Introduction
Chaetocin, a natural small-molecule product produced
by Chaetomium fungal species (1), has repeatedly been reported to be a
promising anticancer agent over the last several years. It has been
reported that chaetocin is an inhibitor of lysine-specific histone
methyltransferases (HMTs), which are the key enzymes that mediate
epigenetic control of gene expression. Chaetocin is also an
inhibitor of the redox enzyme thioredoxin reductase, as it competes
with thioredoxin for binding to thioredoxin reductase, and thus
induces cellular oxidative stress which can eradicate tumor cells
(2). Chaetocin has been shown to
inhibit the viability of various types of cancer, including
melanoma, ovarian and non-small cell lung cancer (3). However, the effects of chaetocin on
human intrahepatic cholangiocarcinoma (ICC) and the related
mechanisms have not yet been reported.
Human cholangiocarcinoma is an epithelial cell
malignancy arising from varying locations within the biliary
system, and is the most common primary malignancy of the biliary
tract (4). It can be classified
into two major categories: ICC and ductal cholangiocarcinoma. ICC
has received increased attention as various studies have shown
marked increases in the morbidity and mortality rates of ICC in
recent years (5). ICC is
characterized by insidious development, late onset of symptoms,
high recurrence rates after surgical resection, and limited
treatment options for the vast majority of patients. Moreover, many
ICC cases are resistant to traditional chemotherapeutics due to the
desmoplastic character of the cancer, the complex tumor
microenvironment and rich genetic heterogeneity (6,7); this
creates further challenges for clinical treatment. Therefore,
effective therapeutic strategies for the treatment of ICC with
minimal side-effects are urgently required.
Induction of apoptosis is considered to be one of
the most effective antitumor strategies. Apoptosis is a type of
cell suicide that is regulated by a series of complex signaling
pathways. Intrinsic or external stimuli induce apoptosis. Oxidative
stress, characterized by high concentrations of intracellular
reactive oxygen species (ROS), is reported to be one of the
intrinsic inducers of apoptosis (8,9).
ROS, which are mainly produced in the mitochondria,
have been reported to induce DNA sequence changes (rearrangements,
deletions, mutations and gene amplifications) and cell apoptosis
(10,11). ROS arrest the cell cycle and
activate different apoptotic pathways, including the apoptosis
signal-regulating kinase (ASK)/c-Jun N-terminal kinase (JNK)
pathways (12–14). Previous findings have revealed that
chaetocin increases the level of ROS and induces cell apoptosis
(15). ROS, which can be produced
in response to various types of cytotoxic stressors, activate ASK-1
directly and, thus, activate JNKs downstream (16). Activated JNKs then directly or
indirectly activate apoptotic signaling pathways, and this
ultimately results in cell apoptosis (17). In the present study, the effect of
chaetocin on human ICC and the associated mechanisms were
investigated.
Materials and methods
Cell culture and reagents
The human ICC cell lines TFK-1 and CCLP-1 (acquired
from the University of Pittsburgh, Pittsburgh, PA, USA) and RBE and
SSP-25 (obtained from Piken University, Japan) were cultured in
RPMI-1640 medium (Gibco, Grand Island, NY, USA) containing 10%
fetal bovine serum (FBS). A normal human intrahepatic bile duct
cell line HIBEC (ScienCell Research Laboratories, San Diego, CA,
USA) was cultured in the same way as mentioned above. HuCCT1 cells
were cultured in Dulbeccos modified Eagles medium (DMEM) (HyClone,
Logan, UT, USA) containing 10% FBS. All cell lines were
supplemented with 100 U/ml of penicillin and 100 µg/ml of
streptomycin and cultured at 37°C with 5% CO2. The cells
were split every 4 days and some of the logarithmically growing
cells were used for all experiments as described below. Chaetocin
(11076 no. 13156; Cayman Chemical Co., Ann Arbor, MI, USA),
SP600125 (no. s1460; Selleck Chemicals, Houston, TX, USA) and
N-acetyl-L-cysteine (NAC) (no. 194603; MP Biomedicals,
Solon, OH, USA) were dissolved in dimethyl sulfoxide for usage.
Cell viability analysis
Cell viability was determined by Cell Counting Kit-8
(CCK-8; Dojindo Laboratories, Kumamoto, Japan). In brief, cells
were digested and cultured in 96-well plates (5×103
cells/well) for 24 h. Then, the cells were placed in fresh medium
(10% fetal) with different concentrations of chaetocin (0, 50, 100,
150 and 200 nM) for 24, 48 and 72 h, respectively. After incubation
for the above times, we replaced each well with CCK-8 at a final
concentration of 10% to co-culture for another 1 h. Cell viability
was determined by its optical density (OD) measured at 450 nm of
absorbance using a microplate reader (Mithras LB 940; Berthold
Technologies, Bad Wildbad, Germany). In some experiments, NAC (5
mM) or SP600125 (50 nM) was used for pretreatment for 1 h before
incubation with chaetocin. Cell viability = (trial group OD - blank
group OD)/(control group OD - blank group OD) × 100%.
Transwell invasion assay
Cell invasion potential was determined using a
Transwell chamber (NY14831; Corning Inc., Corning, New York, NY,
USA). CCLP-1 cells (5×104) in serum-free RPMI-1640
medium with different concentrations of chaetocin (0, 100 or 200
nM) were cultured in the upper chamber, which was coated with
Matrigel (356234; Corning Inc.). The bottom chambers contained 600
µl medium with 10% FBS. Following 24 h of incubation at 37°C with
5% CO2, the cells were fixed using 4% paraformaldehyde
and stained with crystal violet at room temperature for 30 min. The
cells that invaded to the bottom side of the membrane were
photographed at ×100 with a microscope (IX71; Olympus, Tokyo,
Japan). After that, the bottom membrane with crystal violet was
eluted with 33% acetic acid for 30 min and the OD of the acetic
acid was determined at an absorbance rate of 570 nm using a
microplate reader.
Wright-Giemsa staining
On 6-well plates, 1.0×105 cells/well were
seeded and incubated as described above. Solutions of chaetocin (0,
100 and 200 nM) with 10% serum-medium were added to each well, and
then incubated for another 48 h. After being fixed with methanol
and washed with phosphate-buffered saline (PBS), the cells, stained
with Giemsa staining (Xiangya, China), were observed and
photographed using an optical inverted microscope at ×200 (IX71;
Olympus, Tokyo, Japan).
Flow cytometry
The CCLP-1 cells (15×104/well) were
cultured in a 6-well plate as described above. After co-culturing
with chaetocin for 48 h, the cells were harvested, and then treated
using an Annexin V-FITC apoptosis detection kit and propidium
iodide (PI) (BioLegend, Inc., San Diego, CA, USA) according to the
manufacturers protocol. For cell cycle analysis, the cells were
cultured with chaetocin for 24 and 48 h, and then fixed with
alcohol at 4°C. After 12 h of fixation, the cells were stained with
PI (GBC BIO™ Technologies, Guangzhou, China). Finally, stained
cells were analyzed using flow cytometry with FACSCalibur
(Becton-Dickinson, Franklin Lakes, NJ, USA) and FlowJo 7.6.1
software.
Intracellular ROS measurement
The intracellular ROS level was determined using an
ROS detection kit (KeyGen Biotech Co., Ltd., Nanjing, China) using
2′-7′-dichlorodihydrofluorescein diacetate (DCFH-DA). In brief,
after culturing with chaetocin for 24 h in 6 well-plates, the
CCLP-1 cells were washed twice using PBS, and then stained with
DCFH-DA in the dark for 30 min. Cells were then washed and
resuspended in PBS to detect ROS accumulation by flow cytometry
with FACSCalibur and analyzed using FlowJo 7.6.1 software.
Western blot analysis
The CCLP-1 cells were treated with different
concentrations of chaetocin for 48 h in the presence or absence of
SP600125 or NAC. The cells were harvested, and the total proteins
were extracted using the protein extraction kit (KeyGen Biotech
Co., Ltd.). The proteins were separated on SDS-polyacrylamide gels
and transferred to polyvinylidene fluoride membranes (Millipore,
Darmstadt, Germany). Antibodies including phospho-ASK-1,
phospho-JNK (Cell Signaling Technology, Beverly, MA, USA),
phospho-p53, caspase-3, Bcl-2, GAPDH and α-tubulin (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) were used to detected
protein. After being washed, the membranes were incubated with
homologous secondary antibody for 1 h. The signals were detected
with chemiluminescent substrate and photographed using
chemiluminescence immunoassay (Tanon 5200; Tanon Science &
Technology Co., Ltd., Shanghai, China).
In vivo experiment
Twelve nude mice were purchased from the Institute
of Laboratory Animal Sciences (Southern Medical University) and
used for the xenograft model. The mice were housed in controlled
conditions of temperature and humidity with a 12 h light/dark
cycle. The experiment was initiated with 6 week-old mice weighing
20–25 g. CCLP-1 cell suspension in PBS (2×106 cells/ml)
was subcutaneously injected into the right flanks of the nude mice.
When the mouse tumors reached 3–8 mm in size, the experiment was
initiated as the 1st day. Chaetocin was formulated in 3%
physiological saline. The nude mice were then randomly divided into
2 groups and were intraperitoneally injected either with chaetocin
(0.3 mg/kg body weight) or vehicle once every three days for 6
times and terminated on the 18th day. The tumor dimensions were
measured using Vernier calipers once every three days for the
entire life span of the mice. Tumor volumes were calculated using
the formula: a2x b/2 (a is the
width and b is the length of the tumor in mm). The mice were
sacrificed on the 30th day and the tumor mass from each mouse was
dissected and weighed. The experimental mice were treated according
to the standards supported by the Animal Protection Committee of
Southern Medical University.
Statistical analysis
As specified, every experiment was performed at
least three times in triplicate, and the results are presented as
means ± standard errors (SDs). Statistical analysis was performed
by one-way ANOVA or by Student's t-test with SPSS version 18.0
(SPSS, Inc., Chicago, IL, US), and the results were considered
statistically significant at P<0.05.
Results
Chaetocin reduces the viability and
invasive ability of the ICC cells
In order to investigate the effects of chaetocin on
ICC, the viability of different human ICC cell lines was analyzed
using a CCK-8 kit, and the invasive ability of the CCLP-1 cells was
determined using a Transwell invasion assay. As shown in Fig. 1A-C, chaetocin reduced the viability
of all ICC cell lines in a dose- and time-dependent manner. A
significant reduction in cell viability was observed when cells
were treated with 100 nM chaetocin for 48 h. In addition, the
viability of the normal human intrahepatic bile duct HIBEC cell
line was reduced in a concentration- and time-dependent manner, but
HIBEC sensitivity to chaetocin was lower than that of the cancer
cell lines.
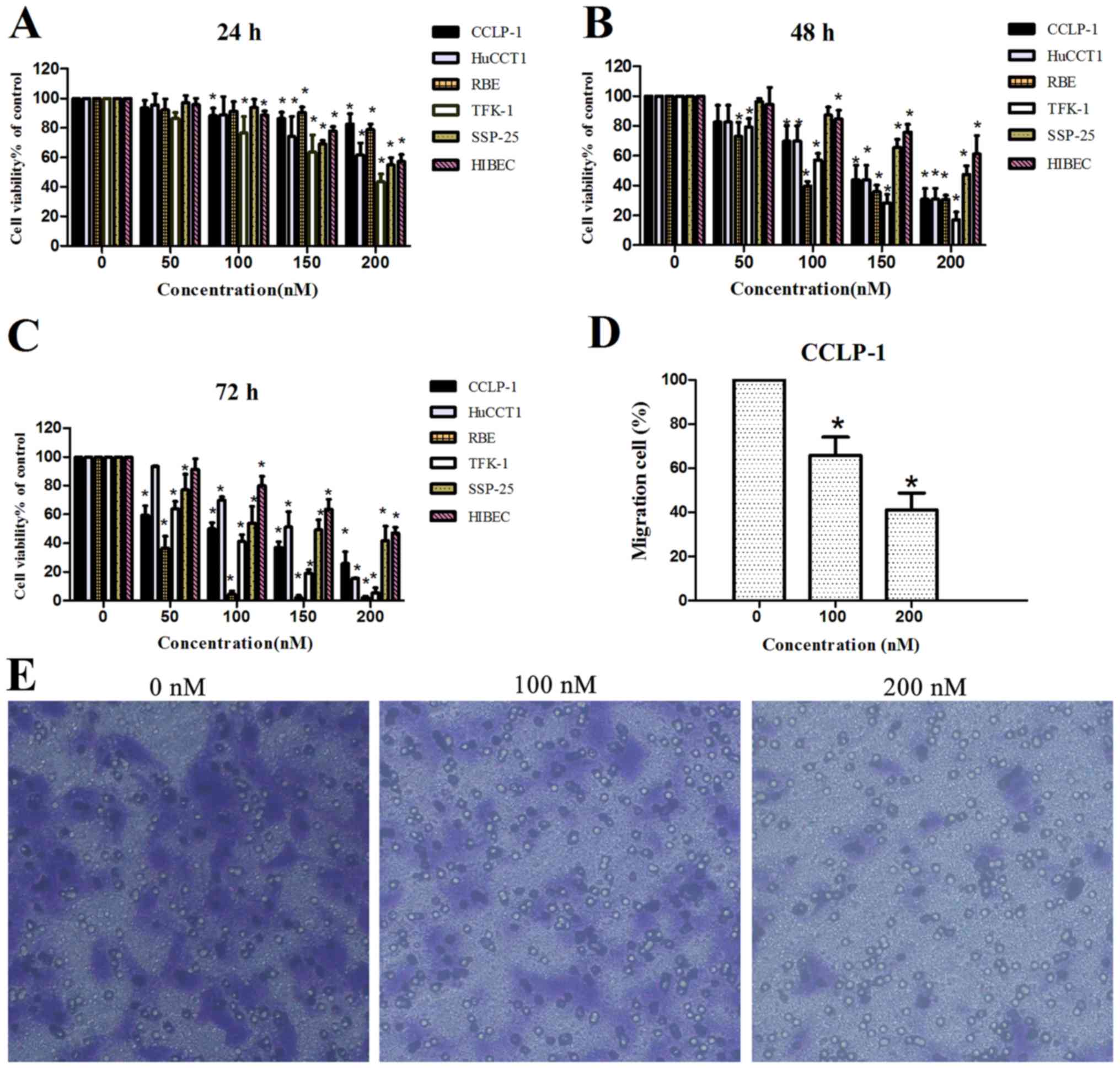 | Figure 1.Effects of chaetocin on cell
viability and invasion. (A-C) Different ICC cell lines CCLP-1,
HuCCT1, RBE, TFK-1 and SSP-25, and normal human intrahepatic bile
duct cell line HIBEC were treated with chaetocin at various
concentrations (0, 50, 100, 150 and 200 nM) for 24, 48 and 72 h,
and then cell viability was detected using CCK-8; *P<0.05
compared with the control group. (D and E) CCLP-1 cells were
treated with various concentrations of chaetocin (0, 100 and 200
nM) for 24 h. The cells that invaded to the bottom side of the
membrane were observed and photographed at a magnification of ×100
via a contrast microscope. Then, the bottom membrane was dyed with
crystal violet and eluted with acetic acid and the OD of the acetic
acid was detected by a microplate reader; *P<0.05 compared with
the control. |
Additionally, the invasive ability of the CCLP-1
cell line was reduced by chaetocin. Considering that the reduced
ICC viability by chaetocin was significant at 48 h, we decided to
observe the results of the Transwell assay after culturing cells
with chaetocin for 24 h. Under microscopic observation
(magnification, ×100), the number of cells that invaded through the
membrane in the control group was markedly higher than that in the
chaetocin-treated group. Quantification by OD detection confirmed
the distinction between the control group and chaetocin-treated
group, indicating that chaetocin inhibited the invasion of CCLP-1
cells (Fig. 1D and E).
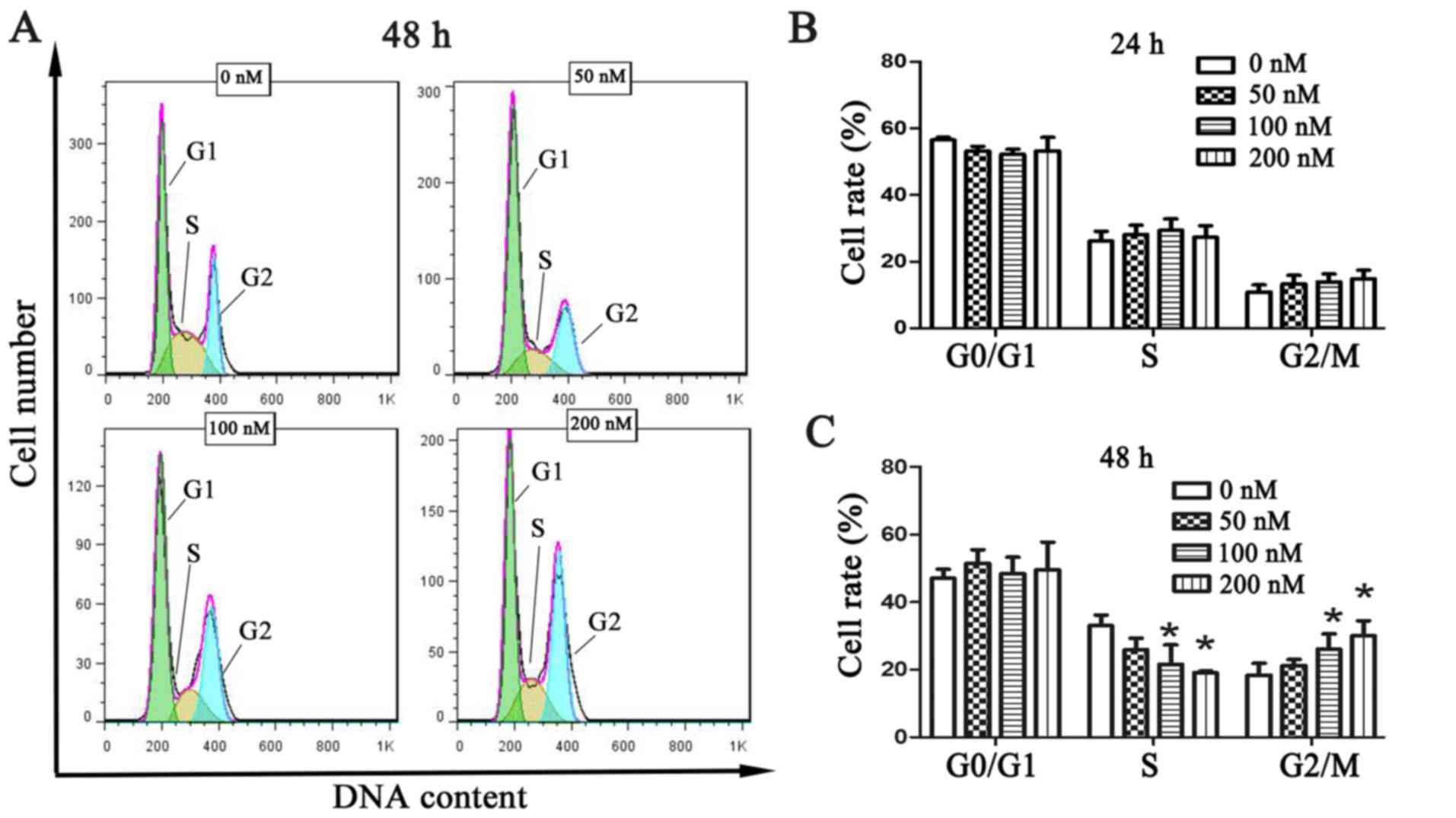
Chaetocin causes cell cycle arrest in
the G2/M phase
To investigate the effect of chaetocin on cell cycle
distribution, the cell cycle of CCLP-1 cells was analyzed by flow
cytometry, using the PI staining method, following treatment with
chaetocin. The results showed that every phase had no statistical
difference at 24 h (Fig. 2B). Yet,
the number of cells in the S phase was significantly decreased and
that in the G2/M phase was increased following chaetocin treatment
at 48 h. The changes were statistically significant. These results
indicated that chaetocin caused cell cycle arrest in the G2/M
phase, inhibiting proper DNA replication (Fig. 2A and C).
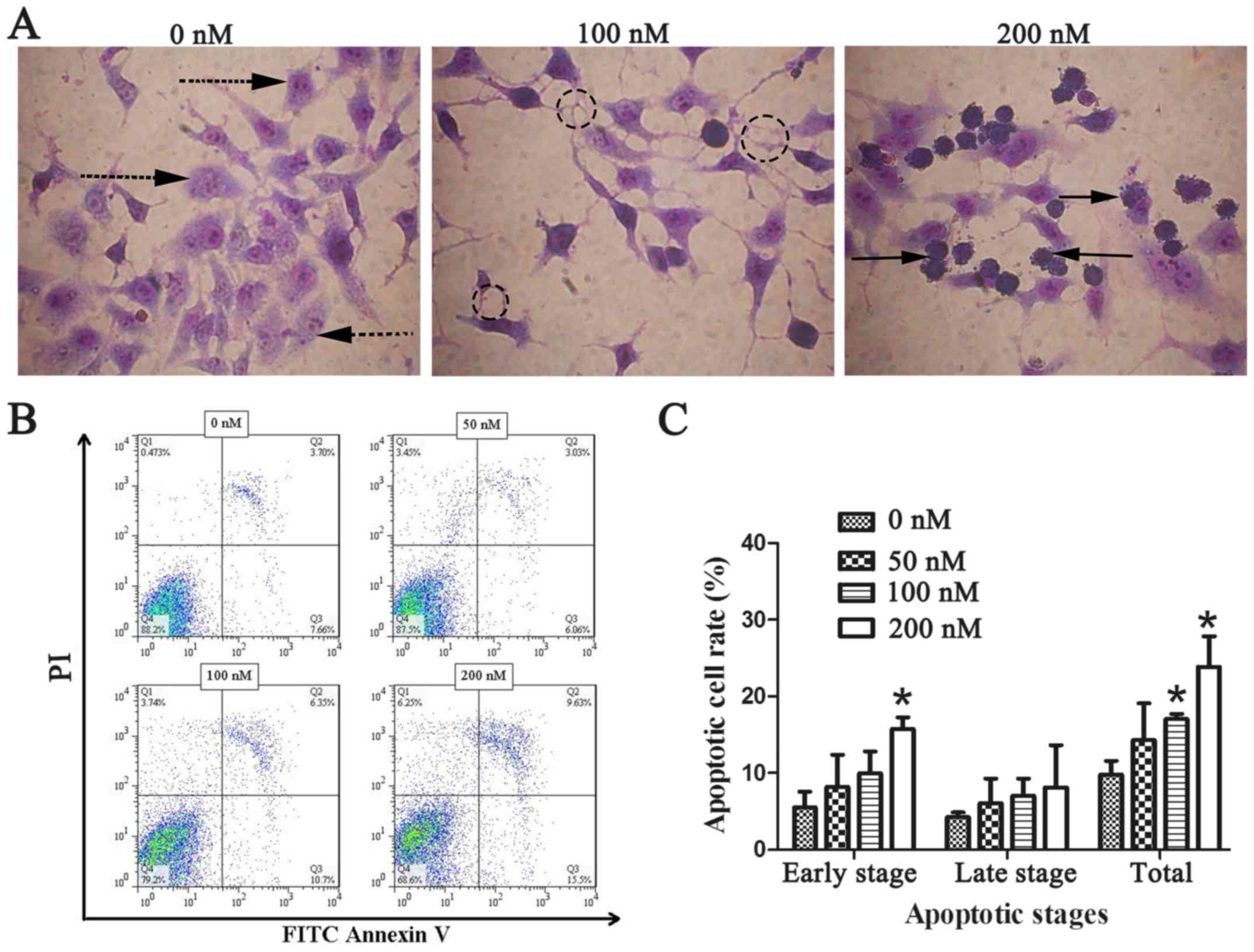
Chaetocin induces CCLP-1 cell
apoptosis
As the previous experiments revealed, chaetocin
exerted an inhibitory effect on ICC cell viability and invasion. We
applied Wright-Giemsa staining and flow cytometry was performed to
examine whether chaetocin induces CCLP-1 cell apoptosis. Under
optical microscopic observation, control group CCLP-1 cells were
plump and densely populated in the visual field. However, the cells
treated with chaetocin were shriveled and flat with reduced numbers
in each visual field. In order to clearly observe the cell
morphology, Giemsa was used to stain the cells. Compared with the
normal morphology of the control cells, the cells of the
chaetocin-treated group exhibited morphological characteristics of
apoptosis, including nuclear pyknosis, sublobe, fragmented shapes,
fringe collection and apoptotic body formation (Fig. 3A). The number of apoptotic bodies
was observably increased with the increasing concentration of
chaetocin. To further ascertain the effects of chaetocin on the
apoptosis of the CCLP-1 cells, apoptosis was detected using Annexin
V-FITC and PI staining. The results (Fig. 3B and C) revealed that the rate of
cell apoptosis was increased in a dose-dependent manner, which was
more evident at the early apoptotic stage.
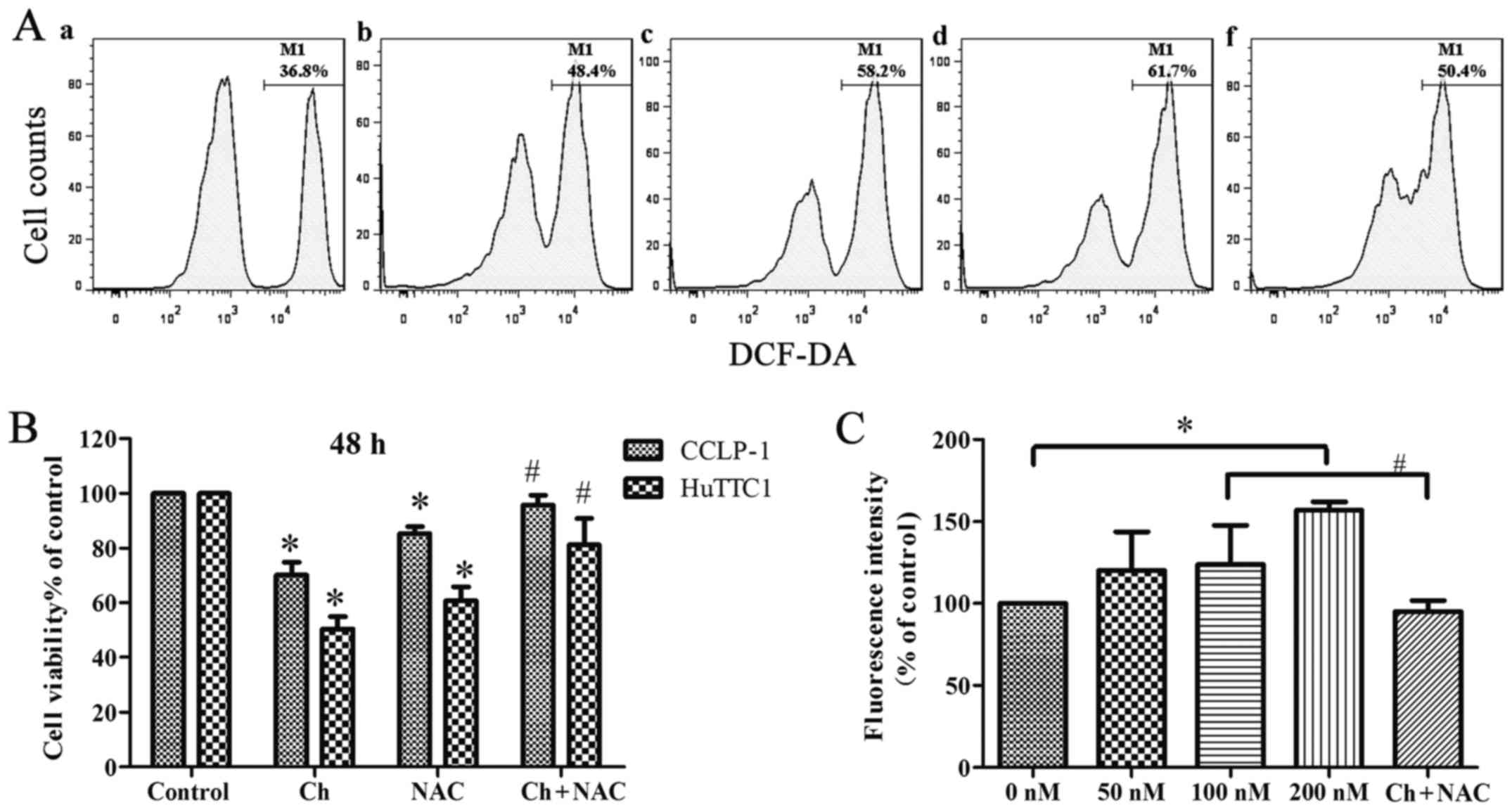
Chaetocin induces oxidative stress in
the CCLP-1 cells
The intracellular ROS generation in CCLP-1 cells was
measured using DCFH-DA. The flow cytometric analysis showed that
the chaetocin-treated cells (at high concentrations) had
significantly higher levels of ROS than the levels noted in the
control cells. However, when cells were cultured with NAC and
chaetocin (100 nM), the intracellular ROS level was less than that
noted in the chaetocin-treated (100 nM) group (Fig. 4A and C). In addition, a low
concentration of chaetocin did not have a significant effect on the
ROS level in the cells, which may be the result of the short
incubation time. The results suggest that chaetocin promotes the
generation of intracellular ROS, leading to oxidative stress.
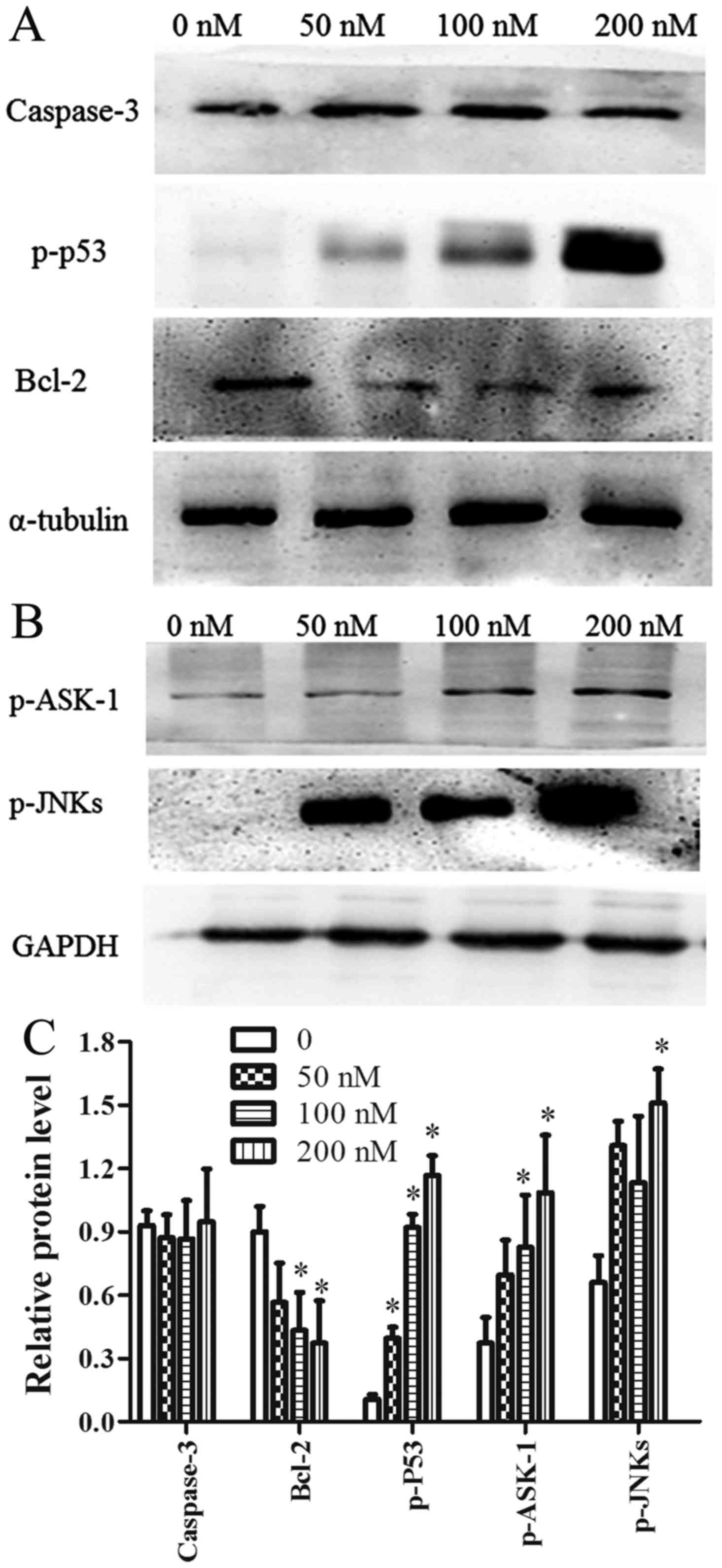
The ASK-1/JNK pathway is involved in
CCLP-1 cell apoptosis
ASK-1 is a member of the mitogen-activated protein
kinase (MAPK) family that can be activated by oxidative stress.
Activated ASK-1 has been reported to activate JNK proteins via
phosphorylation. Therefore, the expression of ASK-1/JNK was
determined using western blot analysis. The results showed that
chaetocin activated ASK-1 and its downstream proteins JNK and p53
in a dose-dependent manner. In addition, the expression level of
Bcl-2 was downregulated in a dose-dependent manner. By contrast,
following chaetocin treatment, the level of caspase-3 exhibited no
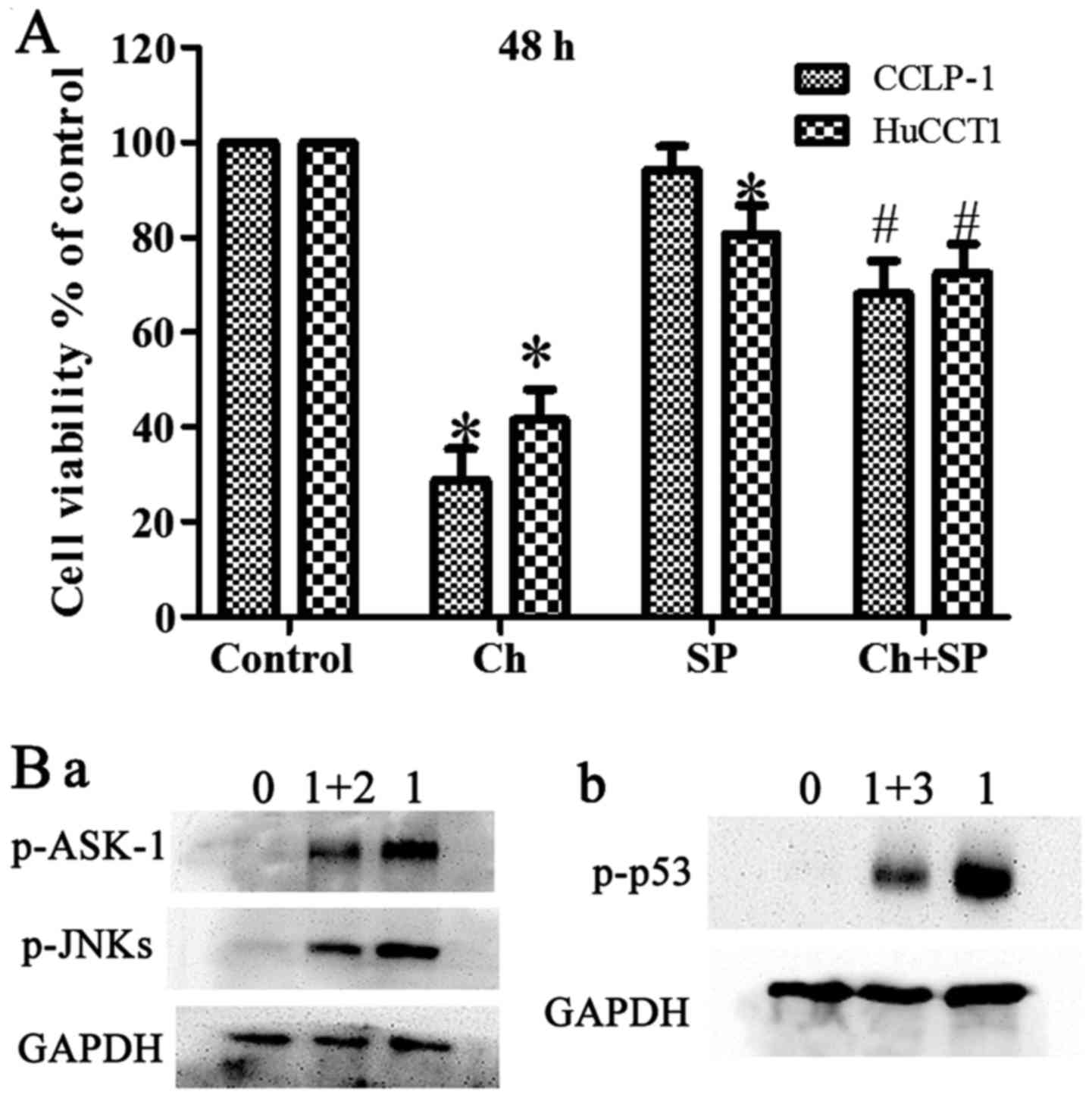
obvious change compared with the control (Fig. 5A-C). Furthermore, pretreatment with
NAC suppressed the chaetocin-induced activation of ASK-1/JNK, which
indicated that ROS have a vital role in the chaetocin-induced
activation of these proteins (Fig.
6B-a). In another experiment, pretreatment with SP600125 (a JNK
inhibitor) attenuated the chaetocin-induced expression of p53
(Fig. 6B-b).
Chaetocin inhibits the growth of
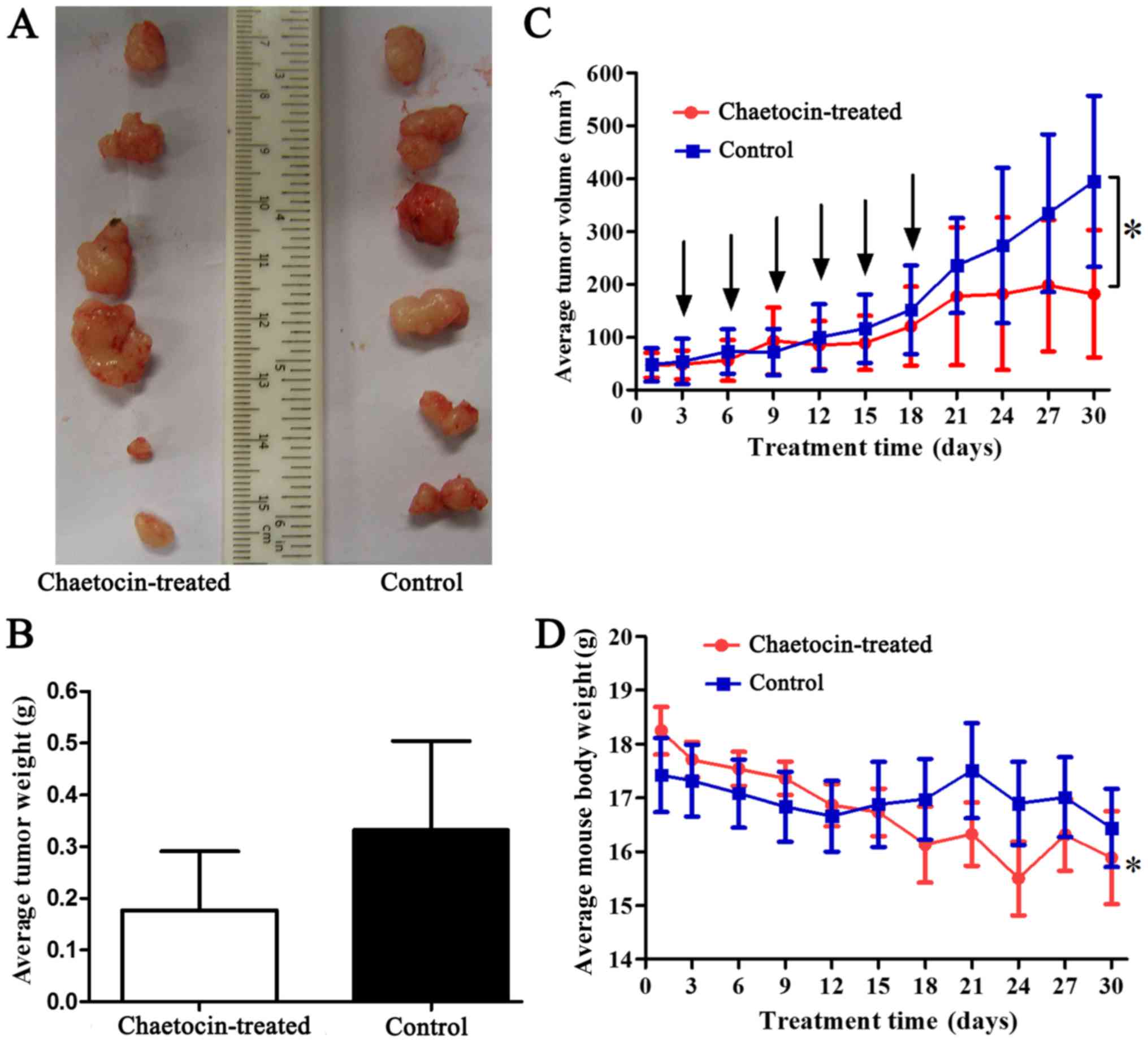
CCLP-1 xenograft tumors in vivo
To detect the antitumor activity of chaetocin in
vivo, human CCLP-1 cholangiocarcinoma xenografts were
established. The results (Fig.
7A-C) showed that the xenografts of the control group grew
rapidly, but growth was reduced by chaetocin treatment in
vivo. Additionally, the average weight of the tumors in the
control group was higher than that of the chaetocin-treated group.
However, there was no statistically significant difference in tumor
weight between the two groups. The difference in tumor weight may
have increased if the duration of the experiment had been extended
(Fig. 7C). Furthermore, at the time
of sacrifice, the average tumor volume of the control group was
significantly higher than that of the chaetocin-treated group.
Therefore, the results indicated that chaetocin inhibited tumor
cell proliferation, although complete regression of the tumor was
not observed. Additionally, the average body weight of the
chaetocin-treated mice was significantly higher at the beginning of
the experiment compared with the weight of the mice at sacrifice.
However, this difference was not observed in the control group
(Fig. 7D).
Discussion
ICC is a treatment-resistant primary liver cancer
with increasing incidence and mortality rates observed worldwide in
recent years (18). For the
majority of patients with advanced ICC, there is no effective or
standard first-line chemotherapy (19). Therefore, it is urgent to identify
effective drugs to treat ICC that have minimal side-effects. In our
previous preliminary drug screening trials, chaetocin was
identified to effectively reduce the viability of RBE cell lines at
low doses (20), indicating that
chaetocin may effectively inhibit the growth of cancer cells with
few side-effects. In vitro experiments in the present study
confirmed that chaetocin reduced the viability of ICC cell lines in
a dose- and time-dependent manner (Fig.
1A-C). In addition, the Transwell chamber assay demonstrated
that chaetocin reduced the invasion of CCLP-1 cells. We
hypothesized that chaetocin may have the same effect on other ICC
cell lines. Our in vivo xenograft tumor model results
(Fig. 7A-C) also confirmed that
chaetocin inhibited ICC tumor growth in mice. The in vitro
and in vivo experiments clearly showed that chaetocin
reduced ICC cell proliferation, but also reduced the viability of
HIBECs, a normal human intrahepatic bile duct cell line, although
HIBEC cells were less sensitive to chaetocin than the cancer cell
lines. In vivo, the bdt weight of the mice tended to be
decreased following chaetocin treatment. The average body weight
change between the early and late stages of the experiment was
statistically significant, whereas the body weights of the control
group were unchanged (Fig. 7D).
Considering these results, the reduced body weight in the
chaetocin-treated group may represent a side-effect of chaetocin
treatment, which should be noted with due attention. Thus, it is
necessary to study the molecular mechanisms that mediate the
effects of chaetocin and identify drugs that could be used in
combination with chaetocin to potentially reduce the required dose
of chaetocin and lessen the associated side-effects.
Cell cycle arrest is a major target of tumor therapy
(21). The uncontrolled
proliferation of tumor cells is due to overexpression of cyclins or
the inactivation of critical cyclin-dependent kinases, which makes
tumor cells unable to stop at predetermined points of the cell
cycle (22,23). This means that arrest of the cell
cycle can inhibit cancer cell proliferation. In the present study,
the results showed that every phase in the CCLP-1 cell cycle had no
change at 24 h. The results at 48 h showed that the percentage of
CCLP-1 cells in the S phase was decreased and that the percentage
of cells in the G2/M phase was significantly increased. This
indicates that chaetocin was able to arrest the cell cycle in the
G2/M phase and decrease DNA replication to inhibit CCLP-1 cell
proliferation. Oxidative stress affects the cell cycle by affecting
the expression of cyclins (24,25).
Considering that the ROS level was not influenced under a low
concentration of chaetocin (Fig.
4A), every phase of the cell cycle of the CCLP-1 cells may not
have been influenced at 24 h. This assumption requires validation
in further experiments.
Apoptosis, a fundamental process essential for the
development and maintenance of tissue homeostasis, is also a major
mechanism used to kill cancer cells (26). Inducing apoptosis is now considered
as one of the most effective strategies for cancer treatment. In
the present study, flow cytometry and observed morphological
changes preliminarily indicated that chaetocin induced the
apoptosis of CCLP-1 cells (Fig.
3A-C). Additionally, expression of p53 (an executor of
apoptosis) was increased by chaetocin (Fig. 5A). This suggests that apoptosis is
one of the mechanisms influenced by chaetocin resulting in reduced
ICC cell viability.
Considering that pretreatment with NAC partially
abrogated the effect of chaetocin on the viability of CCLP-1 cells
(Fig. 4B), we aimed to determine
whether oxidative stress underlied the chaetocin-induced apoptosis
in CCLP-1 cells. ROS, an indicator of oxidative stress, are
produced during normal cellular processes and are present in normal
and cancer cells. At certain concentrations, ROS are required as
critical signaling molecules involved in cell survival and
proliferation (27). However,
oxidative stress occurs when excessive ROS levels overwhelm the
cellular antioxidant system, either through an increase in ROS
concentration or a decrease in the cellular antioxidant capacity.
Oxidative stress induces cell apoptosis and DNA damage (28,29).
The present study showed that the ROS level was higher in the
chaetocin-treated group than that in the control group. These
results indicate that chaetocin can increase the ROS level and
thereby induce oxidative stress in the CCLP-1 cells.
Oxidative stress is an initial signal that can
induce cell apoptosis (30). ASK-1
is one of the proteins most sensitive to oxidative stress. It is
well known that various types of cytotoxic stressors activate ASK-1
by producing excessive ROS, and thus induce apoptosis (17). Under normal conditions, ASK-1 is
inactivated via binding with thioredoxin. ROS can oxidize
thioredoxin and dissociate it from ASK-1. Therefore, when oxidative
stress occurs, ASK-1 becomes activated via dissociation from
thioredoxin and oligomerization into the ASK-1 complex (31,32).
The ASK-1 complex phosphorylates itself and induces the activation
of JNKs (12,33). JNKs also participate in the
regulation of various cellular processes, including cell survival,
proliferation, differentiation and cell death (34). A previous study demonstrated that
chaetocin inhibited energy production and glucose metabolism in
glioma cells in an ROS-JNK-dependent manner (35). Additionally, JNKs are widely
reported to have a close association with ASK-1; therefore, we
hypothesized that, as JNKs are downstream of ASK-1, they may be
involved in the activation of cell apoptosis (36). The potential role of JNKs in
chaetocin-induced apoptosis was investigated. As expected, a CCK-8
assay (Fig. 6A) showed that
SP600125 (a JNK inhibitor) partially abrogated the effect of
chaetocin on ICC cells, and western blotting showed that the
chaetocin-induced expression of p53 (a tumor-suppressor gene) was
reduced following pretreatment with SP600125 (Fig. 6B-b). Furthermore, in our
experiments, the expression levels of phosphorylated ASK-1 and JNKs
were increased by chaetocin treatment (Fig. 5A) and decreased by co-treatment with
chaetocin and NAC (Fig. 6B-a). This
demonstrated that ROS activation of ASK-1/JNK is involved in
chaetocin-induced apoptosis of CCLP-1 cells. Following activation
of JNKs, apoptosis is mediated by two different signaling pathways:
direct and indirect. In the direct pathway, JNKs inhibit Bcl-2, an
anti-apoptotic protein, by phosphorylation at Ser-70 (37). In the indirect pathway, JNKs
phosphorylate and transactivate other transcription factors, such
as p53 (17,38). As expected, our results showed that
inhibition of JNKs decreased p53 phosphorylation (Figs. 5A and 6B-b).
In conclusion, chaetocin suppressed ICC cell
viability and invasion in vitro and tumor growth in
vivo. Furthermore, chaetocin caused CCLP-1 cell apoptosis, cell
cycle arrest and activated the ASK-1/JNK signaling pathways
associated with oxidative stress. In addition, chaetocin reduced
the viability of a normal bile duct cell line. These results may
provide an experimental basis with which to identify new
combinatorial drugs that could be used to reduce the required
dosage of chaetocin.
Acknowledgements
The present study was supported by grants from the
Natural Science Foundation of China (no. 81641110), the Guangdong
Province Natural Science Foundation (no. 2015A030313725), and the
Guangdong Science Province and Technology Program projects
(2012B031800411).
References
|
1
|
Isham CR, Tibodeau JD, Jin W, Xu R, Timm
MM and Bible KC: Chaetocin: A promising new antimyeloma agent with
in vitro and in vivo activity mediated via imposition of oxidative
stress. Blood. 109:2579–2588. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Isham CR, Tibodeau JD, Bossou AR, Merchan
JR and Bible KC: The anticancer effects of chaetocin are
independent of programmed cell death and hypoxia, and are
associated with inhibition of endothelial cell proliferation. Br J
Cancer. 106:314–323. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Teng Y, Iuchi K, Iwasa E, Fujishiro S,
Hamashima Y, Dodo K and Sodeoka M: Unnatural enantiomer of
chaetocin shows strong apoptosis-inducing activity through
caspase-8/caspase-3 activation. Bioorg Med Chem Lett. 20:5085–5088.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Razumilava N and Gores GJ:
Cholangiocarcinoma. Lancet. 383:2168–2179. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Braconi C and Patel T: Cholangiocarcinoma:
New insights into disease pathogenesis and biology. Infect Dis Clin
North Am. 24:871–884, vii. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Blechacz B, Komuta M, Roskams T and Gores
GJ: Clinical diagnosis and staging of cholangiocarcinoma. Nat Rev
Gastroenterol Hepatol. 8:512–522. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sirica AE, Dumur CI, Campbell DJW,
Almenara JA, Ogunwobi OO and Dewitt JL: Intrahepatic
cholangiocarcinoma progression: Prognostic factors and basic
mechanisms. Clin Gastroenterol Hepatol. 7 (Suppl):S68–S78. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Flusberg DA and Sorger PK: Surviving
apoptosis: Life-death signaling in single cells. Trends Cell Biol.
25:446–458. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Matés JM, Segura JA, Alonso FJ and Márquez
J: Oxidative stress in apoptosis and cancer: An update. Arch
Toxicol. 86:1649–1665. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Romero A, Ramos E, Ares I, Castellano V
and Martínez M, Martínez-Larrañaga M, Anadón A and Martínez M:
Oxidative stress and gene expression profiling of cell death
pathways in alpha-cypermethrin-treated SH-SY5Y cells. Arch Toxicol.
91:2151–2164. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang Y, Han J, Zhu CC, Tang F, Cui XS,
Kim NH and Sun SC: Exposure to HT-2 toxin causes oxidative stress
induced apoptosis/autophagy in porcine oocytes. Sci Rep.
6:339042016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zheng R, You Z, Jia J, Lin S, Han S, Liu
A, Long H and Wang S: Curcumin enhances the antitumor effect of
ABT-737 via activation of the ROS-ASK1-JNK pathway in
hepatocellular carcinoma cells. Mol Med Rep. 13:1570–1576. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sekine Y, Hatanaka R, Watanabe T, Sono N,
Iemura S, Natsume T, Kuranaga E, Miura M, Takeda K and Ichijo H:
The Kelch repeat protein KLHDC10 regulates oxidative stress-induced
ASK1 activation by suppressing PP5. Mol Cell. 48:692–704. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kuo PL, Chen CY and Hsu YL:
Isoobtusilactone A induces cell cycle arrest and apoptosis through
reactive oxygen species/apoptosis signal-regulating kinase 1
signaling pathway in human breast cancer cells. Cancer Res.
67:7406–7420. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tibodeau JD, Benson LM, Isham CR, Owen WG
and Bible KC: The anticancer agent chaetocin is a competitive
substrate and inhibitor of thioredoxin reductase. Antioxid Redox
Signal. 11:1097–1106. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tobiume K, Matsuzawa A, Takahashi T,
Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T and
Ichijo H: ASK1 is required for sustained activations of JNK/p38 MAP
kinases and apoptosis. EMBO Rep. 2:222–228. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sinha K, Das J, Pal PB and Sil PC:
Oxidative stress: The mitochondria-dependent and
mitochondria-independent pathways of apoptosis. Arch Toxicol.
87:1157–1180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xie D, Ren Z, Fan J and Gao Q: Genetic
profiling of intrahepatic cholangiocarcinoma and its clinical
implication in targeted therapy. Am J Cancer Res. 6:577–586.
2016.PubMed/NCBI
|
|
19
|
Huang Y, Li X and Zhao Y: Progression of
targeted therapy in advanced cholangiocarcinoma. Chin J Cancer Res.
27:122–127. 2015.PubMed/NCBI
|
|
20
|
Zhou WJ, Zhang JQ, He K, Duan XP, Huang R,
Xia ZL, He JL and Xiang GA: Effects of epigenetic drugs in
intrahepatic cholangiocarcinoma cells. Chin J Exp Surg. 33:662–665.
2016.
|
|
21
|
Wiman KG and Zhivotovsky B: Understanding
cell cycle and cell death regulation provides novel weapons against
human diseases. J Intern Med. 281:483–495. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu G, Kuang S, Wu S, Jin W and Sun C: A
novel polysaccharide from Sargassum integerrimum induces
apoptosis in A549 cells and prevents angiogensis in vitro and in
vivo. Sci Rep. 6:267222016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schwartz GK and Shah MA: Targeting the
cell cycle: A new approach to cancer therapy. J Clin Oncol.
23:9408–9421. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pyo CW, Choi JH, Oh SM and Choi SY:
Oxidative stress-induced cyclin D1 depletion and its role in cell
cycle processing. Biochim Biophys Acta. 1830:5316–5325. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gao L and Williams JL: Nitric
oxide-donating aspirin induces G2/M phase cell cycle
arrest in human cancer cells by regulating phase transition
proteins. Int J Oncol. 41:325–330. 2012.PubMed/NCBI
|
|
26
|
Li S, Dong P, Wang J, Zhang J, Gu J, Wu X,
Wu W, Fei X, Zhang Z, Wang Y, et al: Icariin, a natural flavonol
glycoside, induces apoptosis in human hepatoma SMMC-7721 cells via
a ROS/JNK-dependent mitochondrial pathway. Cancer Lett.
298:222–230. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ray PD, Huang BW and Tsuji Y: Reactive
oxygen species (ROS) homeostasis and redox regulation in cellular
signaling. Cell Signal. 24:981–990. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Duan Y, Gao Y, Zhang J, Chen Y, Jiang Y,
Ji J, Zhang J, Chen X, Yang Q, Su L, et al: Mitochondrial aldehyde
dehydrogenase 2 protects gastric mucosa cells against DNA damage
caused by oxidative stress. Free Radic Biol Med. 93:165–176. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fan XY, Chen XY, Liu YJ, Zhong HM, Jiang
FL and Liu Y: Oxidative stress-mediated intrinsic apoptosis in
human promyelocytic leukemia HL-60 cells induced by organic
arsenicals. Sci Rep. 6:298652016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kwon YH, Bishayee K, Rahman A, Hong JS,
Lim SS and Huh SO: Morus alba accumulates reactive oxygen
species to initiate apoptosis via FOXO-caspase 3-dependent pathway
in neuroblastoma cells. Mol Cells. 38:630–637. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hayakawa R, Hayakawa T, Takeda K and
Ichijo H: Therapeutic targets in the ASK1-dependent stress
signaling pathways. Proc Jpn Acad Ser B Phys Biol Sci. 88:434–453.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Madan E, Gogna R, Kuppusamy P, Bhatt M,
Mahdi AA and Pati U: SCO2 induces p53-mediated apoptosis by
Thr845 phosphorylation of ASK-1 and dissociation of the
ASK-1-Trx complex. Mol Cell Biol. 33:1285–1302. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tobiume K1, Matsuzawa A, Takahashi T,
Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T and
Ichijo H: ASK1 is required for sustained activations of JNK/p38 MAP
kinases and apoptosis. EMBO Rep. 2:222–228. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ki YW, Park JH, Lee JE, Shin IC and Koh
HC: JNK and p38 MAPK regulate oxidative stress and the inflammatory
response in chlorpyrifos-induced apoptosis. Toxicol Lett.
218:235–245. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dixit D, Ghildiyal R, Anto NP and Sen E:
Chaetocin-induced ROS-mediated apoptosis involves ATM-YAP1 axis and
JNK-dependent inhibition of glucose metabolism. Cell Death Dis.
5:e12122014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Mantzaris MD, Bellou S, Skiada V, Kitsati
N, Fotsis T and Galaris D: Intracellular labile iron determines
H2O2-induced apoptotic signaling via
sustained activation of ASK1/JNK-p38 axis. Free Radic Biol Med.
97:454–465. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kelkel M, Cerella C, Mack F, Schneider T,
Jacob C, Schumacher M, Dicato M and Diederich M: ROS-independent
JNK activation and multisite phosphorylation of Bcl-2 link diallyl
tetrasulfide-induced mitotic arrest to apoptosis. Carcinogenesis.
33:2162–2171. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shi Y, Nikulenkov F, Zawacka-Pankau J, Li
H, Gabdoulline R, Xu J, Eriksson S, Hedström E, Issaeva N, Kel A,
et al: ROS-dependent activation of JNK converts p53 into an
efficient inhibitor of oncogenes leading to robust apoptosis. Cell
Death Differ. 21:612–623. 2014. View Article : Google Scholar : PubMed/NCBI
|