Introduction
Non-small cell lung cancer (NSCLC) is the most
common type of lung cancer and the 5-year relative survival rate is
less than 20% (1,2). Dysregulated activation of receptor
tyrosine kinases (RTKs) and their downstream signaling molecules is
closely associated with NSCLC growth and progression. Among RTKs,
epidermal growth factor receptor (EGFR) is highly expressed or
constitutively activated in NSCLC patients with poor prognosis
(3), suggesting the rational
strategy and pharmacological efficacy of EGFR-targeted therapeutics
in the treatment of NSCLC. However, inhibition of RTKs including
EGFR in the clinic often leads to recurrent and metastatic
phenotypes of NSCLC. In addition, anti-angiogenic therapy such as
inhibition of vascular endothelial growth factor-A (VEGF-A)/VEGF
receptor-2 (VEGFR-2) signaling pathways shows a transient
therapeutic effect and subsequent development of NSCLC resistance
(4–6). Thus, a further understanding of the
molecular mechanism of RTK-mediated signaling pathways in NSCLC
growth and progression in the tumor microenvironment is a
prerequisite for the identification of therapeutic targets and the
development of highly effective anticancer drugs.
Broussonetia kazinoki (B. kazinoki)
has been used as a traditional medicine for the treatment of
blurred vision and inflammatory and infectious diseases as well as
a raw material for paper production in Northeastern Asia including
Korea. Previous investigations have demonstrated that B.
kazinoki extract and its bioactive components such as flavan
derivatives have anti-diabetic, anti-allergic, anti-inflammatory
and antitumor properties (7–12). We
recently reported that an ethanolic extract of B. kazinoki
and marmesin regulate VEGF-A-induced endothelial cell fates in
vitro and angiogenic sprouting ex vivo (13,14).
Marmesin, a furanocoumarin component isolated from a variety of
plants including Peucedanum japonicum, Dystaenia
takeshimana, Feronia limonia and Ferula lutea as
well as B. kazinoki, has been reported to exert a variety of
pharmacological functions such as anti-inflammatory,
anti-hepatotoxic, anti-angiogenic and antitumor activities
(14–18). However, the effects and molecular
mechanisms of marmesin on NSCLC cell responses have never been
elucidated, to date. In the present study, we report for the first
time the regulatory effects and molecular mechanisms of marmesin on
NSCLC cell fates and tumor-derived angiogenic responses.
Materials and methods
Cell culture conditions
Human NSCLC cell lines (A549 and H1299) were
obtained from the American Type Culture Collection (ATCC; Manassas,
VA, USA) and were grown in 10% fetal bovine serum-Dulbecco's
modified Eagle's medium (FBS-DMEM) (HyClone Laboratories, Logan,
UT, USA). Human umbilical vein endothelial cells (HUVECs) were
purchased from Lonza (Walkersville, MD, USA) and used between
passages 4 and 6 for all experiments. Cells were cultured in
EGM-2® BulletKit media, according to the manufacturer's
instructions (Lonza).
Reagents
Marmesin was isolated in an ethyl acetate fraction
partitioned from the ethanolic extract of B. kazinoki. The
following pharmacological agents and antibodies were purchased from
commercial sources: anti-phospho-Src (Y416), anti-Src,
anti-phospho-MEK (S217/S221), anti-MEK, anti-phospho-ERK
(T202/Y204), anti-phospho-Akt (S473),
anti-phospho-p70S6K (T421/S424) and phospho-pRb (S780)
(Cell Signaling Technology, Beverly, MA, USA); anti-ERK, anti-Akt,
anti-p70S6K, anti-VEGFR-2, anti-integrin α3,
anti-integrin β1, anti-ILK, anti-Cdk4, anti-Cdk2, anti-actin
antibodies, and mouse and rabbit IgG-horseradish peroxidase
conjugates (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Cell viability and proliferation
assay
Subconfluent A549 and H1299 cells, plated on 6-well
plates (5×104 cells/well; SPL Life Sciences,
Gyeonggi-do, Korea), were serum-starved for 24 h in basal DMEM to
synchronize cells in the G1/G0 phase of the
cell cycle, and pretreated with marmesin (0.1–10 µM) for 30 min
prior to 10% FBS stimulation for 24 h. In some experiments,
quiescent cells were pretreated with marmesin (10 µM) for 30 min,
followed by 10% FBS stimulation for 12 h. After stimulation, cells
were thoroughly rinsed with phosphate-buffered saline (PBS; pH 7.4)
to remove any residual marmesin, and further incubated with 10% FBS
for another 12 h until the end of the 24 h time point. Cell
viability was determined by a Muse™ Cell Analyzer using cell count
and viability assay kit (Merck Millipore, Billerica, MA, USA), and
the cell proliferation was quantified as previously described
(19). The results from triplicate
determinations (mean ± standard deviation) are presented as the
percentage of viable cells of total cell count or the fold-increase
of the untreated controls.
Western blot analysis
Quiescent cells were pretreated with marmesin for 30
min, followed by 10% FBS stimulation for different time points, as
indicated. Cells were rinsed twice with ice-cold PBS and lysed by
incubation in 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 10% glycerol,
1% Triton X-100, 1 mM EDTA, 100 µg/ml AEBSF, 10 µg/ml aprotinin, 1
µg/ml pepstatin A, 0.5 µg/ml leupeptin, 80 mM β-glycerophosphate,
25 mM sodium fluoride and 1 mM sodium orthovanadate for 30 min at
4°C. Cell lysates were clarified at 13,000 × g for 20 min at 4°C,
and the supernatants were subjected to western blotting as
previously described (20). All
western blot analyses are representative of at least three
independent experiments. Bands of interest were integrated and
quantified by the use of National Institutes of Health (NIH) ImageJ
version 1.34s software.
Cell invasion assay
The upper side of the Transwell insert (6.5-mm
diameter insert, 8-µm pore size) (Corning Costar, Inc., Corning,
NY, USA) was coated with 50 µl of 1 mg/ml Matrigel® (BD
Biosciences, Bedford, MA, USA) diluted in serum-free DMEM. Aliquots
(100 µl) of cells (5×105 cells/ml) resuspended in
serum-free DMEM were added to the upper compartment of the
Matrigel-coated Transwell and 600 µl of serum-free DMEM was added
to the lower compartment. After serum starvation for 2 h, the cells
were pretreated with marmesin (10 µM) for 30 min, followed by 10%
FBS stimulation for 16 h. The inserts were fixed with methanol and
using a cotton-tipped swab the non-invasive cells were removed from
the top of the membrane. After staining with 0.04% Giemsa staining
solution (Sigma-Aldrich Co., St. Louis, MO, USA), the numbers of
invasive cells (mean ± standard deviation) were determined from six
different fields using ×200 objective magnification (21,22).
RNA purification and reverse
transcriptase-polymerase chain reaction
Total RNA was purified with PureHelix™ RNA
extraction solution (Nanohelix Co., Daejeon, Korea). Integrity of
RNA was checked by agarose gel electrophoresis and ethidium bromide
staining. One microgram of RNA was used as template for each
reverse transcriptase (RT)-mediated polymerase chain reaction (PCR)
using First Strand cDNA Synthesis kit (BioAssay Co., Ltd., Daejeon,
Korea). Primers for PCR were synthesized by Bioneer Corporation
(Daejeon, Korea). Primer sequences were as follows: MMP-2 forward,
5′-GCTCAGATCCGTGGTGAGAT-3′ and reverse, 5′-GGTGCTGGCTGAGTAGATCC-3′;
VEGFR-2 forward, 5′-TGCCTACCTCACCTGTTTCCT-3′ and reverse,
5′-TACACGGTGGTGTCTGTGTCA-3′; VEGF-A forward,
5′-TCGGGCCTCCGAAACCATGA-3′ and reverse, 5′-CCTGGTGAGAGATCTGGTTC-3′;
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) forward,
5′-GAAGGTGAAGGTCGGAGTC-3′ and reverse, 5′-GAAGATGGTGATGGGATTTC-3′.
Bands of interest were integrated and quantified by the use of NIH
ImageJ version 1.34s software.
VEGF enzyme-linked immunosorbent assay
(ELISA)
Quiescent cells were pretreated with marmesin (10
µM) for 30 min, followed by 10% FBS stimulation for 12 h. Cells
were washed with PBS to remove any residual marmesin, and then
further stimulated with 10% FBS for another 12 h. ELISA assay was
performed to measure the concentration of VEGF in the conditioned
media using VEGF ELISA kit (R&D Systems, Minneapolis, MN, USA),
according to the manufacturer's instructions. Secreted VEGF levels
were analyzed at 450 nm using BioTek Synergy Mx microplate reader
(BioTek Instruments, Winooski, VT, USA).
Tube formation assay
Tube formation assay was performed to examine the
ability of conditioned media from marmesin-treated NSCLC cells to
regulate angiogenic responses in vitro. Quiescent NSCLC
cells were pretreated with marmesin (10 µM) for 30 min, followed by
10% FBS stimulation for 12 h. Cells were washed with PBS to remove
any residual marmesin, and then further stimulated with 10% FBS for
another 12 h. After stimulation, conditioned media were collected.
Quiescent HUVECs (4×104 cells/ml) were added to
Matrigel®-coated plates and treated with conditioned
media for 9 h. Tube formation was observed with an Olympus CKX41
inverted microscope (CAchN 10/0.25php objective) and ToupTek
Toupview software (version ×36, 3.5.563; Hangzhou ToupTek Photonics
Co., Zhejiang, China).
Statistical analysis
Statistical analysis was performed using Student's
t-test, and was based on at least three different experiments. The
results were considered to be statistically significant at
P<0.05.
Results
Marmesin inhibits NSCLC cell
proliferation
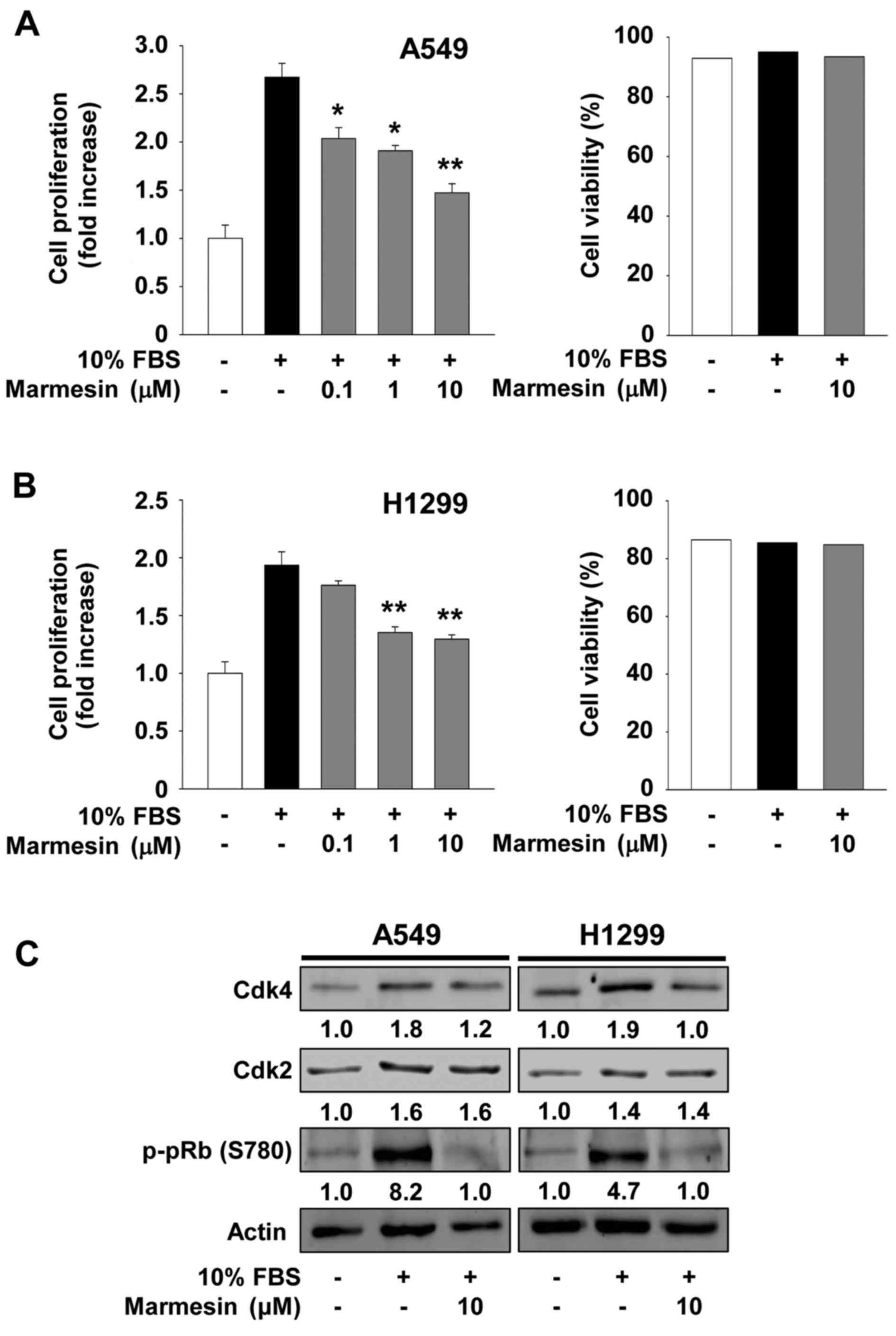
We first investigated the ability of marmesin to
regulate cell proliferation in p53 wild-type A549 and p53-deficient
H1299 NSCLC cells (Fig. 1A and B).
Marmesin treatment inhibited mitogen-stimulated cell proliferation
in a dose-dependent manner and did not alter cell viability and
morphology at the highest concentration used in the present study,
indicating that marmesin-mediated inhibition of cell proliferation
was not mediated by induction of apoptosis or cytotoxicity. Based
on these findings, we next analyzed the changes in the cell cycle
regulatory proteins in the marmesin-treated NSCLC cells (Fig. 1C). Marmesin treatment markedly
suppressed mitogen-induced expression of cyclin-dependent kinase 4
(Cdk4), but not Cdk2, to levels observed in the untreated controls,
resulting in inhibition of pRb phosphorylation in both NSCLC cell
lines. Marmesin has previously been reported to inhibit
proliferation by downregulation of Cdk4, Cdk2 and cyclin D in
VEGF-A-treated HUVECs (14).
Although the molecular mechanism of marmesin in regulating cell
cycle-related proteins appears slightly different in cell types,
these findings demonstrate the antiproliferative activity of
marmesin against various types of cells, independently of p53
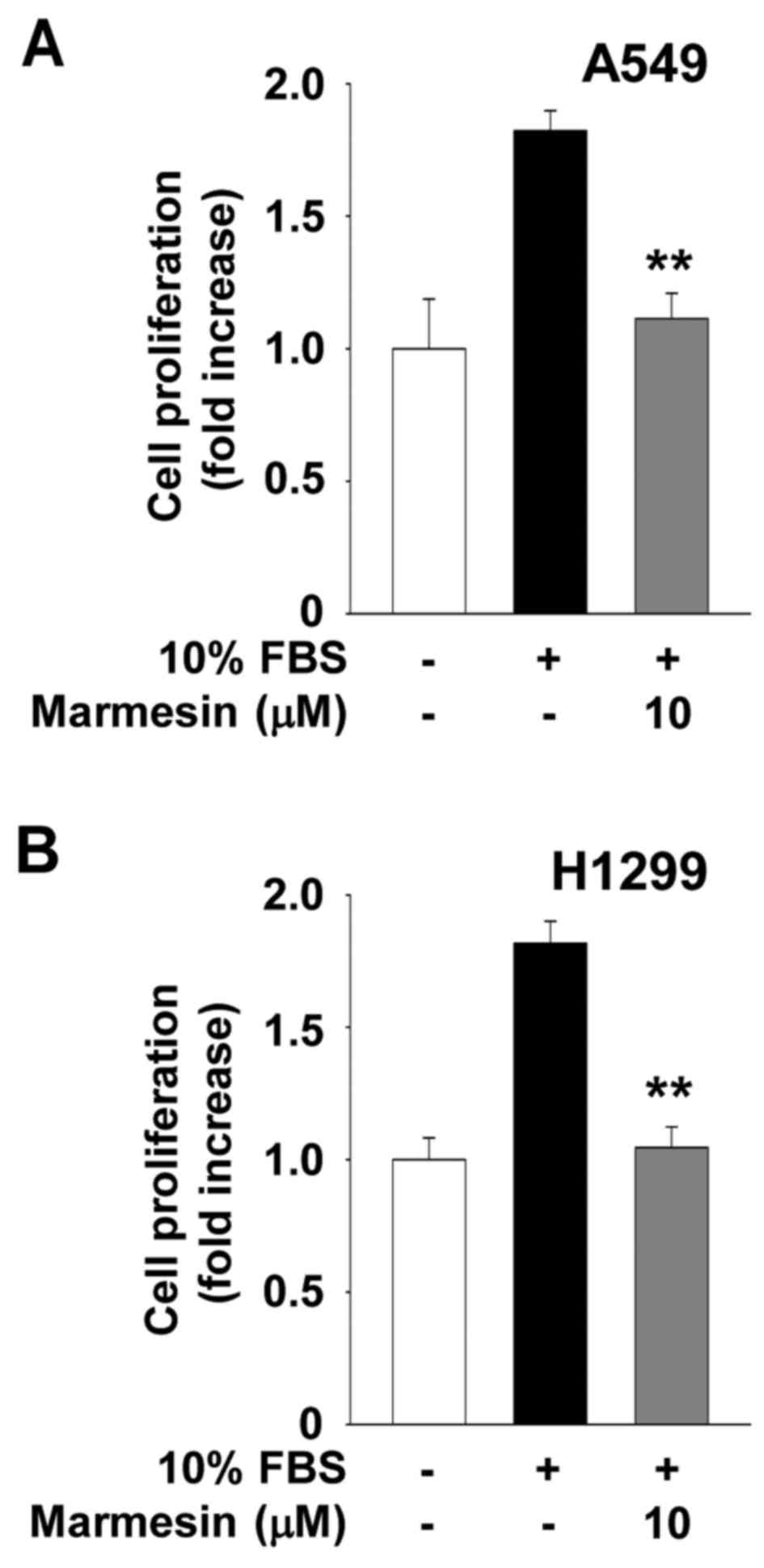
expression status. In addition, the inhibitory effect of marmesin
on NSCLC cell proliferation was not changed after the withdrawal of
marmesin at the 12 h time point, and was sustained up to 24 h,
suggesting that this effect may be irreversible until the end of
this experiment (Fig. 2).
Marmesin inhibits NSCLC cell
invasion
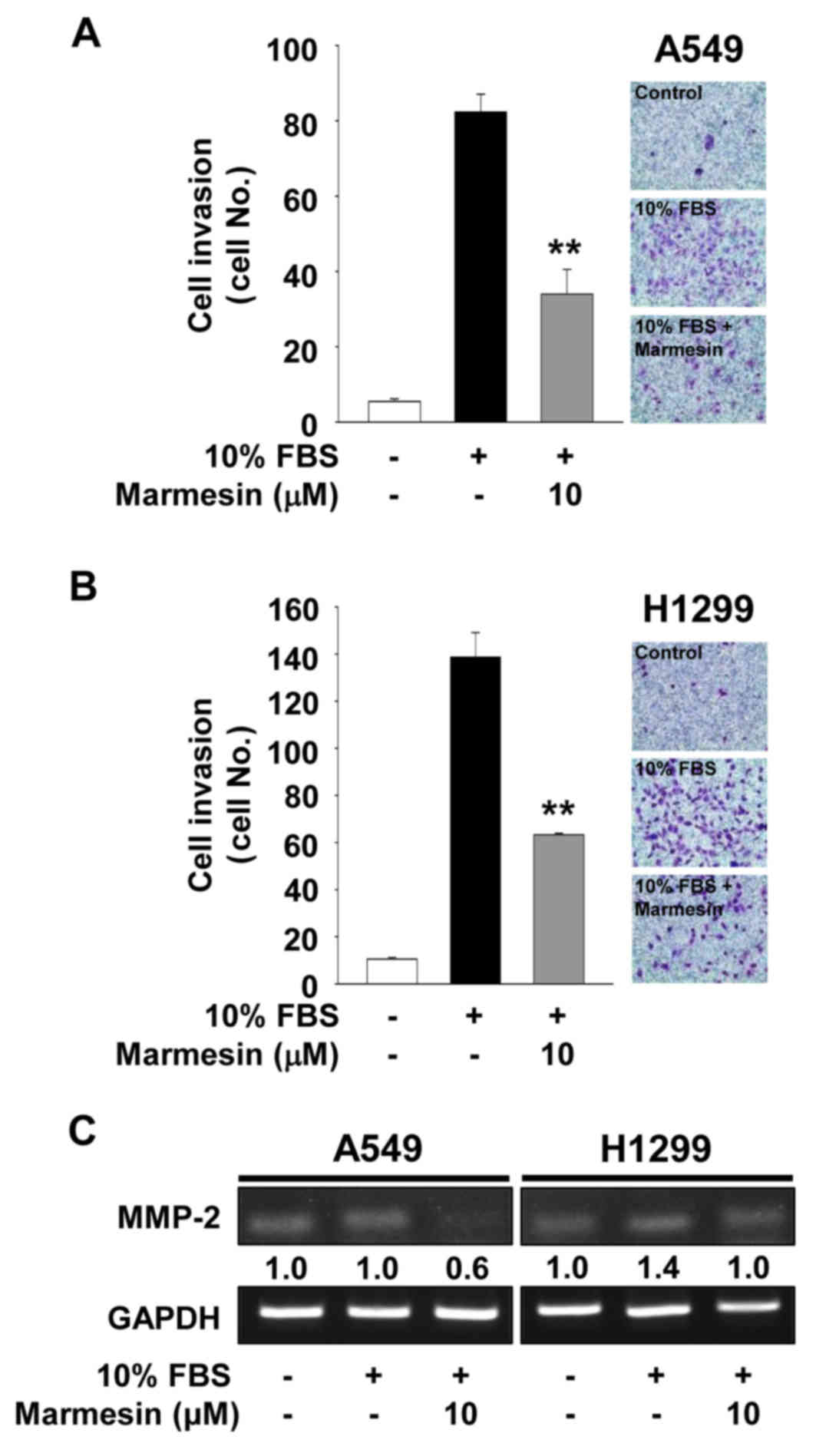
We next examined the effect of marmesin on cell
invasion in the A549 and H1299 cells. Marmesin treatment markedly
blocked mitogen-stimulated cell invasion (Fig. 3A and B). The regulatory pattern of
marmesin on NSCLC cell invasion was very similar to that on NSCLC
cell migration (data not shown). Based on these findings, we
analyzed the changes in expression of matrix metalloproteinases
(MMPs) in the marmesin-treated NSCLC cells. Expression and activity
of MMPs have been known to modulate cell migration, invasion and
angiogenesis by degrading extracellular matrix components and cell
surface molecules (23–26). As shown in Fig. 3C, marmesin treatment suppressed
mitogen-induced expression of MMP-2 in both cell lines. In
contrast, the levels of tissue inhibitor of metalloproteinases-2,
an endogenous inhibitor of MMPs, were not altered in the mitogen-
or marmesin-treated NSCLC cells (data not shown) (27–29).
Collectively, these findings suggest that inhibition of cell
invasion by marmesin is mediated, at least in part, through the
suppression of MMP-2 expression (Fig.
3C).
Marmesin-mediated inhibition of NSCLC
cell proliferation and invasion is mediated through inactivation of
mitogen-stimulated signaling pathways and downregulation of cell
surface signaling molecules
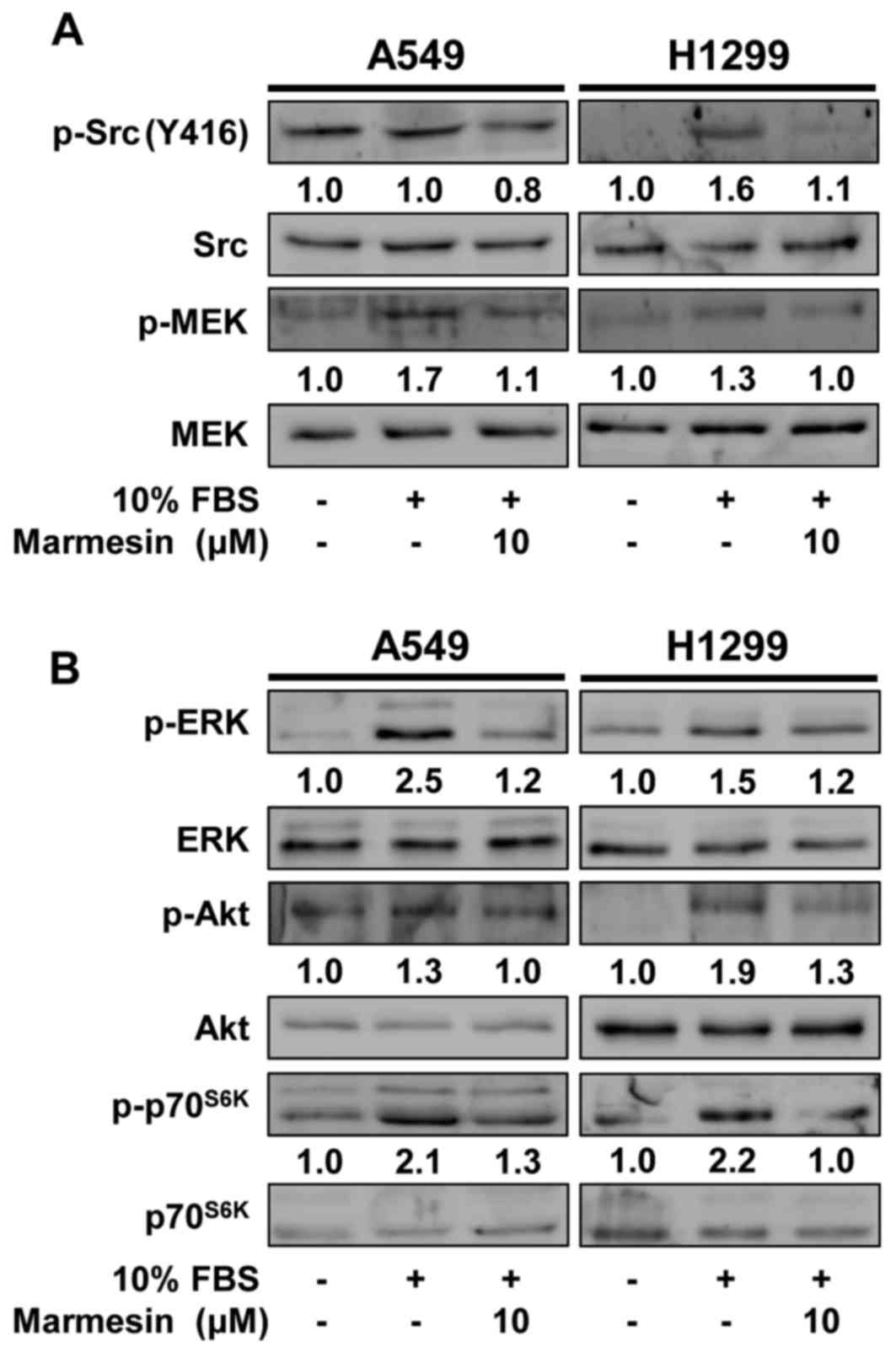
To elucidate the molecular mechanisms and
therapeutic targets of marmesin in regulating NSCLC cell
proliferation and invasion, we examined the changes in activation
of mitogen-stimulated signaling pathways including Src kinase,
mitogen-activated protein kinase (MEK), extracellular
signal-regulated kinase (ERK), Akt and p70S6 kinase
(p70S6K) in marmesin-treated NSCLC cells (14,30).
As expected, mitogenic stimulation significantly increased the
phosphorylation of MEK, ERK, Akt and p70S6K, as compared
with unstimulated controls (Fig.
4). In contrast, marmesin treatment markedly inhibited
mitogen-stimulated phosphorylation of Src, MEK, ERK, Akt and
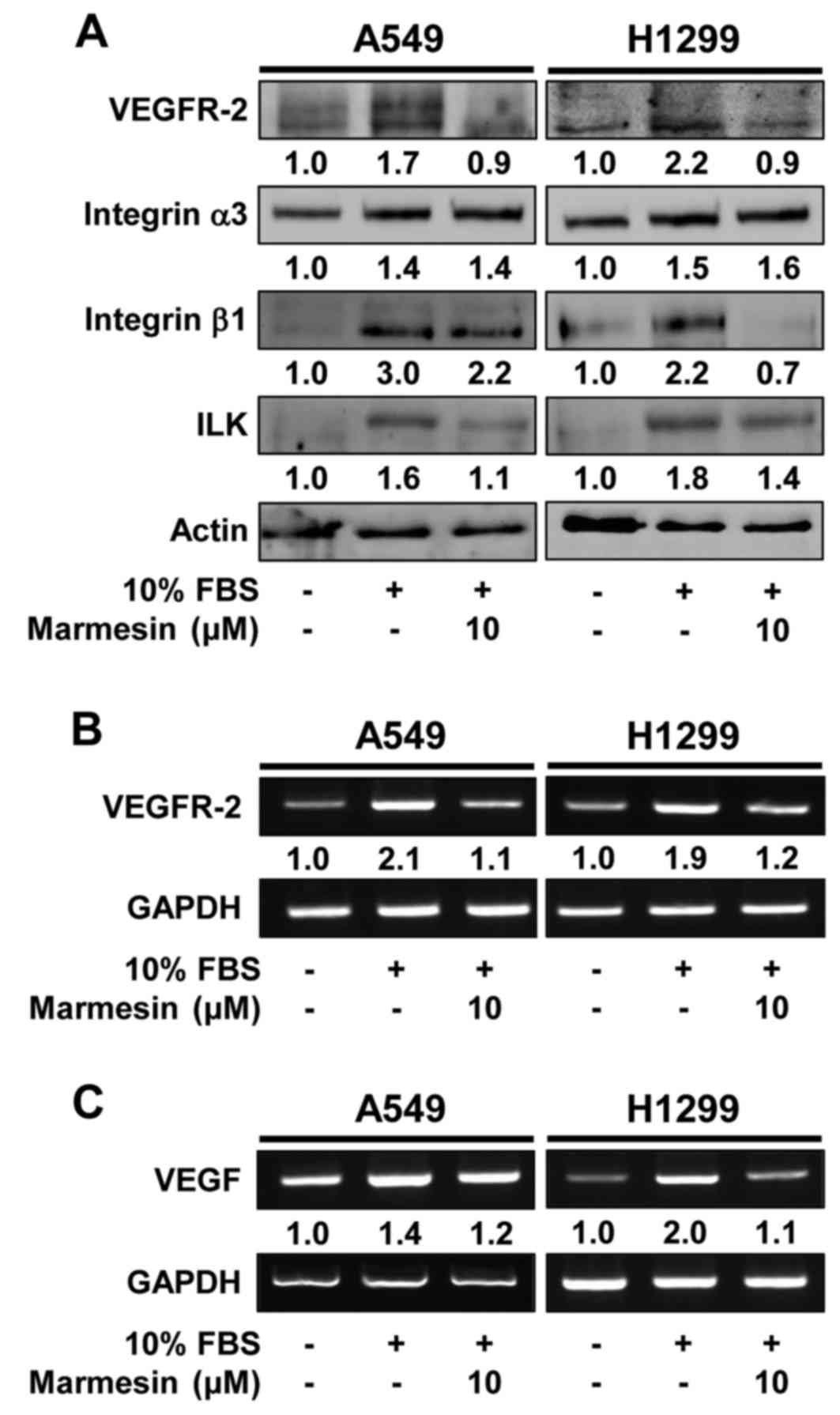
p70S6K in both NSCLC cells. Moreover, marmesin treatment
markedly suppressed mitogen-induced expression of cell signaling
molecules such as VEGFR-2, integrin β1 and integrin-linked kinase
(ILK), and a key angiogenic factor VEGF, which play important roles
in cancer growth and progression associated with angiogenesis
(Fig. 5) (31–33).
In addition to direct antitumor activity, these findings suggest
the possibility that marmesin may regulate angiogenic responses
through inhibition of VEGF expression and secretion in NSCLC
cells.
Marmesin inhibits endothelial cell
tube formation by downregulation of NSCLC-derived VEGF
secretion
In the tumor microenvironment cancer cells secrete a
variety of biological molecules including cytokines and growth
factors, which play important roles in cellular responses such as
proliferation, migration, invasion and angiogenesis (5,34).
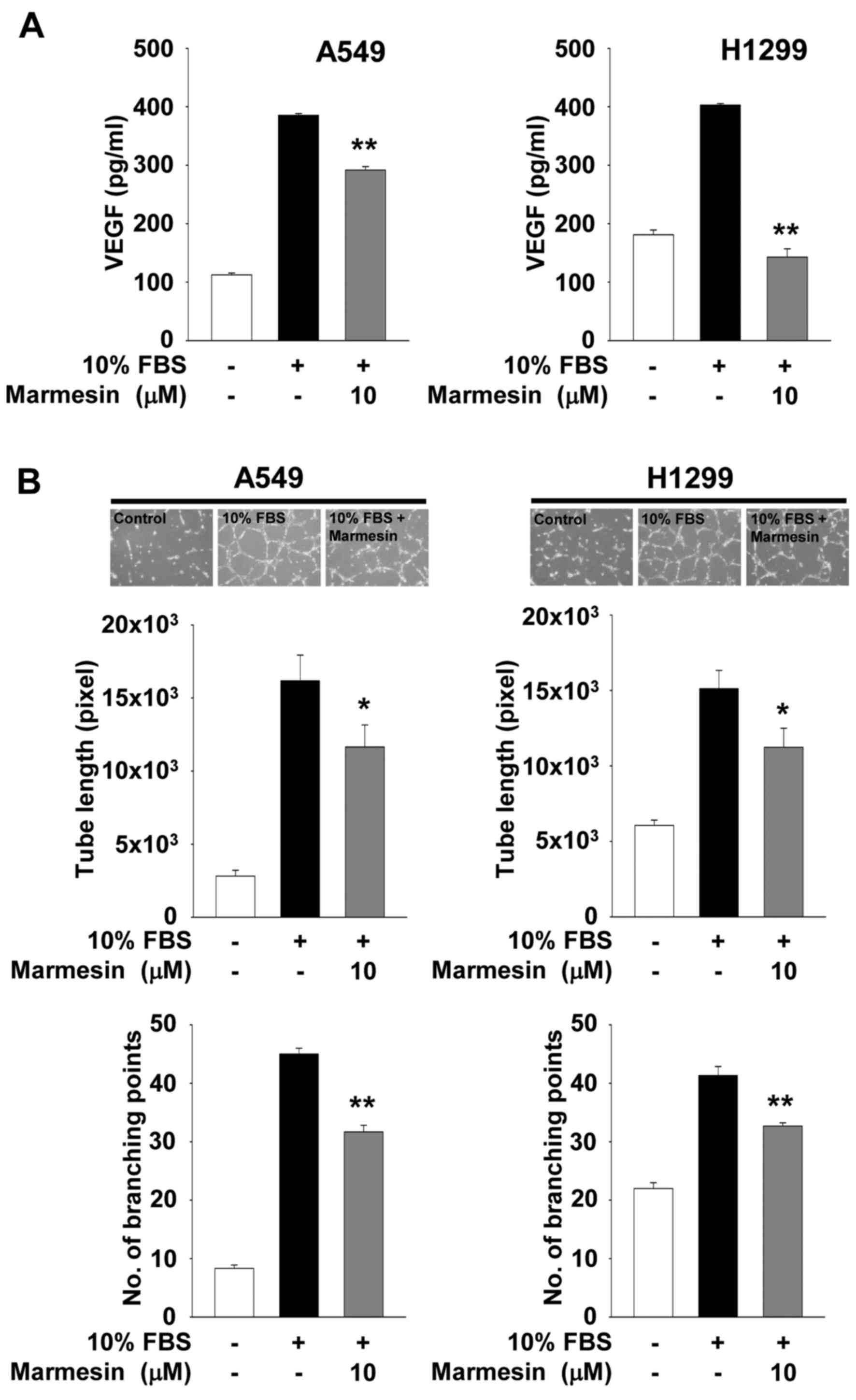
Based on inhibitory effect of marmesin on VEGF expression, we thus
analyzed the secreted levels of VEGF in conditioned media collected
from marmesin-treated NSCLC cells. As shown in Fig. 6A, marmesin treatment significantly
inhibited the secretion of VEGF from both NSCLC cell lines in
response to mitogenic stimulation. H1299 cells were found to be
more responsive to marmesin-mediated inhibition of VEGF secretion,
as compared with A549 cells. These findings are similar to the
patterns of VEGF transcription levels in marmesin-treated NSCLC
cells (Fig. 5C), demonstrating that
marmesin inhibits VEGF secretion through downregulation of VEGF
mRNA expression. To determine whether secreted biomolecules
including VEGF from marmesin-treated NSCLC cells affect the
cellular fate of adjacent and/or other cells, we next performed
in vitro angiogenesis assay using conditioned media from
NSCLC cells treated with or without marmesin (Fig. 6B). The conditioned media from
marmesin-treated NSCLC cells significantly inhibited the formation
of capillary-like structures by HUVECs. Although the types and
levels of biomolecules secreted from marmesin-treated NSCLC cells
remain to be further identified, these observations suggest that
marmesin-mediated inhibition of VEGF expression and secretion in
NSCLC cells may be one of the major factors in the modulation of
tumor-derived angiogenesis (Fig.
6B).
Discussion
Dysregulated activation of RTKs and/or cross-talk
between RTKs and integrins have been known to play important roles
in cancer growth, progression and poor prognosis in human lung
cancer (3,4,20,31,34).
Therefore, highly activated RTKs or integrins are considered as key
targets of antitumor agents in clinical trials and use. Numerous
investigations indicate that drugs targeting RTKs are more
effective than conventional therapeutics for the treatment of lung
cancer. However, molecular-targeted therapy in the clinic
eventually develops recurrent and metastatic lung cancers,
suggesting that the identification of key molecular targets in
RTK/integrin signaling pathways is absolutely required for the
development of effective therapeutic strategies and agents to treat
aggressive types of lung cancer.
Marmesin has been known to exert antitumor activity
against several types of cancer cells including colon cancer
(18). Recently, we reported that
marmesin, a coumarin component isolated from Broussonetia
kazinoki, inhibits VEGF-A-induced endothelial cell responses
in vitro and angiogenic sprouting ex vivo (14). These findings led us to investigate
the effects of marmesin on lung cancer cell fate and lung cancer
cell-derived angiogenesis. In the present study, we showed that
marmesin inhibited proliferation and invasion of NSCLC cells,
independently of p53 expression status. Antiproliferative activity
of marmesin appeared to be irreversible, since withdrawal of
marmesin did not reverse or affect cell proliferation in the NSCLC
cells. In addition, marmesin suppressed VEGF expression and
secretion in NSCLC cells, leading to inhibition of capillary-like
structure formation of HUVECs. The mechanism of these effects
involved inactivation of mitogenic signaling pathways such as Src,
MEK, ERK, Akt and p70S6K, and downregulation of VEGF,
VEGFR-2, integrin β1, ILK and MMP-2. In conclusion, these findings
provide important insights into the regulatory roles and
therapeutic potential of marmesin in NSCLC, and warrant preclinical
evaluation and development of marmesin as a potent antitumor agent
for the treatment of NSCLC associated with pathological angiogenic
responses.
Acknowledgements
The present study was supported by the R&D
Program for Forestry Technology (S121313L070100) through the Korea
Forest Service, and by the Basic Science Research Program
(2014R1A1A2058015) through the National Research Foundation of
Korea, Ministry of Education.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen Z, Fillmore CM, Hammerman PS, Kim CF
and Wong KK: Non-small-cell lung cancers: A heterogeneous set of
diseases. Nat Rev Cancer. 14:535–546. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sharma SV, Bell DW, Settleman J and Haber
DA: Epidermal growth factor receptor mutations in lung cancer. Nat
Rev Cancer. 7:169–181. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huang Y and Carbone DP: Mechanisms of and
strategies for overcoming resistance to anti-vascular endothelial
growth factor therapy in non-small cell lung cancer. Biochim
Biophys Acta. 1855:193–201. 2015.PubMed/NCBI
|
|
5
|
Carmeliet P and Jain RK: Principles and
mechanisms of vessel normalization for cancer and other angiogenic
diseases. Nat Rev Drug Discov. 10:417–427. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ellis LM and Hicklin DJ: VEGF-targeted
therapy: Mechanisms of anti-tumour activity. Nat Rev Cancer.
8:579–591. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cha JY, Kim YT, Kim HS and Cho YS:
Antihyperglycemic effect of stem bark powder from paper mulberry
(Broussonetia kazinoki Sieb.) in type 2 diabetic Otsuka Long-Evans
Tokushima fatty rats. J Med Food. 11:499–505. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bae UJ, Jang HY, Lim JM, Hua L, Ryu JH and
Park BH: Polyphenols isolated from Broussonetia kazinoki prevent
cytokine-induced β-cell damage and the development of type 1
diabetes. Exp Mol Med. 47:e1602015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee JK, Ha H, Lee HY, Park SJ, Jeong SL,
Choi YJ and Shin HK: Inhibitory effects of heartwood extracts of
Broussonetia kazinoki Sieb on the development of atopic dermatitis
in NC/Nga mice. Biosci Biotechnol Biochem. 74:1802–1806. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ryu JH, Ahn H and Jin Lee H: Inhibition of
nitric oxide production on LPS-activated macrophages by kazinol B
from Broussonetia kazinoki. Fitoterapia. 74:350–354. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kim HS, Lim J, Lee DY, Ryu JH and Lim JS:
Kazinol C from Broussonetia kazinoki activates AMP-activated
protein kinase to induce antitumorigenic effects in HT-29 colon
cancer cells. Oncol Rep. 33:223–229. 2015.PubMed/NCBI
|
|
12
|
Jung YC, Han S, Hua L, Ahn YH, Cho H, Lee
CJ, Lee H, Cho YY, Ryu JH, Jeon R, et al: Kazinol-E is a specific
inhibitor of ERK that suppresses the enrichment of a breast cancer
stem-like cell population. Biochem Biophys Res Commun. 470:294–299.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cho YR, Kim JH, Kim JK, Ahn EK, Ko HJ, In
JK, Lee SJ, Bae GU, Kim YK, Oh JS, et al: Broussonetia kazinoki
modulates the expression of VEGFR-2 and MMP-2 through the
inhibition of ERK, Akt and p70S6K-dependent signaling
pathways: Its implication in endothelial cell proliferation,
migration and tubular formation. Oncol Rep. 32:1531–1536.
2014.PubMed/NCBI
|
|
14
|
Kim JH, Kim JK, Ahn EK, Ko HJ, Cho YR, Lee
CH, Kim YK, Bae GU, Oh JS and Seo DW: Marmesin is a novel
angiogenesis inhibitor: Regulatory effect and molecular mechanism
on endothelial cell fate and angiogenesis. Cancer Lett.
369:323–330. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen IS, Chang CT, Sheen WS, Teng CM, Tsai
IL, Duh CY and Ko FN: Coumarins and antiplatelet aggregation
constituents from formosan Peucedanum japonicum. Phytochemistry.
41:525–530. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim JS, Kim JC, Shim SH, Lee EJ, Jin W,
Bae K, Son KH, Kim HP, Kang SS and Chang HW: Chemical constituents
of the root of Dystaenia takeshimana and their anti-inflammatory
activity. Arch Pharm Res. 29:617–623. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jain M, Kapadia R, Jadeja RN, Thounaojam
MC, Devkar RV and Mishra SH: Hepatoprotective activity of Feronia
limonia root. J Pharm Pharmacol. 64:888–896. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Znati M, Ben Jannet H, Cazaux S, Souchard
JP, Skhiri F Harzallah and Bouajila J: Antioxidant, 5-lipoxygenase
inhibitory and cytotoxic activities of compounds isolated from the
Ferula lutea flowers. Molecules. 19:16959–16975. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
In JK, Kim JK, Oh JS and Seo DW:
5-Caffeoylquinic acid inhibits invasion of non-small cell lung
cancer cells through the inactivation of p70S6K and Akt
activity: Involvement of p53 in differential regulation of
signaling pathways. Int J Oncol. 48:1907–1912. 2016.PubMed/NCBI
|
|
20
|
Yoon HJ, Cho YR, Joo JH and Seo DW:
Knockdown of integrin α3β1 expression induces proliferation and
migration of non-small cell lung cancer cells. Oncol Rep.
29:662–668. 2013.PubMed/NCBI
|
|
21
|
Lee HN, Joo JH, Oh JS, Choi SW and Seo DW:
Regulatory effects of Siegesbeckia glabrescens on non-small cell
lung cancer cell proliferation and invasion. Am J Chin Med.
42:453–463. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Joo JH, Hong SS, Cho YR and Seo DW:
10-Gingerol inhibits proliferation and invasion of MDA-MB-231
breast cancer cells through suppression of Akt and
p38MAPK activity. Oncol Rep. 35:779–784. 2016.PubMed/NCBI
|
|
23
|
Bourboulia D and Stetler-Stevenson WG:
Matrix metalloproteinases (MMPs) and tissue inhibitors of
metalloproteinases (TIMPs): Positive and negative regulators in
tumor cell adhesion. Semin Cancer Biol. 20:161–168. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kessenbrock K, Plaks V and Werb Z: Matrix
metalloproteinases: Regulators of the tumor microenvironment. Cell.
141:52–67. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Overall CM and Kleifeld O: Tumour
microenvironment - opinion: Validating matrix metalloproteinases as
drug targets and anti-targets for cancer therapy. Nat Rev Cancer.
6:227–239. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vandenbroucke RE and Libert C: Is there
new hope for therapeutic matrix metalloproteinase inhibition? Nat
Rev Drug Discov. 13:904–927. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Seo DW, Li H, Guedez L, Wingfield PT, Diaz
T, Salloum R, Wei BY and Stetler-Stevenson WG: TIMP-2 mediated
inhibition of angiogenesis: An MMP-independent mechanism. Cell.
114:171–180. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Stetler-Stevenson WG: Tissue inhibitors of
metalloproteinases in cell signaling: Metalloproteinase-independent
biological activities. Sci Signal. 1:re62008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim HJ, Cho YR, Kim SH and Seo DW:
TIMP-2-derived 18-mer peptide inhibits endothelial cell
proliferation and migration through cAMP/PKA-dependent mechanism.
Cancer Lett. 343:210–216. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lemmon MA and Schlessinger J: Cell
signaling by receptor tyrosine kinases. Cell. 141:1117–1134. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Desgrosellier JS and Cheresh DA: Integrins
in cancer: Biological implications and therapeutic opportunities.
Nat Rev Cancer. 10:9–22. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lee HN, Kim JK, Kim JH, Lee SJ, Ahn EK, Oh
JS and Seo DW: A mechanistic study on the anti-cancer activity of
ethyl caffeate in human ovarian cancer SKOV-3 cells. Chem Biol
Interact. 219:151–158. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim SH, Cho YR, Kim HJ, Oh JS, Ahn EK, Ko
HJ, Hwang BJ, Lee SJ, Cho Y, Kim YK, et al: Antagonism of
VEGF-A-induced increase in vascular permeability by an integrin
α3β1-Shp-1-cAMP/PKA pathway. Blood. 120:4892–4902. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Claesson-Welsh L and Welsh M: VEGFA and
tumour angiogenesis. J Intern Med. 273:114–127. 2013. View Article : Google Scholar : PubMed/NCBI
|