Introduction
Oral squamous cell carcinoma (OSCC) is the most
prevalent cancer occurring in the oral cavity and is the sixth most
common cancer in Asia and the eleventh worldwide (1,2). The
majority of OSCC patients are typically diagnosed when the cancer
develops to an advanced stage, which confers a decrease of the
5-year survival rate to 20% (1). In
this advanced-stage cancer, the phenomenon of multidrug resistance
(MDR) is the most critical problem in chemotherapy, leading to
therapeutic failure and disease recurrence (3). MDR can be attributed to multiple
factors, such as upregulation of the ABC transporter family,
aberrant apoptosis, change in DNA damage repair, miRNA regulation
and cancer stem cell regulation (4). To circumvent the drug resistance of
cancer cells, simultaneous activation of different cell-death
pathways, such as apoptosis, autophagy and necroptosis, could be
principally applicable (5–8). Hence, investigation of novel
anticancer agents targeting contemporary non-apoptotic and
apoptotic mechanisms may be a great step forward for
multidrug-resistant OSCC treatment.
Apoptosis and autophagy are catalytic processes
essential for maintaining cell populations and tissue homeostasis.
Apoptosis can be initiated through mitochondrial-mediated and death
receptor-mediated pathways. Both pathways lead to activation of a
cascade of proteolytic caspases, thereby cleaving cytosolic and
nuclear proteins, resulting in nuclear fragmentation and apoptotic
death (9). Apoptosis is well-known
programmed cell death type-1, which has been utilized as a
cell-death mechanism to eliminate cancer cells by a number of
anticancer compounds (10).
Meanwhile, autophagy is a conserved catabolic process whereby
damaged cellular constituents are sequestered into double-membrane
vesicles known as autophagosomes, which are subsequently degraded
by lysosomal machinery (11).
During autophagy, the formation of autophagosomes is processed by a
conversion of microtubule-associated protein 1 light chain 3 (LC3-I
to LC3-II). When the process progresses to the lysosomal
degradation step, the cellular p62/SQSTM1 (p62) level is then
degraded, and this whole process is referred to as autophagic flux
(12). Autophagy plays a pro-death
role or pro-survival role by being a ‘double-edged sword’. The
pro-death role of autophagy occurs when autophagy is excessively
activated, and this has shed light on its anticancer potential
(13). The potential of these two
cell death pathways to effectively eliminate multidrug-resistant
cancer cells has been demonstrated (5,6,14).
Various stress conditions can trigger these
cell-death pathways. Endoplasmic reticulum (ER) stress with the
consequent unfolded protein response (UPR) is one of the stress
responses that confer apoptosis and autophagy (15). Components involved in ER stress and
UPR signaling, such as inositol-requiring protein-1 and
transcription factor C/EBP homologous protein (CHOP), modulate the
mitogen-activated protein kinase (MAPK) signaling pathway,
particularly p38 MAPK and JNK, which are important mediators
promoting cell death (16,17). Accumulating studies have
demonstrated that activated p38 MAPK and JNK are involved in
apoptosis and/or autophagy in various cancer cell lines (18–20).
Collective evidence has revealed that carbazole
alkaloids exhibit a wide range of anticancer activities
demonstrated both in vitro and in vivo (21–23),
leading research in carbazole alkaloids to become more attractive
in the pharmacological field in the last few decades (24). Isomahanine, a bioactive carbazole
alkaloid abundantly found in Murraya koenigii, and its
isomer mahanine are capable of inducing apoptosis in human leukemia
cells via a caspase-dependent pathway (22). Relevant to multidrug-resistant
cancers, mahanine can enhance the chemosensitivity of colon and
cervical cancer cells to the anticancer drugs cisplatin and
5-fluorouracil (25,26). Carbazole derivatives have been
documented to upregulate autophagy and sensitize glioblastoma cells
to anticancer drugs (27). However,
the function of isomahanine in multidrug-resistant OSCC, along with
the underlying mechanisms, have not yet been investigated.
In the present study, we aimed to investigate the
cytotoxic mechanisms of isomahanine via apoptosis and autophagy in
our established multidrug-resistant OSCC cell line (CLS-354/DX). We
found that CLS-354/DX cells exhibited overexpression of multidrug
resistance-associated protein 1 (MRP1) protein and a low
responsiveness to conventional anticancer drugs. Isomahanine
exerted an effective cytotoxic effect against CLS-354/DX cells
regardless of their resistance compared to the parental cell line
(CLS-354/WT). The compound induced ER stress and simultaneously
triggered apoptosis and autophagic flux through the p38 MAPK
signaling pathway. This finding supports the potential use of
isomahanine to circumvent MDR in OSCC cells.
Materials and methods
Preparation of isomahanine
The bioactive carbazole alkaloid, i.e., isomahanine,
was isolated and purified from the CH2Cl2
extract of M. koenigii leaves by repeated silica gel and
Sephadex LH-20 column chromatography. The spectroscopic data of
isomahanine were consistent with the reported values (28). The purified carbazole alkaloid was
dissolved in dimethyl sulfoxide (DMSO) prior to conducting
experiments.
Reagents and antibodies
cis-diamminedichloroplatinum(II) (cisplatin),
camptothecin, DMSO, 3-MA, 4-phenylbutyric acid (4-PBA), chloroquine
(CQ) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) were purchased from Sigma-Aldrich Corp. (St. Louis,
MO, USA). Benzyloxycarbonyl-ValAla-Asp (OMe) and fluoromethylketone
(z-VAD-fmk) were purchased from InvivoGen (San Diego, CA, USA).
Luminita™ Chemiluminescent HRP substrate was purchased from EMD
Millipore (Billerica, MA, USA). All primary antibodies (rabbit),
HRP-conjugated secondary antibodies (anti-rabbit) and MAPK
inhibitors (U0126, SB203580 and SP600125) were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA).
Cell lines and culture
Human OSCC CLS-354 cells (CLS Cell Lines Service
GmbH, Eppelheim, Germany) at passage no. 30–40 were cultured in
RPMI-1640 medium supplemented with 10% fetal bovine serum (Biochrom
GmbH, Berlin, Germany), 1% penicillin/streptomycin and 2 mM stable
L-glutamine (PAA Laboratories GmbH, Pasching, Austria). The cancer
cell line was maintained in an atmosphere of 95% humidity and 5%
CO2 at 37̊C. CLS-354 cells, as parental cells, were oral
epithelial cancer cells exhibiting epithelial-like shape, namely
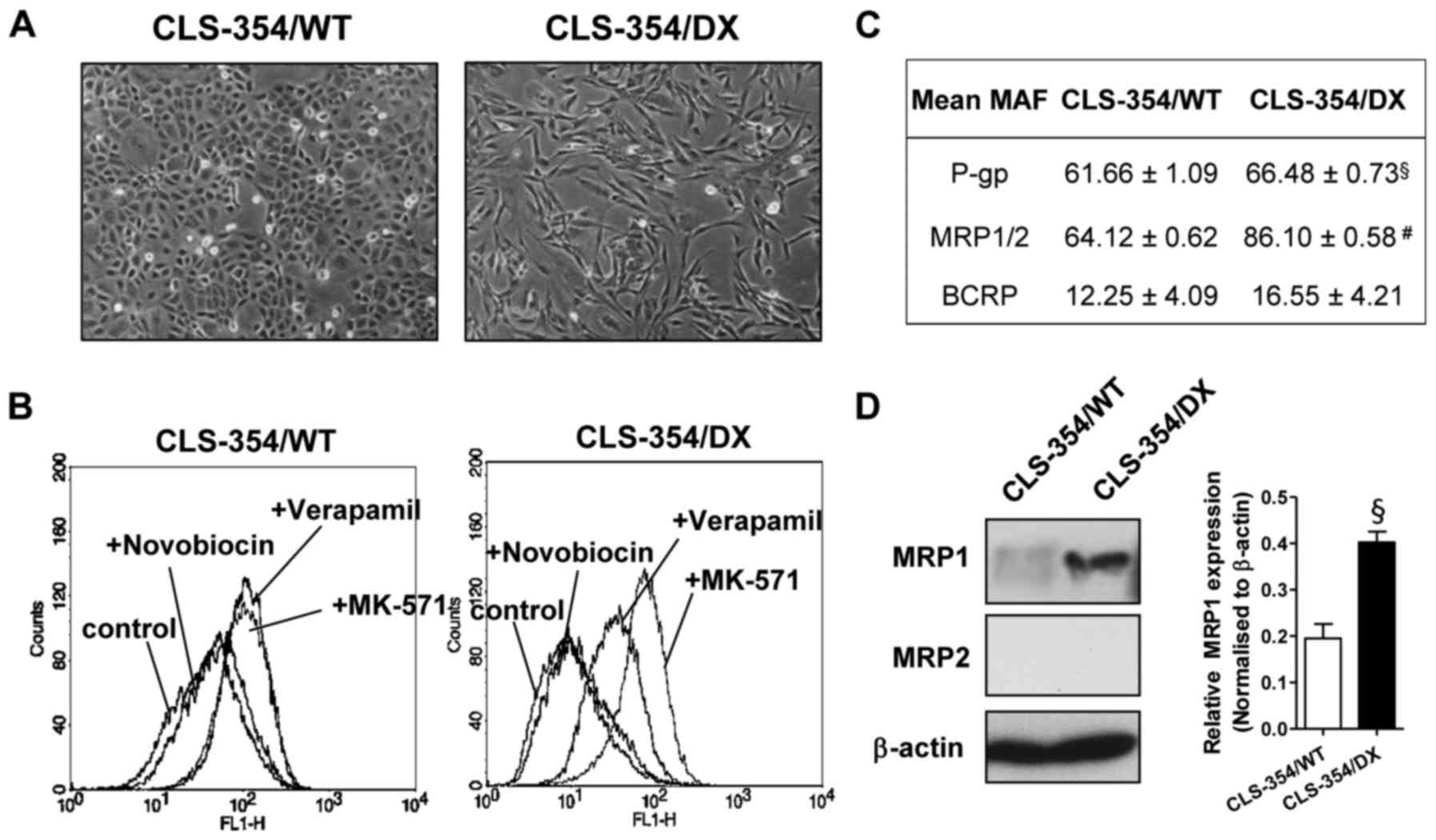
CLS-354/WT (Fig. 1A, left). The
mesenchymal phenotype with the elongated shape was isolated by
differential trypsinization as previously described (29). The cell line was named as CLS-354/DX
(Fig. 1A, right).
MDR assay
Cancer cells (8×105) were seeded onto a
60-mm tissue culture dish and allowed to grow for 36 h. Cells
(2×105 cells/sample) were collected, washed with
phosphate-buffered saline (PBS), and subjected to MDR assay using
eFluxx-ID® Green MDR assay kit (Enzo Life Sciences,
Inc., Farmingdale, NY, USA) according to the manufacturer's
instructions. The cells were analyzed immediately by flow
cytometry. The mean fluorescence intensity (MFI) values were
analyzed using CellQuest™ Pro (BD Biosciences, San Jose, CA, USA).
The MDR activity factor (MAF) for each transporter was calculated
according to the formula: MAF = 100 × (MFIwith inhibitor
- MFI control)/MFIwith inhibitor.
Cell viability assay
Cells (1.5×104 cells/well) were seeded
into 96-well plates and grown for 48 h. Cells were treated with
various concentrations of cisplatin (10–25 µM) and camptothecin
(0.3–15 µM) or isomahanine (10–25 µM) for 24 h or for various
time-points. After treatment, cell viability was assessed by MTT
assay. The half-maximal inhibitory concentration (IC50)
was calculated from concentration-response curves following a 24-h
treatment. The resistance index (RI) was defined as the ratio of
the IC50 value of the resistant cell line to the
IC50 value of the parental cell line of each drug.
Annexin V-FITC/PI staining assay
Cells (4×105 cells/well seeded on a
6-well plate) were incubated with isomahanine for 0, 6, 12 and 24
h. The cells were harvested, washed with PBS, and stained with
Annexin V-FITC and Annexin V-FITC/PI according to the
manufacturer's instructions (Roche Diagnostics Deutschland GmbH,
Mannheim, Germany). Fluorescence intensity (FL-1 and FL-2) was
immediately determined by flow cytometry. For each measurement, at
least 15,000 cells were counted. The percentages of Annexin V-FITC-
and Annexin V-FITC/PI-positive cells were analyzed as apoptotic
cells using CellQuest™ Pro (BD Biosciences).
Analysis of GFP-LC3B puncta
CLS-354/DX cells (2×104 cells/well seeded
on a 24-well plate) were transduced with BacMam LC3B-GFP viral
particles (MIO=30 plaque-forming units/cell), according to the
manufacturer's instructions of the Premo™ Autophagy Sensor kit
(Invitrogen, Carlsbad, CA, USA). After 24 h, the cells were treated
with 20 µM isomahanine or 50 µM CQ. After a 16-h incubation, green
fluorescent signals were monitored and imaged using a Zeiss Axio
Vert.A1 inverted microscope (magnification, ×40). Cells with >5
GFP-LC3B puncta were considered autophagy-positive. The percentage
of cells with punctate GFP-LC3B/total GFP-LC3B-positive cells was
determined.
Western blot analysis
After isomahanine treatment, the cells were
harvested and lysed in lysis buffer containing protease inhibitors.
Protein samples (50 µg) were then prepared in Laemmli sample
buffer. The proteins were separated on 8–12.5% SDS-PAGE gels and
transferred to a polyvinylidene fluoride (PVDF) membrane. The
membrane was blocked with 5% non-fat dry milk, washed with a
mixture of Tris-buffered saline and Tween-20, and probed with
primary antibodies (1:250-1:1,000) at 4̊C overnight. The blot was
then probed with the HRP-conjugated secondary antibody (1:5,000) at
room temperature for 1 h. The proteins were visualized using the
chemiluminescent detection system.
Statistical analysis
All results are expressed as the mean ± SEM. Data
were obtained from at least 3 independent experiments. Significant
differences among the different groups were considered at a p-value
<0.05, using Student's t-test or one-way ANOVA. The statistical
analyses were performed using GraphPad Prism 6 (GraphPad Software,
Inc., La Jolla, CA, USA).
Results
CLS-354 cells develop a
multidrug-resistant phenotype
The multidrug-resistant OSCC cell line was
spontaneously generated from the parental human OSCC cell line
CLS-354 (passage no. 27), which is epithelial-like in shape
(CLS-354/WT) (Fig. 1A, left). After
being cultured for >10 passages, some cells became elongated and
mesenchymal-like. These mesenchymal-like cells were isolated by
differential adhesion (29) and
named CLS-354/DX (Fig. 1A, right).
These two cell lines had similar growth characteristics. The
population doubling times of CLS-354/WT and CLS-354/DX were
31.25±0.64 and 29.17±1.20 h, respectively, which were not
significantly different.
To investigate the drug-resistance profiling, the
activity of a particular multidrug-resistant transporter [P-gp,
MRP1/2 and breast cancer resistance protein (BCRP)] was assessed
using eFluxx-ID® Green probe. The probe is a hydrophilic
fluorescent dye, which is trapped within the cells unless actively
pumped out by these transporters and is detectable by flow
cytometry. As shown in Fig. 1B,
CLS-354/WT cells exhibited an equal increase of fluorescent signals
in the presence of a P-gp inhibitor (verapamil) and an MRP1/2
inhibitor (MK-571) in comparison to the control (Fig. 1B, left). We clearly found a higher
fluorescent signal from the CLS-354/DX cells in the presence of
verapamil and MK-571 when compared with the control (Fig. 1B, right). This result indicated the
essential roles of P-gp and MRP1/2 in these cells. The comparative
analysis of MDR between the CLS-354/WT and CLS-354/DX cell lines
was estalished by calculating the MAF values. The MAF values of
P-gp and MRP1/2 in CLS-354/DX cells were 66.48±0.73 and 86.10±0.58,
respectively, which were significantly greater than those of the
CLS-354/WT cells (Fig. 1C). As a
result, the highest MAF value of MRP1/2 was observed in the
CLS-354/DX cells (Fig. 1C). Hence,
the expression levels of the MRP1 and MRP2 proteins in these cell
lines were examined by western blotting. We found that the MRP1
expression level in the CLS-354/DX cells was significantly greater
than that in the CLS-354/WT cell line (Fig. 1D), while MRP2 was undetectable in
both cell lines. These results indicated that the CLS-354/DX cells
developed the multidrug-resistant phenotype via MRP1
overexpression.
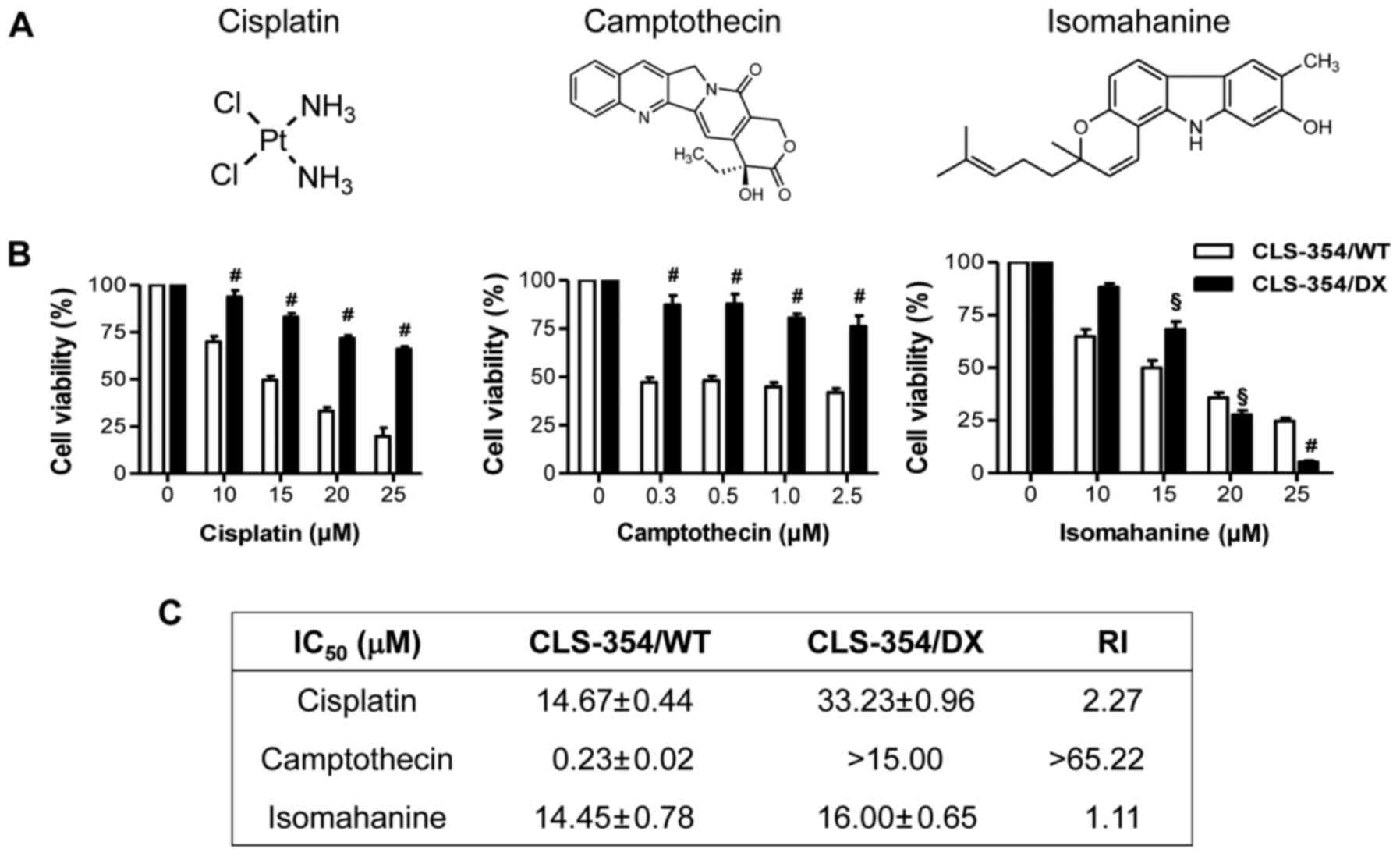
Isomahanine has potential cytotoxicity
against CLS-354/DX cells
The responsiveness to cisplatin, a platinum-based
anticancer drug and camptothecin, an anticancer alkaloid (Fig. 2A, left and middle), was investigated
in the CLS-354/WT and CLS-354/DX cells following 24-h treatments
using the cell viability assay. The results showed that the
response of CLS-354/DX cells to cisplatin and camptothecin was
significantly lower than that of CLS-354/WT cells (Fig. 2B, left and middle). The
IC50 values for CLS-354/DX cell line to cisplatin and
camptothecin were 33.23±0.96 and >15 µM, respectively, which
were 2 times and >65 times, respectively, greater than those of
the CLS-354/WT cells as indicated by the RI (Fig. 2C). These data strongly support the
contribution of MDR in CLS-354/DX cells. Notably, isomahanine
(Fig. 2A, right) at a concentration
starting from 20 µM effectively suppressed the viability of
CLS-354/DX cells without relevance of resistance (Fig. 2B, right). The IC50 values
observed in both cell lines were not significantly different, with
an RI value of 1.1 (Fig. 2C).
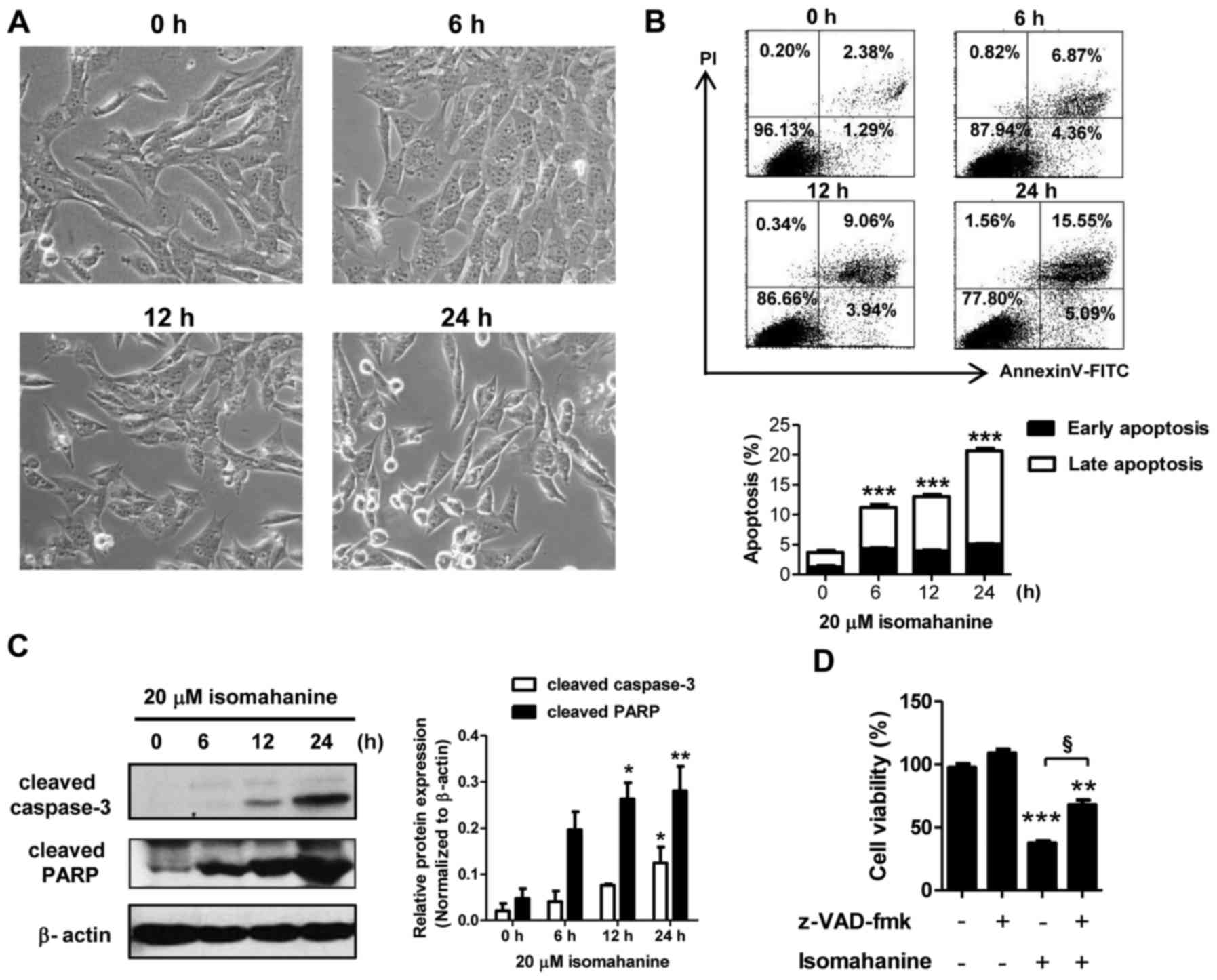
Isomahanine induces apoptosis in
CLS-354/DX cells through a caspase-dependent pathway
The mechanism of cell death was further examined in
the CLS-354/DX cells. Cell morphology was preliminarily observed in
a time-course study following isomahanine treatment (20 µM) for 0,
6, 12 and 24 h. As shown in Fig.
3A, the cells after exposure to isomahanine for 6 h presented
small cytoplasmic vacuoles. After 12 h, the cells detached and
shrank in size. Eventually, several cells became round in shape and
cell death followed after 24-h treatment. Hence, apoptosis was
further confirmed by flow cytometric analysis as well as western
blot analysis of caspase-3 and poly(ADP-ribose) polymerase (PARP).
Isomahanine time-dependently induced early apoptotic and late
apoptotic cells to 5.09 and 15.55%, respectively (Fig. 3B, upper panel). Total apoptosis
significantly increased in a time-dependent manner, while the
maximal induction was increased to 20% following the 24-h treatment
(Fig. 3B, lower panel). Isomahanine
activated cleavage of caspase-3 and PARP in a time-dependent
manner, and the greatest level of increase was observed upon 24-h
treatment (Fig. 3C). To confirm
caspase-dependent apoptotic death, the cells were treated with
isomahanine along with z-VAD-fmk, a pan-caspase inhibitor. The
inhibition of caspase activity significantly rescued
isomahanine-induced cell death (Fig.
3D). Thus, isomahanine induced apoptotic death via a
caspase-dependent pathway.
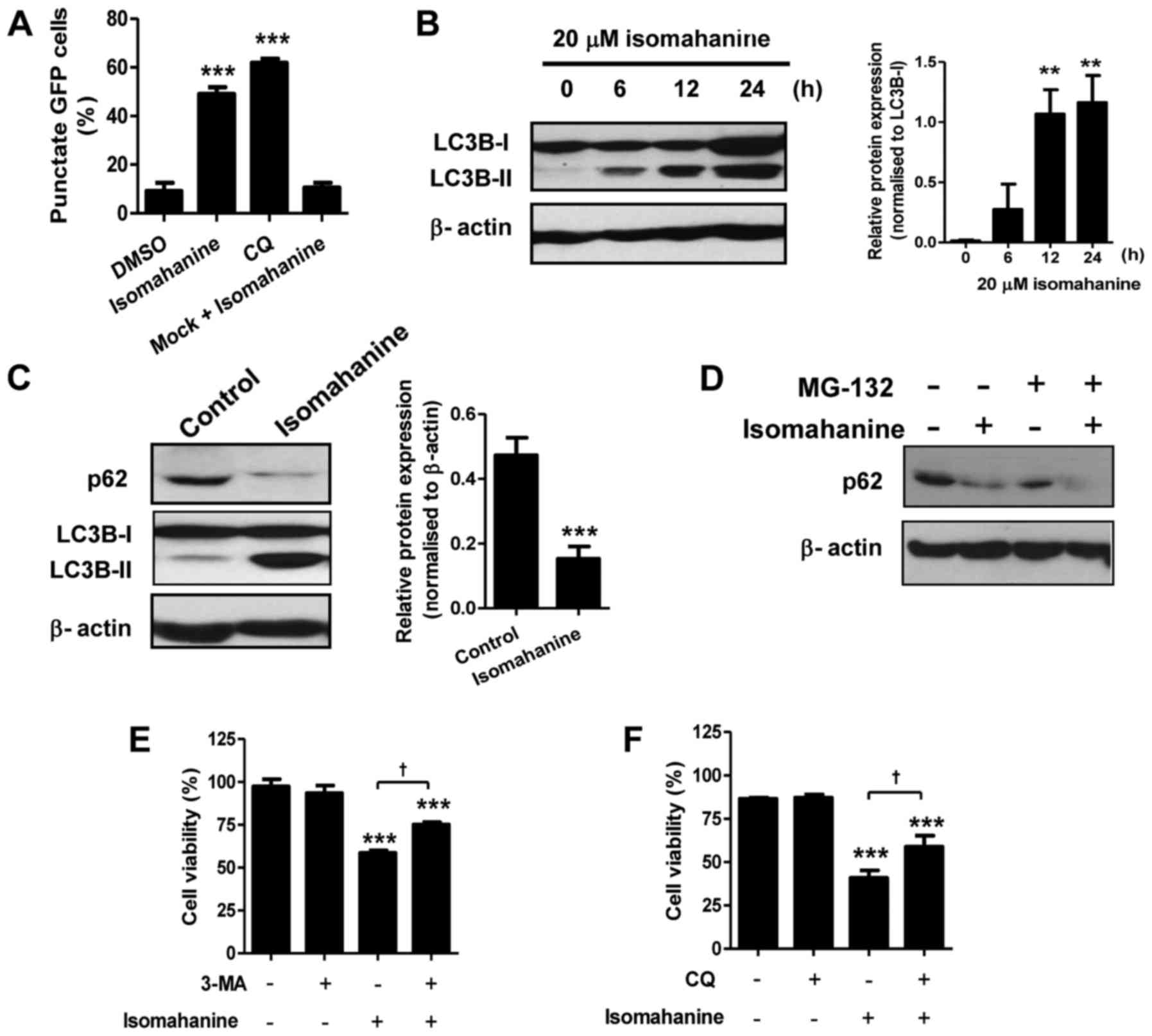
Isomahanine induces autophagic flux in
CLS-354/DX cells
We hypothesized that autophagy may be involved in
cell death upon the morphological observations. We determined
autophagic induction by analysis of GFP-LC3B puncta. Cells treated
with isomahanine displayed a punctate pattern of GFP-LC3B localized
in the perinuclear region, indicating autophagic induction.
Isomahanine-induced GFP-LC3B puncta formation by 50%, which was
comparable to that of CQ (~60%), a positive control (Fig. 4A). We further performed western
blotting of microtubule-associated protein 1B-light chain 3
(LC3B-II), which is an autophagosomal marker. Isomahanine induced
the expression of LC3B-II in a time-dependent manner, and the
expression of LC3B-II/LC3B-I ratio significantly increased after
12-h treatment (Fig. 4B). Thus,
isomahanine induced autophagy in CLS-354/DX cells.
An increase in the number of autophagosomes may be
due to activation or inhibition of autophagic flux. During
autophagy, the cellular level of the p62 protein is typically
degraded, and its decreased level serves as an indicator of
autophagic flux (14). We further
examined the expression of p62 with isomahanine treatment.
Isomahanine markedly decreased the expression level of p62 in
contrast with LC3B-II after 24 h, indicating autophagic flux
(Fig. 4C). We also excluded the
effect of proteasomal degradation activity on the p62 protein using
MG-132, a proteasome inhibitor. The result revealed that
isomahanine decreased the level of p62 as usual, while MG-132 did
not inhibit its decrease (Fig. 4D).
To assess the contribution of autophagy to cell death, the effect
of autophagy inhibitors on cell viability was determined. Results
revealed that the autophagy inhibitors 3-MA and CQ significantly
inhibited isomahanine-induced cell death in the CLS-354/DX cells
(Fig. 4E-F). Collectively, these
findings suggest that autophagic flux induced by isomahanine was
involved in the cytotoxic effects of the CLS-354/DX cells.
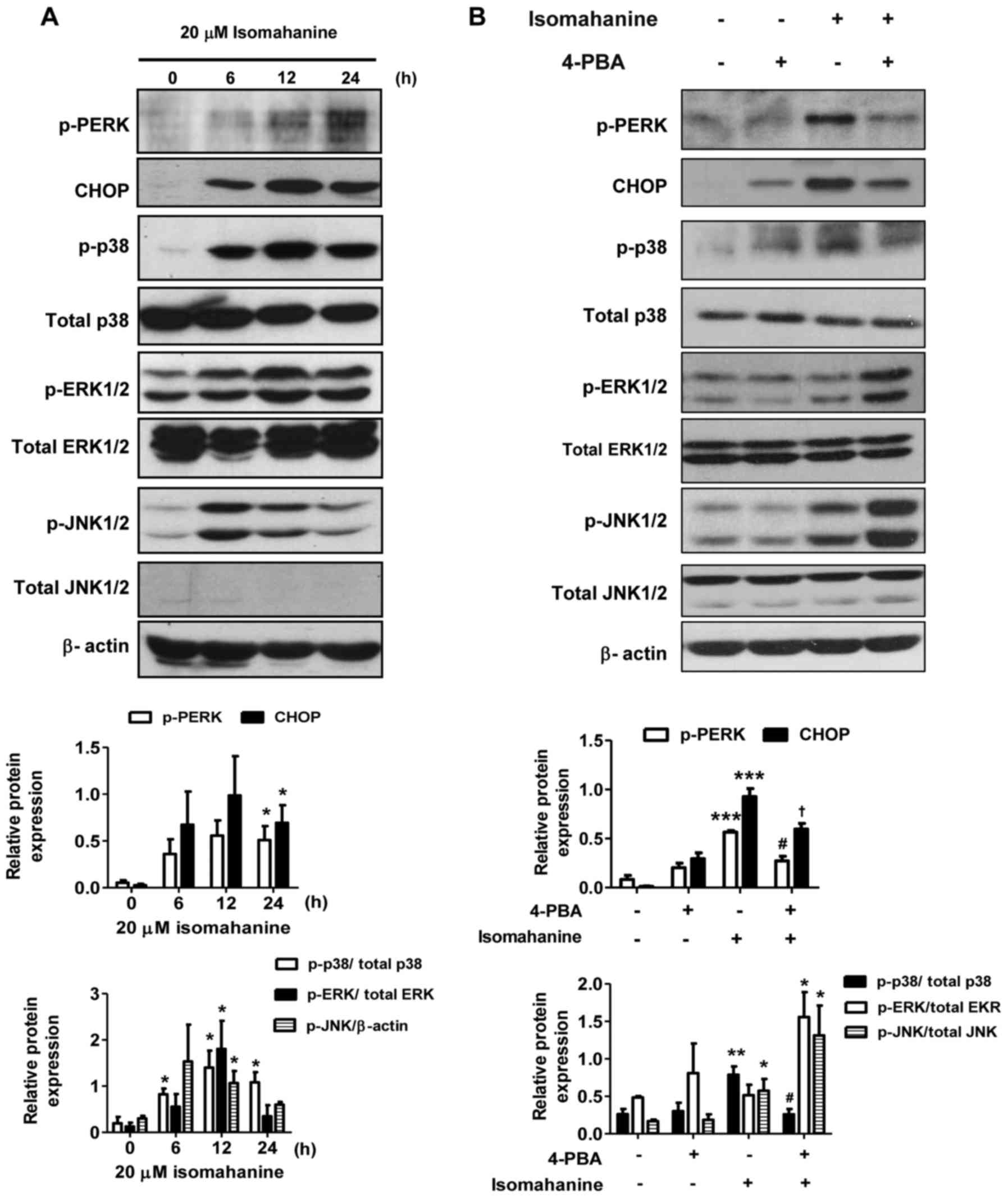
Isomahanine induces ER stress and MAPK
activation in CLS-354/DX cells
To further elucidate the cell-death mechanisms
induced by isomahanine, we examined the effect of this compound on
the activation of ER stress and MAPK phosphorylation. As shown in
Fig. 5A, isomahanine activated the
phosphorylation of protein kinase PNA (PKR)-like ER kinase (PERK),
an ER stress sensor, in a time-dependent manner. Concomitantly, the
compound induced the expression of the transcription factor CHOP,
which is a marker of UPR (Fig. 5A).
Activation of the MAPK family members, p38, ERK1/2 and JNK1/2 was
also determined. The phosphorylation of p38, ERK1/2 and JNK1/2 was
markedly upregulated after exposure to isomahanine for 6 h. At 12 h
of treatment, the phosphorylation of p38 and ERK1/2 was strongly
activated, while their activation slightly decreased after 24 h
(Fig. 5A). JNK1/2 was activated to
a maximal level at 6 h and decreased after 12 h (Fig. 5A). Next, we applied 4-PBA, an
inhibitor of ER stress, to demonstrate the relation between ER
stress and MAPK pathway. As shown in Fig. 5B, the activated PERK and CHOP
proteins were significantly decreased in the presence of 4-PBA. ER
stress inhibition protected against isomahanine-activated p38 MAPK
phosphorylation. Meanwhile, the activation of ERK1/2 and JNK1/2
were concomitantly increased by 4-PBA (Fig. 5B). Collectively, these results
suggest that ER stress mediated the activation of p38 MAPK, but not
that of ERK1/2 and JNK1/2.
Isomahanine induces p38 MAPK-mediated
apoptosis and autophagy in CLS-354/DX cells
Activation of MAPK in response to ER stress can
promote cell death by apoptosis and autophagy. To study the role of
MAPK in apoptosis- and autophagy-induced cell death, we assessed
the cell viability with isomahanine treatment in the presence of
inhibitors of p38 MAPK (SB203580), ERK (U0126) and JNK (SP600125).
As shown in Fig. 6A, SB203580 and
U0126 significantly suppressed isomahanine-induced cell death,
implying that p38 MAPK and ERK participate in cell death. The
function of p38 MAPK and ERK in the induction of apoptosis and
autophagy was further examined using inhibitors SB203580 and U0126.
Isomahanine activated caspase-3, LC3B-II and CHOP and concomitantly
decreased the expression level of p62 as usual. SB203580 inhibited
MAPKAPK-2, a downstream effector of p38 MAPK (Fig. 6B). The blockade of p38 MAPK
completely inhibited isomahanine-activated caspase-3, LC3B-II and
p62, but SB203580 did not affect CHOP expression (Fig. 6C). U0126 markedly inhibited ERK
phosphorylation (Fig. 6B), and this
attenuated only isomahanine-activated caspase-3 (Fig. 6C). These data indicated that p38
MAPK was an important cytotoxic mediator mediating both apoptosis
and autophagic flux in response to isomahanine. Meanwhile, the
activation of ERK, in part, regulated apoptosis under the tested
conditions. As CHOP was not affected by MAPK inhibition, ER stress
may be an upstream regulator involved in this activation. The
overall cytotoxic mechanism of isomahanine against CLS-354/DX cells
is illustrated in Fig. 6D.
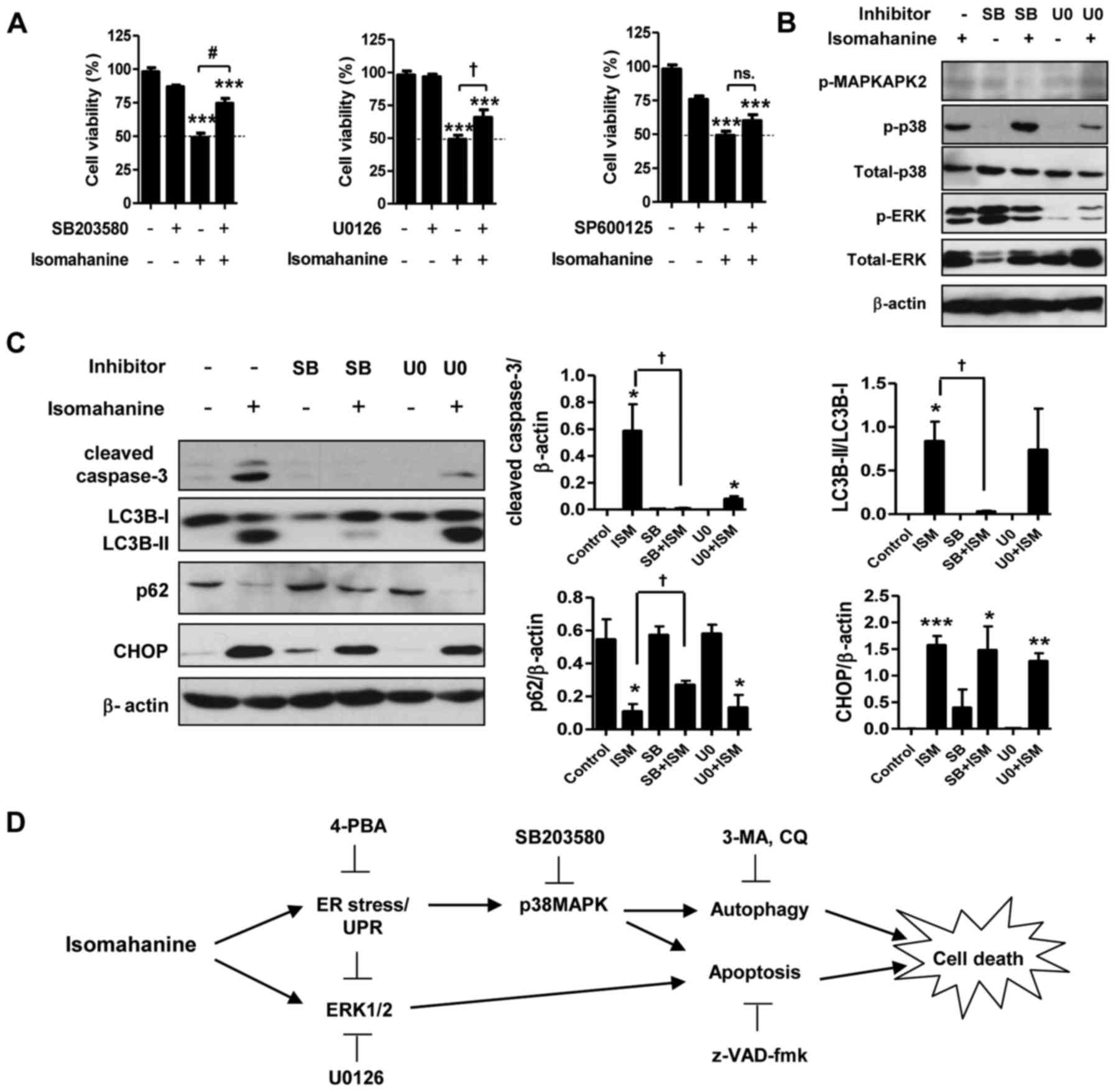 | Figure 6.p38 MAPK mediates autophagy- and
apoptosis-induced cell death in isomahanine-treated CLS-354/DX
cells. (A) Cell viability of CLS-354/DX cells was assessed by MTT
assay after treatment with isomahanine (20 µM) in the absence or
presence of inhibitors against p38 (20 µM SB203580) (left), ERK (10
µM U0126), and JNK (25 µM SP600125) for 24 h. Data are expressed as
mean ± SEM of 4 independent experiments; ***p<0.001 vs. the
control without any treatment; †p<0.05,
#p<0.001 vs. the isomahanine treatment alone. Cells
were treated with isomahanine (ISM) (20 µM) for 24 h in the absence
or presence of SB203580 (SB) (20 µM) and U0126 (U0) (10 µM). After
treatment, (B) expression of MAPKs (MAPKAPK2, p38 and ERK1/2) was
determined by western blotting, and (C) expression of apoptosis,
autophagy, and ER stress markers was determined by western blot
(left) and densitometric analyses of relative expression level
(right). Data are expressed as mean ± SEM of 3 independent
experiments; *p<0.05, **p<0.01, ***p<0.001 vs. the control
without any treatment; †p<0.05 vs. isomahanine
treatment alone. (D) The proposed cytotoxic mechanism of
isomahanine in CLS-354/DX cells. 3-MA, 3-methyladenine; 4-PBA,
4-phenylbutyric acid; CQ, chloroquine; UPR, unfolded protein
response. |
Discussion
MDR in advanced OSCC is associated with disease
recurrence and poor patient survival (30,31).
To overcome multidrug-resistant cancer, identification of effective
therapeutic regimens suitable for drug resistance in different
tumors by targeting different cell-death pathways, particularly
apoptosis and autophagy, has recently received attention (5–8,32).
Herein, we isolated a carbazole alkaloid, namely, isomahanine from
M. koenigii leaves. The cytotoxic activity of this compound
against the multidrug-resistant OSCC cell line CLS-354/DX was
examined. We discovered that isomahanine (20 µM) exerted
cytotoxicity against CLS-354/DX cells regardless of resistance
after stopping treatment. However, some cells were able to recover
their proliferation following treatment with low concentrations of
isomahanine (10 µM). These cells may acquire resistant phenotypes
due to a pro-survival autophagy; hence, the confirmation process
may take a period of time from several months to a year (33). Isomahanine cytotoxicity was due to
the induction of apoptosis and autophagic cell death in CLS-354/DX
cells. This provided the notion that isomahanine may be useful for
eliminating multidrug-resistant cancer cells.
CLS-354/DX cells spontaneously developed MDR through
increased efflux activities of MRP and P-gp, increased expression
of the MRP1 protein and elevated IC50 and RI values to
cisplatin and camptothecin in comparison to the parental CLS-354/WT
cells (4). Overexpression of MRP1
indicates intrinsic drug resistance, which confers resistance to a
wide range of anticancer drugs, including camptothecin (34) and cisplatin (35). High expression of MRP1 was observed
in tongue carcinoma, which was consequently associated with
cisplatin resistance, suggesting the existence of this protein in
head and neck cancer samples (36).
A slightly increased MDR1 activity observed in CLS-354/DX cells may
be due to MRP1, which has a significantly overlapping resistance
profile with P-gp (37).
Isomahanine triggered CLS-354/DX cells to undergo
apoptosis via the caspase-dependent pathway. Our observation was
consistent with an earlier study that reported ROS-mediated
caspase-dependent apoptosis in leukemia cells (21). Disruption of mitochondrial function
may be a contributing effect to apoptosis. The release of
cytochrome c into the cytosol can lead to formation of
apoptosomes and activation of caspase-3 through a
mitochondrial-dependent pathway (38,39).
Next, we clearly demonstrated that isomahanine activated autophagic
flux, which contributed to the cytotoxicity against CLS-354/DX
cells. Accumulating data have shown that plant alkaloids
selectively targeted autophagic cell death against
multidrug-resistant cancer cells via different mechanisms, such as
activation of AMP-activated protein kinase (40), ER stress, inhibition of the Akt/mTOR
pathway (41) and induction of
autophagic flux (42). Although the
role of autophagy in cancer is still controversial as autophagy can
either drive cell survival or cell death, pro-death is likely
effective in particular multidrug-resistant cancer cells (40,42).
Hence, the isomahanine-induced autophagic pathway may circumvent
conventional drug resistance in cancer.
In our experimental setting, ER stress was activated
following isomahanine treatment as shown by the increased levels of
p-PERK and CHOP. ER stress is a crucial stress signal caused by
increased accumulation of unfolded proteins within the ER. From a
structural point of view, isomahanine may disrupt protein folding
in the ER, similar to the effect of plant alkaloid
ellipticine-induced ER stress (43). Planar structure of the carbazole
moiety and lipophilic property of isomahanine may contribute to ER
disruption. Excessive or prolonged ER stress response can further
initiate cell death pathways (15).
Three canonical MAPKs (ERK1/2, JNK and p38) are recognized to be
activated in response to ER stress-induced cell death (16). We found that p38 MAPK and ERK1/2 are
involved in isomahanine-induced apoptosis. During ER stress,
inositol-requiring protein-1 activates apoptosis signal-regulating
kinase 1, which is essential for the activation of p38 MAPK and JNK
(16,44). Isomahanine-induced p38 MAPK not only
triggered apoptosis but also regulated autophagic flux. This may be
due to the fact that autophagy requires p38 MAPK at the
sequestration step during autophagosome formation induced by
nutrient deprivation (45). This
mechanism is consistent with previous studies on human gingival
fibroblasts and human tongue squamous cell carcinoma cells
(45,46). p38 MAPK can act as an important
mediator of apoptotic and autophagic cell death induced by
isomahanine. ER stress induced or decreased ERK1/2 activation with
different kinetics. Short-term induction of ER stress (<6 h)
resulted in a decrease in ERK phosphorylation, while long-lasting
activation (>10 h) induced ERK phosphorylation in hepatocellular
carcinoma cells (47). In addition,
we demonstrated that the MEK inhibitor rescued isomahanine-induced
cell death and decreased cleavage of caspase-3. ERK-dependent
apoptosis can be activated by mitochondrial cytochrome c
release or caspase-8 activation, permanent cell cycle arrest, or
autophagic vacuolization, depending on the different cellular
contexts (48).
In conclusion, the present study elucidated the
cytotoxic mechanism involved in the apoptosis and autophagy induced
by isomahanine in multidrug-resistant OSCC cells. Isomahanine was
capable of inducing ER stress, which may regulate apoptosis as well
as autophagic cell death via p38 MAPK. The simultaneous autophagic
cell death with apoptosis induced by isomahanine may provide a
novel anticancer strategy to circumvent MDR in cancer.
Acknowledgements
The present study was supported by Strategic
Scholarships Fellowships Frontier Research Networks for the Ph.D.
Sandwich Program Doctoral Degree from the Office of the Higher
Education Commission (OHEC), Thailand (06/2556), Walailak
University (WU56113 and WU59201), Walailak University Fund for
Graduate Studentship (27/2556 and WU55603), the Thailand Research
Fund (DBG5980003), and Centre of Excellence for Innovation in
Chemistry, OHEC. S.C. was partially supported by Structural and
Computational Biology Research Group, Special Task Force for
Activating Research (STAR), Faculty of Science,
Rachadaphiseksomphot Endowment Fund, Chulalongkorn University, the
Thailand Research Fund (TRF) (TRG5880222 and IRG 5780008), and the
Institute for the Promotion of Teaching Science and Technology
(IPST) under the Research Fund for DPST Graduate with First
Placement (07/2557). We would like to thank Enago (https://www.enago.com/] for English language editing
and reviewing of this manuscript.
Glossary
Abbreviations
Abbreviations:
|
3-MA
|
3-methyladenine
|
|
4-PBA
|
4-phenylbutyric acid
|
|
BCRP
|
breast cancer resistance protein
|
|
CHOP
|
transcription factor C/EBP homologous
protein
|
|
CQ
|
chloroquine
|
|
ER
|
endoplasmic reticulum
|
|
LC3B
|
microtubule-associated protein light
chain 3B
|
|
MAPK
|
mitogen-activated protein kinases
|
|
MDR
|
multidrug resistance
|
|
MRP
|
multidrug resistance-associated
protein
|
|
OSCC
|
oral squamous cell carcinoma
|
|
p62
|
p62/SQSTM1
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
PERK
|
protein kinase PNA (PKR)-like ER
kinase
|
|
P-gp
|
P-glycoprotein
|
|
UPR
|
unfolded protein response
|
References
|
1
|
Rao SV Krishna, Mejia G, Roberts-Thomson K
and Logan R: Epidemiology of oral cancer in Asia in the past decade
- an update (2000–2012). Asian Pac J Cancer Prev. 14:5567–5577.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Soerjomataram I, Ervik M,
Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and
Bray F: GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality
Worldwide: IARC CancerBase No. 11 (Internet). International Agency
for Research on Cancer; Lyon, France: 2014, doi:10.1002/ijc.29210
PMID:25220842. Accessed October, 9, 2014.
|
|
3
|
Wang C, Liu XQ, Hou JS, Wang JN and Huang
HZ: Molecular mechanisms of chemoresistance in oral cancer. Chin J
Dent Res. 19:25–33. 2016.PubMed/NCBI
|
|
4
|
Wu Q, Yang Z, Nie Y, Shi Y and Fan D:
Multi-drug resistance in cancer chemotherapeutics: Mechanisms and
lab approaches. Cancer Lett. 347:159–166. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Meschini S, Condello M, Marra M, Formisano
G, Federici E and Arancia G: Autophagy-mediated chemosensitizing
effect of the plant alkaloid voacamine on multidrug resistant
cells. Toxicol In Vitro. 21:197–203. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chow MJ, Licona C, Pastorin G, Mellitzer
G, Ang WH and Gaiddon C: Structural tuning of organoruthenium
compounds allows oxidative switch to control ER stress pathways and
bypass multidrug resistance. Chem Sci. 7:4117–4124. 2016.
View Article : Google Scholar
|
|
7
|
Kumar P, Zhang DM, Degenhardt K and Chen
ZS: Autophagy and transporter-based multi-drug resistance. Cells.
1:558–575. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xuan Y and Hu X: Naturally-occurring
shikonin analogues - a class of necroptotic inducers that
circumvent cancer drug resistance. Cancer Lett. 274:233–242. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gewies A: Introduction to apoptosis.
ApoReview. 1–26. 2003.
|
|
10
|
Millimouno FM, Dong J, Yang L, Li J and Li
X: Targeting apoptosis pathways in cancer and perspectives with
natural compounds from mother nature. Cancer Prev Res. 7:1081–1107.
2014. View Article : Google Scholar
|
|
11
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang XJ, Chen S, Huang KX and Le WD: Why
should autophagic flux be assessed? Acta Pharmacol Sin. 34:595–599.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen N and Karantza V: Autophagy as a
therapeutic target in cancer. Cancer Biol Ther. 11:157–168. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiao JW and Wen F: Tanshinone IIA acts via
p38 MAPK to induce apoptosis and the down-regulation of ERCC1 and
lung-resistance protein in cisplatin-resistant ovarian cancer
cells. Oncol Rep. 25:781–788. 2011.PubMed/NCBI
|
|
15
|
Xu C, Bailly-Maitre B and Reed JC:
Endoplasmic reticulum stress: Cell life and death decisions. J Clin
Invest. 115:2656–2664. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Darling NJ and Cook SJ: The role of MAPK
signalling pathways in the response to endoplasmic reticulum
stress. Biochim Biophys Acta. 1843:2150–2163. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Y, Wang JW, Xiao X, Shan Y, Xue B,
Jiang G, He Q, Chen J, Xu HG, Zhao RX, et al: Piperlongumine
induces autophagy by targeting p38 signaling. Cell Death Dis.
4:e8242013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma K, Zhang C, Huang MY, Li WY and Hu GQ:
Cinobufagin induces autophagy-mediated cell death in human
osteosarcoma U2OS cells through the ROS/JNK/p38 signaling pathway.
Oncol Rep. 36:90–98. 2016.PubMed/NCBI
|
|
20
|
Li JP, Yang YX, Liu QL, Pan ST, He ZX,
Zhang X, Yang T, Chen XW, Wang D, Qiu JX, et al: The
investigational Aurora kinase A inhibitor alisertib (MLN8237)
induces cell cycle G2/M arrest, apoptosis, and autophagy
via p38 MAPK and Akt/mTOR signaling pathways in human breast cancer
cells. Drug Des Devel Ther. 9:1627–1652. 2015.PubMed/NCBI
|
|
21
|
Roy MK, Thalang VN, Trakoontivakorn G and
Nakahara K: Mechanism of mahanine-induced apoptosis in human
leukemia cells (HL-60). Biochem Pharmacol. 67:41–51. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ito C, Itoigawa M, Nakao K, Murata T,
Tsuboi M, Kaneda N and Furukawa H: Induction of apoptosis by
carbazole alkaloids isolated from Murraya koenigii. Phytomedicine.
13:359–365. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sarkar S, Dutta D, Samanta SK,
Bhattacharya K, Pal BC, Li J, Datta K and Mandal C and Mandal C:
Oxidative inhibition of Hsp90 disrupts the super-chaperone complex
and attenuates pancreatic adenocarcinoma in vitro and in vivo. Int
J Cancer. 132:695–706. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shaikh MS, Karpoormath R, Thapliyal N,
Rane RA, Palkar MB, Faya AM, Patel HM, Alwan WS, Jain K and
Hampannavar GA: Current perspective of natural alkaloid carbazole
and its derivatives as antitumor agents. Anticancer Agents Med
Chem. 15:1049–1065. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Das R, Bhattacharya K, Sarkar S, Samanta
SK, Pal BC and Mandal C: Mahanine synergistically enhances
cytotoxicity of 5-fluorouracil through ROS-mediated activation of
PTEN and p53/p73 in colon carcinoma. Apoptosis. 19:149–164. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Das R, Bhattacharya K, Samanta SK, Pal BC
and Mandal C: Improved chemosensitivity in cervical cancer to
cisplatin: Synergistic activity of mahanine through STAT3
inhibition. Cancer Lett. 351:81–90. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen CM, Syu JP, Way TD, Huang LJ, Kuo SC,
Lin CT and Lin CL: BC3EE2,9B, a synthetic carbazole derivative,
upregulates autophagy and synergistically sensitizes human GBM8901
glioblastoma cells to temozolomide. Int J Mol Med. 36:1244–1252.
2015.PubMed/NCBI
|
|
28
|
Reisch J, Goj O, Wickramasinghe A, Herath
B and Henkel G: Carbazole alkaloids from seeds of Murraya koenigii.
Phytochemistry. 31:2877–2879. 1992. View Article : Google Scholar
|
|
29
|
Chamulitrat W, Schmidt R, Chunglok W, Kohl
A and Tomakidi P: Epithelium and fibroblast-like phenotypes derived
from HPV16 E6/E7-immortalized human gingival keratinocytes
following chronic ethanol treatment. Eur J Cell Biol. 82:313–322.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim JW, Park Y, Roh JL, Cho KJ, Choi SH,
Nam SY and Kim SY: Prognostic value of glucosylceramide synthase
and P-glycoprotein expression in oral cavity cancer. Int J Clin
Oncol. 21:883–889. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Friedrich RE, Punke C and Reymann A:
Expression of multi-drug resistance genes (mdr1, mrp1, bcrp) in
primary oral squamous cell carcinoma. In Vivo. 18:133–147.
2004.PubMed/NCBI
|
|
32
|
Saraswathy M and Gong S: Different
strategies to overcome multidrug resistance in cancer. Biotechnol
Adv. 31:1397–1407. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Harada K, Ferdous T and Ueyama Y:
Establishment of 5-fluorouracil-resistant oral squamous cell
carcinoma cell lines with epithelial to mesenchymal transition
changes. Int J Oncol. 44:1302–1308. 2014.PubMed/NCBI
|
|
34
|
Yajima T, Ochiai H, Uchiyama T, Takano N,
Shibahara T and Azuma T: Resistance to cytotoxic
chemotherapy-induced apoptosis in side population cells of human
oral squamous cell carcinoma cell line Ho-1-N-1. Int J Oncol.
35:273–280. 2009.PubMed/NCBI
|
|
35
|
Negoro K, Yamano Y, Fushimi K, Saito K,
Nakatani K, Shiiba M, Yokoe H, Bukawa H, Uzawa K, Wada T, et al:
Establishment and characterization of a cisplatin-resistant cell
line, KB-R, derived from oral carcinoma cell line, KB. Int J Oncol.
30:1325–1332. 2007.PubMed/NCBI
|
|
36
|
Zhang B, Liu M, Tang HK, Ma HB, Wang C,
Chen X and Huang HZ: The expression and significance of MRP1, LRP,
TOPOIIβ, and BCL2 in tongue squamous cell carcinoma. J Oral Pathol
Med. 41:141–148. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kathawala RJ, Gupta P, Ashby CR Jr and
Chen ZS: The modulation of ABC transporter-mediated multidrug
resistance in cancer: A review of the past decade. Drug Resist
Updat. 18:1–17. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Roy MK, Thalang VN, Trakoontivakorn G and
Nakahara K: Mahanine, a carbazole alkaloid from Micromelum minutum,
inhibits cell growth and induces apoptosis in U937 cells through a
mitochondrial dependent pathway. Br J Pharmacol. 145:145–155. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sinha S, Pal BC, Jagadeesh S, Banerjee PP,
Bandyopadhaya A and Bhattacharya S: Mahanine inhibits growth and
induces apoptosis in prostate cancer cells through the deactivation
of Akt and activation of caspases. Prostate. 66:1257–1265. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Law BY, Mok SW, Chan WK, Xu SW, Wu AG, Yao
XJ, Wang JR, Liu L and Wong VK: Hernandezine, a novel AMPK
activator induces autophagic cell death in drug-resistant cancers.
Oncotarget. 7:8090–8104. 2016.PubMed/NCBI
|
|
41
|
Hasanain M, Bhattacharjee A, Pandey P,
Ashraf R, Singh N, Sharma S, Vishwakarma AL, Datta D, Mitra K and
Sarkar J: α-Solanine induces ROS-mediated autophagy through
activation of endoplasmic reticulum stress and inhibition of
Akt/mTOR pathway. Cell Death Dis. 6:e18602015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Meschini S, Condello M, Calcabrini A,
Marra M, Formisano G, Lista P, De Milito A, Federici E and Arancia
G: The plant alkaloid voacamine induces apoptosis-independent
autophagic cell death on both sensitive and multidrug resistant
human osteosarcoma cells. Autophagy. 4:1020–1033. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hägg M, Berndtsson M, Mandic A, Zhou R,
Shoshan MC and Linder S: Induction of endoplasmic reticulum stress
by ellipticine plant alkaloids. Mol Cancer Ther. 3:489–497.
2004.PubMed/NCBI
|
|
44
|
Tobiume K, Matsuzawa A, Takahashi T,
Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T and
Ichijo H: ASK1 is required for sustained activations of JNK/p38 MAP
kinases and apoptosis. EMBO Rep. 2:222–228. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kim DS, Kim JH, Lee GH, Kim HT, Lim JM,
Chae SW, Chae HJ and Kim HR: p38 Mitogen-activated protein kinase
is involved in endoplasmic reticulum stress-induced cell death and
autophagy in human gingival fibroblasts. Biol Pharm Bull.
33:545–549. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pan ST, Qin Y, Zhou ZW, He ZX, Zhang X,
Yang T, Yang YX, Wang D, Qiu JX and Zhou SF: Plumbagin induces
G2/M arrest, apoptosis, and autophagy via p38 MAPK- and
PI3K/Akt/mTOR-mediated pathways in human tongue squamous cell
carcinoma cells. Drug Des Devel Ther. 9:1601–1626. 2015.PubMed/NCBI
|
|
47
|
Dai R, Chen R and Li H: Cross-talk between
PI3K/Akt and MEK/ERK pathways mediates endoplasmic reticulum
stress-induced cell cycle progression and cell death in human
hepatocellular carcinoma cells. Int J Oncol. 34:1749–1757.
2009.PubMed/NCBI
|
|
48
|
Cagnol S and Chambard JC: ERK and cell
death: Mechanisms of ERK-induced cell death - apoptosis, autophagy
and senescence. FEBS J. 277:2–21. 2010. View Article : Google Scholar : PubMed/NCBI
|