Introduction
Hepatocellular carcinoma (HCC) is the one of the
most common human types of cancer with a high mortality rate
worldwide (1). To date, surgical
resection is still the best option for early-stage patients.
However, most patients at the time of diagnosis are in the late
stages. Thus, chemotherapy and radiation therapy are the main
therapies for patients who are not suitable candidates for surgery
(2). Doxorubicin (DOX) is a widely
used chemotherapy agent for the clinical treatment of liver cancer.
However, during clinical treatment, the development of drug
resistance limits the application of DOX (3,4). Thus,
it is necessary to investigate the underlying mechanism of drug
resistance of HCC to DOX.
Studies suggest that the occurrence of
epithelial-mesenchymal transition (EMT) has an intimate correlation
with the process of tumor resistance (5–8). EMT
is a special physiological process whereby epithelial cells
transform into motile mesenchymal cells, with the downregulation of
epithelial characteristics (E-cadherin), the enhancement of
mesenchymal characteristics (vimentin, N-cadherin, Snail and Slug)
and increased abilities of invasion and migration.
Connexins (Cxs) have been revealed to be proteins
which form gap junction channels and mediate the communication
between cells (9–11). The intercellular communication is
necessary for managing various pathological processes of the human
body, such as cell proliferation, differentiation, tissue
homeostasis and wound healing (12). Cxs decrease significantly during the
process of carcinogenesis and when cancer cells acquire drug
resistance. Studies have indicated that Cx genes are
tumor-suppressor genes (13–15)
and that recovering or increasing Cx expression is a new strategy
that may be used for cancer therapy.
In the present study, we found that the
DOX-resistant human liver cancer HepG2 cell line (HepG2/DOX)
exhibited EMT characteristics and that the expression level of Cx32
was gradually decreased after HepG2 cells became resistant to DOX.
We also demonstrated that Cx32 regulated EMT in HepG2 and HepG2/DOX
cells. By detecting the expression levels of Cx32, E-cadherin and
vimentin in HCC and the corresponding paracancerous tissues, the
results revealed that the expression levels of Cx32 and E-cadherin
were clearly decreased in HCC tissues when compared with the levels
in the corresponding paracancerous tissues, while the expression of
vimentin was significantly enhanced in the HCC tissues. Moreover,
the expression of Cx32 had a strong correlation with the expression
of E-cadherin and vimentin. These results suggest that Cx32 is an
important regulator of DOX-induced EMT in HCC.
Materials and methods
Immunohistochemical samples
HCC and paracancerous tissue specimens were
collected from 40 HCC patients who underwent surgical resection at
the Hepatobiliary Surgical Department of The First Affiliated
Hospital of Bengbu Medical College (Bengbu, China) from February
2014 to June 2015. Paracancerous tissue was the non-cancerous liver
tissue located 3–5 cm from the primary tumor. No patients received
radiotherapy, chemotherapy, immunotherapy or other related
treatment before surgery. For all patients, complete clinical and
pathological data were obtained. All specimens were fixed with
neutral formalin solution, paraffin-embedded and continuously
sectioned to a 5-µm thickness. The specimen collection protocol was
approved by the Medical Ethics Committee of the Bengbu Medical
College.
Immunohistochemistry
Specimens were embedded in 10% paraffin and cut by a
microtome into 5-µm sections. All paraffin sections were routinely
deparaffinized and rehydrated in xylene, and graded ethanol
solutions. Endogenous peroxidase activity was blocked by 0.3%
H2O2 in methanol for 20 min. Then, the
sections were incubated with the primary antibodies [Cx32 (1:100),
E-cadherin (1:400) and vimentin (1:100); Cell Signaling Technology,
Danvers, MA, USA] at 4̊C overnight. Subsequently, the sections were
exposed to the biotin-labeled secondary antibody for 20 min.
Immunostaining was visualized with 3,3-diaminobenzidine (DAB) and
hematoxylin counterstaining. Phosphate-buffered saline (PBS)
instead of a primary antibody was used as the negative control.
Positive staining was observed and images were captured with an
optical microscope. The stained cells were scored as follows:
negative (−), stained cells <5%; positive (+), stained cells
5–10%; positive (++), stained cells >10–50%; positive (+++),
stained cells >50–75%; positive (++++), stained cells
>75%.
Materials
The HepG2 cell line was obtained from Shanghai Bai
Li Biological Technology Co., Ltd. (Shanghai, China). Cell culture
reagents were obtained from Invitrogen (Carlsbad, CA, USA). DOX and
dimethyl sulfoxide (DMSO) were obtained from Sigma-Aldrich (St.
Louis, MO, USA). Antibodies for E-cadherin, vimentin, β-actin and
the secondary antibodies were obtained from Cell Signaling
Technology. shRNA-Cx32, pcDNA-Cx32 and Lipofectamine™ 2000 were
obtained from Shanghai GenePharma Co., Ltd. (Shanghai, China).
MTT assay
Cell viability was evaluated by an MTT assay as
previously described (16). The
cell inhibition rate was calculated to determine the standard
pharmacological activity at OD490 nm. Briefly, cells were harvested
and seeded in 96-well plates. Various concentrations of DOX were
added into the wells followed by 24 h of incubation (at 37̊C with
5% CO2). Then, 20 µl of 5 mg/ml MTT was added into each
well after the removal of the culture medium. Next, the cells were
incubated for an additional 4 h. Subsequently, 150 µl of DMSO was
used after discarding the culture medium. Plates were placed on a
plate shaker for 10 min and the absorbance was assessed by a
microplate reader at 490 nm.
Western blotting
Cell proteins were isolated using protein lysis
buffer at 4̊C. Equal amounts of cell protein samples were separated
by SDS-polyacrylamide (10%) gel electrophoresis, and then
transferred to polyvinylidene fluoride (PVDF) membranes. Then, the
membranes were blocked using 5% bovine serum albumin in
Tris-buffered saline Tween-20 (TBST) for 2 h. Subsequently, the
membranes were incubated with the specific antibodies overnight,
followed by incubation with a secondary antibody for 2 h. Finally,
the protein bands were exposed using ECL-detecting reagents.
Transwell assay
After Transwell chambers in 24-well plates were
coated with 1 mg/ml of Matrigel matrix or not, cells were harvested
and 5×105 cells/ml were seeded into the Transwell
chambers. The culture medium was removed after 24 h of incubation
(at 37̊C with 5% CO2). Then, the cells were fixed with
4% paraformaldehyde for 15 min and stained with 1% crystal violet
for another 15 min. Subsequently, the inner-cells were wiped off
with a swab and the cells were counted in random fields using a
light microscope.
Cell transfection
Cells were randomly divided into 3 groups: the
control, the negative control (transfected with an empty plasmid)
and the shRNA-Cx32 or pcDNA-Cx32 group (transfected with Cx32-gene
plasmids). According to the manufacturers instructions, following
incubation of cells in 6-well plates for 24 h, shRNA-Cx32 or
pcDNA-Cx32 genes were transfected into HepG2 or HepG2/DOX cells
using Lipofectamine 2000, and then the cells were co-cultured with
serum-free opti-MEM medium for 6 h. Subsequently, the old culture
was discarded and was replaced with normal culture medium.
Statistical analysis
An unpaired Students t-test with SigmaPlot 10.0
software (Jandel Scientific, San Rafael, CA, USA) was used to
assess the statistical data. Data are presented as the means ± SEM.
Differences with P<0.05 are considered to have statistical
significance. SPSS 17.0 was used to analyze the relationship
between Cx32, E-cadherin and vimentin.
Results
Expression levels of Cx32, E-cadherin
and vimentin in HCC specimens, and the corresponding paracancerous
tissues
Forty patients (31 males and 9 females) were
enrolled in the present study. The expression levels of Cx32,
E-cadherin and vimentin in HCC tissues and adjacent non-tumorous
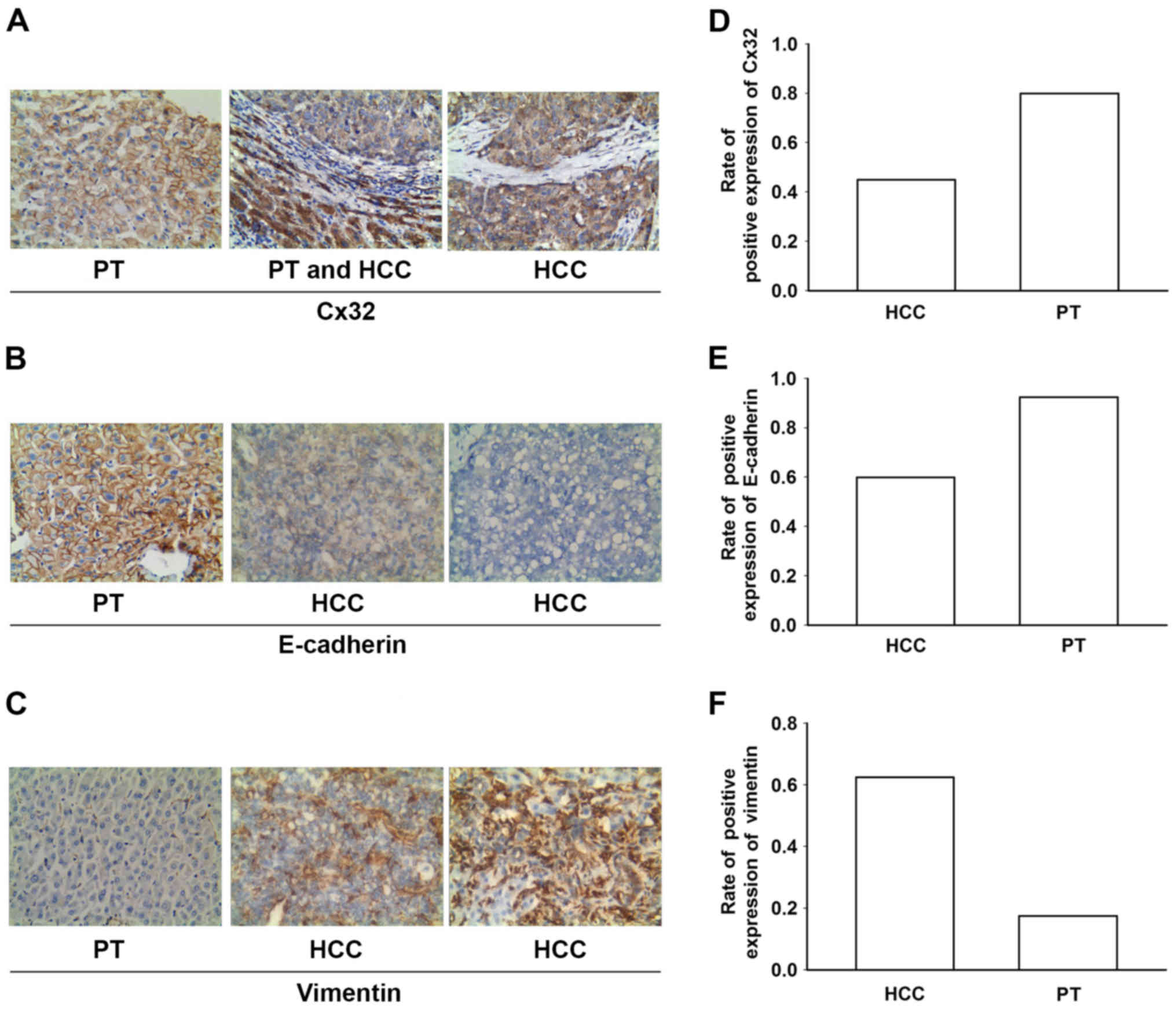
liver samples were examined using immunohistochemistry (Fig. 1A-C). The results revealed that the
expression levels of Cx32 and E-cadherin (an epithelial marker)
were significantly decreased in the HCC tissues when compared to
levels in the corresponding paracancerous tissues, while the
expression of vimentin (a mesenchymal marker) was enhanced in the
HCC specimens (Fig. 1D-F; Table I). HCC samples (55 and 40%) showed
decreased Cx32 and E-cadherin expression respectively, while 62.5%
of the HCC samples showed positive expression of vimentin.
 | Table I.Positive rates of Cx32, E-cadherin and
vimentin in the HCC and paracancerous tissues. |
Table I.
Positive rates of Cx32, E-cadherin and
vimentin in the HCC and paracancerous tissues.
|
| HCC tissues
(n=40) | Paracancerous tissues
(n=40) |
|
|
|---|
|
|
|
|
|
|
|---|
| Variant | − | + | ++ | +++ | ++++ | % | − | + | ++ | +++ | ++++ | % | Fisher | P-value |
|---|
| Cx32 | 22 | 11 | 1 | 6 | 0 | 45.0 | 8 | 2 | 1 | 24 | 5 | 80.0 | 32.207 |
<0.001a |
| E-cadherin | 16 | 11 | 8 | 5 | 0 | 60.0 | 3 | 5 | 17 | 9 | 6 | 92.5 | 46.567 |
<0.001a |
| Vimentin | 15 | 3 | 14 | 5 | 3 | 62.5 | 33 | 4 | 3 | 0 | 0 | 17.5 | 11.908 | 0.046a |
Spearman correlation analysis was used to observe
the relationship between Cx32, E-cadherin and vimentin in the HCC
samples. Table II shows that there
is a strong correlation between Cx32, E-cadherin and vimentin in
HCC tissues. The expression level of E-cadherin was positively
correlated with the expression of Cx32, while the expression of
vimentin was negatively correlated with the expression of Cx32.
| Table II.Correlation analysis of Cx32,
E-cadherin and vimentin in the HCC samples. |
Table II.
Correlation analysis of Cx32,
E-cadherin and vimentin in the HCC samples.
|
| Cx32 |
|---|
|
|
|
|---|
| Variables | Spearman
indices | P-value |
|---|
| E-cadherin |
0.812 |
<0.001a |
| Vimentin | −0.502 |
0.001a |
HepG2/DOX cell line is
established
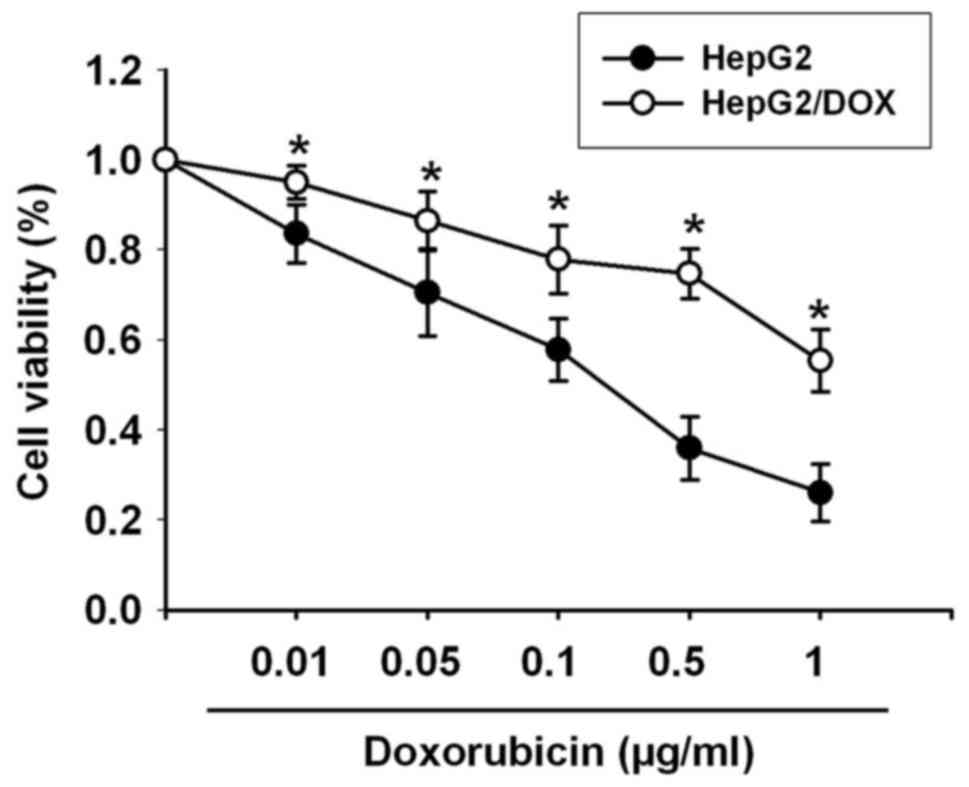
The HepG2/DOX cell line was induced via serially
increasing DOX concentrations. An MTT assay was used to investigate
the cytotoxicity of DOX in HepG2 cells (sensitive to DOX) and
HepG2/DOX (resistant to DOX) cells. The results in Fig. 2 showed that the IC50
value of DOX in the HepG2 cells was 0.182 µg/ml, while the
IC50 value of DOX in the HepG2/DOX cells was 2.058
µg/ml. Thus, the HepG2/DOX cells demonstrated an 11.31-fold higher
resistance to DOX than the HepG2 cells.
HepG2/DOX cells undergo EMT
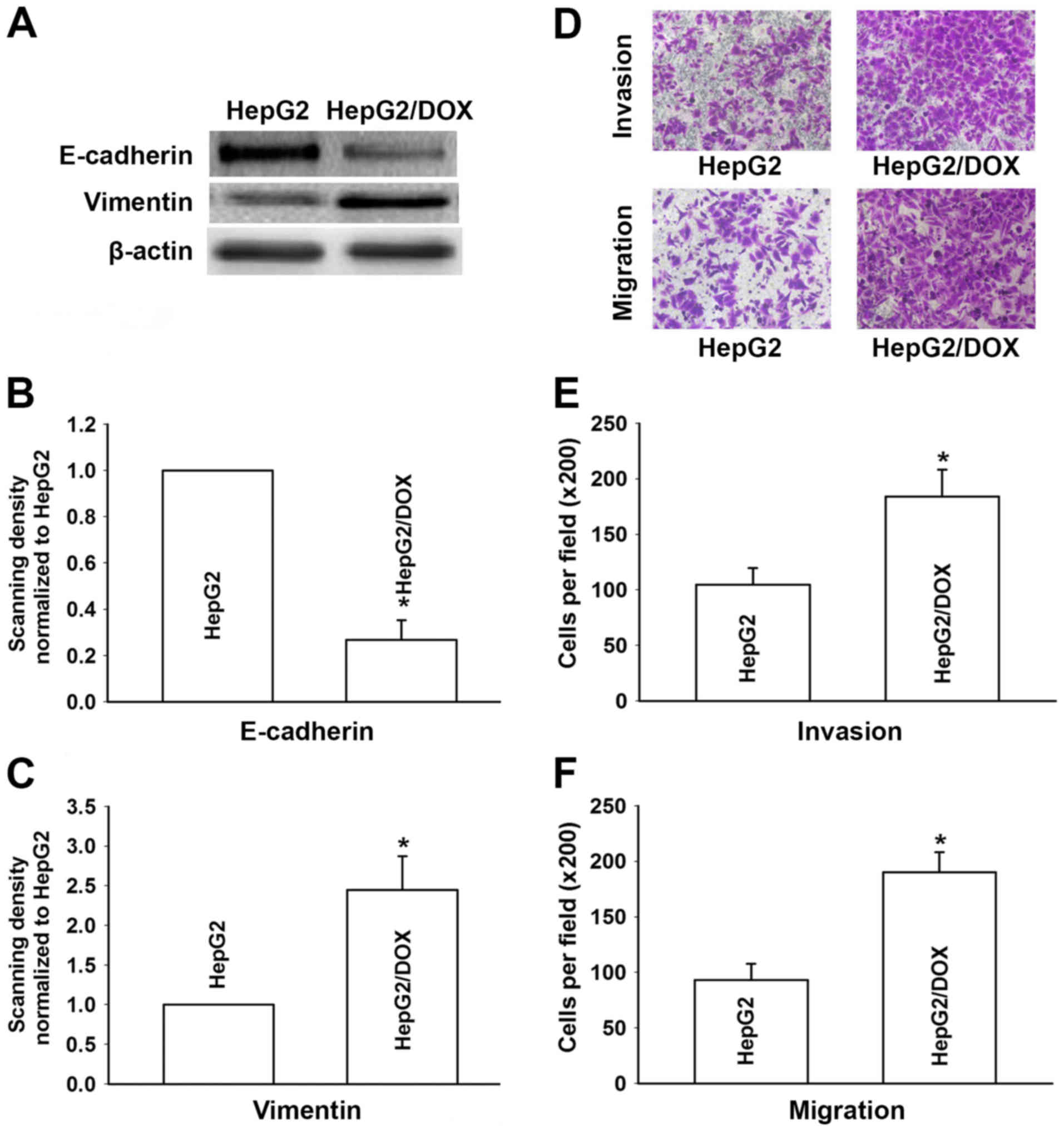
The expression of major markers of EMT in the HepG2
cells and HepG2/DOX cells were observed by western blotting.
Results revealed that epithelial marker E-cadherin was
significantly decreased in the HepG2/DOX cells compared with that
in the HepG2 cells. In contrast, the expression level of
mesenchymal marker vimentin was clearly increased in the HepG2/DOX
cells (Fig. 3A-C). In order to
further observe the changes of EMT in HepG2/DOX cells, the invasive
and migratory abilities of the HepG2/DOX and HepG2 cells were
detected by Transwell assays. Fig.
3D-F revealed that compared to the HepG2 cells, the HepG2/DOX
cells demonstrated higher invasion and migration potential. This
result suggests that acquired resistance could enhance the invasion
and migration abilities of HepG2 cells. All the aforementioned
results demonstrated that the HepG2/DOX cells acquired an EMT
phenotype.
Acquired resistance of DOX decreases
the expression of Cx32
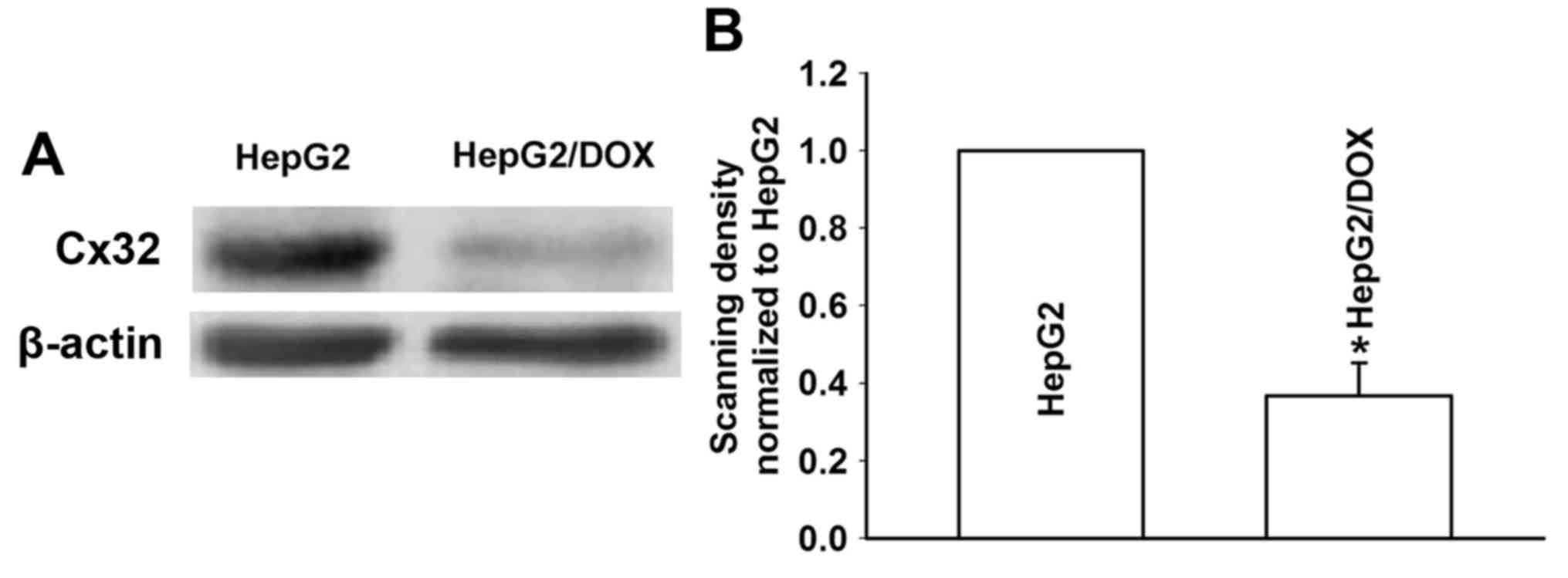
We investigated the expression of Cx32 by western
blotting in HepG2 and HepG2/DOX cells. As shown in Fig. 4, the HepG2 cells showed a
significantly increased expression level of Cx32 compared with that
in the HepG2/DOX cells. This result demonstrated that Cx32 may play
an important role in the mechanism of acquired resistance of HepG2
cells to DOX.
Cx32 regulates EMT in HCC
Inhibition of Cx32 induces EMT in HepG2
cells
The aforementioned results showed that the HepG2/DOX
cells initiated EMT with decreased expression of Cx32. Then, we
observed the effect of Cx32 on DOX-induced EMT in two ways:
inhibition of Cx32 expression with shRNA-Cx32 in HepG2 cells, and
overexpression of Cx32 with pcDNA-Cx32 in HepG2/DOX cells.
First, we decreased the expression of Cx32 in HepG2
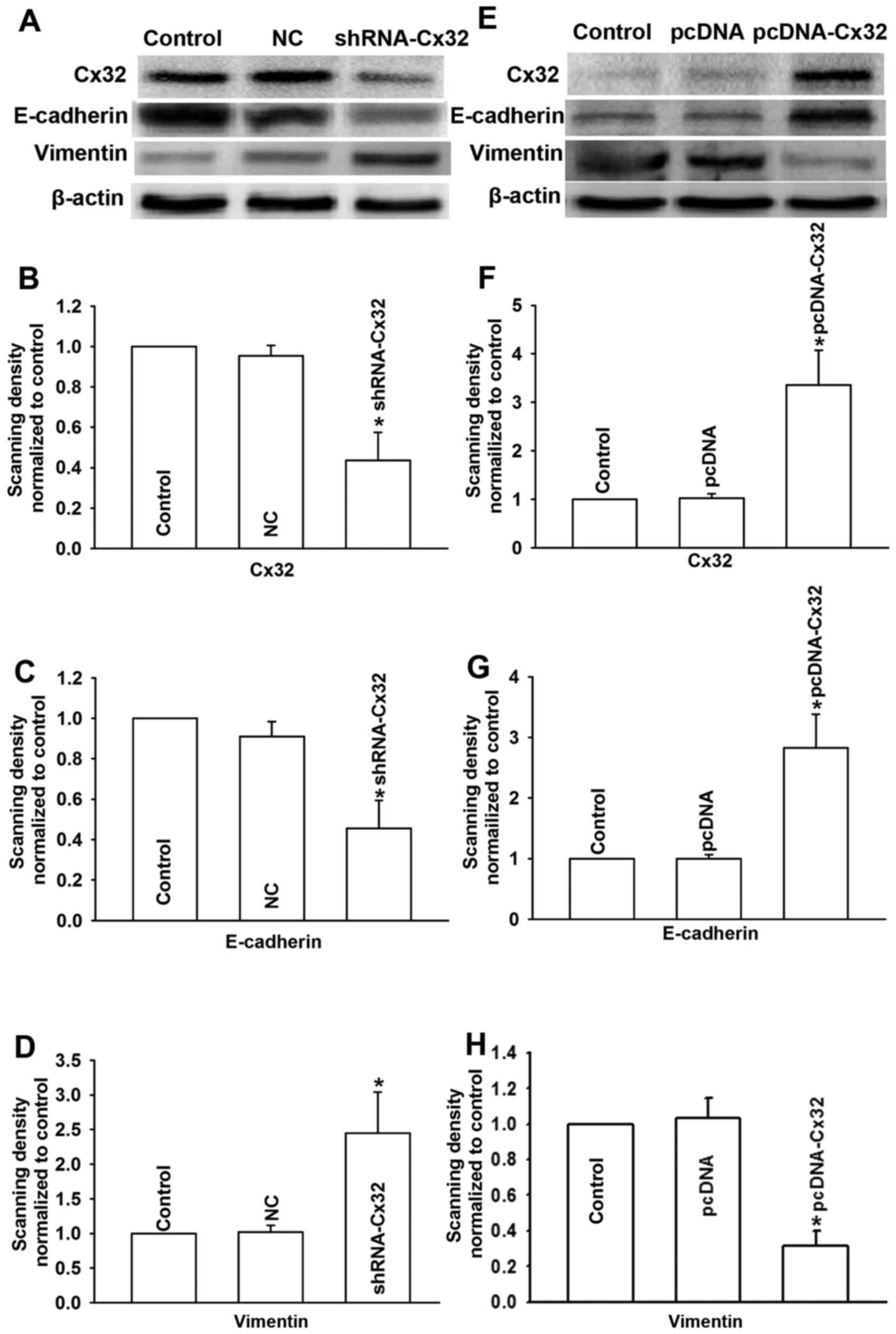
cells by shRNA-Cx32. As shown in Fig.
5A and B, after transfecting the HepG2 cells with shRNA-Cx32,
the expression of Cx32 was significantly decreased in the
shRNA-Cx32 group when compared with the control or negative control
group. Then, to explore the effect of Cx32 on EMT in HepG2 cells,
we also detected the expression of EMT markers (E-cadherin and
vimentin) by western blotting as well as the invasion and migration
abilities of the cells by Transwell assays. The results in Fig. 5A and C demonstrated that the
expression of E-cadherin in the shRNA-Cx32 group was clearly
decreased when compared with the control or negative control group.
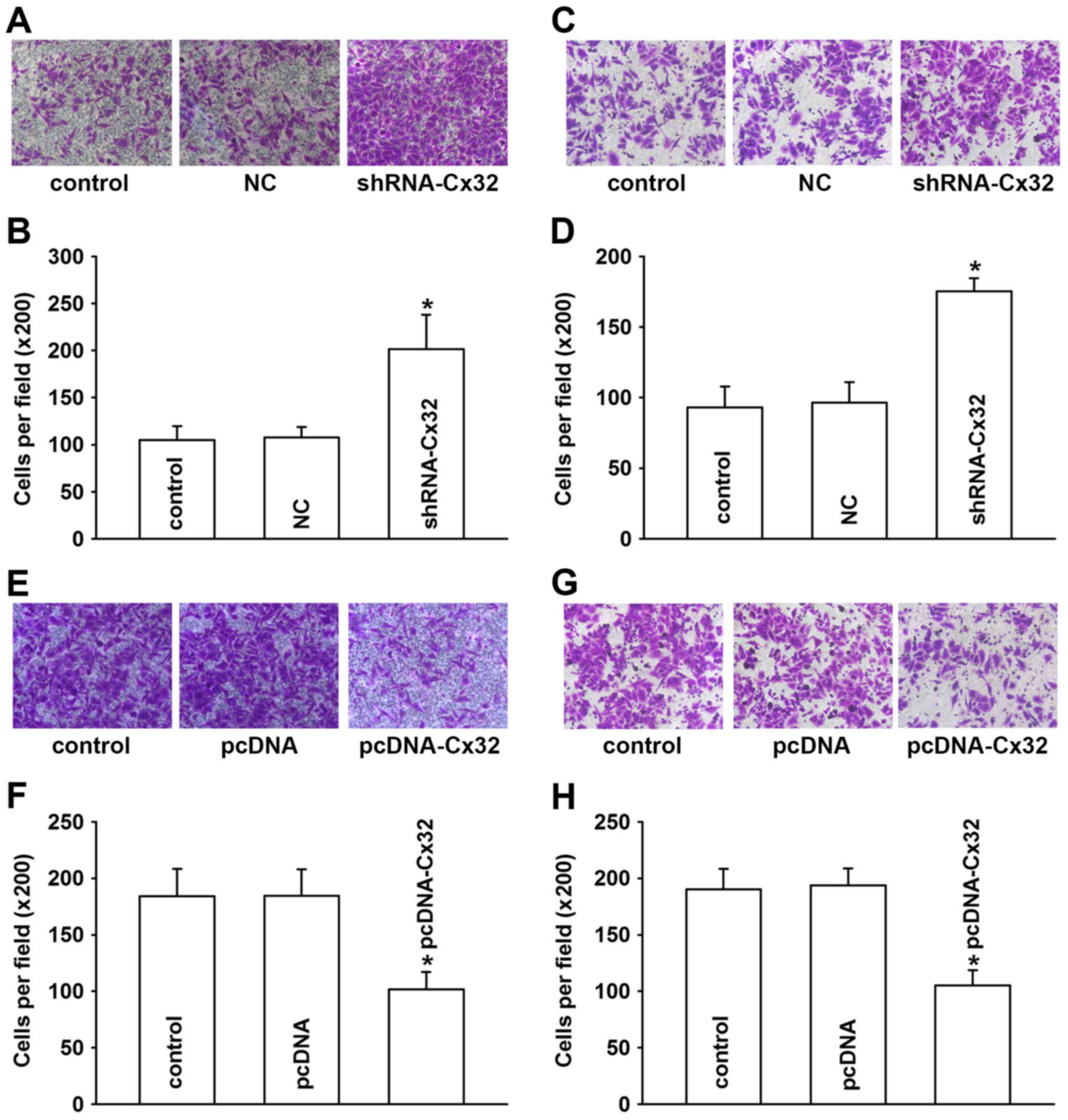
Inversely, vimentin was significantly enhanced (Fig. 5A and D). The results of Transwell
assays also revealed that the HepG2 cells acquired enhanced
invasive and migratory abilities after the inhibition of the
expression of Cx32 (Fig. 6A and D).
These results suggest that downregulation of Cx32 by shRNA-Cx32
induced EMT in the HepG2 cells, which indicated that Cx32 is partly
responsible for the initiation of EMT in HepG2 cells.
Overexpression of Cx32 converts EMT to
mesenchymal-epithelial transition (MET) in HepG2/DOX cells
To further observe the role of Cx32 in the mechanism
of DOX-induced EMT, we used a Cx32 gene fragment to overexpress
Cx32 in HepG2/DOX cells which were resistant to DOX. After
HepG2/DOX cells were transfected with pcDNA-Cx32 for 24 h, the
expression level of Cx32 was detected by western blotting. The
result revealed that the pcDNA-Cx32 group showed significantly
increased expression of Cx32 when compared to that of the control
or the negative control group (Fig. 5E
and F). To explore the effect of the upregulation of Cx32 on
EMT in HepG2/DOX cells, EMT markers E-cadherin and vimentin as well
as the invasion and migration abilities of the cells were also
examined. As shown in Fig. 5E and
G, E-cadherin was significantly enhanced in the pcDNA-Cx32
group, while vimentin was obviously decreased (Fig. 5E and H). The invasion and migration
abilities also decreased after transfecting HepG2/DOX cells with
Cx32 (Fig. 6E-H). These findings
indicate that overexpression of Cx32 reverses EMT to MET in
HepG2/DOX cells.
Discussion
In recent years, studies have focused on the role of
EMT in the acquired resistance of chemotherapeutics drugs (16,17–20),
and acquisition of EMT has recently been proposed as an important
mechanism for drug resistance in cancer cells. For example,
cisplatin-resistance in lung cancer cells, cisplatin-resistance in
cervical cancer cells, crizotinib-resistant anaplastic lymphoma
kinase (ALK)-positive lung cancer cells, gemcitabine-resistant
hepatoma cells, tamoxifen-resistant MCF7 breast cancer cells all
possess EMT characteristics. Recently, targeting EMT has been
considered as a new strategy to combat cancer drug resistance
(21). In the present study, we
also found that during the process of acquisition of DOX
resistance, HepG2/DOX cells exhibited an EMT phenotype.
Cxs have been reported to be the components of
gap-junctional communication channels and important tumor
suppressors (9,10,22).
Recently, 21 connexins and related genes have been found in the
human body (23), among them, Cx43,
Cx26 and Cx32 have the most extensive distribution. Normal
hepatocytes express Cx32 and to a lesser extent Cx26, which
represent ~90 and 5%, respectively, of the total Cx amount in rat
and human livers. Cx32 is uniformly distributed throughout the
liver, while Cx26 is mainly expressed in the periportal acinar area
(24). During the process of
hepatocarcinogenesis, both Cx32 and Cx26 are decreased (24). In the present study, Cx26 was not
detected in the HepG2 and HepG2/DOX cells (data not shown), while
the aforementioned 2 cell lines expressed different levels of Cx32.
Thus, the research focused on the effect of Cx32, but not Cx26, on
DOX-induced EMT. Reports showed that Cx32 demonstrated a
suppressive effect on liver inflammation, fibrosis,
hepatocarcinogenesis and hepatoma cell metastasis (25–28).
Nakashima et al also found that the expression of Cx32 in
hepatocellular carcinoma (HCC) patients was significantly decreased
compared with the non-HCC patients (27). Cx32 overexpression decreased the
malignant phenotype of liver tumors (29, 30). The present study
reveals a new role of Cx32 in the regulation of cancer metastasis
and drug resistance in HCC.
In our previous study (16), we found that cisplatin
(CDDP)-resistant cell line A549/CDDP acquired an EMT phenotype.
Overexpression of Cx43 converted EMT to MET and enhanced the
sensitivity of CDDP to A549/CDDP cells. The results revealed that
Cx43 was correlated with EMT in A549/CDDP cells. The present study
further investigated the relationship between Cx and EMT in
chemotherapy drug-resistant cells. We used HepG2 cells to establish
the DOX-resisistant cell line. The present study demonstrated that
when the HepG2 cells became resistant to DOX, the HepG2 cells
acquired an EMT phenotype, meanwhile, Cx32 was significantly
decreased in the HepG2/DOX cells. We also found that downregulation
of Cx32 by shRNA-Cx32 induced EMT in the HepG2 cells, while
overexpression of Cx32 reversed EMT in the HepG2/DOX cells. Thus,
the results in HCC were consistent with the results in human lung
adenocarcinoma, which suggest that Cx-induced EMT may be
responsible for drug resistance in cancer cells and Cx is an
important target for overcoming drug resistance when cells initiate
EMT.
Cx hemichannels can supply a pathway for cellular
signaling between cells and their extracellular environment
(31–33). The messengers that diffuse through
Cx hemichannels usually include adenosine triphosphate (ATP),
glutamate and glutathione. Studies have shown that both ATP and
glutathione participate in the regulation of EMT in different cells
(34–39). Thus, an inference is that ATP and/or
glutathione may be responsible for Cx32-mediated EMT in HCC.
Studies may be performed in the future to observe the role of ATP
and glutathione in Cx32-mediated EMT in HCC.
In summary, the present study is the first to
establish the role of Cx32 in the occurrence of EMT in liver
carcinoma cells. We demonstrated that the upregulation of the
expression of Cx32 in HepG2/DOX cells inhibited EMT and enhanced
DOX-induced cytotoxicity. Therefore, Cx32 may be a new target to
potentiate DOX cytotoxicity in hepatic carcinoma in the future.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of Anhui (grant no. 1508085QH151) and
the Natural Science Foundation of the Provincial Education
Department of Anhui (no. KJ2015A147), the visiting project of the
Provincial Education Department of Anhui (gxfxZD2016142), the
National Natural Science Foundation of China (no. 81001457), and
the Foundation of Bengbu Medical College (nos. Byycxz1422 and
Byky1407ZD).
References
|
1
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhu AX: Systemic therapy of advanced
hepatocellular carcinoma: How hopeful should we be? Oncologist.
11:790–800. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Asghar U and Meyer T: Are there
opportunities for chemotherapy in the treatment of hepatocellular
cancer? J Hepatol. 56:686–695. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xu F, Wang F, Yang T, Sheng Y, Zhong T and
Chen Y: Differential drug resistance acquisition to doxorubicin and
paclitaxel in breast cancer cells. Cancer Cell Int. 14:5382014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Harada K, Ferdous T and Ueyama Y:
Establishment of 5-fluorouracil-resistant oral squamous cell
carcinoma cell lines with epithelial to mesenchymal transition
changes. Int J Oncol. 44:1302–1308. 2014.PubMed/NCBI
|
|
6
|
Haider M, Zhang X, Coleman I, Ericson N,
True LD, Lam HM, Brown LG, Ketchanji M, Nghiem B, Lakely B, et al:
Epithelial mesenchymal-like transition occurs in a subset of cells
in castration resistant prostate cancer bone metastases. Clin Exp
Metastasis. 33:239–248. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xu H, Pirisi L and Creek KE: Six1
overexpression at early stages of HPV16-mediated transformation of
human keratinocytes promotes differentiation resistance and EMT.
Virology. 474:144–153. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dinicola S, Pasqualato A, Proietti S,
Masiello MG, Palombo A, Coluccia P, Canipari R, Catizone A, Ricci
G, Harrath AH, et al: Paradoxical E-cadherin increase in
5FU-resistant colon cancer is unaffected during
mesenchymal-epithelial reversion induced by γ-secretase inhibition.
Life Sci. 145:174–183. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou JZ and Jiang JX: Gap junction and
hemichannel-independent actions of connexins on cell and tissue
functions - an update. FEBS Lett. 588:1186–1192. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Aasen T: Connexins: Junctional and
non-junctional modulators of proliferation. Cell Tissue Res.
360:685–699. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jiang JX and Gu S: Gap junction- and
hemichannel-independent actions of connexins. Biochim Biophys Acta.
1711:208–214. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Diezmos EF, Bertrand PP and Liu L:
Purinergic signaling in gut inflammation: The role of connexins and
pannexins. Front Neurosci. 10:3112016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cronier L, Crespin S, Strale PO, Defamie N
and Mesnil M: Gap junctions and cancer: New functions for an old
story. Antioxid Redox Signal. 11:323–338. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kandouz M and Batist G: Gap junctions and
connexins as therapeutic targets in cancer. Expert Opin Ther
Targets. 14:681–692. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Trosko JE and Ruch RJ: Gap junctions as
targets for cancer chemoprevention and chemotherapy. Curr Drug
Targets. 0.3:465–482. 2002. View Article : Google Scholar
|
|
16
|
Yu M, Zhang C, Li L, Dong S, Zhang N and
Tong X: Cx43 reverses the resistance of A549 lung adenocarcinoma
cells to cisplatin by inhibiting EMT. Oncol Rep. 31:2751–2758.
2014.PubMed/NCBI
|
|
17
|
Song J and Li Y: miR-25-3p reverses
epithelial-mesenchymal transition via targeting Sema4C in
cisplatin-resistance cervical cancer cells. Cancer Sci. Oct
15–2016.(Epub ahead of print). doi: 10.1111/cas.13104.
|
|
18
|
Gao HX, Yan L, Li C, Zhao LM and Liu W:
miR-200c regulates crizotinib-resistant ALK-positive lung cancer
cells by reversing epithelial-mesenchymal transition via targeting
ZEB1. Mol Med Rep. 14:4135–4143. 2016.PubMed/NCBI
|
|
19
|
Wu Q, Wang R, Yang Q, Hou X, Chen S, Hou
Y, Chen C, Yang Y, Miele L, Sarkar FH, et al: Chemoresistance to
gemcitabine in hepatoma cells induces epithelial-mesenchymal
transition and involves activation of PDGF-D pathway. Oncotarget.
4:1999–2009. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hiscox S, Jiang WG, Obermeier K, Taylor K,
Morgan L, Burmi R, Barrow D and Nicholson RI: Tamoxifen resistance
in MCF7 cells promotes EMT-like behaviour and involves modulation
of β-catenin phosphorylation. Int J Cancer. 118:290–301. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Du B and Shim JS: Targeting
epithelial-mesenchymal transition (EMT) to overcome drug resistance
in cancer. Molecules. 21:212016. View Article : Google Scholar
|
|
22
|
Kay CW, Ursu D, Sher E and King AE: The
role of Cx36 and Cx43 in 4-aminopyridine-induced rhythmic activity
in the spinal nociceptive dorsal horn: An electrophysiological
study in vitro. Physiol Rep. 4:42016. View Article : Google Scholar
|
|
23
|
Unger VM, Kumar NM, Gilula NB and Yeager
M: Three-dimensional structure of a recombinant gap junction
membrane channel. Science. 283:1176–1180. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Maes M, Decrock E, Cogliati B, Oliveira
AG, Marques PE, Dagli ML, Menezes GB, Mennecier G, Leybaert L,
Vanhaecke T, et al: Connexin and pannexin (hemi)channels in the
liver. Front Physiol. 4:4052014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
De Maio A, Gingalewski C, Theodorakis NG
and Clemens MG: Interruption of hepatic gap junctional
communication in the rat during inflammation induced by bacterial
lipopolysaccharide. Shock. 14:53–59. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sagawa H, Naiki-Ito A, Kato H, Naiki T,
Yamashita Y, Suzuki S, Sato S, Shiomi K, Kato A, Kuno T, et al:
Connexin 32 and luteolin play protective roles in non-alcoholic
steatohepatitis development and its related hepatocarcinogenesis in
rats. Carcinogenesis. 36:1539–1549. 2015.PubMed/NCBI
|
|
27
|
Nakashima Y, Ono T, Yamanoi A, El-Assal
ON, Kohno H and Nagasue N: Expression of gap junction protein
connexin32 in chronic hepatitis, liver cirrhosis, and
hepatocellular carcinoma. J Gastroenterol. 39:763–768. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao B, Zhao W, Wang Y, Xu Y, Xu J, Tang
K, Zhang S, Yin Z, Wu Q and Wang X: Connexin32 regulates hepatoma
cell metastasis and proliferation via the p53 and Akt pathways.
Oncotarget. 6:10116–10133. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang J, Ichikawa A and Tsuchiya T: A novel
function of connexin 32: Marked enhancement of liver function in a
hepatoma cell line. Biochem Biophys Res Commun. 307:80–85. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Muramatsu A, Iwai M, Morikawa T, Tanaka S,
Mori T, Harada Y and Okanoue T: Influence of transfection with
connexin 26 gene on malignant potential of human hepatoma cells.
Carcinogenesis. 23:351–358. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Decrock E, Vinken M, De Vuyst E, Krysko
DV, Herde DK, Vanhaecke T, Vandenabeele P, Rogiers V and Leybaert
L: Connexin-related signaling in cell death: To live or let die?
Cell Death Differ. 16:524–536. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chandrasekhar A and Bera AK: Hemichannels:
Permeants and their effect on development, physiology and death.
Cell Biochem Funct. 30:89–100. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kar R, Batra N, Riquelme MA and Jiang JX:
Biological role of connexin intercellular channels and
hemichannels. Arch Biochem Biophys. 524:2–15. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Unahabhokha T, Chanvorachote P, Sritularak
B, Kitsongsermthon J and Pongrakhananon V: Gigantol Inhibits
Epithelial to Mesenchymal Process in Human Lung Cancer Cells. Evid
Based Complement Alternat Med. 2016:45616742016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Park SJ, Choi YS, Lee S, Lee YJ, Hong S,
Han S and Kim BC: BIX02189 inhibits TGF-β1-induced lung cancer cell
metastasis by directly targeting TGF-β type I receptor. Cancer
Lett. 381:314–322. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhou Q, Abraham AD, Li L, Babalmorad A,
Bagby S, Arcaroli JJ, Hansen RJ, Valeriote FA, Gustafson DL,
Schaack J, et al: Topoisomerase IIα mediates TCF-dependent
epithelial-mesenchymal transition in colon cancer. Oncogene.
35:4990–4999. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang HJ, Zhu J and Zheng GY: Role of
glutathione and other antioxidants in the inhibition of apoptosis
and mesenchymal transition in rabbit lens epithelial cells. Genet
Mol Res. 13:7149–7156. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang J, Qiu M, Ma Y, Bu Y, Yang L and
Tang X: Advanced oxidation protein products induce
epithelial-to-mesenchymal transition in cultured human proximal
tubular epithelial cells via oxidative stress. Nan Fang Yi Ke Da
Xue Xue Bao. 34:659–663. 2014.(In Chinese). PubMed/NCBI
|
|
39
|
Ryoo IG, Shin DH, Kang KS and Kwak MK:
Involvement of Nrf2-GSH signaling in TGFβ1-stimulated
epithelial-to-mesenchymal transition changes in rat renal tubular
cells. Arch Pharm Res. 38:272–281. 2015. View Article : Google Scholar : PubMed/NCBI
|