The major problem in the fight against cancer is
metastatic disease and growing resistance to available therapies.
Therefore, it is important to understand the mechanisms responsible
for the emergence and development of tumors to establish novel
molecular-based strategies to enable a more successful destruction
of aggressive tumor disease.
One of the well-known factors connected with tumor
growth and metastasis is the MET receptor (1). The MET tyrosine kinase receptor
together with its ligand, hepatocyte growth factor (HGF) also known
as scatter factor (SF), was identified to play a key role during
embriogenesis (2–5). At the early stages of development, HGF
and MET, are expressed in endoderm and mesoderm and act in an
autocrine manner (2). Later, during
organogenesis, MET is expressed in epithelial cells of many organs
(liver, kidney, lung and skin), whereas HGF in mesenchymal cells
(2). Moreover, MET is expressed in
some myoblasts and neuronal precursors, and contributes to the
development of muscular and nervous structures (2,5).
Crucial role of MET during embryogenesis was confirmed in
experiments with knockout mice that died in utero at E15 (4). HGF/MET axis is also important in the
process of skin, liver and kidney regeneration (6,7).
Ligand-induced MET activation leads to
phosphorylation of tyrosine residues (Tyr1230, Tyr1234 and Tyr1235)
in the kinase domain of the receptor and allows binding of effector
proteins, such as Gab1, Grb2, Shc, PI3K, Src, STAT3 or PLCγ
(8). It activates mainly the
RAS-MAPK and PI3K-AKT pathways leading to pleiotropic biological
effects on various target cells including the induction of cell
proliferation, morphogenetic transformation, cell motility and
invasiveness under both normal and pathological conditions
(9–11). The last findings also indicate that
MET is able to act through c-Abl and p-38-MAPK to induce p53
phosphorylation and promotes cell survival (12).
The MET receptor has been postulated as an essential
factor responsible for the functional cancer stem cell phenotype in
some tumors and as a CSC factor is believed to be responsible for
therapy resistance. The present review provides examples that MET
may be a potential cancer stem cell factor responsible for drug
resistance and tumor relapse.
In the early 1990s it was shown that mouse and human
cell lines with overexpression of HGF and/or MET become tumorigenic
and metastatic in nude mice and the level of MET and HGF directly
correlates with invasiveness and metastatic process (27). Nowadays, it is well documented that
deregulation of MET expression and activity is characteristic for
multiple cancer types and is a key event underlying tumor
progression and metastasis (1). A
large number of studies show that HGF and/or MET are frequently
expressed in human carcinomas and in other types of solid tumors
and in their metastases (1). MET
overexpression has been demonstrated in a variety of tumors,
including lung, breast, ovary, cervical, kidney, colon, thyroid,
liver, gastric carcinomas, glioma and osteosarcoma (28–41).
Activating point mutations of MET occur in sporadic and
inherited human renal carcinomas, hepatocellular carcinomas and
several other cancer types (33,35,42).
Moreover, in case of MET and/or its ligand HGF,
overexpression or misexpression often correlates with poor
prognosis (1,31,33,37).
MET was shown to be more frequently amplified in advanced stage of
colorectal and gastric cancers suggesting its role in the
metastatic process of malignant progression (33,43,44).
It was demonstrated for human head and neck cancers that activating
mutations of MET are clonally selected during the process of
metastasis and its level increased from 2% in the primary tumors to
50% in the metastases (45).
Interestingly enough, MET expression may vary within the same
tumor. As Pennacchietti and colleagues showed (46) both in carcinoma and sarcoma cells
hypoxia promotes the expression of met protooncogene and hypoxic
areas overexpress the MET receptor leading to activation of
invasive growth (46). It was also
shown that MET-positive cells within glioblastoma are located close
to the nearest blood vessels (47).
MET positive cells co-express glioblastoma stem cell markers, CD133
and CD15, compared with MET-negative cells. Moreover, MET
expression was efficient in inducing tumor formation regardless of
CD133 expression (47). CD133
glycoprotein has been widely used to purify hematopoietic stem and
progenitor cells and it was shown to define a subpopulation of
brain tumor cells with significantly increased capacity for tumor
initiation in xenograft models (48,49).
The authors suggest that MET signaling was responsible for
glioblastoma stem cell maintenance, migration and resistance to
radiation (47).
High MET expression pattern is currently associated
with increased tumor growth rate and metastasis, poor prognosis and
resistance to radiotherapy (57–59).
MET overexpression has been postulated as a prognostic factor in
lung (60,61), breast (62), head and neck (63), gastric (64), ovarian (65) and clear cell renal cell carcinoma
(66). MET overexpression is also
associated with poor prognosis and tumor invasiveness in
glioblastoma patients (67,68). It has been demonstrated that
enhanced level of MET in primary colorectal cancer may predict
tumor invasion and metastatic process (69). High MET protein level and its
activation, resulting from MET amplification, have been
reported as associated with a poor prognosis in colorectal and
gastric cancers (33,44,64).
It was also shown that MET overexpression was significantly
associated with worse 3- and 5-year overall survival,
progression-free survival and distant metastases in cervical cancer
patients (70). Similar results
were obtained after a follow-up of 50 months for multiple myeloma
patients, where high MET mRNA expression characterized a worse
progression-free and overall survival (71). Moreover, co-expression of the MET
receptor together with CD47 was proposed as a novel prognostic
factor for survival of patients suffering from luminal breast
cancer (72). Another study
proposed the MET receptor as independent predictor of decreased
5-year survival of patients with invasive ductal breast carcinoma
(62). Similar results were
obtained by the Edakuni group (73)
and showed correlation between co-expression of HGF and MET in
breast cancer, histologic grade and reduced patient survival
(73). All these examples highlight
MET as a prognostic factor whose presence and activity is important
for the overall survival and development of metastatic disease in
tumor patients.
The HGF-MET pathway has been proven to be an
attractive drug target for antitumor therapies. Several monoclonal
antibodies or small molecules targeting HGF or MET have been
discovered and used in monotherapy, in combination with other
targeted therapy or with chemotherapy (13). Despite encouraging results involving
the use of MET inhibitors in the laboratory and in clinical trials,
as well as in studies with other RTK inhibitors, it has been
suggested that resistance will develop even in the subset of
cancers that initially derive clinical benefits (14,15).
Several possible mechanisms of resistance to MET inhibitors such
as, MET point mutations, amplification or MET gene
overexpression, activation of MET parallel pathways or
amplification of the KRAS gene, have been described
(74–76). Cepero and colleagues (74) established cell lines resistant to
long-term treatment with MET inhibitors and showed that prolonged
exposure to increasing doses of c-MET inhibitors leads to
amplification, overexpression and activation of wild-type
MET and KRAS in gastric cell lines. Furthermore, they
observed strong activation of the mitogen-activated protein kinase
(MAPK) pathway (74).
Another mechanism of resistance showed that cells
developed resistance by acquired mutation in the MET activation
loop or activated epidermal growth factor receptor pathway due to
increased expression of transforming growth factor α (75). Two other studies showed that
overexpression of HER family members in gastric carcinoma cells and
non-small cell lung cancer cells are responsible for acquired
resistance to MET kinase inhibitors (76,77).
The authors concluded that cells carrying high MET copy number will
undergo an oncogenic switch that will create an ERBB tyrosine
kinase dependency (76,77).
A recent study revealed the acquisition of secondary
resistance to MET monoclonal antibodies. In a very elegant study of
Martin and coworkers (78),
MET-addicted lung cancer cells continuously treated with MET
monoclonal antibody became resistant to treatment, as a result of
an increase of MET gene copy number and MET overexpression.
However, MET antibody resistant cells were sensitive to
MET-specific small tyrosine kinase inhibitors (TKIs) and acquired
drug-dependence. Moreover, cells resistant to MET TKIs can still be
sensitive to treatment with the antibody. The authors suggest that
a discontinuous, combined treatment by antibodies and chemical
kinase inhibitors may increase the clinical response and bypass
resistance to anti-MET targeted therapies through synergistic
effect on tumor cells (78). The
results demonstrate that despite the acquired resistance to one
type of inhibitors, it is possible to use another type and achieve
good therapeutic effects. Furthermore, these results show the
importance of MET as a therapeutic target.
HGF-MET axis was also shown to be involved in
resistance to anti-VEGR therapy. In tumors resistant to inhibitor
of VEGF pathway, sunitinib, after treatment with highly selective
MET inhibitor, PF-04217903, together with sunitinib, tumor growth
was inhibited (82). The study on
renal cell carcinoma model demonstrated that the MET receptor is
involved in sunitinib acquired resistance (83). Combined treatment with the VEGF and
MET inhibitors induced prolonged survival and inhibited tumor
growth in mice giving hope for potential therapeutic use in the
clinical treatment (83).
In light of these data MET seems to be a very good
target for tumors resistant to tyrosine kinase targeted therapies.
However, the activity and function of the receptor depend on the
cell type and heterogeneity of tumors. Recent studies connect the
presence of the MET receptor with cancer stem cell phenotype.
It has been demonstrated that the MET receptor is
expressed in stem/progenitor cells in various types of adult normal
tissues and maintains stem cell properties. The MET receptor was
considered as a putative pancreatic stem/progenitor cell marker in
adult mouse pancreas (84,85). In the developing liver, cells
expressing MET can form stem cell colonies in vitro and
migrate and differentiate into liver parenchymal cells and
cholangiocytes when they were transplanted into the spleen or liver
of mice subjected to liver injury (86). The essential role of the HGF/MET
axis in hepatocyte-mediated liver regeneration, was shown by
Ishikawa and colleagues with the use of MET knockout mice (87). In the liver, the MET receptor
supported survival, proliferation, sphere formation and
differentiation properties of oval cells (87). Another study showed that MET, in
cardiac stem cells and early committed cells, is responsible for
proliferation, survival, migration and regeneration of the
infracted myocardium and improvement of ventricular function
(88).
Recently, the MET receptor has been postulated as an
essential factor responsible for the functional CSCs phenotype in
some tumors. It was reported that MET expression was associated
with glioblastoma stem cells (GSCs) identified by prospective
isolation from fresh tumors (47)
or with neurospheres endowed with specific genetic/molecular
features (91). Furthermore, MET
was considered to play a central role in maintaining CSC
populations in human glioblastoma multiforme (GBM), suggesting a
link between MET signaling and CSCs (91,92).
Other studies, on GBM cell subpopulations, showed that only cells
expressing high level of MET retained clonogenic, tumorigenic and
radioresistant properties, features of CSCs (47,91).
The authors demonstrated pivotal role of MET in supporting the pool
of GBM SCs (47). They used freshly
isolated patient-derived GBM cells and provided evidence suggesting
that MET plays critical role in SC maintenance, migration and
resistance to radiation (47).
Subpopulation with high MET level displayed enhanced kinetics
growth and was highly tumorigenic in vivo as well (47,91).
Moreover, only small population of GBM cells has been shown to be
positive for the MET receptor and to contain amplification of
MET, independent of other RTKs (93,94).
The study by Li et al (95)
involved MET as a novel, functional, stem cell marker for
pancreatic adenocarcinoma. The authors identified the population
with a high expression of MET and suggested that the receptor
regulates SCs proliferation, cell renewal and has the ability to
form tumors in NOD/SCID mice (95).
This study also showed that the use of the MET inhibitor or small
hairpin RNAs in pancreatic adenocarcinoma significantly inhibited
tumor sphere formation and self-renewal capacity (95). In pancreatic tumors established in
NOD SCID mice, MET inhibition decreased tumor growth, reduced the
population of CSCs and prevented the development of metastases
(95). The study of Sun and Wang
(96) on human head and neck
squamous cell carcinoma (HNSCC) demonstrated that MET expressing
cells have the capacity for self-renewal (96). Furthermore, the MET receptor was
responsible for tumor formation and metastatic process in NOD/SCID
mice and cisplatin resistance (96). It was also shown that the HGF/MET
axis regulates stem- like phenotype in human prostate cancer
(97). The study of Gastaldi and
coworkers (98) emphasized the role
of MET in breast tumorigenesis. The authors showed that MET acts as
a critical regulator of luminal cell proliferation and
differentiation in the context of murine mammary morphogenesis
(98). Moreover, the authors
presented that MET is preferentially expressed in luminal
progenitors and its activation stimulates clonogenic activity in
vitro, confers repopulating potential in vivo and
promotes aberrant branching morphogenesis (98). Table
I summarizes the data with reference to MET expression
correlated with cancer stem cell phenotype.
Our study on rhabdomyosarcoma showed that silencing
of the MET receptor stimulates tumor cell differentiation and
activation of MET signaling may be the cause of its development and
progression (99,100). We have also demonstrated that
cervical cancer cells depend on sustained MET activity for their
growth and survival and downregulation of MET decreased tumor
growth and forced tumor differentiation in vivo (101). Our observation on cervical cancer
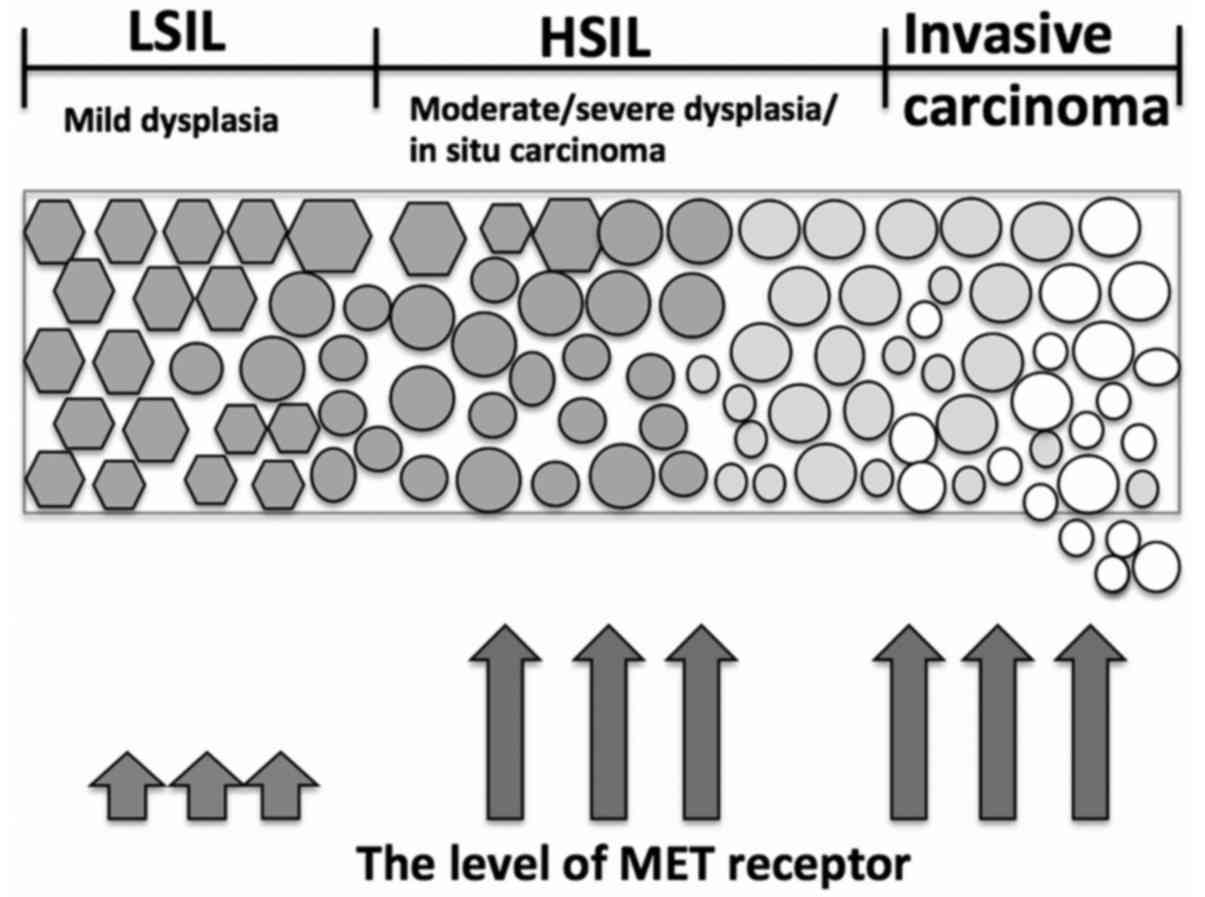
patient samples revealed that low level of MET accompanied
low-grade squamous intraepithelial lesion, whereas increased
heavily in high-grade squamous intraepithelial lesion and invasive
carcinoma (101) (Fig. 1).
The MET receptor not only supports stem-like
phenotype of cancer cells but also affects the expression and
activity of stem cell markers. It has been shown that MET signaling
can regulate glioma subpopulations and expand the pool of stem-like
cells. The study of Li and colleagues (92) revealed that MET positively
correlates with stem cell marker expression and the neoplastic stem
cell phenotype in glioblastoma neurospheres, as well as in clinical
glioblastoma specimens. MET expression and activation influences
the expression of reprogramming transcription factors known to
support embryonic stem cells, Sox2, Klf4, c-Myc, Oct4 and Nanog,
known to induce stem-like properties in differentiated cells
(92,102). Moreover, MET enhances stem cell
characteristics of neurosphere formation and neurosphere cell
self-renewal (92). The MET
receptor supports the GBM SC phenotype by involving an endogenous
dynamic mechanism analogous to cellular reprogramming (92). It was shown that MET-positive cells
expressed high levels of stemness transcriptional regulators, Oct4,
Nanog and Klf4, when compared to MET-negative cells and the
activation of MET signaling increases the expression of the Oct4,
Nanog and Klf4 (103). The
expression returned to basal levels in response to MET inhibition
(103). It was also shown that MET
induces a stem-like phenotype in prostate cancer and is expressed
together with stem-like markers CD49b and CD49f and (97). Another study reported that
cabozantinib, a novel inhibitor of MET, downregulated CSC markers,
SOX2 and CD133, induced apoptosis and increased efficacy of
gemcitabine, currently used in standard therapy for advanced
pancreatic cancer (104).
In our study, we have reported that blocking of the
MET receptor could influence expression and function of the
chemokine CXCR4 receptor in rhabdomyosarcoma and cervical carcinoma
cells (99,101). Cells with decreased MET expression
had impaired intracellular signaling and chemotaxis toward SDF-1
gradient, a ligand of the CXCR4 receptor, which was in accordance
with decreased expression of CXCR4 (99,101).
CXCR4 overexpression and hyperactivation was shown for the first
time to correlate with the metastatic ability of breast cancer
cells (105). Since that time, the
SDF-1-CXCR4 axis has been shown to be involved in the regulation of
metastasis to organs that highly express SDF-1 (e.g., lymph nodes,
lungs, liver and bones) (106). It
was postulated that cancer stem cells and trafficking of normal
stem cells involve similar mechanisms regulated partially by CXCR4
(107).
Growing evidence shows that CSCs are responsible for
resistance to conventional therapies, and thus, are the most likely
cause of tumor recurrence (22). It
has been postulated that stemness features of CSCs would allow them
to escape conventional antitumor therapy and maintain minimal
residual disease, leading to tumor relapse (23). It has been shown for breast cancer
(108) and glioma (21) that CSCs survived after radiation,
repaired their damaged DNA more efficiently than their non-CSC
counterparts and began the process of self-renewal (21,108).
Recently, it has been shown, in samples from patient tumors, that
CSC marker expression is associated with a poor clinical outcome
and may have prognostic value (24–26,109).
All the observations are clinically appealing
because combined treatment with an EGFR and MET inhibitor,
specifically in patients with evidence of MET amplification at
baseline, may lead to extended progression and better outcome.
A study showed that MET amplification together with
EMT, and stem cell-like features are observed in non-small cell
lung cancer cells with acquired resistance to Afatinib, an EGFR-TKI
(118). It was also demonstrated
that resistance of non-small lung cancer patients to EGFR
inhibitors is due to EGFR T790M mutation and MET amplification
(119). Moreover, the patients
acquired resistance to the MET receptor inhibitors used as a
therapeutic approach in clinical trials. The mechanism of the
resistance involved ABCB1 overexpression, which was associated with
CSC properties and EMT (119).
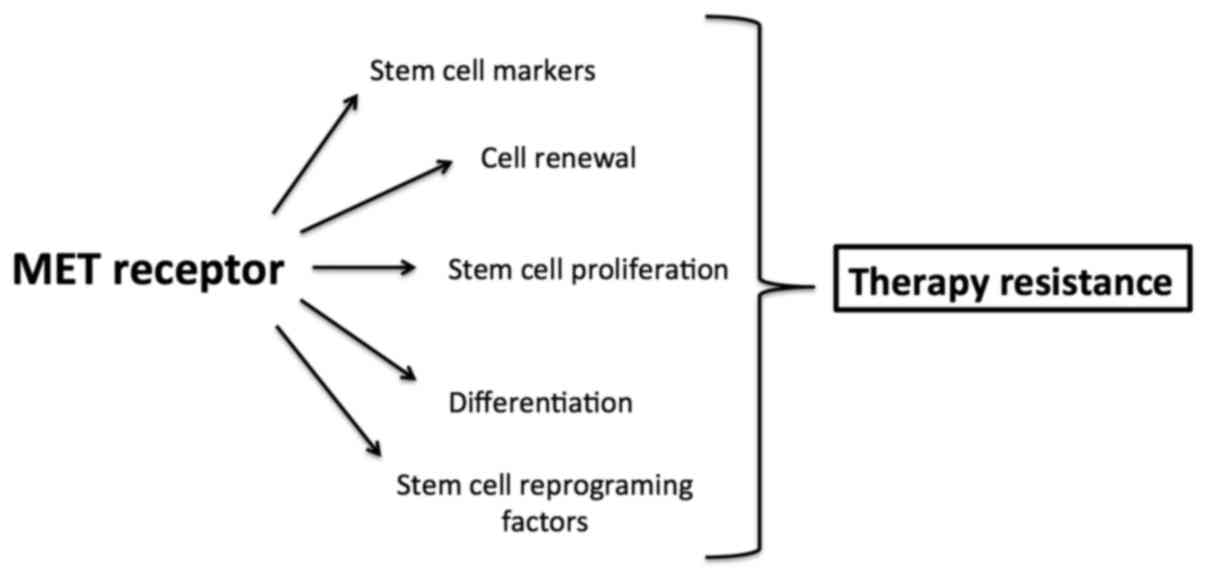
Taken together, MET involved in enhancing and
maintaining cancer stem cell properties may be responsible for
resistance to antitumor therapy (Fig.
2).
Targeted therapies with compounds inhibiting a
specific target molecule opened a new direction in the treatment of
cancer. The development of targeted therapies requires the
identification of good targets that are known to play a key role in
tumor cell growth and survival and are more effective and less
toxic than previous standards of care involving cytotoxic therapies
(120). Targeted therapy relies on
the concept of ‘oncogene addiction’ that reveals a possible
‘Achilles heel’ of cancer cells, wherein they depend on a single
oncogenic pathway for sustained proliferation and/or survival
(121,122). This means that the inhibition of a
single pathway, gene or protein to which they are addicted results
in the inhibition of their growth or even their death (121). Unfortunately, targeted
therapeutics in cancer has not yet met the high expectations of
patients and physicians because some patients relapsed following
treatment with specific inhibitors as a result of acquired
resistance mechanisms (120,123). CSCs have been shown to be largely
responsible for chemoresistant phenotypes in various tumors, thus,
the development of new, targeted, effective therapies has become
focused on identifying factors that drive and sustain CSCs. The CSC
hypothesis predicts that only therapies that efficiently eliminate
population of CSCs are able to induce long-term response and stop
tumor recurrence. The activation of the MET receptor axis has been
directly implicated in acquiring chemoresistance, maintaining
clonogenicity and ability to self-renew in various tumor cell
populations. In the light of our knowledge MET seems to have two
faces: acts as a promising factor for developing personalized
cancer therapy and as a factor responsible for cancer stem cell
properties and therapy resistance.
The present study was supported by a grant from the
National Science Centre (no. 2013/09/D/NZ5/00249) to K.M. The
Faculty of Biochemistry, Biophysics and Biotechnology of the
Jagiellonian University is a beneficiary of the structural funds
from the European Union and the Polish Ministry of Science and
Higher Education (grants nos. POIG.02.01.00-12-064/08 and
02.02.00-00-014/08) and is a partner of the Leading National
Research Center (KNOW) supported by the Ministry of Science and
Higher Education. I am very grateful to Mikolaj Przywara for
language editing, proofreading and suggestions.
|
1
|
Birchmeier C, Birchmeier W, Gherardi E and
Vande Woude GF: Met, metastasis, motility and more. Nat Rev Mol
Cell Biol. 4:915–925. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Andermarcher E, Surani MA and Gherardi E:
Co-expression of the HGF/SF and c-met genes during early mouse
embryogenesis precedes reciprocal expression in adjacent tissues
during organogenesis. Dev Genet. 18:254–266. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schmidt C, Bladt F, Goedecke S, Brinkmann
V, Zschiesche W, Sharpe M, Gherardi E and Birchmeier C: Scatter
factor/hepatocyte growth factor is essential for liver development.
Nature. 373:699–702. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Uehara Y, Minowa O, Mori C, Shiota K, Kuno
J, Noda T and Kitamura N: Placental defect and embryonic lethality
in mice lacking hepatocyte growth factor/scatter factor. Nature.
373:702–705. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Maina F, Hilton MC, Ponzetto C, Davies AM
and Klein R: Met receptor signaling is required for sensory nerve
development and HGF promotes axonal growth and survival of sensory
neurons. Genes Dev. 11:3341–3350. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Borowiak M, Garratt AN, Wüstefeld T,
Strehle M, Trautwein C and Birchmeier C: Met provides essential
signals for liver regeneration. Proc Natl Acad Sci USA.
101:10608–10613. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huh CG, Factor VM, Sánchez A, Uchida K,
Conner EA and Thorgeirsson SS: Hepatocyte growth factor/c-met
signaling pathway is required for efficient liver regeneration and
repair. Proc Natl Acad Sci USA. 101:4477–4482. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Furlan A, Kherrouche Z, Montagne R, Copin
MC and Tulasne D: Thirty years of research on met receptor to move
a biomarker from bench to bedside. Cancer Res. 74:6737–6744. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kermorgant S, Aparicio T, Dessirier V,
Lewin MJ and Lehy T: Hepatocyte growth factor induces colonic
cancer cell invasiveness via enhanced motility and protease
overproduction. Evidence for PI3 kinase and PKC involvement.
Carcinogenesis. 22:1035–1042. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Weidner KM, Sachs M and Birchmeier W: The
Met receptor tyrosine kinase transduces motility, proliferation,
and morphogenic signals of scatter factor/hepatocyte growth factor
in epithelial cells. J Cell Biol. 121:145–154. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Trusolino L, Bertotti A and Comoglio PM:
MET signalling: Principles and functions in development, organ
regeneration and cancer. Nat Rev Mol Cell Biol. 11:834–848. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Furlan A, Stagni V, Hussain A, Richelme S,
Conti F, Prodosmo A, Destro A, Roncalli M, Barilà D and Maina F:
Abl interconnects oncogenic Met and p53 core pathways in cancer
cells. Cell Death Differ. 18:1608–1616. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vigna E and Comoglio PM: Targeting the
oncogenic Met receptor by antibodies and gene therapy. Oncogene.
34:1883–1889. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Engelman JA and Settleman J: Acquired
resistance to tyrosine kinase inhibitors during cancer therapy.
Curr Opin Genet Dev. 18:73–79. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sierra JR, Cepero V and Giordano S:
Molecular mechanisms of acquired resistance to tyrosine kinase
targeted therapy. Mol Cancer. 9:752010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lapidot T, Sirard C, Vormoor J, Murdoch B,
Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA and
Dick JE: A cell initiating human acute myeloid leukaemia after
transplantation into SCID mice. Nature. 367:645–648. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Clarke MF, Dick JE, Dirks PB, Eaves CJ,
Jamieson CH, Jones DL, Visvader J, Weissman IL and Wahl GM: Cancer
stem cells - perspectives on current status and future directions:
AACR Workshop on cancer stem cells. Cancer Res. 66:9339–9344. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
OBrien CA, Pollett A, Gallinger S and Dick
JE: A human colon cancer cell capable of initiating tumour growth
in immunodeficient mice. Nature. 445:106–110. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ricci-Vitiani L, Lombardi DG, Pilozzi E,
Biffoni M, Todaro M, Peschle C and De Maria R: Identification and
expansion of human colon-cancer-initiating cells. Nature.
445:111–115. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen J, Li Y, Yu TS, McKay RM, Burns DK,
Kernie SG and Parada LF: A restricted cell population propagates
glioblastoma growth after chemotherapy. Nature. 488:522–526. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q,
Hjelmeland AB, Dewhirst MW, Bigner DD and Rich JN: Glioma stem
cells promote radioresistance by preferential activation of the DNA
damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kreso A and Dick JE: Evolution of the
cancer stem cell model. Cell Stem Cell. 14:275–291. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ghiaur G, Gerber J and Jones RJ: Concise
review: Cancer stem cells and minimal residual disease. Stem Cells.
30:89–93. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Maeda S, Shinchi H, Kurahara H, Mataki Y,
Maemura K, Sato M, Natsugoe S, Aikou T and Takao S: CD133
expression is correlated with lymph node metastasis and vascular
endothelial growth factor-C expression in pancreatic cancer. Br J
Cancer. 98:1389–1397. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Vogler T, Kriegl L, Horst D, Engel J,
Sagebiel S, Schäffauer AJ, Kirchner T and Jung A: The expression
pattern of aldehyde dehydrogenase 1 (ALDH1) is an independent
prognostic marker for low survival in colorectal tumors. Exp Mol
Pathol. 92:111–117. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zeppernick F, Ahmadi R, Campos B, Dictus
C, Helmke BM, Becker N, Lichter P, Unterberg A, Radlwimmer B and
Herold-Mende CC: Stem cell marker CD133 affects clinical outcome in
glioma patients. Clin Cancer Res. 14:123–129. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rong S, Segal S, Anver M, Resau JH and
Woude GF Vande: Invasiveness and metastasis of NIH 3T3 cells
induced by Met-hepatocyte growth factor/scatter factor autocrine
stimulation. Proc Natl Acad Sci USA. 91:4731–4735. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tokunou M, Niki T, Eguchi K, Iba S, Tsuda
H, Yamada T, Matsuno Y, Kondo H, Saitoh Y, Imamura H, et al: c-MET
expression in myofibroblasts: Role in autocrine activation and
prognostic significance in lung adenocarcinoma. Am J Pathol.
158:1451–1463. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tsao MS, Liu N, Chen JR, Pappas J, Ho J,
To C, Viallet J, Park M and Zhu H: Differential expression of
Met/hepatocyte growth factor receptor in subtypes of non-small cell
lung cancers. Lung Cancer. 20:1–16. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Olivero M, Rizzo M, Madeddu R, Casadio C,
Pennacchietti S, Nicotra MR, Prat M, Maggi G, Arena N, Natali PG,
et al: Overexpression and activation of hepatocyte growth
factor/scatter factor in human non-small-cell lung carcinomas. Br J
Cancer. 74:1862–1868. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lengyel E, Prechtel D, Resau JH, Gauger K,
Welk A, Lindemann K, Salanti G, Richter T, Knudsen B, Woude GF
Vande, et al: C-Met overexpression in node-positive breast cancer
identifies patients with poor clinical outcome independent of
Her2/neu. Int J Cancer. 113:678–682. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Di Renzo MF, Olivero M, Katsaros D,
Crepaldi T, Gaglia P, Zola P, Sismondi P and Comoglio PM:
Overexpression of the Met/HGF receptor in ovarian cancer. Int J
Cancer. 58:658–662. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Di Renzo MF, Olivero M, Giacomini A, Porte
H, Chastre E, Mirossay L, Nordlinger B, Bretti S, Bottardi S,
Giordano S, et al: Overexpression and amplification of the met/HGF
receptor gene during the progression of colorectal cancer. Clin
Cancer Res. 1:147–154. 1995.PubMed/NCBI
|
|
34
|
Natali PG, Prat M, Nicotra MR, Bigotti A,
Olivero M, Comoglio PM and Di Renzo MF: Overexpression of the
met/HGF receptor in renal cell carcinomas. Int J Cancer.
69:212–217. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Schmidt L, Duh FM, Chen F, Kishida T,
Glenn G, Choyke P, Scherer SW, Zhuang Z, Lubensky I, Dean M, et al:
Germline and somatic mutations in the tyrosine kinase domain of the
MET proto-oncogene in papillary renal carcinomas. Nat Genet.
16:68–73. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Knowles LM, Stabile LP, Egloff AM,
Rothstein ME, Thomas SM, Gubish CT, Lerner EC, Seethala RR, Suzuki
S, Quesnelle KM, et al: HGF and c-Met participate in paracrine
tumorigenic pathways in head and neck squamous cell cancer. Clin
Cancer Res. 15:3740–3750. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ramirez R, Hsu D, Patel A, Fenton C,
Dinauer C, Tuttle RM and Francis GL: Over-expression of hepatocyte
growth factor/scatter factor (HGF/SF) and the HGF/SF receptor
(cMET) are associated with a high risk of metastasis and recurrence
for children and young adults with papillary thyroid carcinoma.
Clin Endocrinol (Oxf). 53:635–644. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Soman NR, Correa P, Ruiz BA and Wogan GN:
The TPR-MET oncogenic rearrangement is present and expressed in
human gastric carcinoma and precursor lesions. Proc Natl Acad Sci
USA. 88:4892–4896. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Koochekpour S, Jeffers M, Rulong S, Taylor
G, Klineberg E, Hudson EA, Resau JH and Woude GF Vande: Met and
hepatocyte growth factor/scatter factor expression in human
gliomas. Cancer Res. 57:5391–5398. 1997.PubMed/NCBI
|
|
40
|
Ferracini R, Di Renzo MF, Scotlandi K,
Baldini N, Olivero M, Lollini P, Cremona O, Campanacci M and
Comoglio PM: The Met/HGF receptor is over-expressed in human
osteosarcomas and is activated by either a paracrine or an
autocrine circuit. Oncogene. 12:1697–1705. 1996.PubMed/NCBI
|
|
41
|
Di Renzo MF, Poulsom R, Olivero M,
Comoglio PM and Lemoine NR: Expression of the Met/hepatocyte growth
factor receptor in human pancreatic cancer. Cancer Res.
55:1129–1138. 1995.PubMed/NCBI
|
|
42
|
Ma PC, Tretiakova MS, MacKinnon AC,
Ramnath N, Johnson C, Dietrich S, Seiwert T, Christensen JG,
Jagadeeswaran R, Krausz T, et al: Expression and mutational
analysis of MET in human solid cancers. Genes Chromosomes Cancer.
47:1025–1037. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zeng ZS, Weiser MR, Kuntz E, Chen CT, Khan
SA, Forslund A, Nash GM, Gimbel M, Yamaguchi Y, Culliford AT IV, et
al: c-Met gene amplification is associated with advanced stage
colorectal cancer and liver metastases. Cancer Lett. 265:258–269.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tsugawa K, Yonemura Y, Hirono Y, Fushida
S, Kaji M, Miwa K, Miyazaki I and Yamamoto H: Amplification of the
c-met, c-erbB-2 and epidermal growth factor receptor gene in human
gastric cancers: Correlation to clinical features. Oncology.
55:475–481. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Di Renzo MF, Olivero M, Martone T, Maffe
A, Maggiora P, Stefani AD, Valente G, Giordano S, Cortesina G and
Comoglio PM: Somatic mutations of the MET oncogene are selected
during metastatic spread of human HNSC carcinomas. Oncogene.
19:1547–1555. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pennacchietti S, Michieli P, Galluzzo M,
Mazzone M, Giordano S and Comoglio PM: Hypoxia promotes invasive
growth by transcriptional activation of the met protooncogene.
Cancer Cell. 3:347–361. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Joo KM, Jin J, Kim E, Ho Kim K, Kim Y, Gu
Kang B, Kang YJ, Lathia JD, Cheong KH, Song PH, et al: MET
signaling regulates glioblastoma stem cells. Cancer Res.
72:3828–3838. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Singh SK, Clarke ID, Terasaki M, Bonn VE,
Hawkins C, Squire J and Dirks PB: Identification of a cancer stem
cell in human brain tumors. Cancer Res. 63:5821–5828.
2003.PubMed/NCBI
|
|
49
|
Bidlingmaier S, Zhu X and Liu B: The
utility and limitations of glycosylated human CD133 epitopes in
defining cancer stem cells. J Mol Med (Berl). 86:1025–1032. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Comoglio PM, Giordano S and Trusolino L:
Drug development of MET inhibitors: Targeting oncogene addiction
and expedience. Nat Rev Drug Discov. 7:504–516. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Benvenuti S, Lazzari L, Arnesano A, Li
Chiavi G, Gentile A and Comoglio PM: Ron kinase
transphosphorylation sustains MET oncogene addiction. Cancer Res.
71:1945–1955. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Boccaccio C and Comoglio PM: The MET
oncogene in glioblastoma stem cells: Implications as a diagnostic
marker and a therapeutic target. Cancer Res. 73:3193–3199. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Corso S, Migliore C, Ghiso E, De Rosa G,
Comoglio PM and Giordano S: Silencing the MET oncogene leads to
regression of experimental tumors and metastases. Oncogene.
27:684–693. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lennerz JK, Kwak EL, Ackerman A, Michael
M, Fox SB, Bergethon K, Lauwers GY, Christensen JG, Wilner KD,
Haber DA, et al: MET amplification identifies a small and
aggressive subgroup of esophagogastric adenocarcinoma with evidence
of responsiveness to crizotinib. J Clin Oncol. 29:4803–4810. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lutterbach B, Zeng Q, Davis LJ, Hatch H,
Hang G, Kohl NE, Gibbs JB and Pan BS: Lung cancer cell lines
harboring MET gene amplification are dependent on Met for growth
and survival. Cancer Res. 67:2081–2088. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Smolen GA, Sordella R, Muir B, Mohapatra
G, Barmettler A, Archibald H, Kim WJ, Okimoto RA, Bell DW, Sgroi
DC, et al: Amplification of MET may identify a subset of cancers
with extreme sensitivity to the selective tyrosine kinase inhibitor
PHA-665752. Proc Natl Acad Sci USA. 103:2316–2321. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
De Bacco F, Luraghi P, Medico E, Reato G,
Girolami F, Perera T, Gabriele P, Comoglio PM and Boccaccio C:
Induction of MET by ionizing radiation and its role in
radioresistance and invasive growth of cancer. J Natl Cancer Inst.
103:645–661. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Matsui S, Osada S, Tomita H, Komori S,
Mori R, Sanada Y, Takahashi T, Yamaguchi K and Yoshida K: Clinical
significance of aggressive hepatectomy for colorectal liver
metastasis, evaluated from the HGF/c-Met pathway. Int J Oncol.
37:289–297. 2010.PubMed/NCBI
|
|
59
|
Navab R, Liu J, Seiden-Long I, Shih W, Li
M, Bandarchi B, Chen Y, Lau D, Zu YF, Cescon D, et al:
Co-overexpression of Met and hepatocyte growth factor promotes
systemic metastasis in NCI-H460 non-small cell lung carcinoma
cells. Neoplasia. 11:1292–1300. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cai YR, Zhang HQ, Qu Y, Mu J, Zhao D, Zhou
LJ, Yan H, Ye JW and Liu Y: Expression of MET and SOX2 genes in
non-small cell lung carcinoma with EGFR mutation. Oncol Rep.
26:877–885. 2011.PubMed/NCBI
|
|
61
|
Masuya D, Huang C, Liu D, Nakashima T,
Kameyama K, Haba R, Ueno M and Yokomise H: The tumour-stromal
interaction between intratumoral c-Met and stromal hepatocyte
growth factor associated with tumour growth and prognosis in
non-small-cell lung cancer patients. Br J Cancer. 90:1555–1562.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ghoussoub RA, Dillon DA, DAquila T, Rimm
EB, Fearon ER and Rimm DL: Expression of c-met is a strong
independent prognostic factor in breast carcinoma. Cancer.
82:1513–1520. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Qian CN, Guo X, Cao B, Kort EJ, Lee CC,
Chen J, Wang LM, Mai WY, Min HQ, Hong MH, et al: Met protein
expression level correlates with survival in patients with
late-stage nasopharyngeal carcinoma. Cancer Res. 62:589–596.
2002.PubMed/NCBI
|
|
64
|
Nakajima M, Sawada H, Yamada Y, Watanabe
A, Tatsumi M, Yamashita J, Matsuda M, Sakaguchi T, Hirao T and
Nakano H: The prognostic significance of amplification and
overexpression of c-met and c-erb B-2 in human gastric carcinomas.
Cancer. 85:1894–1902. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Sawada K, Radjabi AR, Shinomiya N, Kistner
E, Kenny H, Becker AR, Turkyilmaz MA, Salgia R, Yamada SD, Woude GF
Vande, et al: c-Met overexpression is a prognostic factor in
ovarian cancer and an effective target for inhibition of peritoneal
dissemination and invasion. Cancer Res. 67:1670–1679. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Gibney GT, Aziz SA, Camp RL, Conrad P,
Schwartz BE, Chen CR, Kelly WK and Kluger HM: c-Met is a prognostic
marker and potential therapeutic target in clear cell renal cell
carcinoma. Ann Oncol. 24:343–349. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Nabeshima K, Shimao Y, Sato S, Kataoka H,
Moriyama T, Kawano H, Wakisaka S and Koono M: Expression of c-Met
correlates with grade of malignancy in human astrocytic tumours: An
immunohistochemical study. Histopathology. 31:436–443. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kong DS, Song SY, Kim DH, Joo KM, Yoo JS,
Koh JS, Dong SM, Suh YL, Lee JI, Park K, et al: Prognostic
significance of c-Met expression in glioblastomas. Cancer.
115:140–148. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Takeuchi H, Bilchik A, Saha S, Turner R,
Wiese D, Tanaka M, Kuo C, Wang HJ and Hoon DS: c-MET expression
level in primary colon cancer: A predictor of tumor invasion and
lymph node metastases. Clin Cancer Res. 9:1480–1488.
2003.PubMed/NCBI
|
|
70
|
Refaat T, Donnelly ED, Sachdev S, Parimi
V, El Achy S, Dalal P, Farouk M, Berg N, Helenowski I, Gross JP, et
al: c-Met overexpression in cervical cancer, a prognostic factor
and a potential molecular therapeutic target. Am J Clin Oncol. Jun
10–2015.(Epub ahead of print). View Article : Google Scholar
|
|
71
|
Rocci A, Gambella M, Aschero S, Baldi I,
Trusolino L, Cavallo F, Gay F, Larocca A, Magarotto V, Omedè P, et
al: MET dysregulation is a hallmark of aggressive disease in
multiple myeloma patients. Br J Haematol. 164:841–850. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Baccelli I, Stenzinger A, Vogel V,
Pfitzner BM, Klein C, Wallwiener M, Scharpff M, Saini M,
Holland-Letz T, Sinn HP, et al: Co-expression of MET and CD47 is a
novel prognosticator for survival of luminal breast cancer
patients. Oncotarget. 5:8147–8160. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Edakuni G, Sasatomi E, Satoh T, Tokunaga O
and Miyazaki K: Expression of the hepatocyte growth factor/c-Met
pathway is increased at the cancer front in breast carcinoma.
Pathol Int. 51:172–178. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Cepero V, Sierra JR, Corso S, Ghiso E,
Casorzo L, Perera T, Comoglio PM and Giordano S: MET and KRAS gene
amplification mediates acquired resistance to MET tyrosine kinase
inhibitors. Cancer Res. 70:7580–7590. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Qi J, McTigue MA, Rogers A, Lifshits E,
Christensen JG, Jänne PA and Engelman JA: Multiple mutations and
bypass mechanisms can contribute to development of acquired
resistance to MET inhibitors. Cancer Res. 71:1081–1091. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Corso S, Ghiso E, Cepero V, Sierra JR,
Migliore C, Bertotti A, Trusolino L, Comoglio PM and Giordano S:
Activation of HER family members in gastric carcinoma cells
mediates resistance to MET inhibition. Mol Cancer. 9:1212010.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
McDermott U, Pusapati RV, Christensen JG,
Gray NS and Settleman J: Acquired resistance of non-small cell lung
cancer cells to MET kinase inhibition is mediated by a switch to
epidermal growth factor receptor dependency. Cancer Res.
70:1625–1634. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Martin V, Corso S, Comoglio PM and
Giordano S: Increase of MET gene copy number confers resistance to
a monovalent MET antibody and establishes drug dependence. Mol
Oncol. 8:1561–1574. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Bean J, Brennan C, Shih JY, Riely G, Viale
A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, et al: MET
amplification occurs with or without T790M mutations in EGFR mutant
lung tumors with acquired resistance to gefitinib or erlotinib.
Proc Natl Acad Sci USA. 104:20932–20937. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Shattuck DL, Miller JK, Carraway KL III
and Sweeney C: Met receptor contributes to trastuzumab resistance
of Her2-overexpressing breast cancer cells. Cancer Res.
68:1471–1477. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Shojaei F, Lee JH, Simmons BH, Wong A,
Esparza CO, Plumlee PA, Feng J, Stewart AE, Hu-Lowe DD and
Christensen JG: HGF/c-Met acts as an alternative angiogenic pathway
in sunitinib-resistant tumors. Cancer Res. 70:10090–10100. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Ciamporcero E, Miles KM, Adelaiye R,
Ramakrishnan S, Shen L, Ku S, Pizzimenti S, Sennino B, Barrera G
and Pili R: Combination strategy targeting VEGF and HGF/c-met in
human renal cell carcinoma models. Mol Cancer Ther. 14:101–110.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Teng C, Guo Y, Zhang H, Zhang H, Ding M
and Deng H: Identification and characterization of label-retaining
cells in mouse pancreas. Differentiation. 75:702–712. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Oshima Y, Suzuki A, Kawashimo K, Ishikawa
M, Ohkohchi N and Taniguchi H: Isolation of mouse pancreatic ductal
progenitor cells expressing CD133 and c-Met by flow cytometric cell
sorting. Gastroenterology. 132:720–732. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Kamiya A, Gonzalez FJ and Nakauchi H:
Identification and differentiation of hepatic stem cells during
liver development. Front Biosci. 11:1302–1310. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Ishikawa T, Factor VM, Marquardt JU, Raggi
C, Seo D, Kitade M, Conner EA and Thorgeirsson SS: Hepatocyte
growth factor/c-met signaling is required for stem-cell-mediated
liver regeneration in mice. Hepatology. 55:1215–1226. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Urbanek K, Rota M, Cascapera S, Bearzi C,
Nascimbene A, De Angelis A, Hosoda T, Chimenti S, Baker M, Limana
F, et al: Cardiac stem cells possess growth factor-receptor systems
that after activation regenerate the infarcted myocardium,
improving ventricular function and long-term survival. Circ Res.
97:663–673. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Chmielowiec J, Borowiak M, Morkel M,
Stradal T, Munz B, Werner S, Wehland J, Birchmeier C and Birchmeier
W: c-Met is essential for wound healing in the skin. J Cell Biol.
177:151–162. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Nicoleau C, Benzakour O, Agasse F, Thiriet
N, Petit J, Prestoz L, Roger M, Jaber M and Coronas V: Endogenous
hepatocyte growth factor is a niche signal for subventricular zone
neural stem cell amplification and self-renewal. Stem Cells.
27:408–419. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
De Bacco F, Casanova E, Medico E,
Pellegatta S, Orzan F, Albano R, Luraghi P, Reato G, DAmbrosio A,
Porrati P, et al: The MET oncogene is a functional marker of a
glioblastoma stem cell subtype. Cancer Res. 72:4537–4550. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Li Y, Li A, Glas M, Lal B, Ying M, Sang Y,
Xia S, Trageser D, Guerrero-Cázares H, Eberhart CG, et al: c-Met
signaling induces a reprogramming network and supports the
glioblastoma stem-like phenotype. Proc Natl Acad Sci USA.
108:9951–9956. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Snuderl M, Fazlollahi L, Le LP, Nitta M,
Zhelyazkova BH, Davidson CJ, Akhavanfard S, Cahill DP, Aldape KD,
Betensky RA, et al: Mosaic amplification of multiple receptor
tyrosine kinase genes in glioblastoma. Cancer Cell. 20:810–817.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Szerlip NJ, Pedraza A, Chakravarty D, Azim
M, McGuire J, Fang Y, Ozawa T, Holland EC, Huse JT, Jhanwar S, et
al: Intratumoral heterogeneity of receptor tyrosine kinases EGFR
and PDGFRA amplification in glioblastoma defines subpopulations
with distinct growth factor response. Proc Natl Acad Sci USA.
109:3041–3046. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Li C, Wu JJ, Hynes M, Dosch J, Sarkar B,
Welling TH, di Magliano M Pasca and Simeone DM: c-Met is a marker
of pancreatic cancer stem cells and therapeutic target.
Gastroenterology. 141:2218–2227.e5. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Sun S and Wang Z: Head neck squamous cell
carcinoma c-Met+ cells display cancer stem cell
properties and are responsible for cisplatin-resistance and
metastasis. Int J Cancer. 129:2337–2348. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
van Leenders GJ, Sookhlall R, Teubel WJ,
de Ridder CM, Reneman S, Sacchetti A, Vissers KJ, van Weerden W and
Jenster G: Activation of c-MET induces a stem-like phenotype in
human prostate cancer. PLoS One. 6:e267532011. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Gastaldi S, Sassi F, Accornero P, Torti D,
Galimi F, Migliardi G, Molyneux G, Perera T, Comoglio PM, Boccaccio
C, et al: Met signaling regulates growth, repopulating potential
and basal cell-fate commitment of mammary luminal progenitors:
Implications for basal-like breast cancer. Oncogene. 32:1428–1440.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Miekus K, Lukasiewicz E, Jarocha D, Sekula
M, Drabik G and Majka M: The decreased metastatic potential of
rhabdomyosarcoma cells obtained through MET receptor downregulation
and the induction of differentiation. Cell Death Dis. 4:e4592013.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Skrzypek K, Kusienicka A, Szewczyk B,
Adamus T, Lukasiewicz E, Miekus K and Majka M: Constitutive
activation of MET signaling impairs myogenic differentiation of
rhabdomyosarcoma and promotes its development and progression.
Oncotarget. 6:31378–31398. 2015.PubMed/NCBI
|
|
101
|
Miekus K, Pawlowska M, Sekuła M, Drabik G,
Madeja Z, Adamek D and Majka M: MET receptor is a potential
therapeutic target in high grade cervical cancer. Oncotarget.
6:10086–10101. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Takahashi K and Yamanaka S: Induction of
pluripotent stem cells from mouse embryonic and adult fibroblast
cultures by defined factors. Cell. 126:663–676. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Jun HJ, Bronson RT and Charest A:
Inhibition of EGFR induces a c-MET-driven stem cell population in
glioblastoma. Stem Cells. 32:338–348. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Hage C, Rausch V, Giese N, Giese T,
Schönsiegel F, Labsch S, Nwaeburu C, Mattern J, Gladkich J and Herr
I: The novel c-Met inhibitor cabozantinib overcomes gemcitabine
resistance and stem cell signaling in pancreatic cancer. Cell Death
Dis. 4:e6272013. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Müller A, Homey B, Soto H, Ge N, Catron D,
Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, et al:
Involvement of chemokine receptors in breast cancer metastasis.
Nature. 410:50–56. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Zlotnik A, Burkhardt AM and Homey B:
Homeostatic chemokine receptors and organ-specific metastasis. Nat
Rev Immunol. 11:597–606. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Kucia M, Reca R, Miekus K, Wanzeck J,
Wojakowski W, Janowska-Wieczorek A, Ratajczak J and Ratajczak MZ:
Trafficking of normal stem cells and metastasis of cancer stem
cells involve similar mechanisms: Pivotal role of the SDF-1-CXCR4
axis. Stem Cells. 23:879–894. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Phillips TM, McBride WH and Pajonk F: The
response of CD24−/low/CD44+ breast
cancer-initiating cells to radiation. J Natl Cancer Inst.
98:1777–1785. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Martin TA and Jiang WG: Evaluation of the
expression of stem cell markers in human breast cancer reveals a
correlation with clinical progression and metastatic disease in
ductal carcinoma. Oncol Rep. 31:262–272. 2014.PubMed/NCBI
|
|
110
|
Bardelli A, Corso S, Bertotti A, Hobor S,
Valtorta E, Siravegna G, Sartore-Bianchi A, Scala E, Cassingena A,
Zecchin D, et al: Amplification of the MET receptor drives
resistance to anti-EGFR therapies in colorectal cancer. Cancer
Discov. 3:658–673. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Turke AB, Zejnullahu K, Wu YL, Song Y,
Dias-Santagata D, Lifshits E, Toschi L, Rogers A, Mok T, Sequist L,
et al: Preexistence and clonal selection of MET amplification in
EGFR mutant NSCLC. Cancer Cell. 17:77–88. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Yano S, Wang W, Li Q, Matsumoto K,
Sakurama H, Nakamura T, Ogino H, Kakiuchi S, Hanibuchi M, Nishioka
Y, et al: Hepatocyte growth factor induces gefitinib resistance of
lung adenocarcinoma with epidermal growth factor
receptor-activating mutations. Cancer Res. 68:9479–9487. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Wilson TR, Fridlyand J, Yan Y, Penuel E,
Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, et al:
Widespread potential for growth-factor-driven resistance to
anticancer kinase inhibitors. Nature. 487:505–509. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Straussman R, Morikawa T, Shee K,
Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J,
Frederick DT, et al: Tumour micro-environment elicits innate
resistance to RAF inhibitors through HGF secretion. Nature.
487:500–504. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Luraghi P, Reato G, Cipriano E, Sassi F,
Orzan F, Bigatto V, De Bacco F, Menietti E, Han M, Rideout WM III,
et al: MET signaling in colon cancer stem-like cells blunts the
therapeutic response to EGFR inhibitors. Cancer Res. 74:1857–1869.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Vermeulen L, De Sousa E, Melo F, van der
Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M,
Merz C, Rodermond H, et al: Wnt activity defines colon cancer stem
cells and is regulated by the microenvironment. Nat Cell Biol.
12:468–476. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Kim KH, Seol HJ, Kim EH, Rheey J, Jin HJ,
Lee Y, Joo KM, Lee J and Nam DH: Wnt/β-catenin signaling is a key
downstream mediator of MET signaling in glioblastoma stem cells.
Neuro Oncol. 15:161–171. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Hashida S, Yamamoto H, Shien K, Miyoshi Y,
Ohtsuka T, Suzawa K, Watanabe M, Maki Y, Soh J, Asano H, et al:
Acquisition of cancer stem cell-like properties in non-small cell
lung cancer with acquired resistance to afatinib. Cancer Sci.
106:1377–1384. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Sugano T, Seike M, Noro R, Soeno C, Chiba
M, Zou F, Nakamichi S, Nishijima N, Matsumoto M, Miyanaga A, et al:
Inhibition of ABCB1 overcomes cancer stem cell-like properties and
acquired resistance to MET inhibitors in non-small cell lung
cancer. Mol Cancer Ther. 14:2433–2440. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Figlin RA, Kaufmann I and Brechbiel J:
Targeting PI3K and mTORC2 in metastatic renal cell carcinoma: New
strategies for overcoming resistance to VEGFR and mTORC1
inhibitors. Int J Cancer. 133:788–796. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Weinstein IB: Cancer. Addiction to
oncogenes - the Achilles heal of cancer. Science. 297:63–64. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Sharma SV and Settleman J: Oncogene
addiction: Setting the stage for molecularly targeted cancer
therapy. Genes Dev. 21:3214–3231. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Viedma-Rodríguez R, Baiza-Gutman L,
Salamanca-Gómez F, Diaz-Zaragoza M, Martínez-Hernández G,
Esparza-Garrido R Ruiz, Velázquez-Flores MA and Arenas-Aranda D:
Mechanisms associated with resistance to tamoxifen in estrogen
receptor-positive breast cancer (Review). Oncol Rep. 32:3–15.
2014.PubMed/NCBI
|