Introduction
Pseudolaric acid B (PAB) is a diterpene acid
isolated from the root and trunk bark of Pseudolarix
kaempferi Gordon (Pinaceae), known as ‘Tu-Jin-Pi’ in
Chinese. PAB has been used to treat dermatological fungal
infections. PAB was found to exert potent inhibitory activity on
cell growth in vitro in various tumor cell lines (1–6)
through cell cycle arrest, apoptosis or autophagy. However, to date
the role of PAB in neuroglial cells is not clear.
Human neuroglioma is one of the most common
malignant intracranial tumors in neurosurgery, and accounts for
more than 50% of all brain cancer cases, and is recently diagnosed
in a younger population (7,8). The pathogenesis of neuroglioma is
related to multiple processes affected by dozens of regulatory
factors (9); however, our knowledge
concerning the underlying factors regulating neuroglioma
progression remains unknow, and clinically effective drugs to treat
neuroglioma are urgently required.
The DNA damage response, caused by a variety of
stimuli, arrests the cell cycle to allow damage repair or direct
cell apoptosis (10). Apoptosis, as
one type of antitumor mechanism, has been a focus of numerous
studies for antitumor drug development (11,12).
In mammalian cells, the MAPK pathway is involved in cell
proliferation or cell death: stress-activated protein
kinase/c-Jun-N-terminal kinase (Sapk/Jnk), p38 kinase and
extracellular signal-regulated kinase (Erk) (13). Generally, Erk protein promotes
inflammation, apoptosis, growth, differentiation, oncogenic
transformation, while Jnk and p38 are implicated in growth,
differentiation and development (14,15).
Cell cycle arrest is important after eukaryotic normal cell cycle
progression is affected. These regulatory pathways are commonly
referred to as cell cycle checkpoints (16). Cells are arrested at cell cycle
checkpoints temporarily to allow for: i) cellular damage to be
repaired; ii) the dissipation of an exogenous cellular stress
signal; or iii) the availability of essential growth factors,
hormones or nutrients.
In the present study, we showed that PAB inhibited
neuroglioma cell proliferation and cell migration through
influencing tubulin aggregation. Meanwhile, PAB further induced DNA
damage response to induce cell cycle arrest and cell death, which
may provide a strategy to increase the antitumor effect of PAB.
Materials and methods
Materials
PAB, which was purchased from the National Institute
for the Control of Pharmaceutical and Biological Products (Beijing,
China), was dissolved in dimethyl sulfoxide (DMSO) to make a stock
solution. DMSO concentration was maintained at <0.01% in all the
cell cultures, and did not exert any detectable effect on cell
growth or cell death. Propidium iodode (PI),
phalloidin-tetramethylrhodamine B isothiocyanate, Hoechst 33258 and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
were purchased from Sigma Chemical Co. (St. Louis, MO, USA). TRIzol
reagent and SuperScript™ III RT-PCR kit were purchased from
Invitrogen (Carlsbad, CA, USA). Power SYBR-Green PCR Master Mix was
purchased from ABI (Vernon, CA, USA). Caffeine was purchased from
Shanghai YuanYe Biological Technology Co., Ltd. (Shanghai, China).
The following mouse or rabbit antibodies were used in the western
blot analyses: anti-CDK2 (Cell Signaling Technology, Inc., Danvers,
MA, USA), anti-cyclin E1, anti-cyclin A2 (both from ProteinTech
Group, Chicago, IL, USA), anti-CDK6, anti-cyclin D (both from Cell
Signaling Technology, Inc.), anti-CDK1 (Boster Biological
Technology Co., Ltd., Wuhan, China), anti-cyclin B1 (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), anti-JNK1 and 2,
anti-MAPK14 (p38) (both from Boster Biological Technology Co.,
Ltd.), anti-ERK1/2 (ProteinTech Group), anti-histone H3 (Amersham
Pharmacia Biotech, Piscataway, NJ, USA), anti-γ-h2ax (Cell
Signaling Technology, Inc.), anti-actin, and alkaline phosphatase
labeled-secondary antibodies (both obtained from Santa Cruz
Biotechnology, Inc.).
Cell culture
The human glioblastoma A172 (CRL-1620™) cell line
was obtained from the American Type Culture Collection (ATCC;
Manassas, VA, USA), and was cultured in Dulbeccos modified Eagles
medium (DMEM) (HyClone, Logan, UT, USA) supplemented with 10% fetal
calf serum (FCS), 2 mM glutamine (both from Gibco-BRL, Grand
Island, NY, USA), penicillin (100 U/ml) and streptomycin (100
µg/ml), and maintained at 37°C with CO2 in a humidified
atmosphere.
Cell growth inhibition test
Inhibition of cell growth was determined by MTT
assay. A172 cells (1.0×104 cells/well) were seeded into
96-well culture plates (Nunc, Roskilde, Denmark). After 24 h of
incubation, different concentrations of PAB were added to the
plates, and 0 µM PAB was used as a control, and other doses as
samples. Following incubation, cell growth was measured at
different time points by addition of 20 µl MTT (5 mg/ml) at 37°C
for 2 h, and DMSO (150 µl) was added to dissolve the formazan
crystals. Absorbance was measured at 492 nm with an enzyme-linked
immunosorbent assay plate reader (Bio-Rad, Hercules, CA, USA). The
percentage of inhibition was calculated as follows:
Cell death (%) = [A492 (control) -
A492(sample)]/A492 (control) × 100%
Microtubule aggregation by
fluorescence staining
A172 cells (5×105) were placed on a
coverslip in a 6-well plate. After 24 h of cell culture, they were
treated with 4 µM PAB for 24 or 48 h, and then washed with PBS,
fixed in 3.7% formaldehyde, and then rinsed three times in 1X PBS.
Next, 0.8% Triton X-100 was added for 15 min, and 5 µg/ml
phalloidin-tetramethylrhodamine B isothiocyanate staining was
performed for 40 min, followed by one rinse in 1X PBS and then
staining with 5 mg/l Hoechst 33258 for 30 min. The intensity of red
staining was measured by fluorescence microscopy at excitation
wavelength 584 nm with emission filter 607 nm (Leica, Nusslich,
Germany). Nuclear changes were observed by fluorescence microscopy
at excitation wavelength 350 nm with emission filter 460 nm
(Leica).
Cell migration
A172 cells were cultured for 24 h, and then a line
was scratched randomly with a pipette tip. Lines with the same
width were chosen, and the width was recorded at 0, 12, 24, 36 and
48 h after control medium or PAB treatment by phase contrast
microscopy (Leica).
Flow cytometric analysis of cell cycle
distribution
A172 cells were cultured for 24 h, and then
collected at 0, 12, 24, 36 and 48 h after control medium or PAB
treatment. Collected A172 cells (1.0×106) were harvested
and rinsed with PBS. The cell pellets were fixed in 70% ethanol at
4°C overnight. After washing twice with PBS, the cells were stained
with 1.0 ml of PI solution containing PI 50 mg/l, RNase A 1 g/l and
0.1% Triton X-100 in sodium citrate 3.8 mmol/l, followed by
incubation on ice in the dark for 30 min. The samples were analyzed
by a FACScan flow cytometer (Becton-Dickinson, Franklin Lakes, NJ,
USA).
Quantitative real-time PCR
RNA was extracted from cells treated with control
medium and PAB using TRIzol reagent (Gibco-BRL, Rockville, MD, USA)
and isolated as specified by the manufacturer. The RNA was treated
with DNAse (DNase I-RNase-Free; Ambion, Foster City, CA, USA) to
remove any contaminating DNA; 200 ng of total RNA was
reverse-transcribed with oligo-dT primers using the High Capacity
cDNA RT kit (Applied Biosystems, Foster City, CA, USA) in a 20 µl
cDNA reaction, as specified by the manufacturer. For quantitative
PCR, the template cDNA was added to a 20 µl reaction with
SYBR-Green PCR Master Mix (Applied Biosystems) and 0.2 µM of the
primers was added (primers are listed in Table I). Amplification was carried out
using an ABI PRISM 7000 for 40 cycles under the following
conditions: initial denaturation at 95°C for 10 min, plus 40 cycles
of 95°C for 15 sec, followed by 60°C for 1 min. The fold changes
were calculated relative to GAPDH using the ΔΔCt method for target
gene mRNA analysis.
 | Table I.Primers for real-time PCR. |
Table I.
Primers for real-time PCR.
| Primer pair | Role | Forward sequence
5′-3′ | Reverse sequence
5′-3′ |
|---|
| Cyclin E | Real-time |
TCAGGGTATCAGTGGTGCGA |
CAAATCCAAGCTGTCTCTGTG |
| CDK2 | Real-time |
CTCCTGGGCTCGAAATATTATTCCACAG |
CCGGAAGAGCTGGTCAATCTCAGA |
| Cyclin A | Real-time |
GCATGTCACCGTTCCTCCTT |
CAGGGCATCTTCACGCTCTAT |
| CDK6 | Real-time |
GGTCAGGTTGTTTGATGTGTGC |
TATCCTTTATGGTTTCAGTGGG |
| Cyclin D | Real-time |
CTACTACCGCCTCACACGCTTC |
TCCTCCTCCTCTTCCTCCTCCT |
| CDK1 | Real-time |
TCAAGTGGTAGCCATGAAAAAA |
TAACCTGGAATCCTGCATAAGC |
| Cyclin B1 | Real-time |
TGGCCTCACAAAGCACATGA |
GCTGTGCCAGCGTGCTAATC |
| GAPDH | Real-time |
GCAAATTCCATGGCACCGT |
TCGCCCCACTTGATTTTGG |
Observation of morphologic changes by
light microscopy
A172 cells (5×105 cells/well) were
cultured into a 6-well plate for 12 h. Then, the cells were treated
with 4 µM PAB and/or caffeine for 0, 12, 24, 36 and 48 h, and
morphologic changes were observed by phase contrast microscopy
(Leica).
Western blot analysis of protein
expression
A172 cells (1×106) were cultured in a
25-ml culture bottle for the indicated time, and then were treated
with 4 µM PAB for the indicated time. Both adherent and floating
cells were collected and frozen at −80°C. Western blot analysis was
performed for total proteins as described previously (5). Protein expression was detected using
primary polyclonal antibody (1:1,000) followed by a corresponding
AP-conjugated secondary antibody diluted 1:1,000. Proteins were
visualized using NBT and BCIP.
Statistical analysis
All data represent at least three independent
experiments, and are expressed as mean ± SD. P-values <0.001
were considered to represent statistically significant
differences.
Results
Inhibitory effect of PAB on A172
cells
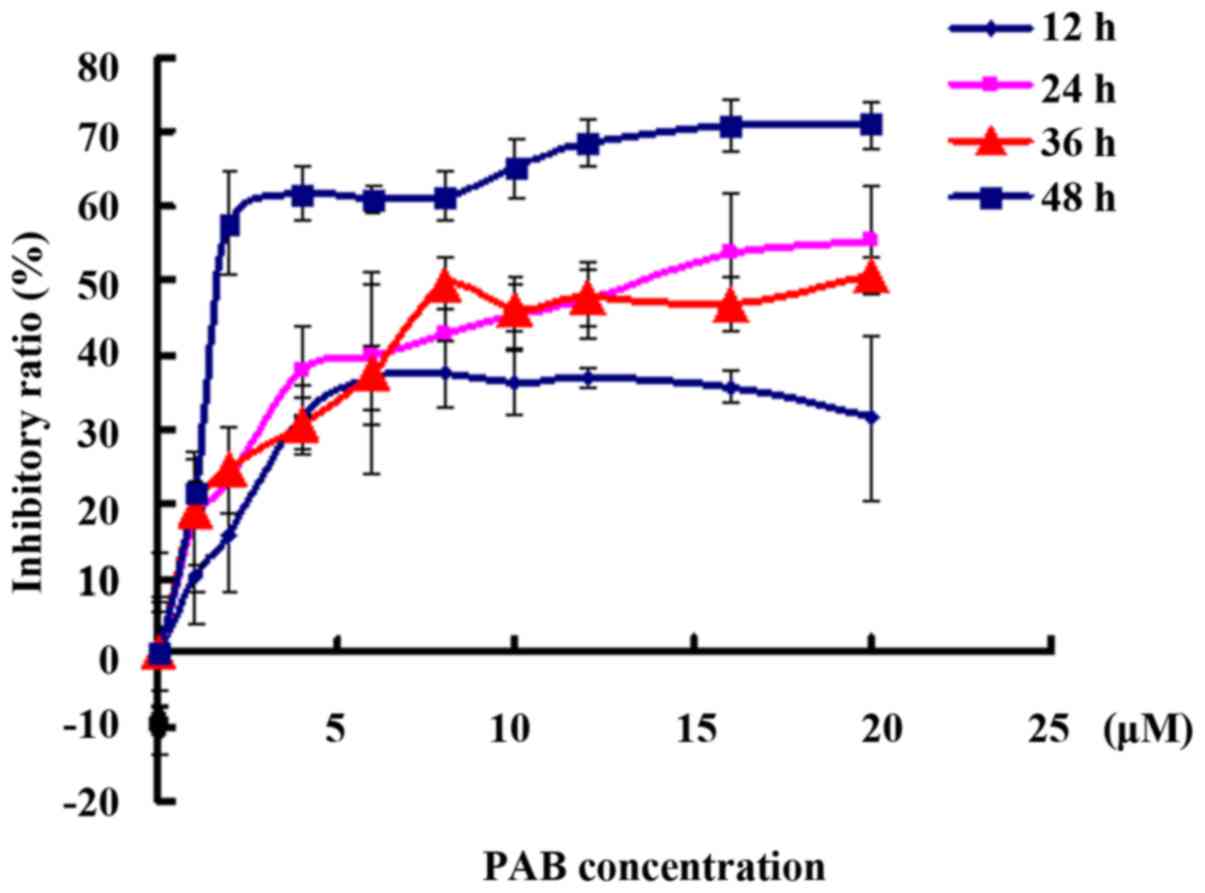
To detect the inhibitory effect of PAB on
neuroglioma cell proliferation, MTT assay was carried out. PAB
treatment had an inhibitory effect on the A172 cells in a time- and
dose-dependent manner (Fig. 1). In
addition, 4 µM PAB inhibited cell growth with an inhibitory ratio
of 40% at 48 h, as found in previous studies (5,6). Thus,
we chose 4 µM PAB in the following experiments.
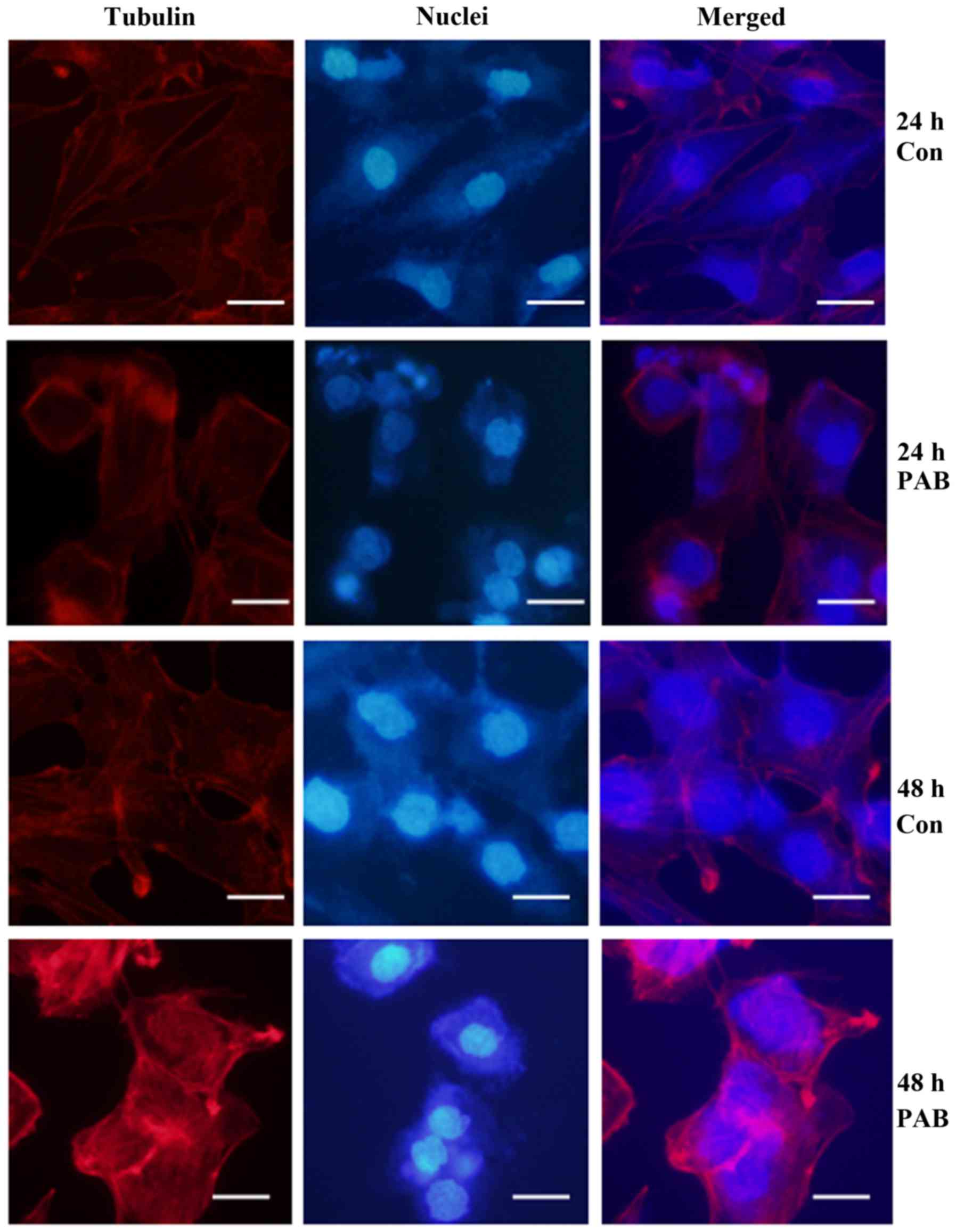
PAB promotes the aggregation of
microtubule fiber
PAB exerts its function by influencing microtubule
fiber aggregation (17). We found
that PAB treatment promoted the uneven aggregation of microtubule
fibers with more obvious red staining, while control cells showed
even aggregation of microtubule fibers (Fig. 2). Therefore, PAB influenced cell
microtubule fiber to exert its function.
PAB inhibits cell migration
Tumor cells with a high ability for migration are
difficult to treat. In the migration assay, we confirmed that A172
cells had a high migration ability since with increasing time the
scratch wound line disappeared. Meanwhile, after PAB treatment, the
scratch wound line that remained was wider than that of the control
cells (Fig. 3). Therefore, A172
cells had a high ability of migration, and PAB inhibited the cell
migration of these cells.
PAB induces G2/M cell cycle
arrest
It was observed that from 12 h, PAB treatment
induced obvious G2/M cell cycle arrest. At 24 h, in the control
group, the percentage of cells in the G2/M phases was 3.70±1.47%,
while after PAB treatment it was 45.82±0.64%; at 36 h in the
control group, the percentage of cells was 2.97±1.10%, while after
PAB treatment it was 64.83±4.27%; at 48 h in the control group the
percentage of cells was 3.45±1.29%, while after PAB treatment it
was 56.98±2.14% (Fig. 4A).
Therefore, PAB induced G2/M arrest. In addition, at 24 h after PAB
treatment the percentage of cells in the G0/G1 phase was
4.50±3.98%, while at 36 h after PAB treatment the percentage of
G0/G1 cells was 8.39±1.46%, and at 48 h after PAB treatment the
percentage of S phase cells (34.65±2.82%) was increased compared to
the PAB group at 36 h (26.77±3.36%). Therefore, PAB induced cell
cycle slippage from G2/M into G0/G1 and then into S phase. To
further investigate the mechanism underlying the effect on the cell
cycle by PAB, cyclin B1 and CDK1 expression was detected owning to
the fact that the cyclin B1/CDK1 complex regulates G2/M process. It
was found that from 12 h, the expression of cyclin B1 was increased
after PAB treatment compared to the control treatment, and the
expression of CDK1 was increased from 24 h, while at 48 h, the
expression of CDK1 and cyclin B1 after PAB treatment was not
increased compared to the control treatment (Fig. 4B). Therefore, PAB by regulating
cyclin B1/CDK1 complex expression induced cell cycle arrest at G2/M
until 36 h, and at 48 h cyclin B1/CDK1 complex expression was
consistent with the cell cycle slippage. To explain the expression
alteration of the cyclin B1/CDK1 complex, the transcription of this
complex was analyzed. It was found that the mRNA level of cyclin B1
in the PAB group was 5.39 times higher than that in the control
group after a 12-h treatment (P<0.01), while at 48 h, there was
no obvious difference. For CDK1, at 12 and 48 h after PAB
treatment, there was no difference with the control treatment
(Fig. 4C). Therefore, PAB regulated
protein expression through different mechanisms, partly through the
transcriptional level.
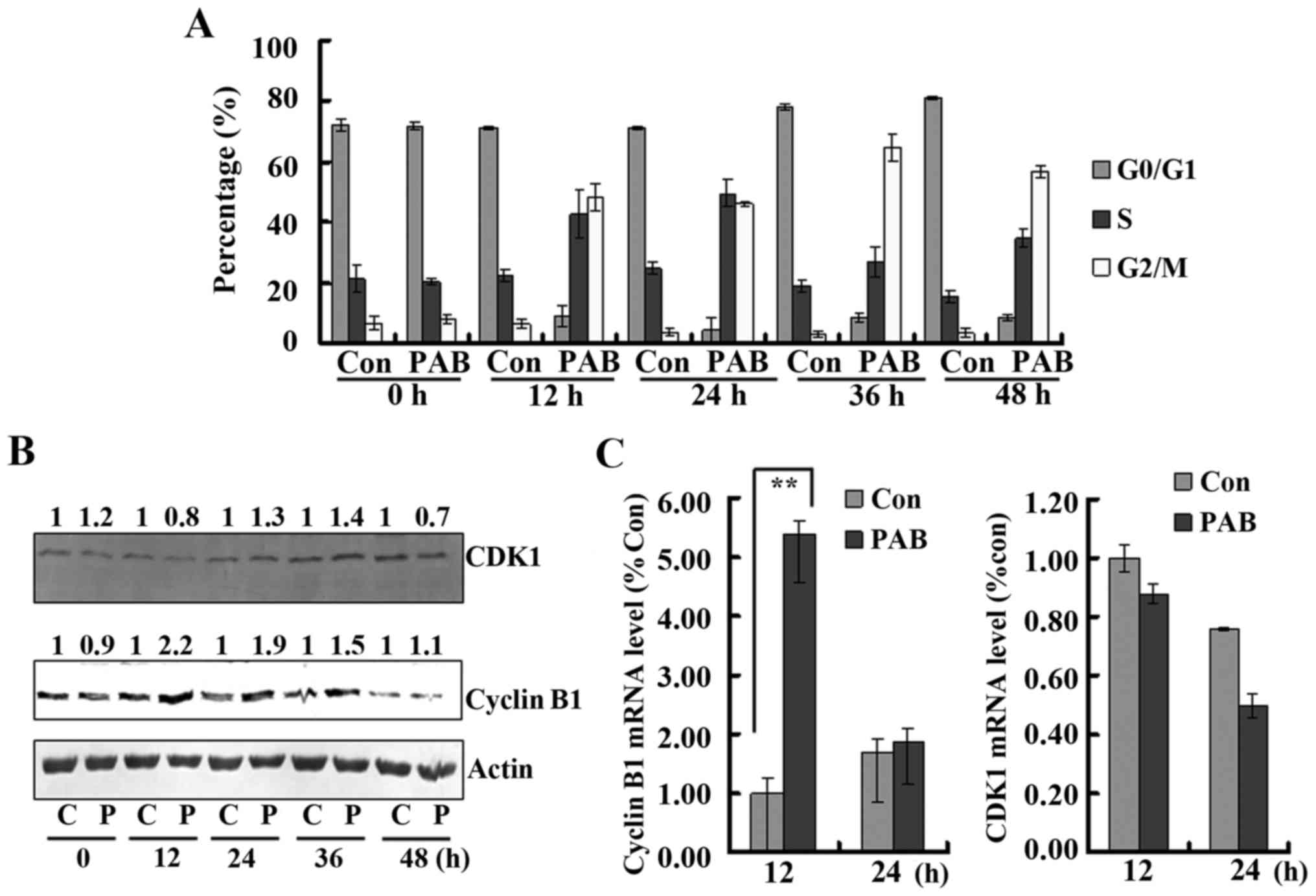 | Figure 4.Pseudolaric acid B (PAB) promotes cell
cycle arrest. (A) At 12, 24, 36 and 48 h, PAB induced obvious cell
cycle arrest. Flow cytometric analysis showed that from 12 to 48 h,
4 µM PAB induced obvious cell cycle arrest. Mean ± SD, n=3. (B) PAB
regulated cyclin B1 and CDK1 expression. At 12 and 48 h after PAB
treatment, the cells were analyzed and western blotting was
performed for protein expression. Actin is shown as a loading
control. The number above each western blot band is provided, which
were standardized using actin and normalized to 1.0 in the control
group. C, control; P, PAB. (C) At 12 and 48 h after PAB treatment,
intracellular cyclin B1 and CDK1 mRNA levels were detected in the
control group (con) or PAB treatment group by real-time
quantitative PCR. The results were standardized using GAPDH mRNA as
a control and normalized to 1.0 in the control group. The results
represent the mean ± SD of three independent experiments;
**P<0.01. |
Meanwhile, the effect of PAB on S phase and
G0/G1-related protein expression was observed. It was found that in
regards to S phase-related proteins, PAB decreased the expression
of cyclin E at 24 and 36 h after PAB treatment, but at 48 h PAB
treatment increased the expression of cyclin E, and PAB decreased
the expression of CDK2 from 12 to 36 h after PAB treatment, but at
48 h, PAB treatment had no effect on CDK2 expression (Fig. 5A), which was consistent with the
slippage into S phase at 48 h after PAB treatment. However, PAB
treatment had no effect on the expression of cyclin A from 12 to 48
h compared to control treatment (Fig.
5A), indicating that PAB regulated cell cycle progression,
possibly independent of cyclin A expression. In addition, we also
assessed the transcription of them to explain the alteration at the
protein level, and we found that the mRNA level of CDK2 was
decreased after PAB treatment at 12 h compared to the control
treatment, and the mRNA level of cyclin E was increased after PAB
treatment at 48 h compared to the control treatment, which could
partly explain the alteration of CDK2 and cyclin E (Fig. 5C). For G0/G1-related protein
expression, it was found that at 36 h after PAB treatment, the
expression of cyclin D was increased, but at 48 h, the expression
of cyclin D was decreased (Fig.
5B), which was consistent with the slippage into S phase at 36
h, and then into S phase at 48 h after PAB treatment. The
expression of CDK6 was not affected when compared to the control
treatment (Fig. 5B), indicating
that PAB regulated cyclin D expression to regulate the activity of
cyclin D and CDK6 complex. In addition, at 12 and 48 h, PAB did not
affect the mRNA levels of cyclin D and CDK6 compared to the levels
noted in the control treatment group (Fig. 5C). Therefore, PAB regulates protein
expression, but not at the level of transcription.
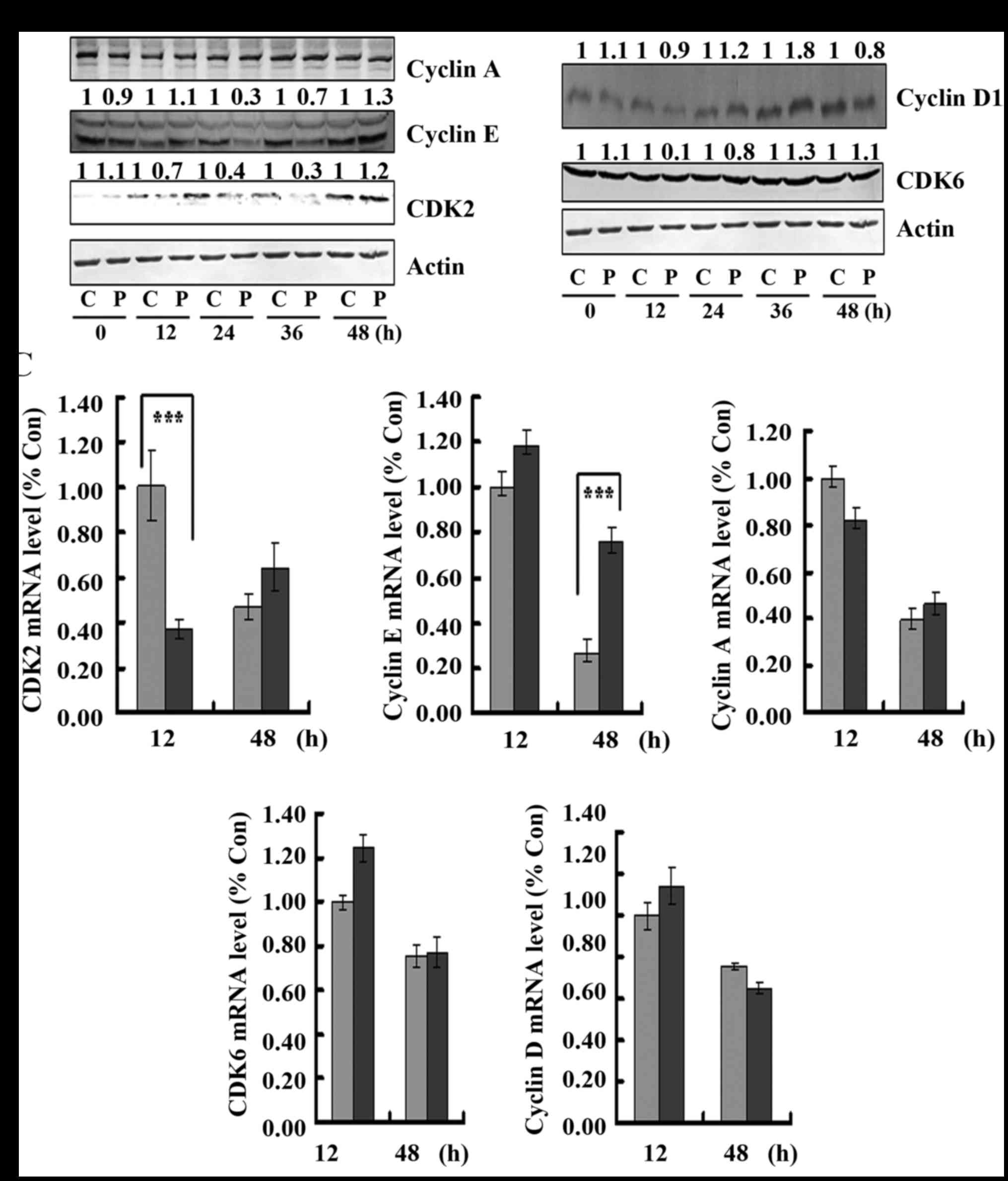 | Figure 5.Pseudolaric acid B (PAB) regulates
expression of S and G0/G1 phase-related proteins. (A and B) At 12
and 36 h after PAB treatment, the cells were analyzed and western
blotting was performed for cyclin A1, cyclin E1, CDK2, cyclin D1
and CDK6 protein expression. Actin is shown as a loading control.
The number above each western blot band is provided, which was
standardized using actin and normalized to 1.0 in the control
group. C, control; P, PAB. (C) At 12 and 48 h after PAB treatment,
intracellular cyclin A1 and E1, CDK2, cyclin D1 and CDK6 mRNA
levels were detected in the control group (Con) or PAB treatment
group by real-time quantitative PCR. The results were standardized
using GAPDH mRNA as a control and normalized to 1.0 in the control
group. The results represent the mean ± SD of three independent
experiments; ***P<0.001. |
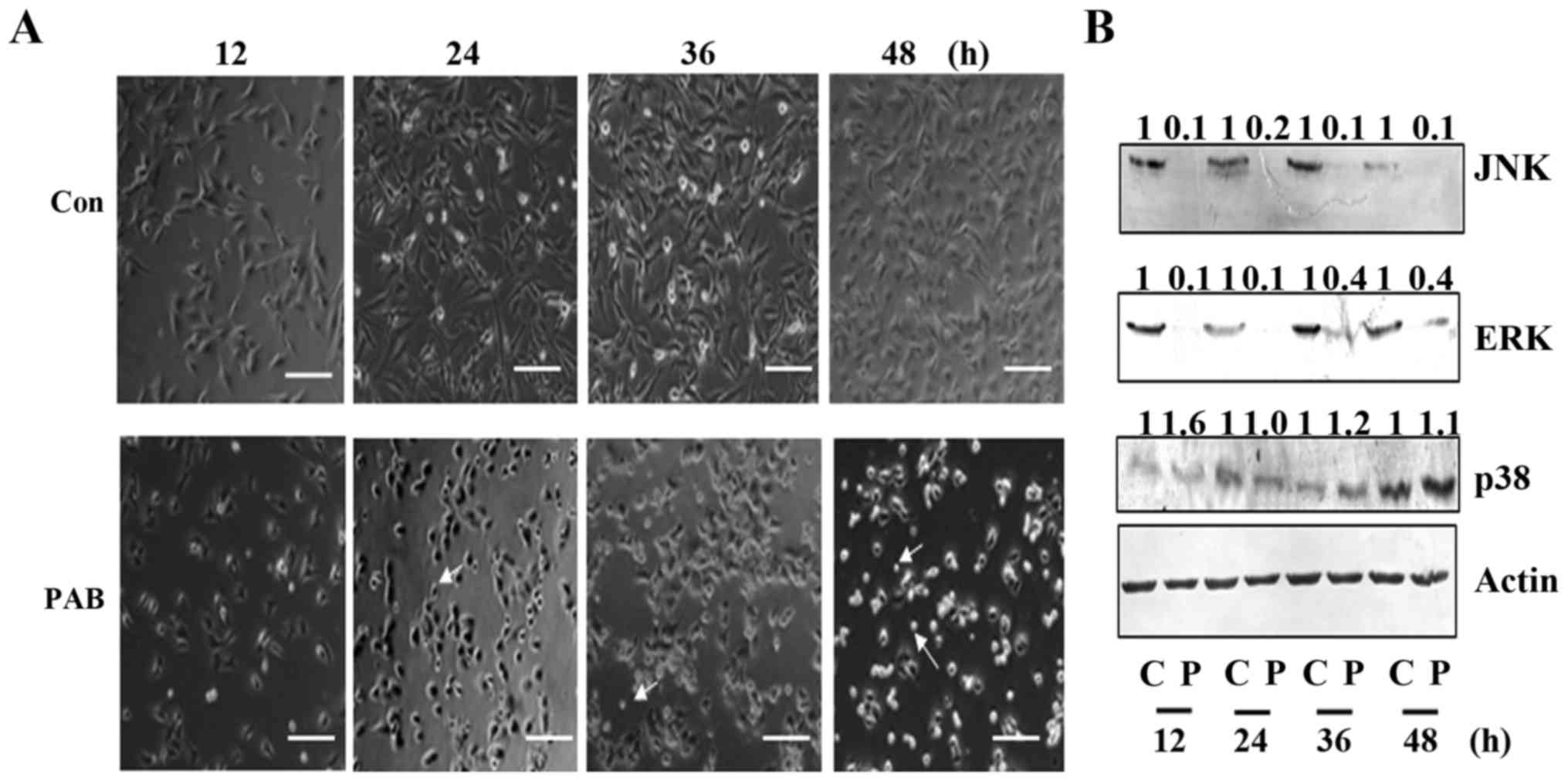
PAB induces cell death through MAPK
protein
To further confirm the mechanism underlying the
inhibition of cell proliferation by PAB, in the present study we
confirmed the effect of PAB on cell death. From morphologic
analysis, it was found that from 12 h, compared to the control
group, the cell number was decreased by PAB treatment. Meanwhile it
was found that in the PAB treatment group, the number of floating
cells was increased compared to the number noted in the control
group, indicating that PAB induced cell death (Fig. 6A). Then, we further analyzed the
mechanism of cell death, and it was confirmed that from 12 to 48 h,
the expression levels of JNK and ERK were decreased by PAB
treatment compared to these levels in the control treatment group,
while the expression of p38 was increased by PAB (Fig. 6B). Therefore, by regulating the
expression of MAPK, PAB induced cell death in the A172 cells.
PAB induces cell death through the DNA
damage response
Cell death and cell cycle arrest usually are
downstream of the DNA damage response (10). In the present study, we found that
PAB induced cell death and cell cycle arrest, and we aimed to
ascertain whether PAB induces a DNA damage response. Thus, the
expression of γH2AX, which is the marker of DNA damage response,
was determined. It was found that PAB treatment increased the
expression of γH2AX at 12, 24, 36 and 48 h compared to the
expression levels noted in the control treatment group (Fig. 7A). Then, we analyzed the effect of
caffeine on the inhibitory ratio of PAB, and it was found that at
24 h, caffeine treatment decreased the inhibitory ratio from
35.90±5.67 to 26.38±2.52%, and at 48 h caffeine treatment decreased
the inhibitory ratio from 44.12±3.56 to 21.79±1.89%. Therefore, PAB
activated the DNA damage response to induce apoptosis (Fig. 7B). Subsequently, caffeine was used
to inhibit the DNA damage response, and it was found that 2 mM
caffeine treatment inhibited cell death induced by PAB at 24 h and
48 h compared to that noted in the PAB treatment group (Fig. 7C).
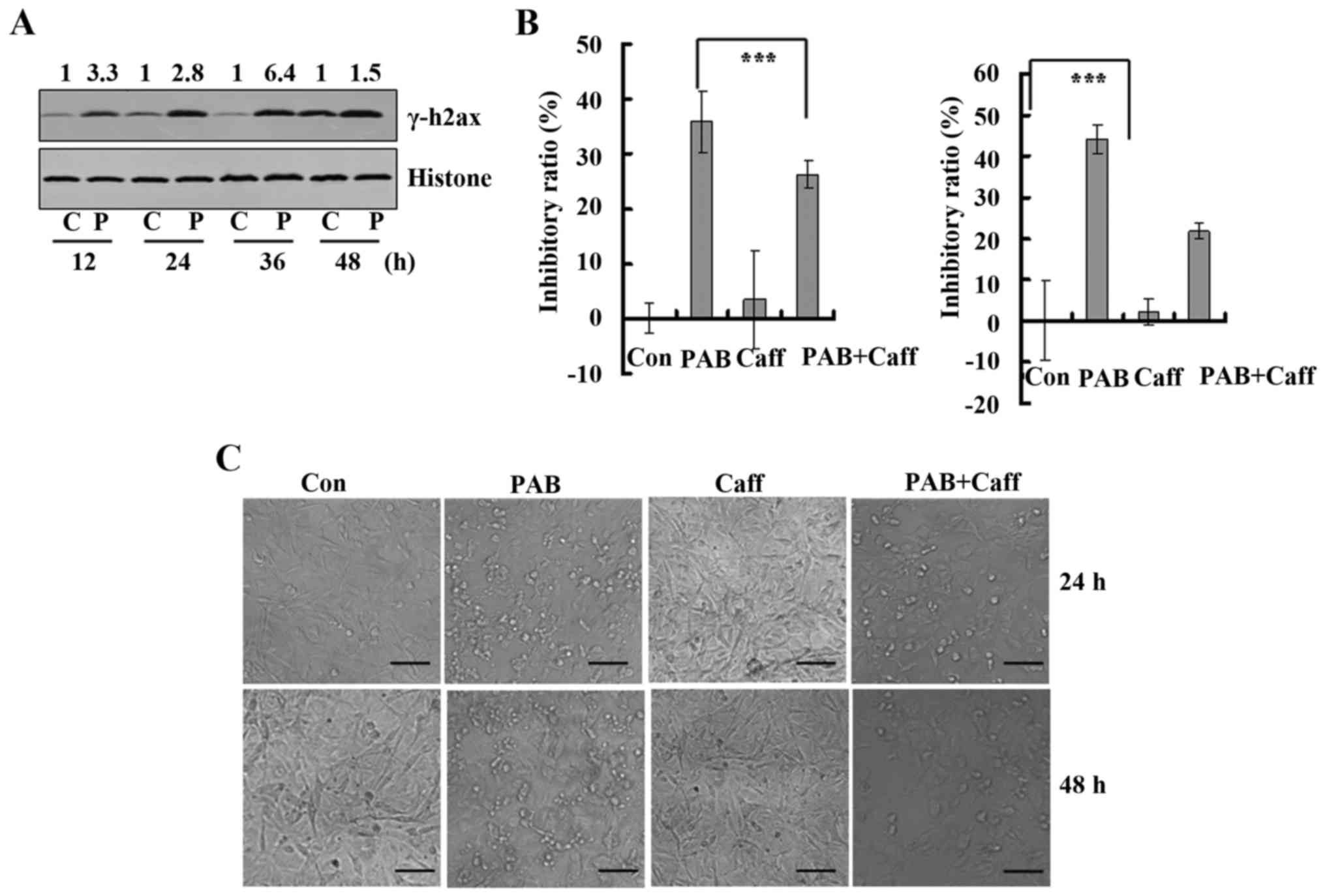 | Figure 7.Pseudolaric acid B (PAB) induces DNA
damage response for cell death. (A) The expression of γ-H2AX
protein after 0, 12, 24, 36 and 48 h of PAB treatment. Histone is
shown as a loading control. The number above each western blot band
is provided, which was standardized using histone and normalized to
1.0 in the control group. Data are representative of three
individual experiments (n=3). (B) The effect of caffeine, an
inhibitor of DNA damage response, on cell death induced by PAB in
A172 cells at 24 and 48 h as evaluated by MTT method. Mean ± SD,
n=3; ***P<0.001. (C) After 24 and 48 h of PAB and/or 2 mM
caffeine treatment morphological changes were visualized in A172
cells under phase contrast microscopy; Scale bar, 50 µm. C,
control; P, PAB; Caff, caffeine. |
Discussion
Glioblastoma is the most common cancer of the brain
and displays an increasing incidence. Despite major advances in the
field, there is no curative therapy for glioblastoma, to date
(18). Pseudolaric acid B (PAB), a
diterpene acid isolated from the root and trunk bark of
Pseudolarix kaempferi Gordon was found to exert antitumor
effect on different cell lines (1–6), and
in the present study we explored the role of PAB in
glioblastoma.
Firstly, we found that PAB inhibited glioblastoma
cell growth in a time- and dose-dependent manner, indicating that
PAB had potential anti-glioblastoma function. Similar to the role
of PAB in other cell lines (17,19,20),
PAB mainly affected microtubule aggregation in glioblastoma to
inhibit cancer cell growth. It has been reported that glioblastoma
cell migration and invasion occur at multiple stages of cancer
progression and are a clinical obstacle for therapy. Thus,
suppression of glioblastoma cell migration and invasion may provide
an effective therapeutic strategy (21). The present study confirmed that PAB
inhibited the migration of glioblastoma, which is another advantage
of PAB in the course of antitumor therapy.
Cell cycle arrest is an important mechanism by which
to inhibit cell growth. We found that from 12 h, PAB treatment
obviously increased the percentage of cells in the G2/M phase
compared to the percentage in the control treatment group,
indicating that PAB had the ability to induce cell cycle arrest to
inhibit cell growth. In addition, PAB treatment increased the
expression of cyclin B1/CDK1 complex expression, which was
consistent with the profile of G2/M arrest. Meanwhile, PAB
treatment increased the mRNA level of cyclin B1 at 12 h, indicating
that PAB affected cyclin B1 transcription to regulate protein
expression, and PAB did not affect CDK1 transcription, indicating
that PAB regulated protein expression through different mechanisms.
Meanwhile it was noted that at 36 h after PAB treatment, the
percentage of G0/G1 phase cells was increased compared to 24 h
after PAB treatment, and at 48 h after PAB treatment, the
percentage of S phase cells was increased compared to 36 h after
PAB treatment, indicating that at 36 h, cell cycle slippage into
G0/G1 occurred, and at 48 h cell cycle slippage into S occurred.
Furthermore, expression levels of cell cycle-related G0/G1 and S
phase proteins were observed, and it was found that at 36 h after
PAB treatment, the expression of cyclin D1 was increased compared
to that in the control treatment group, and at 48 h, the expression
of cyclin E and CDK2 was increased compared to levels in the
control treatment group, which was consistent with the results of
cell cycle slippage.
Cell death is another mechanism of inhibiting cell
growth. In the present study, based on morphologic analysis, we
found that PAB treatment promoted cell death with increasing time,
and PAB treatment inhibited the expression of JNK and ERK, and
increased the expression of p38 compared to levels in the control
treatment group. Therefore, PAB regulated corresponding protein
expression to induce cell death. Meanwhile DNA damage response is
commonly activated to promote cell death. We found that PAB
treatment increased the expression of γ-H2AX, which is the marker
of DNA damage response. Therefore, PAB treatment activated the DNA
damage response. In addition, caffeine was used by us to inhibit
the DNA damage response. We found that caffeine reversed the effect
of PAB to protect cells from cell death, and correspondingly to
decrease the inhibitory ratio; thus PAB activated the DNA damage
response to induce cell death, which provided a target to increase
the antitumor effect of PAB. In conclusion, PAB is a candidate for
anti-glioblastoma treatment.
Acknowledgements
The present study was supported by funding from the
National Natural Science Foundation of China (no. 81301416), the
Postdoctoral Science Foundation of China (nos. 2014M561302 and
2015T80299), the Norman Bethune Program of Jilin University (no.
2015202), the Jilin Provincial Science and Technology Department
(nos. 20140204004YY and 20160414025GH), and the Department of Human
Resources and Social Security of Jilin Province (no. 2016014).
References
|
1
|
Gong X, Wang M, Tashiro S, Onodera S and
Ikejima T: Involvement of JNK-initiated p53 accumulation and
phosphorylation of p53 in pseudolaric acid B induced cell death.
Exp Mol Med. 38:428–434. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gong XF, Wang MW, Tashiro S, Onodera S and
Ikejima T: Pseudolaric acid B induces apoptosis through p53 and
Bax/Bcl-2 pathways in human melanoma A375-S2 cells. Arch Pharm Res.
28:68–72. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pan DJ, Li ZL, Hu CQ, Chen K, Chang JJ and
Lee KH: The cytotoxic principles of Pseudolarix kaempferi:
Pseudolaric acid-A and -B and related derivatives. Planta Med.
56:383–385. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yu J, Li X, Tashiro S, Onodera S and
Ikejima T: Bcl-2 family proteins were involved in pseudolaric acid
B-induced autophagy in murine fibrosarcoma L929 cells. J Pharmacol
Sci. 107:295–302. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yu JH, Cui Q, Jiang YY, Yang W, Tashiro S,
Onodera S and Ikejima T: Pseudolaric acid B induces apoptosis,
senescence, and mitotic arrest in human breast cancer MCF-7. Acta
Pharmacol Sin. 28:1975–1983. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yu JH, Wang HJ, Li XR, Tashiro S, Onodera
S and Ikejima T: Protein tyrosine kinase, JNK, and ERK involvement
in pseudolaric acid B-induced apoptosis of human breast cancer
MCF-7 cells. Acta Pharmacol Sin. 29:1069–1076. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shakur SF, Bit-Ivan E, Watkin WG, Merrell
RT and Farhat HI: Multifocal and multicentric glioblastoma with
leptomeningeal gliomatosis: A case report and review of the
literature. Case Rep Med. 2013:1326792013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Walker C, Baborie A, Crooks D, Wilkins S
and Jenkinson MD: Biology, genetics and imaging of glial cell
tumours. Br J Radiol. 84:S90–S106. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Parsons DW, Jones S, Zhang X, Lin JC,
Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et
al: An integrated genomic analysis of human glioblastoma
multiforme. Science. 321:1807–1812. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lozano G and Elledge SJ: p53 sends
nucleotides to repair DNA. Nature. 404:24–25. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li FF, Yi S, Wen L, He J, Yang LJ, Zhao J,
Zhang BP, Cui GH and Chen Y: Oridonin induces NPM mutant protein
translocation and apoptosis in NPM1c+ acute myeloid leukemia cells
in vitro. Acta Pharmacol Sin. 35:806–813. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Qi M, Yao G, Fan S, Cheng W, Tashiro S,
Onodera S and Ikejima T: Pseudolaric acid B induces mitotic
catastrophe followed by apoptotic cell death in murine fibrosarcoma
L929 cells. Eur J Pharmacol. 683:16–26. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Klekotka PA, Santoro SA, Wang H and Zutter
MM: Specific residues within the alpha 2 integrin subunit
cytoplasmic domain regulate migration and cell cycle progression
via distinct MAPK pathways. J Biol Chem. 276:32353–32361. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yoon S and Seger R: The extracellular
signal-regulated kinase: Multiple substrates regulate diverse
cellular functions. Growth Factors. 24:21–44. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hartwell LH and Weinert TA: Checkpoints:
Controls that ensure the order of cell cycle events. Science.
246:629–634. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu J, Ren P, Zhong T, Wang Y, Yan M, Xue
B, Li R, Dai C, Liu C, Chen G, et al: Pseudolaric acid B inhibits
proliferation in SW579 human thyroid squamous cell carcinoma. Mol
Med Rep. 12:7195–7202. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu X, Chong Y, Tu Y, Liu N, Yue C, Qi Z,
Liu H, Yao Y, Liu H, Gao S, et al: CRM1/XPO1 is associated with
clinical outcome in glioma and represents a therapeutic target by
perturbing multiple core pathways. J Hematol Oncol. 9:1082016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tong YG, Zhang XW, Geng MY, Yue JM, Xin
XL, Tian F, Shen X, Tong LJ, Li MH, Zhang C, et al: Pseudolarix
acid B, a new tubulin-binding agent, inhibits angiogenesis by
interacting with a novel binding site on tubulin. Mol Pharmacol.
69:1226–1233. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wong VK, Chiu P, Chung SS, Chow LM, Zhao
YZ, Yang BB and Ko BC: Pseudolaric acid B, a novel
microtubule-destabilizing agent that circumvents multidrug
resistance phenotype and exhibits antitumor activity in vivo. Clin
Cancer Res. 11:6002–6011. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu B, Dong H, Lin X, Yang X, Yue X, Yang
J, Li Y, Wu L, Zhu X, Zhang S, et al: RND3 promotes Snail 1 protein
degradation and inhibits glioblastoma cell migration and invasion.
Oncotarget. 7:82411–82423. 2016.PubMed/NCBI
|