Introduction
Glioblastoma multiforme (GBM), which is an
astrocytic tumor of neuroepithelial tissue, occurs most often in
the subcortical white matter of the cerebral hemispheres. Tumor
infiltration often extends into the adjacent cortex of the basal
ganglia and becomes substantially large before turning symptomatic
(1). A growing tumor causes an
increase of intracranial pressure and sometimes it leads to
hydrocephaly (2). GBM is also
characterized by high proliferative activity, a large network of
hyperplastic blood vessels and finger-like tentacles which spread
out infiltrating other parts of the brain, rendering its complete
resection very challenging and radiotherapy inefficient (3,4).
Moreover, the blood-brain barrier renders treatment more difficult
and tumor cells found in hypoxic areas become resistant (5,6).
Surgical resection, followed by chemotherapy and radiotherapy is
the mainstay of GBM treatment. However, it offers only limited
survival advantages (7). Gliomas
have been divided into grades I–IV on the basis of the degree of
malignancy by the WHO grading system (8). Grade I gliomas generally behave in a
benign manner whereas grades II–IV are malignant and infiltrative
to the brain. Among astrocytic gliomas, GBM or grade IV glioma is
the most prevalent and aggressive type that poses a unique
challenge for treatment due to its predisposition for invasion and
proliferation. To complicate the therapeutic scenario these tumors
are also highly resistant to conventional therapies (9).
Standard chemotherapy used for GBM involves
temozolimide (TMZ). The cytotoxicity of TMZ is thought to result
from the formation of O6-methylguanine in DNA,
which mispairs with thymine during DNA replication triggering
futile cycles of the mismatch repair and subsequent DNA damage
(10). However, apoptosis occurs
only in few of the treated GBM cells and at least 50% of
TMZ-treated patients do not respond to it (11). This is thought to be due to the
overexpression of O6-methylguanine
methyltransferase (MGMT) and/or lack of a DNA repair pathway in GBM
cells (12). In addition, results
obtained from studies with intrinsic and acquired TMZ-resistant GBM
cells revealed that resistance in GBM is not just mediated by a
single molecular event but by multiple ones (13). The overall 5-year survival after
radiotherapy with concomitant and adjuvant TMZ treatment is only
9.8% (14). Hence, it is important
to explore alternative options for GBM sensitization such as,
combining two or more drugs that have different cytotoxic
mechanisms or targeting alternate pathways resulting in additive or
a synergistic effect.
Indirect evidence from recent studies suggest that
autophagy, a cellular homeostatic and recycling mechanism may be
highly relevant to gliomas (15).
Furthermore, the poor response of GBM to current treatment
modalities, which largely depends on apoptosis, makes it all the
more important to consider autophagy as an alternative death
pathway. Notably, one of the most common genetic alterations
observed in GBM are amplification of EGFR or deletion of PTEN
resulting in activation of the PI3K/Akt/mTOR pathway promoting
survival and drug-resistance (16).
However, clinical studies with small molecule inhibitors of EGFR or
individual inhibitors of PI3K/mTOR have been disappointing
(17,18). A dual inhibitor of PI3K and mTOR has
exhibited some promise in gliomas (19). However, therapies targeting
components of the RTK/PI3K/Akt/mTOR axis typically promote
autophagy, thus playing a cytoprotective role (20). Hence, we assume that combination of
autophagy promoters alongside autophagosome-lysosome fusion
inhibitors could increase cytotoxicity in GBMs by inducing enhanced
autophagic stress. GBM cells with inhibited autophagy may
significantly accumulate a higher number of damaged mitochondria
(due to its reduced clearance by mitophagy) and protein aggregates
which can lead to elevated levels of ROS resulting in enhanced cell
death. In corroboration to the aforementioned, combination of
bafilomycin (a late-stage autophagy inhibitor) with an mTOR
inhibitor was found to enhance glioma cell death (20). However, the outcomes of autophagy
inhibition may depend on the cell type, the combination therapy
explored and other factors, which are yet to be clearly
understood.
Vorinostat (SAHA) is a member of a promising class
of antitumor agents, HDACi, that have the capacity to enhance the
activity of commonly used autophagy inhibitors in tumor therapy.
SAHA is currently being used in the treatment of cutaneous T-cell
lymphoma and under clinical trials for multiple other cancer types
(21,22). HDACi are most frequently known to
induce apoptosis via caspases (23), however, more recently, HDACi such as
SAHA have been reported to act as inhibitors of the mechanistic
target of rapamycin (mTOR) pathway thus increasing autophagy
(24). Autophagy activation has
been frequently demonstrated to inhibit the onset of apoptotic cell
death, however, in certain cases autophagy may have an additive
role in the death process as well (25). More precisely excessive
‘self-eating’ through autophagy or accumulation of autophagosomes
may contribute to cell death. In this context, lysosomotropic
agents such as chloroquine (CQ) are known to increase the lysosomal
pH, which leads to inhibition of the fusion of the autophagosome
with the lysosome, resulting in hyper-accumulation of autophagic
vacuoles which expedite apoptotic cell death (26). Hence, we presumed that a combination
treatment of SAHA and CQ may lead to increased formation of
autophagosomes and suppression of the autophagosome-lysome fusion,
resulting in hyper-accumulation of autophagic vacuoles, ultimately
leading to the enhanced death of GBM cells. Collectively, combining
SAHA therapy with autophagy inhibition can be a promising clinical
approach in GBM treatment.
Materials and methods
Chemicals and reagents
2′,7′-Dichlorofluorescin diacetate (DCFDA; cat. no.
D6883), monodansylcadaverine (MDC; cat. no. D4008),
chloroquine-di-phosphate (CQ; cat. no. C6528), propidium iodide
(PI; cat. no. P4864) were all purchased from Sigma-Aldrich (St.
Louis, MO, USA). N-Acetyl-L-cysteine (NAC; cat. no. 47866) and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
cat. no. 33611) were obtained from Sisco Research Laboratories Pvt.
Ltd. (SRL; Mumbai, India). FITC-conjugated Annexin V (cat. no.
A13199) and Annexin V binding buffer (cat. no. V13246), MitoTracker
Deep Red FM (cat. no. M22426) and LysoTracker Green DND-26 (cat.
no. L7526) were procured from Thermo Fisher Scientific, Inc.
(Waltham, MA, USA). JC-1 (cat. no. sc-364116A) was purchased from
Santa Cruz Biotechnology (Dallas, TX, USA).
Cell culture
The U87MG cell line was obtained from The National
Centre for Cell Science (NCCS; Pune, India) and cultured at 37°C
and 5% CO2, in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% fetal bovine serum (FBS; both from
Invitrogen; Thermo Fisher Scientific, Inc.; cat. no. 26140-079).
Cells were grown to 80% confluency, rinsed in phosphate-buffered
saline (PBS) and placed in fresh medium prior to treatment with
compounds.
Analysis of cytotoxicity
In vitro cytotoxicity was performed following
the methods previously described by Chowdhury et al
(27). Briefly, U87MG cells were
seeded at a density of 8×104 cells in 96-well plates.
After overnight culture of the cells, they were treated with TMZ,
CQ, 3-MA, rapamycin, SAHA, SAHA+CQ for 48 h. Thereafter, ΜΤΤ was
added to each well containing cells and incubated for 4 h. The
formazan crystals formed due to the presence of live cells were
solubilized in dimethyl sulfoxide (DMSO) and readings were obtained
with a spectrophotometer at 570 nm with a differential filter of
630 nm using Multiskan GO microplate spectrophotometer (Thermo
Fisher Scientific, Inc.; cat. no. 51119200). The percentage of
viable cells was calculated using the formula: Viability (%) =
(mean absorbance value of drug-treated cells)/(mean absorbance
value of the control) × 100. A concentration of 0.2% DMSO was found
to be non-toxic and was used for dissolving SAHA, and used as a
control in the cytotoxicity experiments.
Morphological analysis
U87MG cells were seeded at a density of
1×106 cells in 6-well plates and treated with CQ, SAHA
or SAHA+CQ for 48 h. Following the treatment period, images were
captured using an Olympus (CKX41) microscope at a ×20 magnification
(Olympus Corp., Tokyo, Japan).
Apoptosis assay
U87MG cells were seeded at a density of
1×106 cells in a 6-well plate. After overnight culture,
the cells were treated with CQ, SAHA, SAHA+CQ for 48 h.
Post-treatment, the cells were collected and centrifuged at 380 × g
for 5 min at 4°C. The cell pellet was washed with PBS followed by
centrifugation at 380 × g for 5 min at 4°C. The washed cell pellet
was then dissolved in 500 µl of binding buffer. Following this 4 µl
of Annexin V/FITC and 10 µl of PI were added. The samples were then
incubated in the dark for 20 min and then 10,000 events were
acquired in a flow cytometer (CytoFLEX; Beckman Coulter, Brea, CA,
USA). The data was analyzed using CytExpert (Beckman Coulter) and
the percentage of apoptotic cells was calculated and represented in
a bar diagram (27).
Cell cycle analysis
For the analysis of the cells at various phases of
the cell cycle, cells were seeded at a density of 1×105,
grown overnight and exposed to CQ, SAHA, SAHA+CQ for 48 h.
Following incubation with the drugs the cells were collected using
PBS, and thereafter fixed in 70% ethanol for 24 h at −20°C.
Following fixation the cell pellet was re-suspended in PBS and then
propidium iodide (PI; 20 µg/ml) was added to stain the DNA. The
dye-added cell suspension was incubated in the dark for 20 min and
then 10,000 events were acquired in a flow cytometer (CytoFLEX;
Beckman Coulter). The data was analyzed using CytExpert and the
percentage of cells in various phases of the cell cycle was
calculated and represented in a bar diagram (27).
Analysis of fragmented DNA using
DAPI
U87MG cells were seeded at a density of
1×106, grown overnight and exposed to CQ, SAHA, SAHA+CQ
for 48 h. Following treatment for 48 h, the media was removed, the
cells were washed with PBS and methanol was added to fix the cells.
After 20 min of incubation, a PBS wash was performed to remove the
methanol. Then, the cells were stained with DAPI and mounted on
slides. The slides were then visualized under a blue filter using a
fluorescence microscope (28).
Measurement of intracellular ROS
ROS levels were estimated using DCFHDA
(Sigma-Aldrich) which passively enters the cells, where it reacts
with ROS to form the highly fluorescent compound,
dichlorofluorescein (DCF). U87MG cells were seeded at a density of
8×104 in 96-well plates. After overnight culture of the
cells they were treated with CQ, SAHA, SAHA+CQ for 48 h and 5 mM
NAC was added 1 h prior to treatment to inhibit ROS.
Post-treatment, the cells were washed with PBS and then incubated
in 100 µl of working solution of DCFH-DA at 37°C for 45 min.
Following incubation, fluorescence was assessed at a 485 nm
excitation and a 530 nm emission using a microplate reader
(Fluoroskan Ascent™; cat. no. 5210470; Thermo Fisher Scientific,
Inc.) (29).
Measurement of mitochondrial membrane
potential
Flow cytometric analysis of mitochondrial membrane
potential was performed using the JC-1 dye. U87MG cells were seeded
at a density of 1×106 in a 6-well plate. Following
overnight culture, the cells were treated with CQ, SAHA, SAHA+CQ
for 48 h. Post-treatment, the cells were collected in eppendorf
tubes. The cells were centrifuged at 380 × g for 5 min at 4ºC and
the total volume was brought to 1 ml using complete DMEM. The cell
suspension was stained with 2.5 µg/ml of JC-1 dye. Then, the
samples were kept in the dark for 15 min at room temperature (RT).
Following this, the cells were centrifuged at 380 × g using double
the volume of PBS. Finally, the cells were re-suspended in 300 µl
of PBS and analyzed using a flow cytometer (CytoFLEX). The shift
from green to red fluorescence was analyzed using CytExpert
(27).
Monodansylcadaverine (MDC) staining of
autophagic vacuoles
The autofluorescent dye and a specific
autophagolysosomal marker, MDC, was used to analyze the autophagic
process. U87MG cells were seeded at a density of 1×106
cells in a 6-well plate. Following SAHA+CQ treatment, the cells
were incubated for 10 min with 0.05 mM MDC in PBS at 37°C.
Following incubation, the coverslips containing the cells were
washed with PBS and mounted with an antifade mountant (containing
DAPI). Intracellular MDC in the form of punctate dots were analyzed
by fluorescence microscopy. For fluorimetric assessment, following
incubation of the cells with CQ, SAHA, SAHA+CQ and labeling with
MDC for 10 min, the cells were washed with PBS and collected in 10
mM Tris-HCl (pH 8.0) containing 0.1% Triton X-100. Intracellular
MDC was assessed by fluorescence photometry (excitation 380 nm and
emission 525 nm) on a microplate reader (Fluoroskan Ascent™)
(30,31). An increase in MDC fluorescence upon
treatment was expressed as a fold change with respect to the
control.
Immunoblotting
U87MG cells were treated with either CQ or SAHA or
SAHA plus CQ for 48 h. Thereafter, cells were lysed in a modified
RIPA buffer (Sigma-Aldrich; Merck KGaA), and the protein content
was measured using the Bradford reagent. Then, the loading buffer
was added to the lysates followed by heat denaturation (100°C for
10 min) and cooling on ice. Equal concentrations of protein lysates
were loaded in denaturing polyacrylamide gels and thereafter they
were transferred to polyvinylidene fluoride (PVDF) membranes
(Thermo Fisher Scientific, Inc.; cat. no. 88518) for blocking with
5% skimmed milk (HiMedia; Mumbai, India; cat. no. GRM1254). The
blots were probed with anti-LC3 specific primary antibody at
dilution of 1:1,000; (cat. no. 3868; Cell Signaling Technology,
Danvers, MA, USA). β-actin (dilution 1:2,000; cat. no. sc69879;
Santa Cruz Biotechnology) was used as a loading control. The
secondary antibody used was horseradish peroxide-conjugated goat
anti rabbit IgG at dilution of 1:10,000 (cat. no. 7074; Cell
Signaling Technology). The protein intensity was detected using an
enhanced chemiluminescence detection system (Thermo Fisher
Scientific, Inc.). The expression levels were densitometrically
quantified using ImageJ software (National Institutes of Health,
Bethesda, MD, USA) and normalized to the control (27).
Confocal microscopy
U87MG cells were cultured overnight on coverslips
and kept in a 35-mm dish at a density of 10×106
cells/dish and then, the cells were exposed to CQ, SAHA, SAHA+CQ
for 48 h. Post-treatment, the media was removed, the cells were
washed with PBS, MitoTracker Deep Red was added (MTR, 0.5 µM) and
incubation followed for 1 h in humidified air at 37°C. After
incubation, the cells were washed with PBS and methanol was added
to fix the cells. Following 20 min of incubation, a PBS wash was
performed to remove the methanol. Then, the cells were stained with
DAPI and mounted on slides and visualized under a confocal
microscope. Excitation of MTR and DAPI at 644 and 358 nm and
fluorescence emission was assessed at 665 and 461 nm, respectively
(32).
LysoTracker staining of acidic
organelles
The LysoTracker Green DND (Thermo Fisher Scientific,
Inc.) is a fluorescent probe which is used to visualize acidic
organelles in live cells. U87MG cells were cultured overnight on
coverslips and kept in a 35-mm dish at a density of
10×106 cells/dish and then cells were exposed to CQ,
SAHA, SAHA+CQ for 48 h. Post-treatment, the media was removed, the
cells were washed with PBS, LysoTracker Green DND was added (LTG,
0.5 µM) and incubation followed for 20 min in humidified air at
37°C. Following 20 min of incubation, the cells were stained with
DAPI and mounted on slides. The cells were then visualized under a
fluorescence microscope and the intensity of LysoTracker
fluorescence was compared with the untreated control (32).
Statistical analysis
The obtained data were analyzed using the
GraphPrism® software (version 5.01; GraphPad Software,
Inc., La Jolla, CA, USA). The effect of various treatments was
statistically analyzed using one-way ANOVA and Tukey's multiple
comparison test was used for comparison of the control with
different tests. All data points represent the mean of independent
measurements and were represented as the standard error in the form
of bars.
Results
Effect of SAHA and CQ co-treatment on
the sensitization of U87MG glioblastoma cells
GBM is one of the deadliest types of brain
malignancy with inadequate responsiveness to commonly used
therapeutic interventions. Currently, the gold standard treatment
for GBM constitutes surgical resection followed by adjuvant
chemotherapy with TMZ. However, these tumors have been well
documented for their development of resistance to the standard
therapy (33). One of the
anticipated mechanisms for acquisition of therapeutic resistance in
GBM is induction of stress-induced autophagy. Ηowever, the precise
role of autophagy in cancer development, and its role in response
to chemotherapy are highly controversial (9). There are conflicting reports
pertaining to TMZ-induced autophagy in GBM. For example, Kanzawa
et al observed significant autophagy induction following TMZ
treatment which acted as a cell death-inducing phenomenon (34); while, subsequent studies have
reflected a pro-survival response in GBM cells to TMZ (35). Notably, in most cases, GBM cells
when treated with TMZ revealed the onset of apoptosis only after
4–6 days following treatment, where autophagy was an early response
inhibitor of cell death (36). The
temporal delayed action of TMZ has progressively hindered the use
of TMZ in GBM therapy necessitating the search for novel
therapy.
In the present study, we first explored the efficacy
of HDACi, SAHA for GBM therapy. For this purpose, the human GBM
cell line U87MG was selected as the in vitro model system
since these GBM cells are known to show resistance to TMZ treatment
(37). In addition, in the present
study we observed that U87MG cells were highly resistant to TMZ as
it failed to induce any cytotoxic effect up to a 30-µM
concentration and 48 h of exposure (data available upon request).
In contrast, these cells were found to be responsive to SAHA
treatment. The results revealed that SAHA exhibited dose-dependent
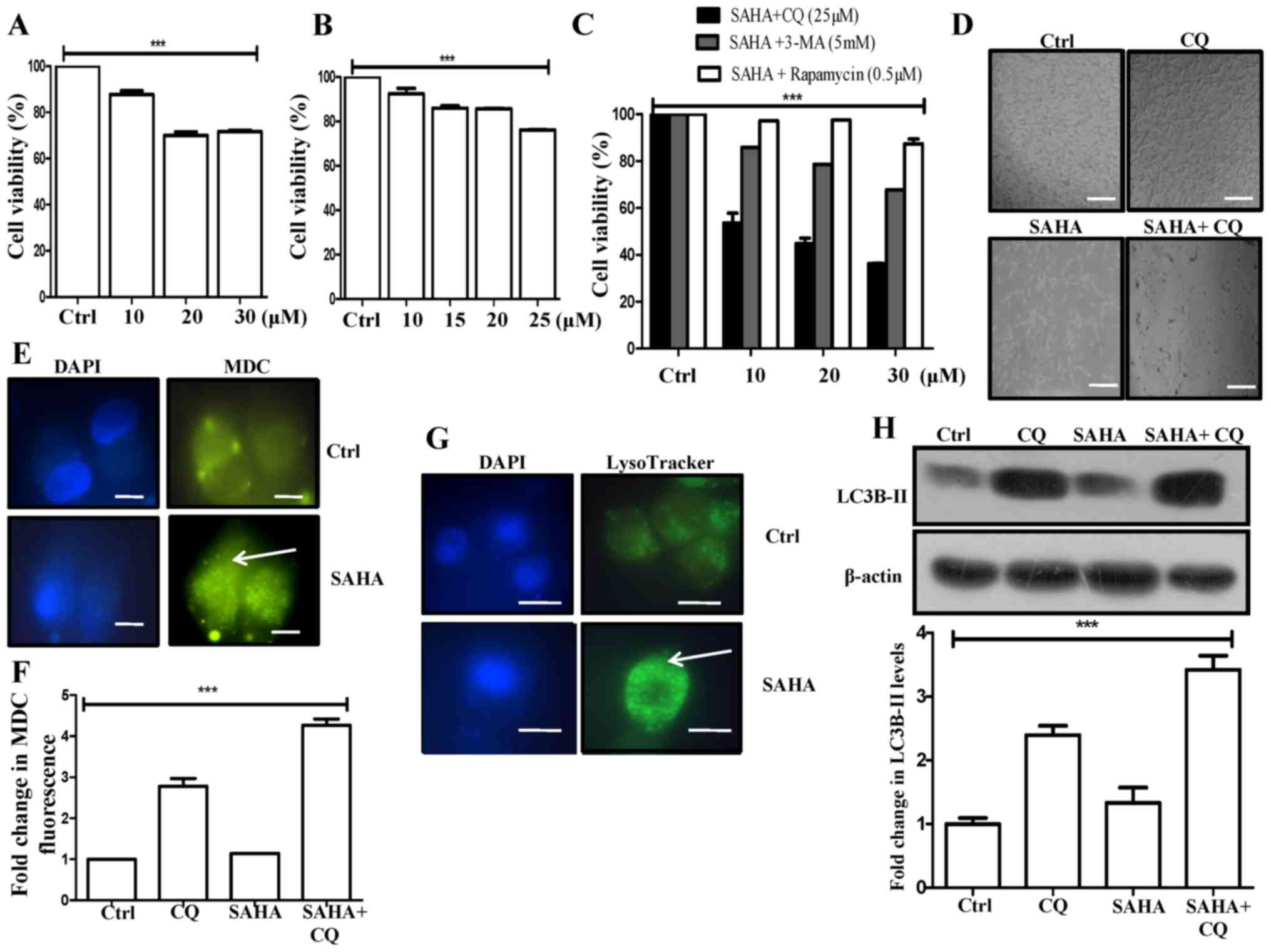
cytotoxicity and reduced the cell viability to ~30% with a 30-µM
concentration (Fig. 1A). Next, we
investigated whether autophagy modulation can increase the
cytotoxic effect of SAHA in the U87MG cells. Recent studies have
revealed that HDACi, such as SAHA can modulate autophagic protein
expression in human cancer cells (38). However, the precise role and the
molecular mechanisms underlying HDACi-mediated autophagy are still
not clear. Moreover, the role of autophagy in cell death remains
controversial and is likely to be context-dependent. We hence
determined whether autophagy modulators could lead
chemotherapy-resistant GBM cells towards cell death. We studied the
activity of different autophagy modulators on the viability of the
U87MG cells and found that while early autophagy inhibitor 3-MA did
not have any significant effect on cell viability (data available
upon request), the lysosomotropic agent- choloroquine (used as the
di-phosphate salt, CQ) exhibited dose-dependent cytotoxicity
(Fig. 1B). Furthermore, rapamycin,
an inhibitor of mTOR signaling, which is often used as an autophagy
activator, did not demonstrate any cytotoxic effect (data available
upon request). Next, we studied the effect of these autophagy
modulators on the SAHA-treated U87MG cells. Notably, co-treatment
with SAHA and CQ exhibited significantly higher cytotoxicity (36%
viability at 30 µM SAHA and 25 µM CQ) than SAHA or CQ alone,
whereas co-treatment with 3-MA or rapamycin along with SAHA had no
significant synergistic effect on cytotoxicity (Fig. 1C). Pronounced morphological
variations were also observed upon SAHA+CQ combination treatment in
the U87MG cells when compared to the control or other treatments
(Fig. 1D).
Based on previous studies, it has been established
that SAHA induces autophagy (39).
In this study, SAHA treatment in U87MG cells exhibited a
significant increase in MDC fluorescent punctate dots (Fig. 1E). MDC stains autophagic vacuoles
and an increase in MDC fluorescence is often correlated with
enhanced autophagy. Additionally, treatment of SAHA+CQ revealed a
3-fold increase in MDC fluorescence in comparison to only SAHA, as
analyzed by fluorimetry (Fig. 1F).
Chloroquine is known to act by increasing lysosomal pH and thereby
inhibiting lysosomal activity (40). MDC on the other hand is known to
label not only acidic lysosomes but also endosomes and
autophagosomes (41); i.e., MDC can
incorporate into membranes based on lipid characteristics
independent of pH (42). Since, CQ
increases the accumulation of autophagosomes or autophagic
vacuoles, we observed an increased MDC fluorescence in CQ-treated
cells that increased even further upon treatment with SAHA plus CQ.
This was likely to be the MDC trapped in the accumulated vacuoles
after CQ treatment. Furthermore, the increased MDC fluorescence
with SAHA+CQ treatment could be attributed to the fact that SAHA
induces autophagy while CQ blocks autophagic flux resulting in
increased accumulation of autophagic vacuoles leading to even more
trapping of MDC in them. Hence, MDC fluorescence was the highest in
the combination treatment, followed by treatment only with CQ, and
then SAHA-treated samples.
On a similar note, SAHA treatment also increased
green punctate dots with stronger fluorescence intensity indicating
an enhanced lysosomal number in comparison to the control, as
analyzed through LysoTracker staining, in GBM cells (Fig. 1G). This is supportive of the view
that SAHA triggers autophagy in U87MG cells. In addition,
microtubule-associated protein light chain 3-II (LC3B-II), an
autophagic marker, exhibited a significant increase in the SAHA+CQ
combination treatment, than only the CQ treatment reflecting the
induction of autophagic flux by administration of SAHA (Fig. 1H). We assumed that inhibition of
autophagosome-lysosome fusion by CQ presumably resulted in an
increased autophagosome accumulation evident from enhanced MDC
fluorescence in the combination treatment (Fig. 1F). Our results revealed that
blocking of SAHA-induced autophagy by inhibitors of autophagic flux
can increase SAHA-mediated cell death preferentially in U87MG
cells. However, upstream autophagic block or autophagy inducers
have limited effect on cell death.
Inhibition of autophagy by CQ
potentiates SAHA-mediated apoptosis
We were thereafter interested in understanding the
mode of cell death by the combination treatment. According to
present literature, the primary antitumor activity of HDACi is
believed to be through induction of apoptosis in a variety of
cancer cells (43). Moreover,
recent studies have also demonstrated that HDACi, such as SAHA can
also induce autophagy in human cancer cells (39). However, currently the role of
HDACi-mediated autophagy in context to apoptosis remains
controversial and is still not clear. We hence analyzed apoptosis
using Annexin V-FITC/PI dye with SAHA and CQ treatment using flow
cytometry. The Annexin V-FITC conjugate binds to phosphotidyl
serine on the cell surface and detects cells in the early stages of
apoptosis and PI binds to fragmented DNA and detects cells that
have undergone cell death, while cells positive for both represents
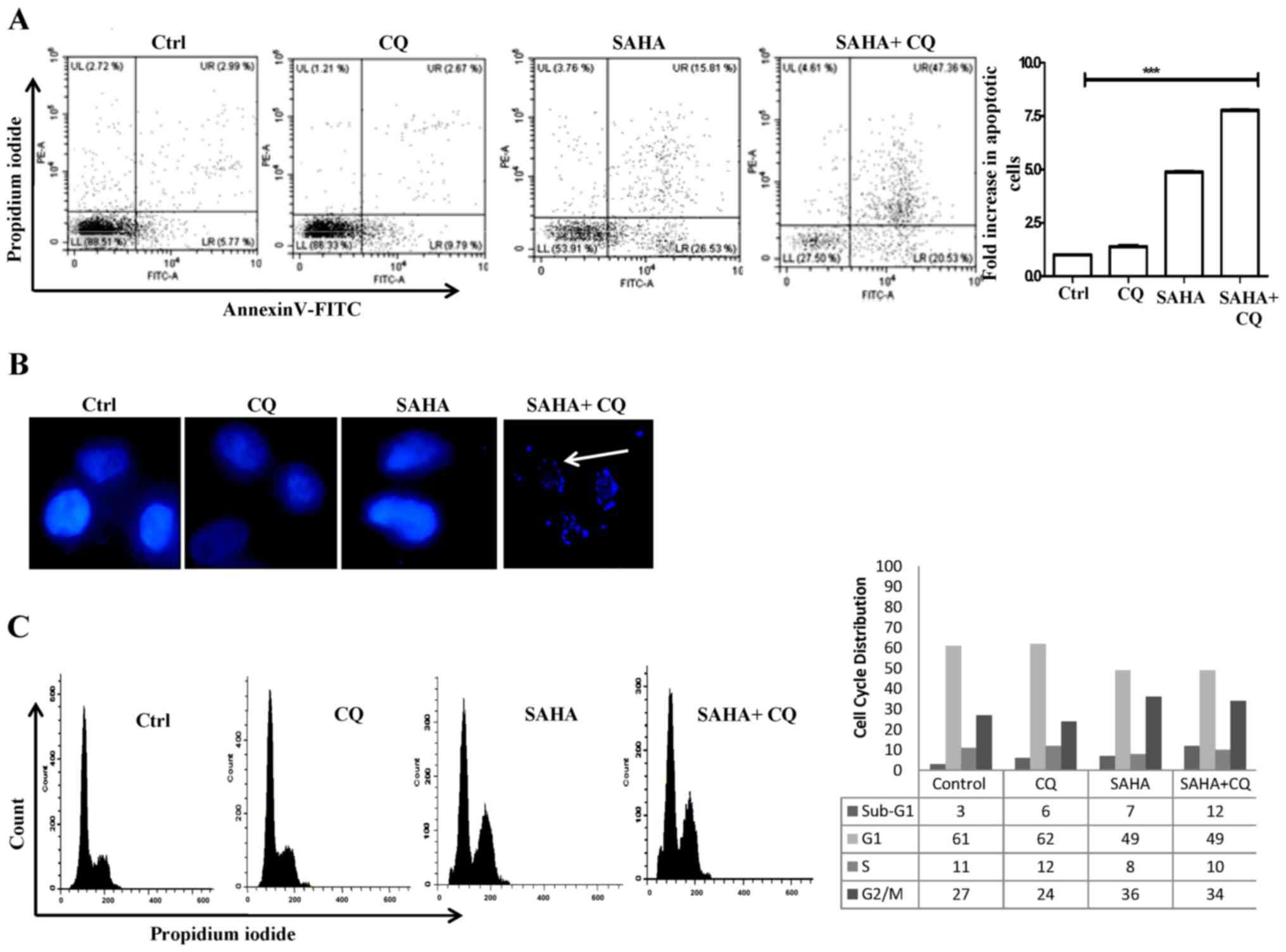
late apoptosis. Upon treatment with only SAHA, a significant
percentage (42.34%; 4.83-fold) of cells were found to undergo
apoptosis when compared to the control (Fig. 2A). This was expected, since SAHA is
known to induce apoptosis. Only CQ treatment failed to exhibit any
significant increase in apoptotic cells. However, cells treated
with SAHA+CQ exhibited a marked increase (67.89%; 7.74-fold to the
control) in apoptotic cells (Fig.
2A). This is indicative of the fact that inhibition of
autophagic flux increases SAHA-induced apoptosis. To confirm cell
death, DAPI staining of the nucleus was also performed to check for
fragmented DNA, as DNA fragmentation is a hallmark of apoptosis.
The presence of fragmented nuclei, indicative of the apoptotic
process, in the SAHA+CQ-treated cells is shown in Fig. 2B. We thereafter examined the effect
of the combination treatment on the cell cycle. SAHA treatment
increased the cell population at the G2/M phase whereas,
with the combination treatment, there was a significant increase in
the sub G1 population of cells from 7 to 12%, which confirmed the
induction of apoptosis (Fig. 2C).
These data revealed that simultaneous induction of autophagosome
formation and inhibition of autophagosome-lysosomal fusion in U87MG
cells by SAHA+CQ treatment was associated with increased apoptotic
cell death compared to only HDACi treatment.
SAHA+CQ increases ROS production
leading to enhanced apoptosis in GBM cells
Autophagy can potentially act as a pro-survival
strategy in response to drug stress by eliminating damaged
intra-cellular mitochondria by a process known as mitophagy
(44). In normal conditions, ROS
generated during mitochondrial oxidative metabolism plays an
important role in the maintenance of cellular homeostasis. However,
cancer cells due to extensive proliferative requirements tend to
generate excess ROS than normal cells. In addition, they utilize
intricate cellular mechanisms to keep a check on ROS levels which
if not can be fatal for the tumor cells. Under stress, cancer cells
can also limit ROS accumulation via an increase in cytoprotective
selective autophagy e.g., mitophagy to facilitate higher
proliferation, metastasis and also resistance against drug
treatment (45). As aforementioned,
we observed that SAHA induces autophagy and when autophagy is
inhibited by CQ autophagosomes presumably accumulate, as observed
through increased MDC fluorescence in the combination treatment. We
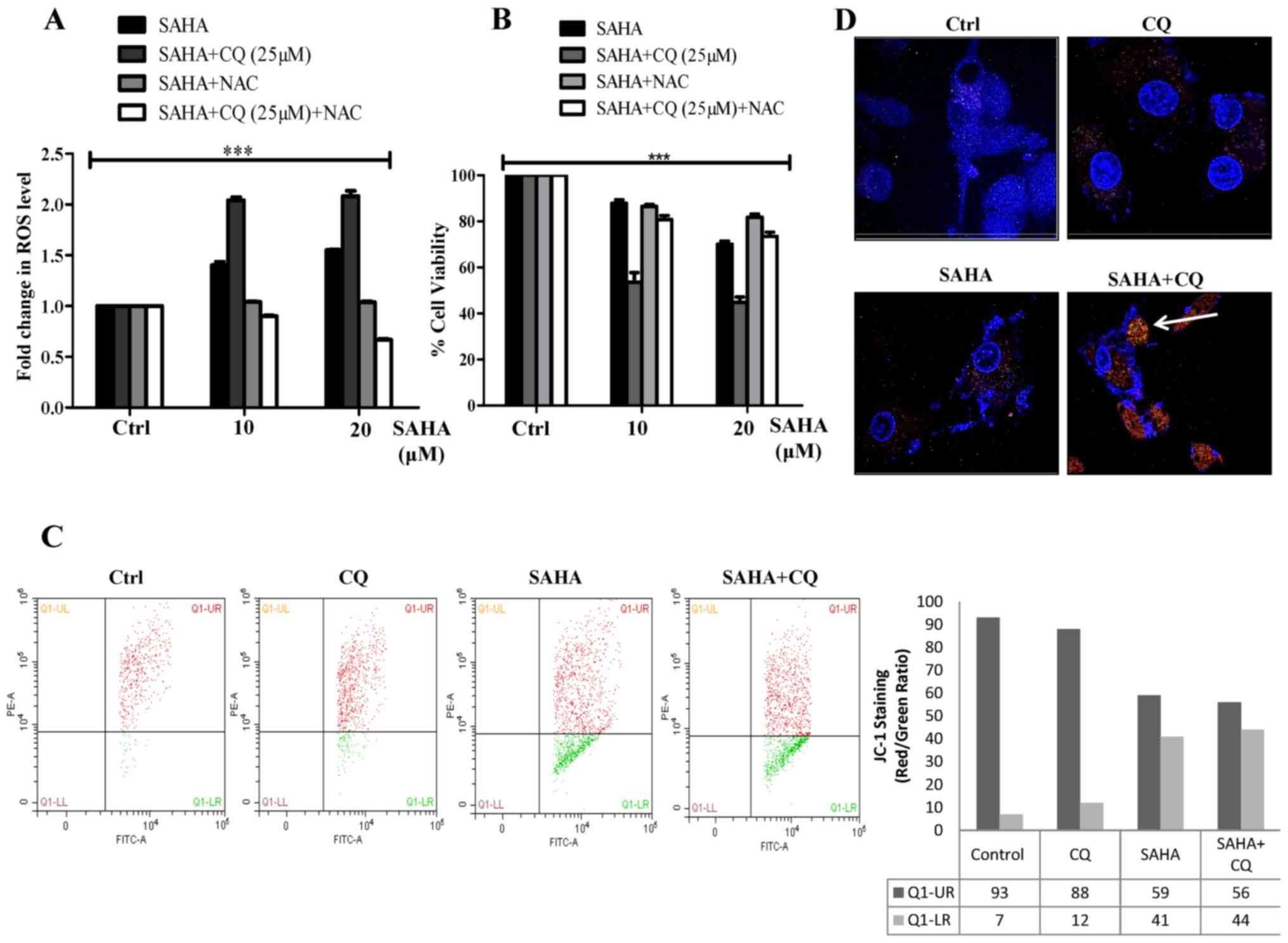
found that co-treatment with SAHA and CQ resulted in a significant
increase in ROS production, maybe due to the inhibition of
autophagic flux (Fig. 3A). SAHA has
been previously demonstrated to simultaneously induce apoptosis by
increasing ROS production and also trigger autophagy (39,46).
Autophagy under this circumstance, presumably acts as a survival
strategy by clearance of damaged mitochondria generated by SAHA
treatment, thus maintaining cellular homeostasis and survival.
However, with the addition of CQ along with SAHA, selective
autophagy is inhibited leading to excess ROS accumulation. Notably,
when ROS was quenched in the SAHA+CQ-treated cells by the ROS
scavenger (N-acetyl cysteine, NAC), cell viability increased
(Fig. 3B) which confirmed that
SAHA+CQ-mediated enhanced cell death was significantly dependent on
ROS. The accumulation of ROS with the combination treatment was
also associated with mitochondrial membrane potential (MMP)
reduction, a prerequisite to cell death. Upon treatment with
SAHA+CQ, not only did the amount of ROS increase but it also led to
increased collapse of MMP as determined by JC-1 assay (Fig. 3C). To further validate the
hypothesis that CQ induces mitochondria accumulation in
SAHA-treated cells, we used confocal microscopy with MitoTracker
Red dye. The results revealed MitoTracker Red indicative of the
number of mitochondria significantly accumulated in the
SAHA-treated cells in which autophagic flux was blocked with CQ
(Fig. 3D). The aforementioned
result along with increased ROS confirm that damaged mitochondria
accumulated in the SAHA+CQ-treated cells lead to increased
apoptosis.
Discussion
For decades temozolomide (TMZ), has been the drug of
choice for glioblastoma multiforme (GBM) (47). Yet, a major hindrance to TMZ therapy
has been the intrinsic or acquired resistance of GBM patients
(48). Recent studies also revealed
that TMZ could induce autophagy in GBM cells. However, TMZ-induced
autophagy also induced a cytoprotective ATP surge in glioma cells
(49,50). Furthermore, in contrast to the
aforementioned, a recent study also reported that rapamycin, an
autophagy activator could enhance glioma cell sensitization in
vitro and in immuno-compromised mice (51). These findings along with reports of
acquired resistance of GBM cells necessitate the design of
alternate therapeutic strategies for effective treatment of GBM.
Similarly, the role of autophagy in GBM is also not clearly
understood as conflicting observations portray both its
cytoprotective as well as pro-apoptotic role.
Recent studies indicate that HDACi such as SAHA, in
addition to their ability to induce apoptosis are also capable of
stimulating autophagy. SAHA has a wide range of effects extending
from inhibition of cell cycle progression, suppression of the
angiogenic effect to the induction of apoptosis in cancer cells
(52). However, in addition to its
role in apoptosis SAHA has also been reported to promote autophagic
cell death, which offers an obvious advantage for therapy against
apoptosis-resistant tumor cells (53,54).
We therefore explored SAHA as a therapeutic agent in combination
with known autophagy modulators. Anti-malarial drugs such as
choloroquine that block autophagic flux by inhibiting lysosomal
acidification, are currently in clinical trials in combination with
TMZ for treatment of GBM (55).
While we are still awaiting the results from these trials, we
wondered about the consequences of simultaneous application of SAHA
and CQ in GBM cells. We also tried increasing the autophagy levels
with mTOR inhibitor, rapamycin, alongside SAHA, realizing that an
enhanced autophagy would lead GBM cells from proliferation towards
cell death. Notably, while the use of rapamycin along with SAHA had
a limited effect on the survival of GBM cells, concurrent treatment
of SAHA+CQ resulted in significant cell death. Moreover, inhibition
of upstream autophagy events with 3-MA also had a reduced effect on
SAHA-induced cytotoxicity in GBM cells suggesting that modulation
of autophagic flux has an enhanced effect than the inhibition of
the early steps of autophagy.
There is an increasing body of evidence suggesting
that in cancer cells, the autophagy-lysosomal pathway integrates
signals from varied upstream events thereby resulting in an
intricate regulatory mechanism which can culminate in cell death or
survival (56,57). However, the roadmap to the ultimate
effect in a particular cancer cell is purely context-dependent and
this knowledge is critical for formulating effective therapeutic
strategies (58–60). Hence, this becomes increasingly
important in the context of glioma cells, which display significant
therapeutic resistance against conventionally used drugs. In the
present study, we revealed that the combination treatment of SAHA
and CQ resulted in an increase of damaged mitochondria accumulation
in U87MG cells leading to a significant increase in ROS levels and
a reduction in mitochondrial membrane potential, triggering
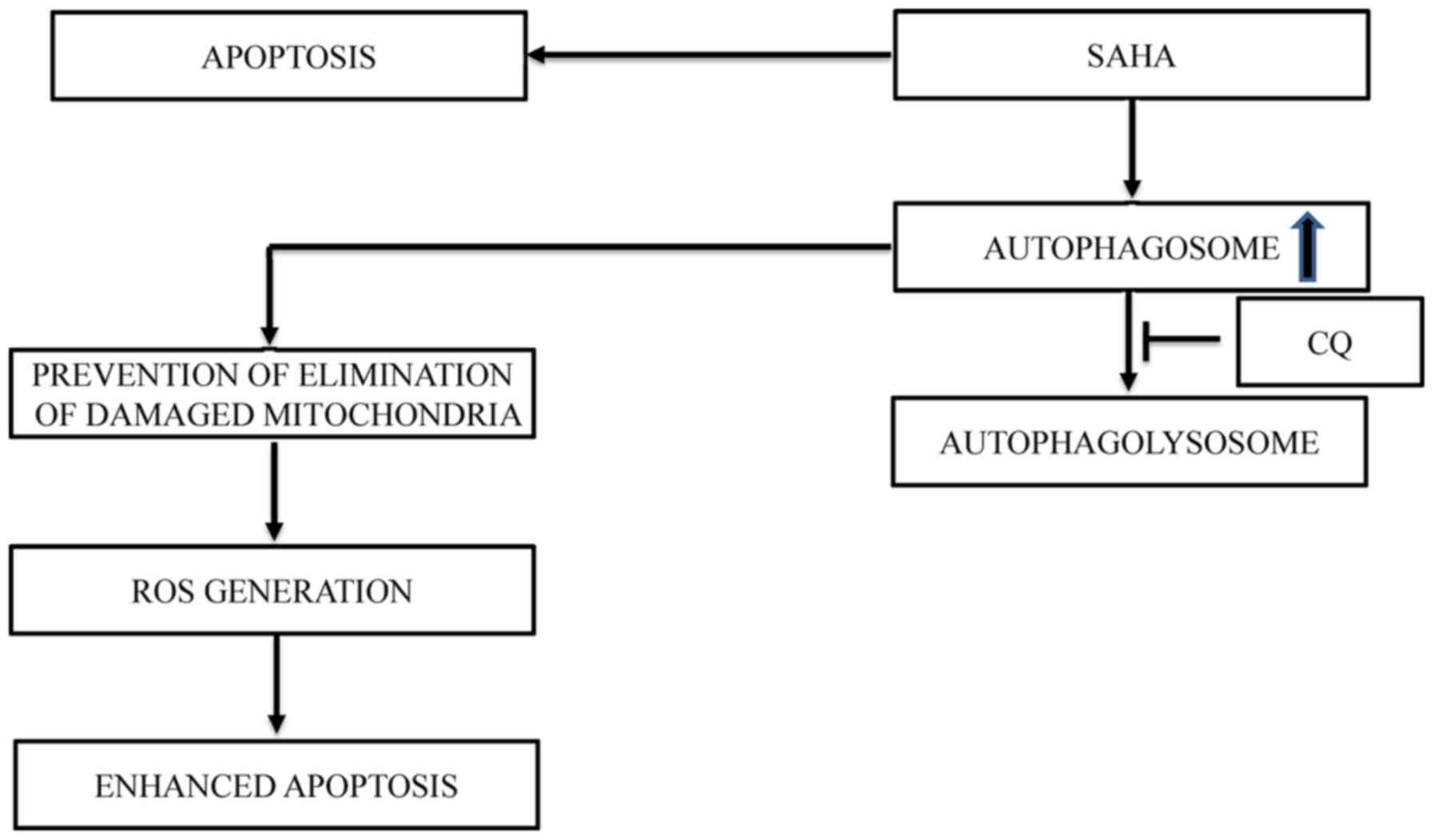
apoptosis. We postulated that the lack of clearance of damaged
mitochondria by mitophagy in these cells post-treatment with SAHA
and CQ could be the cause of the induction of cell death (Fig. 4). We further revealed that
inhibition of autophagy at a late stage, but not at an early stage,
increased the cytotoxic effect of SAHA via apoptosis. Hence, this
study provides cellular and molecular evidence concerning the
combined effect of SAHA and CQ which can be developed as a
therapeutic strategy for glioblastoma treatment in future.
Acknowledgements
We acknowledge BITS-Pilani for providing us with
infrastructural support. LK would like to thank BITS-Pilani for
providing support for his master's thesis.
Funding
The present study was supported in part by the
DST-SERB grant of SM (SERB/LS-77/2013), RC (SB/FT/LS-233/2012) and
the DBT grant of RC (BT/PR/8799/MED/30/1067/2013).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
LK, HS, AS and SM performed and analyzed the
experiments. RC and AR designed and planned the study; RC, AR and
LK wrote the manuscript. All authors read and approved the
manuscript and agree to be accountable for all aspects of the
research in ensuring that the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
SAHA
|
suberanilohydroxamic acid
|
|
GBM
|
glioblastoma multiforme
|
|
TMZ
|
temozolomide
|
|
CQ
|
chloroquine
|
|
ROS
|
reactive oxygen species
|
|
MMP
|
mitochondrial membrane potential
|
|
MDC
|
monodansylcadaverine
|
|
LC3B-II
|
microtubule-associated protein light
chain 3-II
|
References
|
1
|
Iacob G and Dinca EB: Current data and
strategy in glioblastoma multiforme. J Med Life. 2:386–293.
2009.
|
|
2
|
Jung WH, Choi S, Oh KK and Chi JG:
Congenital glioblastoma multiforme-report of an autopsy case. J
Korean Med Sci. 5:225–231. 1990. View Article : Google Scholar
|
|
3
|
Wang R, Chadalavada K, Wilshire J, Kowalik
U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C and
Tabar V: Glioblastoma stem-like cells give rise to tumour
endothelium. Nature. 468:829–833. 2010. View Article : Google Scholar
|
|
4
|
Linkous AG and Yazlovitskaya EM:
Angiogenesis in glioblastoma multiforme: Navigating the maze.
Anticancer Agents Med Chem. 11:712–718. 2011. View Article : Google Scholar
|
|
5
|
Van Tellingen O, Yetkin-Arik B, de Gooijer
MC, Wesseling P, Wurdinger T and de Vries HE: Overcoming the
blood-brain tumor barrier for effective glioblastoma treatment.
Drug Resist Updat. 19:1–12. 2015. View Article : Google Scholar
|
|
6
|
Huang WJ, Chen WW and Zhang X:
Glioblastoma multiforme: Effect of hypoxia and hypoxia inducible
factors on therapeutic approaches. Oncol Lett. 12:2283–2288. 2016.
View Article : Google Scholar
|
|
7
|
Thomas AA, Ernstoff MS and Fadul CE:
Immunotherapy for the treatment of glioblastoma. Cancer J.
18:59–68. 2012. View Article : Google Scholar
|
|
8
|
Jovčevska I, Kočevar N and Komel R: Glioma
and glioblastoma-how much do we (not) know? Mol Clin Oncol.
1:935–941. 2013. View Article : Google Scholar
|
|
9
|
Sui X, Chen R, Wang Z, Huang Z, Kong N,
Zhang M, Han W, Lou F, Yang J, Zhang Q, et al: Autophagy and
chemotherapy resistance: A promising therapeutic target for cancer
treatment. Cell Death Dis. 4:e8382013. View Article : Google Scholar
|
|
10
|
Zhang J, Stevens MF and Bradshaw TD:
Temozolomide: Mechanisms of action, repair and resistance. Curr Mol
Pharmacol. 5:102–114. 2012. View Article : Google Scholar
|
|
11
|
Beier D, Schulz JB and Beier CP:
Chemoresistance of glioblastoma cancer stem cells-much more complex
than expected. Mol Cancer. 10:1282011. View Article : Google Scholar
|
|
12
|
Wilson TA, Karajannis MA and Harter DH:
Glioblastoma multiforme: State of the art and future therapeutics.
Surg Neurol Int. 5:642014. View Article : Google Scholar
|
|
13
|
Haar CP, Hebbar P, Wallace GC IV, Das A,
Vandergrift WA III, Smith JA, Giglio P, Patel SJ, Ray SK and Banik
NL: Drug resistance in glioblastoma: A mini review. Neurochem Res.
37:1192–1200. 2012. View Article : Google Scholar
|
|
14
|
Stupp R, Hegi ME, Mason WP, van den Bent
MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B,
Belanger K, et al: Effects of radiotherapy with concomitant and
adjuvant temozolomide versus radiotherapy alone on survival in
glioblastoma in a randomised phase III study: 5-year analysis of
the EORTC-NCIC trial. Lancet Oncol. 10:459–466. 2009. View Article : Google Scholar
|
|
15
|
Kaza N, Kohli L and Roth KA: Autophagy in
brain tumors: A new target for therapeutic intervention. Brain
Pathol. 22:89–98. 2012. View Article : Google Scholar
|
|
16
|
Cheng CK, Fan QW and Weiss WA: PI3K
signaling in glioma-animal models and therapeutic challenges. Brain
Pathol. 19:112–120. 2009. View Article : Google Scholar
|
|
17
|
Carrasco-García E, Saceda M, Grasso S,
Rocamora-Reverte L, Conde M, Gómez-Martínez A, García-Morales P,
Ferragut JA and Martínez-Lacaci I: Small tyrosine kinase inhibitors
interrupt EGFR signaling by interacting with erbB3 and erbB4 in
glioblastoma cell lines. Exp Cell Res. 317:1476–1489. 2011.
View Article : Google Scholar
|
|
18
|
Rich JN, Reardon DA, Peery T, Dowell JM,
Quinn JA, Penne KL, Wikstrand CJ, Van Duyn LB, Dancey JE, McLendon
RE, et al: Phase II trial of gefitinib in recurrent glioblastoma. J
Clin Oncol. 22:133–142. 2004. View Article : Google Scholar
|
|
19
|
Fan QW, Knight ZA, Goldenberg DD, Yu W,
Mostov KE, Stokoe D, Shokat KM and Weiss WA: A dual PI3 kinase/mTOR
inhibitor reveals emergent efficacy in glioma. Cancer Cell.
9:341–349. 2006. View Article : Google Scholar
|
|
20
|
Fan QW, Cheng C, Hackett C, Feldman M,
Houseman BT, Nicolaides T, Haas-Kogan D, James CD, Oakes SA,
Debnath J, et al: Akt and autophagy cooperate to promote survival
of drug-resistant glioma. Sci Signal. 3:ra812010. View Article : Google Scholar
|
|
21
|
Duvic M, Talpur R, Ni X, Zhang C, Hazarika
P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM and Frankel
SR: Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic
acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood.
109:31–39. 2007. View Article : Google Scholar
|
|
22
|
Munster PN, Thurn KT, Thomas S, Raha P,
Lacevic M, Miller A, Melisko M, Ismail-Khan R, Rugo H, Moasser M
and Minton SE: A phase II study of the histone deacetylase
inhibitor vorinostat combined with tamoxifen for the treatment of
patients with hormone therapy-resistant breast cancer. Br J Cancer.
104:1828–1835. 2011. View Article : Google Scholar
|
|
23
|
Shao Y, Gao Z, Marks PA and Jiang X:
Apoptotic and autophagic cell death induced by histone deacetylase
inhibitors. Proc Natl Acad Sci USA. 101:18030–18035. 2004.
View Article : Google Scholar
|
|
24
|
Hrzenjak A, Kremser ML, Strohmeier B,
Moinfar F, Zatloukal K and Denk H: SAHA induces
caspase-independent, autophagic cell death of endometrial stromal
sarcoma cells by influencing the mTOR pathway. J Pathol.
216:495–504. 2008. View Article : Google Scholar
|
|
25
|
Yonekawa T and Thorburn A: Autophagy and
cell death. Essays Biochem. 55:105–117. 2013. View Article : Google Scholar
|
|
26
|
Kim EL, Wüstenberg R, Rübsam A,
Schmitz-Salue C, Warnecke G, Bücker EM, Pettkus N, Speidel D, Rohde
V, Schulz-Schaeffer W, et al: Chloroquine activates the p53 pathway
and induces apoptosis in human glioma cells. Neuro Oncol.
12:389–400. 2010. View Article : Google Scholar
|
|
27
|
Chowdhury R, Chowdhury S, Roychoudhury P,
Mandal C and Chaudhuri K: Arsenic induced apoptosis in malignant
melanoma cells is enhanced by menadione through ROS generation, p38
signaling and p53 activation. Apoptosis. 14:108–123. 2009.
View Article : Google Scholar
|
|
28
|
Pajaniradje S, Mohankumar K, Pamidimukkala
R, Subramanian S and Rajagopalan R: Antiproliferative and apoptotic
effects of Sesbania grandiflora leaves in human cancer cells.
Biomed Res Int. 2014:4749532014. View Article : Google Scholar
|
|
29
|
Laporte AN, Barrott JJ, Yao RJ, Poulin NM,
Brodin BA, Jones KB, Underhill TM and Nielsen TO: HDAC and
proteasome inhibitors synergize to activate pro-apoptotic factors
in synovial sarcoma. PLoS One. 12:e01694072017. View Article : Google Scholar
|
|
30
|
Mizushima N: Methods for monitoring
autophagy. Int J Biochem Cell Biol. 36:2491–2502. 2004. View Article : Google Scholar
|
|
31
|
Munafó DB and Colombo MI: A novel assay to
study autophagy: Regulation of autophagosome vacuole size by amino
acid deprivation. J Cell Sci. 114:3619–3629. 2001.
|
|
32
|
Rodriguez-Enriquez S, Kim I, Currin RT and
Lemasters JJ: Tracker dyes to probe mitochondrial autophagy
(mitophagy) in rat hepatocytes. Autophagy. 2:39–46. 2006.
View Article : Google Scholar
|
|
33
|
Adamson C, Kanu OO, Mehta AI, Di C, Lin N,
Mattox AK and Bigner DD: Glioblastoma multiforme: A review of where
we have been and where we are going. Expert Opin Investig Drugs.
18:1061–1083. 2009. View Article : Google Scholar
|
|
34
|
Kanzawa T, Bedwell J, Kondo Y, Kondo S and
Germano IM: Inhibition of DNA repair for sensitizing resistant
glioma cells to temozolomide. J Neurosurg. 99:1047–1052. 2003.
View Article : Google Scholar
|
|
35
|
Fu J, Liu ZG, Liu XM, Chen FR, Shi HL,
Pangjesse CS, Ng HK and Chen ZP: Glioblastoma stem cells resistant
to temozolomide-induced autophagy. Chin Med J (Engl).
122:1255–1259. 2009.
|
|
36
|
Wojton J, Elder J and Kaur B: How
efficient are autophagy inhibitors as treatment for glioblastoma?
CNS Oncol. 3:5–7. 2014. View Article : Google Scholar
|
|
37
|
Hegi ME, Liu L, Herman JG, Stupp R, Wick
W, Weller M, Mehta MP and Gilbert MR: Correlation of
O6-methylguanine methyltransferase (MGMT) promoter methylation with
clinical outcomes in glioblastoma and clinical strategies to
modulate MGMT activity. J Clin Oncol. 26:4189–4199. 2008.
View Article : Google Scholar
|
|
38
|
Liu YL, Yang PM, Shun CT, Wu MS, Weng JR
and Chen CC: Autophagy potentiates the anti-cancer effects of the
histone deacetylase inhibitors in hepatocellular carcinoma.
Autophagy. 6:1057–1065. 2010. View Article : Google Scholar
|
|
39
|
Gammoh N, Lam D, Puente C, Ganley I, Marks
PA and Jiang X: Role of autophagy in histone deacetylase
inhibitor-induced apoptotic and nonapoptotic cell death. Proc Natl
Acad Sci USA. 109:6561–6565. 2012. View Article : Google Scholar
|
|
40
|
Guha S, Coffey EE, Lu W, Lim JC, Beckel
JM, Laties AM, Boesze-Battaglia K and Mitchell CH: Approaches for
detecting lysosomal alkalinization and impaired degradation in
fresh and cultured RPE cells: Evidence for a role in retinal
degenerations. Exp Eye Res. 126:68–76. 2014. View Article : Google Scholar
|
|
41
|
Perry CN, Kyoi S, Hariharan N, Takagi H,
Sadoshima J and Gottlieb RA: Novel methods for measuring cardiac
autophagy in vivo. Methods Enzymol. 453:325–342. 2009. View Article : Google Scholar
|
|
42
|
Iwai-Kanai E, Yuan H, Huang C, Sayen MR,
Perry-Garza CN, Kim L and Gottlieb RA: A method to measure cardiac
autophagic flux in vivo. Autophagy. 4:322–329. 2008. View Article : Google Scholar
|
|
43
|
Kim HJ and Bae SC: Histone deacetylase
inhibitors: molecular mechanisms of action and clinical trials as
anti-cancer drugs. Am J Transl Res. 3:166–179. 2011.
|
|
44
|
Gomes LR, Vessoni AT and Menck CFM:
Microenvironment and autophagy cross-talk: Implications in cancer
therapy. Pharmacol Res. 107:300–307. 2016. View Article : Google Scholar
|
|
45
|
Poillet-Perez L, Despouy G,
Delage-Mourroux R and Boyer-Guittaut M: Interplay between ROS and
autophagy in cancer cells, from tumor initiation to cancer therapy.
Redox Biol. 4:184–192. 2015. View Article : Google Scholar
|
|
46
|
Ruefli AA, Ausserlechner MJ, Bernhard D,
Sutton VR, Tainton KM, Kofler R, Smyth MJ and Johnstone RW: The
histone deacetylase inhibitor and chemotherapeutic agent
suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway
characterized by cleavage of Bid and production of reactive oxygen
species. Proc Natl Acad Sci USA. 98:10833–10838. 2001. View Article : Google Scholar
|
|
47
|
Ray S, Bonafede MM and Mohile NA:
Treatment patterns, survival, and healthcare costs of patients with
malignant gliomas in a large US commercially insured population. Am
Health Drug Benefits. 7:140–149. 2014.
|
|
48
|
Zhang J, Stevens MF, Laughton CA,
Madhusudan S and Bradshaw TD: Acquired resistance to temozolomide
in glioma cell lines: Molecular mechanisms and potential
translational applications. Oncology. 78:103–114. 2010. View Article : Google Scholar
|
|
49
|
Fulda S and Kögel D: Cell death by
autophagy: Emerging molecular mechanisms and implications for
cancer therapy. Oncogene. 34:5105–5113. 2015. View Article : Google Scholar
|
|
50
|
Katayama M, Kawaguchi T, Berger M and
Pieper R: DNA damaging agent-induced autophagy produces a
cytoprotective adenosine triphosphate surge in malignant glioma
cells. Cell Death Differ. 14:548–558. 2007. View Article : Google Scholar
|
|
51
|
Voss V, Senft C, Lang V, Ronellenfitsch
MW, Steinbach JP, Seifert V and Kögel D: The pan-Bcl-2 inhibitor
(−)-gossypol triggers autophagic cell death in malignant glioma.
Mol Cancer Res. 8:1002–1016. 2010. View Article : Google Scholar
|
|
52
|
Falkenberg KJ and Johnstone RW: Histone
deacetylases and their inhibitors in cancer, neurological diseases
and immune disorders. Nat Rev Drug Discov. 13:673–691. 2014.
View Article : Google Scholar
|
|
53
|
Lee YJ, Won AJ, Lee J, Jung JH, Yoon S,
Lee BM and Kim HS: Molecular mechanism of SAHA on regulation of
autophagic cell death in tamoxifen-resistant MCF-7 breast cancer
cells. Int J Med Sci. 9:881–993. 2012. View Article : Google Scholar
|
|
54
|
Carew JS, Nawrocki ST, Kahue CN, Zhang H,
Yang C, Chung L, Houghton JA, Huang P, Giles FJ and Cleveland JL:
Targeting autophagy augments the anticancer activity of the histone
deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug
resistance. Blood. 110:313–322. 2007. View Article : Google Scholar
|
|
55
|
Rosenfeld MR, Ye X, Supko JG, Desideri S,
Grossman SA, Brem S, Mikkelson T, Wang D, Chang YC, Hu J, et al: A
phase I/II trial of hydroxychloroquine in conjunction with
radiation therapy and concurrent and adjuvant temozolomide in
patients with newly diagnosed glioblastoma multiforme. Autophagy.
10:1359–1368. 2014. View Article : Google Scholar
|
|
56
|
Das G, Shravage BV and Baehrecke EH:
Regulation and function of autophagy during cell survival and cell
death. Cold Spring Harb Perspect Biol. 4:pii: 008813. 2012.
View Article : Google Scholar
|
|
57
|
Navarro-Yepes J, Burns M, Anandhan A,
Khalimonchuk O, del Razo LM, Quintanilla-Vega B, Pappa A,
Panayiotidis MI and Franco R: Oxidative stress, redox signaling,
and autophagy: Cell death versus survival. Antioxid Redox Signal.
21:66–85. 2014. View Article : Google Scholar
|
|
58
|
Gong C, Song E, Codogno P and Mehrpour M:
The roles of BECN1 and autophagy in cancer are context dependent.
Autophagy. 8:1853–1855. 2012. View Article : Google Scholar
|
|
59
|
White E: Deconvoluting the
context-dependent role for autophagy in cancer. Nat Rev Cancer.
12:401–410. 2012. View Article : Google Scholar
|
|
60
|
White E: The role for autophagy in cancer.
J Clin Invest. 125:42–46. 2015. View Article : Google Scholar
|