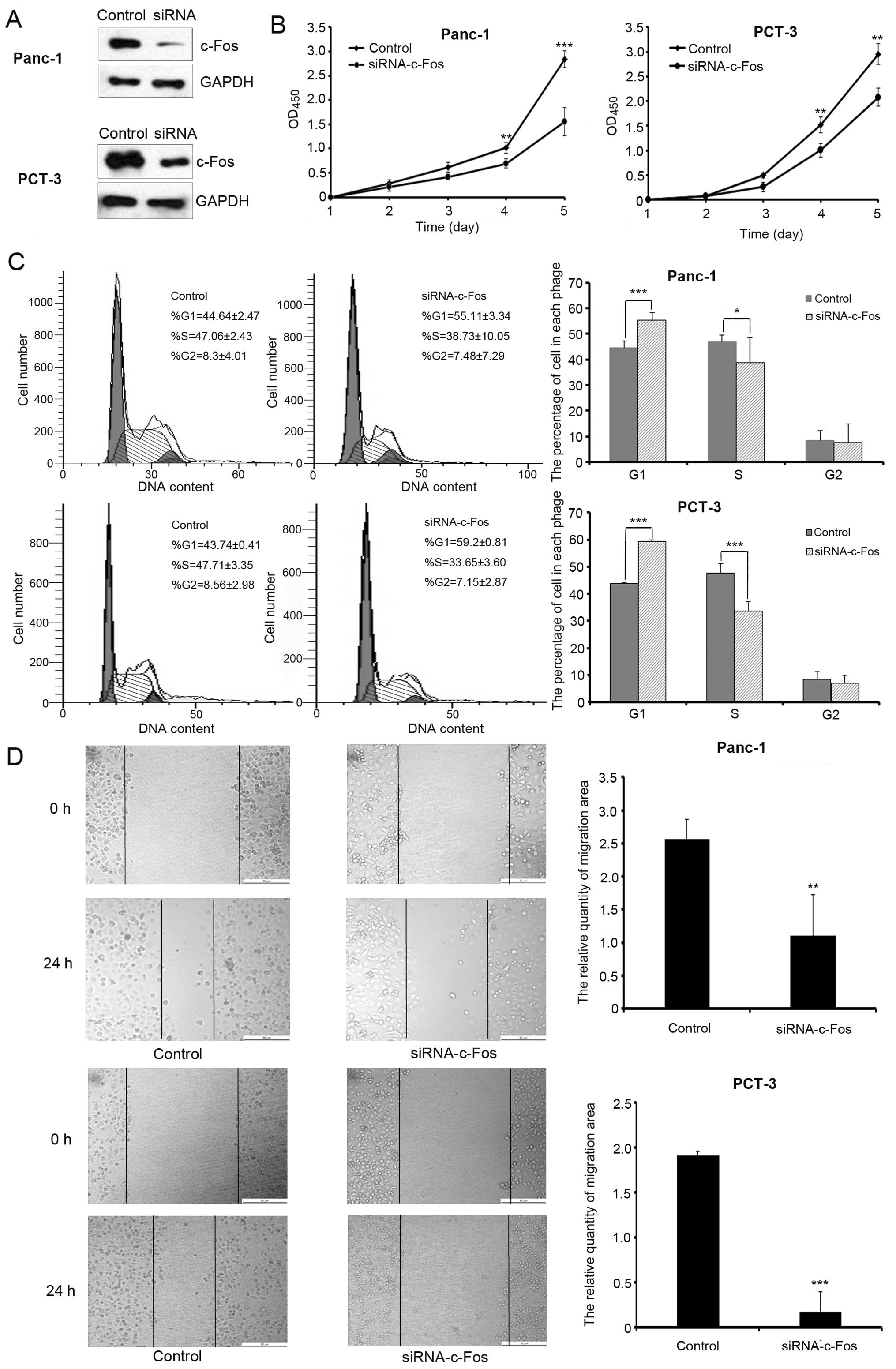
To examine the effects of c-Fos on pancreatic cancer
cell growth, we first suppressed endogenous c-Fos expression in
Panc-1 and PCT-3 cells by transfecting cells with siRNA against
c-Fos (Fig. 1A). The results of the
cell proliferation assay showed that c-Fos knockdown significantly
inhibited cell growth (Fig. 1B).
Flow cytometric analysis was used to test cell cycle progression.
Our results show that the percentage of G1 phase cells was
increased following knockdown of c-Fos expression (Fig. 1C). Wound-healing assay was performed
to examine the effects of c-Fos on migration. Reduction in c-Fos
levels decreased cell migration compared with the control group
(Fig. 1D).
Characterization of the mouse
model
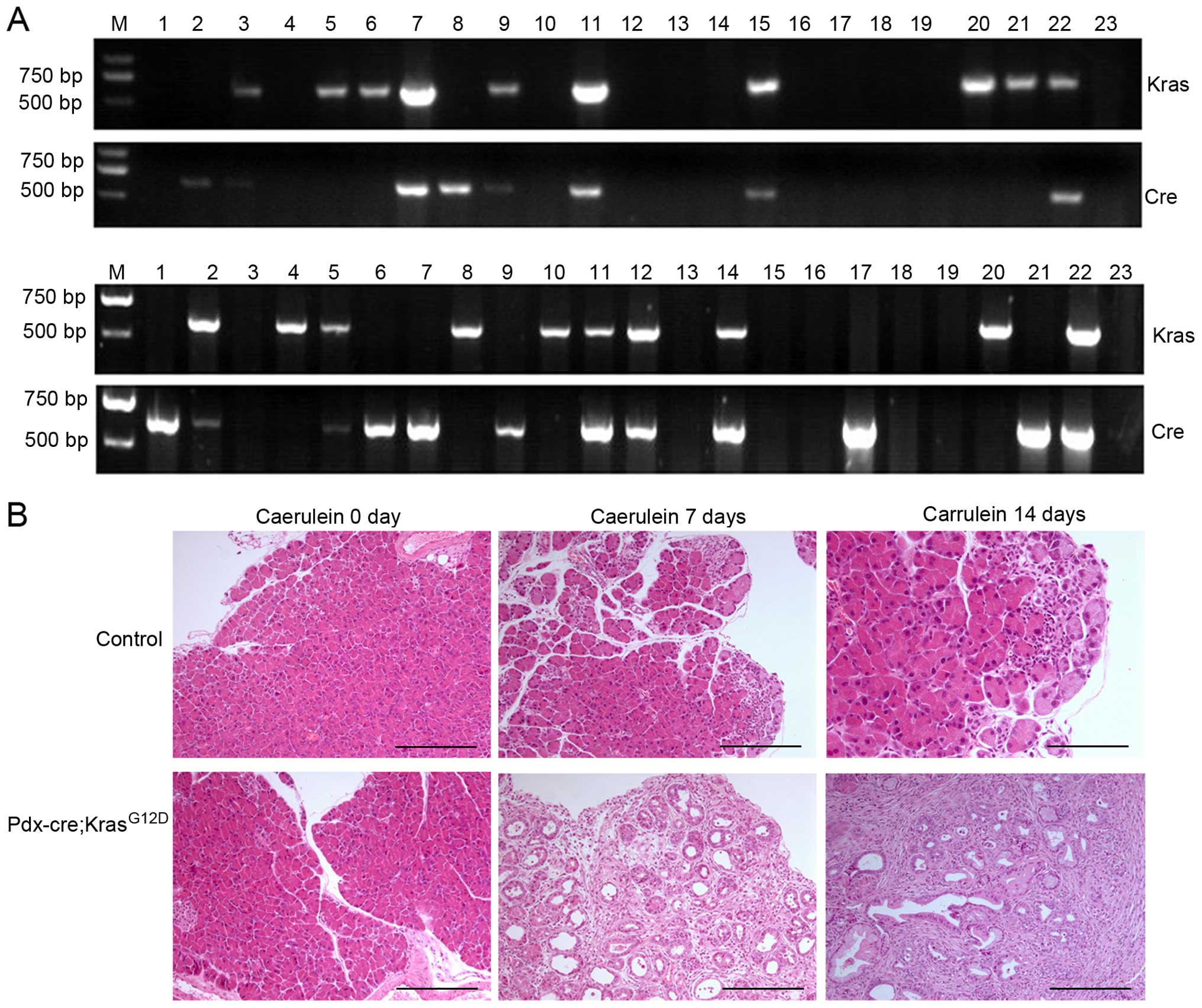
Pdx-cre; KrasG12D (PK) mice are attained
by crossing LSL-KrasG12D mice with Pdx-cre mice. To
identify transgenic mice, DNA was isolated and PCR amplification of
both lox and cre genes indicated that the recombinant
mouse model was established successfully (Fig. 2A). The positive rate for recombinant
mice was <20%. The pancreas from PK mice is larger than their
littermate controls and have more nodules, particularly those
treated by caerulein for a longer period of time.
Immunohistochemistry (IHC) revealed that the pancreas of PK mice
treated by caerulein for 7 days developed PanIN-1 to PanIN-2
lesions, and those treated by caerulein for 14 days developed a
higher degree of PanIN lesions relative to PDAC (Fig. 2B). In contrast, the pancreas from
control mice developed few ductal lesions with the exception of
some inflammatory infiltration after caerulein treatment for 7 or
14 days.
The expression of ERK/c-Fos increased
during PDAC initiation and progression
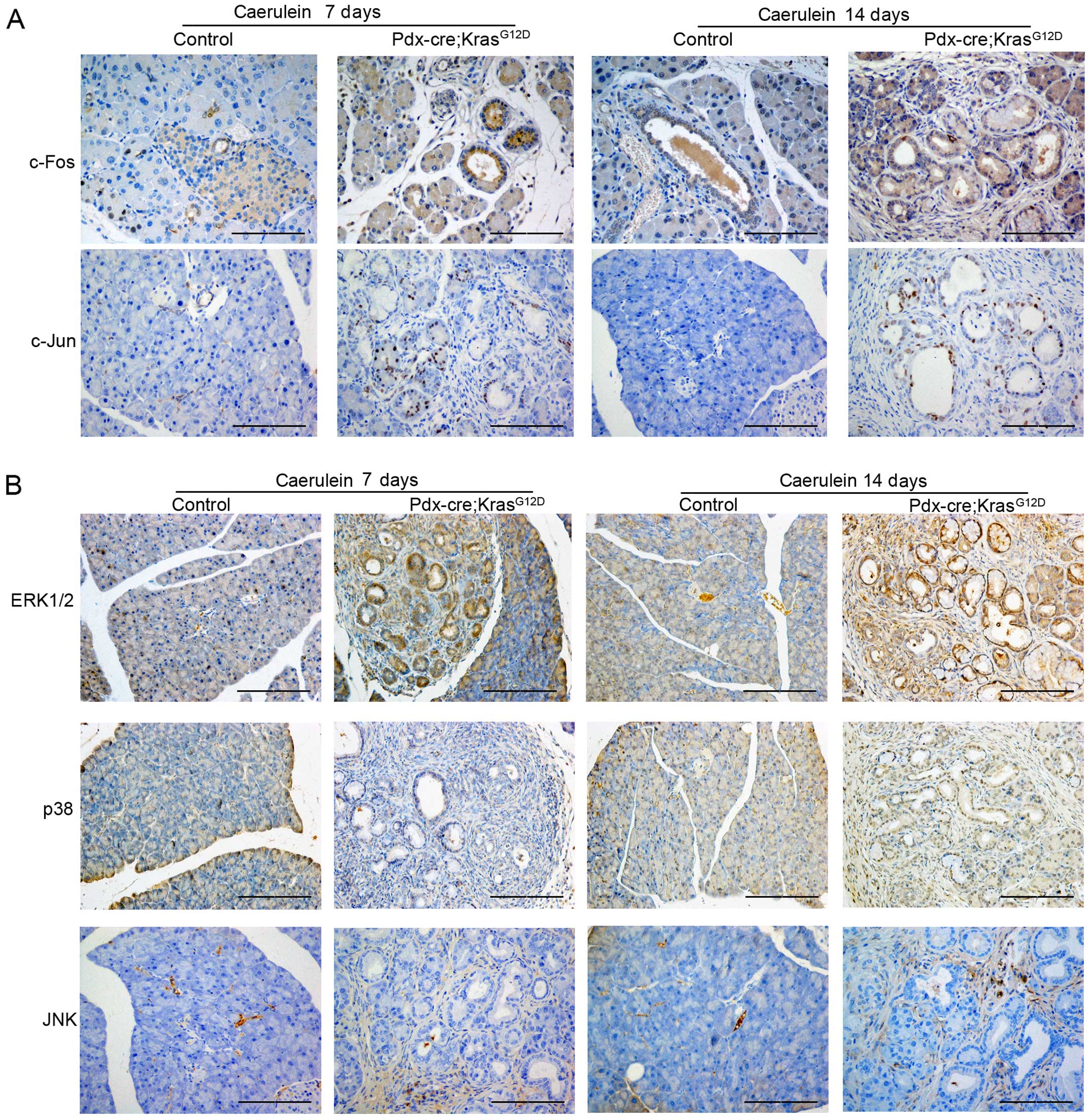
C-Jun and c-Fos are components of the AP-1 complex,
which is involved in numerous cell activities including
proliferation, apoptosis, survival, tumorigenesis and tissue
morphogenesis (12). To
characterize the function of AP-1 in PDAC tumorigenesis, we
examined the expression of both c-Jun and c-Fos in the pancreas of
PK mice. Compared with the control mice, the expression level of
c-Fos, but not c-Jun, was enhanced in PanIN lesions of
caerulein-treated PK mice and progressively increased as pancreatic
lesion stage progressed (Fig. 3A).
AP-1 is reportedly involved in the downstream regulation of mitogen
activated protein kinase (MAPK) (13,14);
therefore, we examined the expression of ERK, JNK and p38 in PK
mice. As shown in Fig. 3B, the
expression of ERK is similar to c-Fos levels, whereas JNK and p38
showed little difference in expression. Therefore, the expression
of ERK/c-Fos increased during PDAC initiation and progression.
ERK/c-Fos is required for PDAC
initiation and progression
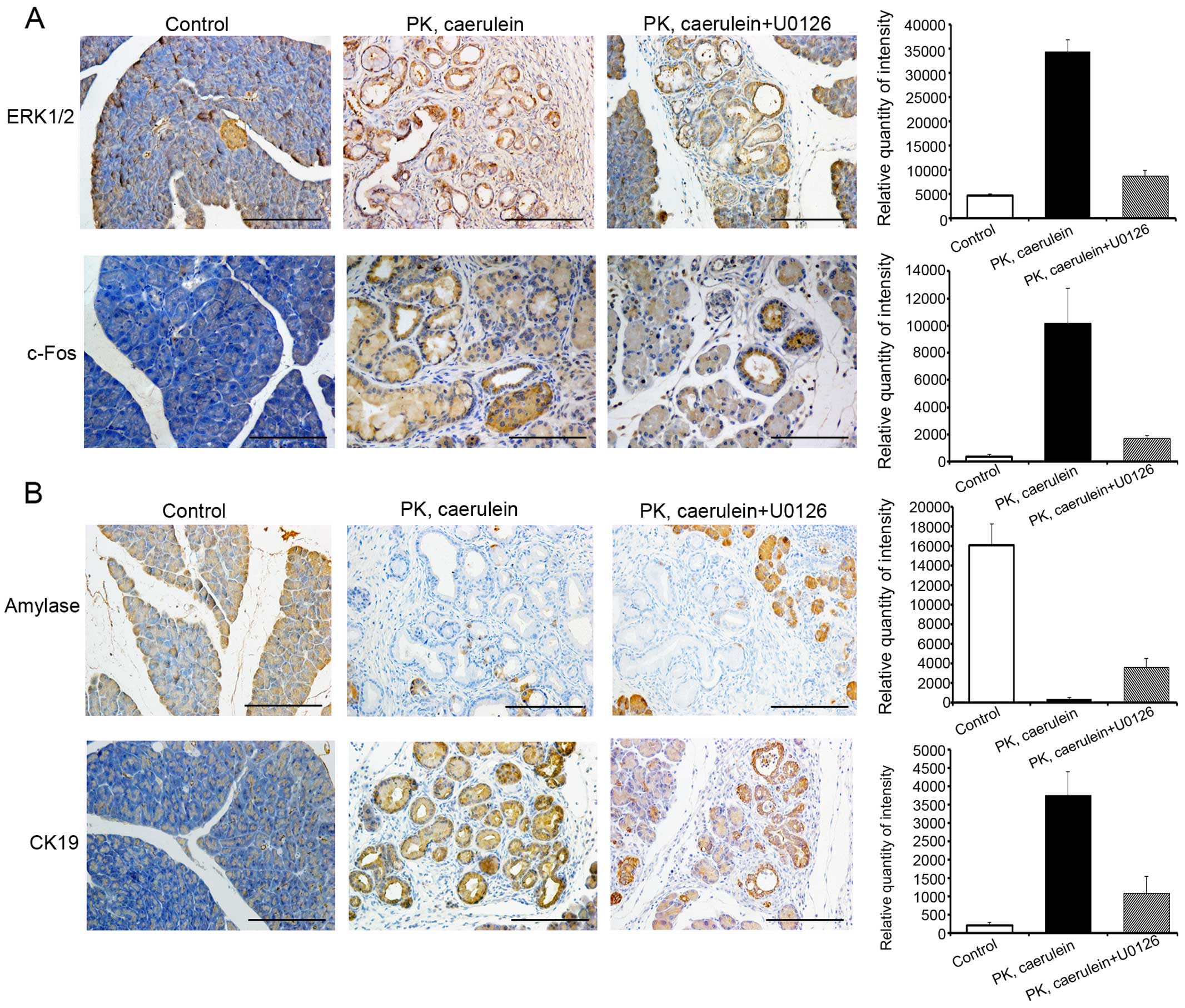
As ERK/c-Fos is highly expressed in pancreatic
cancer, we postulated that ERK/c-Fos is required for tumorigenesis.
To test this hypothesis, we treated 6-week-old PK mice with the ERK
inhibitor, U0126, with or without caerulein for 15 days. U0126
reduced the expression of ERK and c-Fos and suppressed pancreatic
ductal lesions (Fig. 4A).
Furthermore, after 15 days of U0126+caerulein injection, the
exocrine compartment of PK mice was partially replaced by fibrosis
and ductal structures, as evident by the expression level of acinar
marker amylase and duct marker cytokeratin 19 (CK19) (Fig. 4B). In contrast, the pancreas of PK
mice treated with only caerulein displayed markedly more CK
19-positive duct structures and fewer amylase-positive
parenchyma.
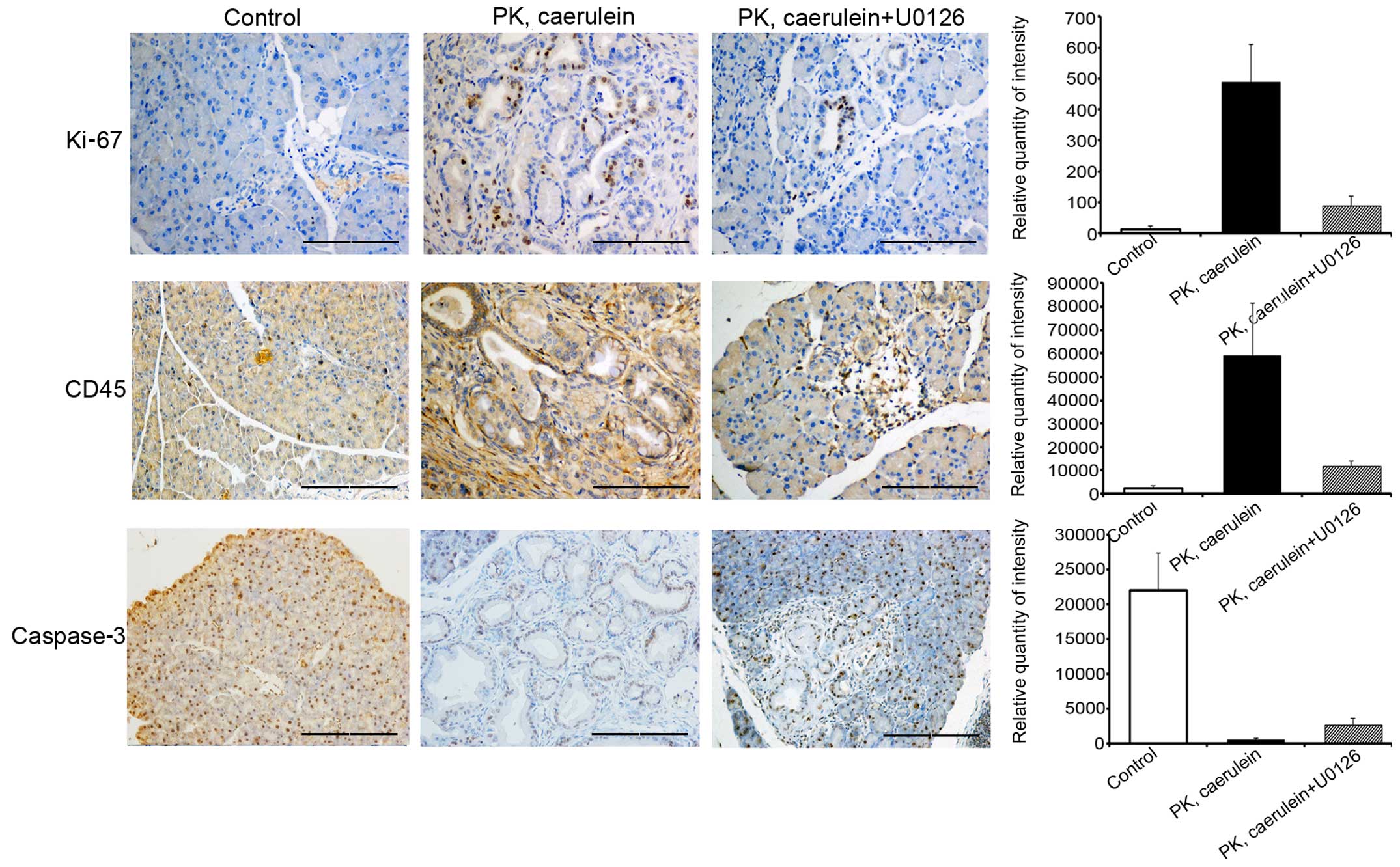
Suppression of ERK/c-Fos attenuates
inflammation and proliferation, but promotes apoptosis
To determine the role of ERK/c-Fos in PanIN
formation and development of PDAC, we tested additional markers of
tumor phenotype. Levels of proliferation marker Ki-67 and
inflammatory marker CD45 expression were substantially higher in
PanIN and PDAC from PK mice compared to control mice, whereas the
apoptosis marker, caspase-3, exhibited the opposite results
(Fig. 5). Immunohistochemistry data
also suggested that U0126 inhibits Ki-67 and CD45 expression and
increased the levels of caspase-3. Therefore, the suppression of
ERK/c-Fos attenuates inflammation and proliferation but promotes
apoptosis to repress PanIN and PDAC progression.
Discussion
PDAC remains a highly lethal disease (15,16).
The posterior location of pancreas, which is in close proximity to
duodenum, common bile duct, celiac plexus, superior mesenteric
artery (SMA), and portal vein, contributes to late diagnosis, as
well as the bothersome symptoms of obstruction of biliary drainage,
including infection, pain, chemotherapy resistance and unresectable
pancreatic cancer (17–20). A genetically engineered mouse model
is an appropriate tool to investigate the pathological mechanisms
and early diagnosis of pancreatic cancer.
The Pdx-cre; KrasG12D (PK) mouse model,
which targets pancreas-specific expression of mutated Kras,
recapitulates the human PanIN-to-PDAC sequence (7). PanIN lesions begin to appear at
approximately 2 months, and high-grade PanINs are observed at 5
months. The progression from PanIN to invasive and metastatic PDAC
occurs over several months, which is not conducive for a research
study. Chronic pancreatitis has been identified as risk factor for
PDAC development in humans and has been shown to significantly
accelerate PanIN and PDAC development in Kras-driven mouse models
(21,22). Acute pancreatitis can progress to
chronic pancreatitis in human patients under certain conditions
(23). Several studies have also
shown that acute pancreatitis markedly accelerates PanIN and PDAC
development in Kras-driven mouse models (24). In response to acute pancreatitis
induced by the cholecystokinin analog caerulein, acini transiently
dedifferentiated into duct-like structures. Mutant Kras compromises
the ability of acinar cells to regenerate following acute
pancreatitis and locks damaged cells in a persistently
dedifferentiated ductal state that can rapidly give rise to PanINs
(25,26). Thus, caerulein-induced pancreatitis
provides a permissive environment for Kras-driven neoplasia. In the
present study, the pancreas of PK mice treated by caerulein for 7
days developed PanIN-1 to PanIN-2 lesions, and PK mice treated by
caerulein for 14 days developed a higher degree of PanIN lesions
relative to PDAC. This improved PK mouse model allowed us to study
PanIN to PDAC development on an accelerated timeline.
MAPKs are a family of serine-threonine protein
kinases involved in many cellular processes including cell
proliferation, differentiation, inflammation and cell death
(27). Activation of several MAPKs,
including ERK, p38 and c-Jun N-terminal kinase (JNK) are able to
stimulate AP-1 (28). The results
in PK mice suggested that the expression of ERK and c-Fos increased
during PanIN formation and progression to PDAC, whereas the level
of p38, JNK and c-Jun remained unchanged. In addition to the
Kras gene, inactivation of other genes are involved in the
pathophysiology of PDAC, including INK4A, TP53,
SMAD4 or BRCA2 (29).
Additionally, several signaling pathways may positively or
negatively modulate the PanIN and PDAC development, including TGFα,
Hedgehog, Notch, EGFR and STAT (30–32).
Here we provided evidence that the ERK/c-Fos pathway is essential
for Kras-driven PDAC. Furthermore, our experiment showed that the
ERK/c-Fos inhibitor U0126 suppressed acinar-ductal metaplasia,
proliferation and inflammation and promoted apoptosis, leading to
inhibition of pancreatic ductal lesions.
In summary, our results demonstrate that c-Fos
induced cell growth, cell cycle and migration in the the pancreatic
cancer cells. The in vivo experiments revealed that the
expression of c-Fos, and the upstream transcription factor ERK,
increased during PanIN formation. Additionally, the ERK/c-Fos
inhibitor, U0126, suppressed the PanIN/PDAC progression initiation
through proliferation, inflammation and apoptosis. Our findings
suggest that the ERK/c-Fos pathway is required for PanIN formation
and progression to PDAC, which will help to characterize the early
prognosis of pancreatic cancer.
Acknowledgements
The present study was supported by grant for the
Research Special Fund for Public Welfare Industry of Health
(201402001), the Research Fund for the Doctoral Program of Higher
Education (20131106110008) and the Research Fund for the Doctoral
Program of Higher Education (20121106120048).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ligat L, Saint-Laurent N, El-Mrani A,
Gigoux V, Al Saati T, Tomasini R, Nigri J, Dejean S, Pont F, Baer
R, et al: Pancreatic preneoplastic lesions plasma signatures and
biomarkers based on proteome profiling of mouse models. Br J
Cancer. 113:1590–1598. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Heestand GM, Murphy JD and Lowy AM:
Approach to patients with pancreatic cancer without detectable
metastases. J Clin Oncol. 33:1770–1778. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shen R, Wang Q, Cheng S, Liu T, Jiang H,
Zhu J, Wu Y and Wang L: The biological features of PanIN initiated
from oncogenic Kras mutation in genetically engineered mouse
models. Cancer Lett. 339:135–143. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hruban RH, Adsay NV, Albores-Saavedra J,
Anver MR, Biankin AV, Boivin GP, Furth EE, Furukawa T, Klein A,
Klimstra DS, et al: Pathology of genetically engineered mouse
models of pancreatic exocrine cancer: Consensus report and
recommendations. Cancer Res. 66:95–106. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
di Magliano MP and Logsdon CD: Roles for
KRAS in pancreatic tumor development and progression.
Gastroenterology. 144:1220–1229. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hingorani SR, Petricoin EF III, Maitra A,
Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD,
Hitt BA, et al: Preinvasive and invasive ductal pancreatic cancer
and its early detection in the mouse. Cancer Cell. 4:437–450. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wagner EF: Bone development and
inflammatory disease is regulated by AP-1 (Fos/Jun). Ann Rheum Dis.
69:(Suppl 1). i86–i88. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ren X, Song W, Liu W, Guan X, Miao F, Miao
S and Wang L: Rhomboid domain containing 1 inhibits cell apoptosis
by upregulating AP-1 activity and its downstream target Bcl-3. FEBS
Lett. 587:1793–1798. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shi R, Peng H, Yuan X, Zhang X, Zhang Y,
Fan D, Liu X and Xiong D: Down-regulation of c-fos by shRNA
sensitizes adriamycin-resistant MCF-7/ADR cells to chemotherapeutic
agents via P-glycoprotein inhibition and apoptosis augmentation. J
Cell Biochem. 114:1890–1900. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jia ZC, Wan YL, Tang JQ, Dai Y, Liu YC,
Wang X and Zhu J: Tissue factor/activated factor VIIa induces
matrix metalloproteinase-7 expression through activation of c-Fos
via ERK1/2 and p38 MAPK signaling pathways in human colon cancer
cell. Int J Colorectal Dis. 27:437–445. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Meng Q and Xia Y: c-Jun, at the crossroad
of the signaling network. Protein Cell. 2:889–898. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lappas M, Riley C, Lim R, Barker G, Rice
GE, Menon R and Permezel M: MAPK and AP-1 proteins are increased in
term pre-labour fetal membranes overlying the cervix: Regulation of
enzymes involved in the degradation of fetal membranes. Placenta.
32:1016–1025. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu X, Li Q, Dowdell K, Fischer ER and
Cohen JI: Varicella-Zoster virus ORF12 protein triggers
phosphorylation of ERK1/2 and inhibits apoptosis. J Virol.
86:3143–3151. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Eferl R and Wagner EF: AP-1: A
double-edged sword in tumorigenesis. Nat Rev Cancer. 3:859–868.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bailey JM, Hendley AM, Lafaro KJ, Pruski
MA, Jones NC, Alsina J, Younes M, Maitra A, McAllister F,
Iacobuzio-Donahue CA, et al: P53 mutations cooperate with oncogenic
Kras to promote adenocarcinoma from pancreatic ductal cells.
Oncogene. 441:1–7. 2015.
|
|
17
|
Han H and Von Hoff DD: SnapShot:
Pancreatic cancer. Cancer Cell. 23:424–424.e1. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stylianopoulos T, Martin JD, Chauhan VP,
Jain SR, Diop-Frimpong B, Bardeesy N, Smith BL, Ferrone CR,
Hornicek FJ, Boucher Y, et al: Causes, consequences, and remedies
for growth-induced solid stress in murine and human tumors. Proc
Natl Acad Sci USA. 109:15101–15108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tabernero J, Chiorean EG, Infante JR,
Hingorani SR, Ganju V, Weekes C, Scheithauer W, Ramanathan RK,
Goldstein D, Penenberg DN, et al: Prognostic factors of survival in
a randomized phase III trial (MPACT) of weekly nab-paclitaxel plus
gemcitabine versus gemcitabine alone in patients with metastatic
pancreatic cancer. Oncologist. 20:143–150. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sherman MH, Yu RT, Engle DD, Ding N,
Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S,
et al: Vitamin D receptor-mediated stromal reprogramming suppresses
pancreatitis and enhances pancreatic cancer therapy. Cell.
159:80–93. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guerra C, Schuhmacher AJ, Cañamero M,
Grippo PJ, Verdaguer L, Pérez-Gallego L, Dubus P, Sandgren EP and
Barbacid M: Chronic pancreatitis is essential for induction of
pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice.
Cancer Cell. 11:291–302. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fukuda A, Wang SC, Morris JP IV, Folias
AE, Liou A, Kim GE, Akira S, Boucher KM, Firpo MA, Mulvihill SJ, et
al: Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma
initiation and progression. Cancer Cell. 19:441–455. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sankaran SJ, Xiao AY, Wu LM, Windsor JA,
Forsmark CE and Petrov MS: Frequency of progression from acute to
chronic pancreatitis and risk factors: A meta-analysis.
Gastroenterology. 149:1490–1500.e1. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Carrière C, Young AL, Gunn JR, Longnecker
DS and Korc M: Acute pancreatitis accelerates initiation and
progression to pancreatic cancer in mice expressing oncogenic Kras
in the nestin cell lineage. PLoS One. 6:e277252011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Morris JP IV, Cano DA, Sekine S, Wang SC
and Hebrok M: Beta-catenin blocks Kras-dependent reprogramming of
acini into pancreatic cancer precursor lesions in mice. J Clin
Invest. 120:508–520. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guerra C and Barbacid M: Genetically
engineered mouse models of pancreatic adenocarcinoma. Mol Oncol.
7:232–247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jafri M, Donnelly B, McNeal M, Ward R and
Tiao G: MAPK signaling contributes to rotaviral-induced
cholangiocyte injury and viral replication. Surgery. 142:192–201.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Garcia MN, Grasso D, Lopez-Millan MB,
Hamidi T, Loncle C, Tomasini R, Lomberk G, Porteu F, Urrutia R and
Iovanna JL: IER3 supports KRASG12D-dependent pancreatic cancer
development by sustaining ERK1/2 phosphorylation. J Clin Invest.
124:4709–4722. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Thompson KN, Whipple RA, Yoon JR, Lipsky
M, Charpentier MS, Boggs AE, Chakrabarti KR, Bhandary L, Hessler
LK, Martin SS, et al: The combinatorial activation of the PI3K and
Ras/MAPK pathways is sufficient for aggressive tumor formation,
while individual pathway activation supports cell persistence.
Oncotarget. 6:35231–35246. 2015.PubMed/NCBI
|
|
31
|
Mace TA, Shakya R, Elnaggar O, Wilson K,
Komar HM, Yang J, Pitarresi JR, Young GS, Ostrowski MC, Ludwig T,
et al: Single agent BMS-911543 Jak2 inhibitor has distinct
inhibitory effects on STAT5 signaling in genetically engineered
mice with pancreatic cancer. Oncotarget. 6:44509–44522.
2015.PubMed/NCBI
|
|
32
|
Reichert M and Rustgi AK: Pancreatic
ductal cells in development, regeneration, and neoplasia. J Clin
Invest. 121:4572–4578. 2011. View
Article : Google Scholar : PubMed/NCBI
|