Introduction
Ovarian carcinoma represents the sixth most common
type of cancer and the fifth leading cause of cancer-associated
deaths in developed countries. It affects ~204,000 women annually
worldwide, and is responsible for ~125,000 deaths (1). Among the drugs currently used in
ovarian cancer therapy are taxanes, a family of natural diterpenes
that bind tubulin and promote microtubule stabilization in the
polymerized state. One of these agents is paclitaxel (PTX), a
natural product present in the Pacific yew, Taxus
brevifolia. By causing microtubule stabilization, PTX blocks
cell cycle progression at the G2/M phase, inhibiting
proliferation and inducing cellular apoptosis (2–4). While
chemotherapy is the most common approach to cancer treatment, its
efficacy often includes drawbacks. One of these is the necessity of
dose reduction in order to avoid unwanted toxicity, which limits
the drug efficacy in monotherapy. Another is the generation of drug
resistance upon prolonged treatment; prolonged PTX treatment may
induce a multi-drug resistance phenotype, which represents a
serious obstacle in ovarian cancer treatment (5–7).
Hence, there is an urgent requirement to implement novel strategies
to potentiate PTX efficacy, allowing a decrease in the effective
dosage and the circumvention of resistance mechanisms.
Polyphenols and phenolic compounds represent a large
collection of molecules present in the plant kingdom. At the low
doses attainable through dietary intake, these compounds exert
multiple protective functions, including against inflammation and
tumorigenesis. Conversely, at high, albeit still pharmacologically
attainable, concentrations, many phenolic compounds may promote the
death of cancer cells, modulating key elements in signal
transduction pathways linked to apoptosis (8). In this regard, gallic acid (GA), a
phenol of natural origin with antioxidant activity, isolated from
Caesalpinia mimosoides, exerts antitumor action in
cholangiocarcinoma cell lines. GA was found to induce cell death in
promyelocytic leukemia HL-60RG cells; morphological and biochemical
studies indicated that the induced cell death occurs via apoptosis
(9,10). Concerning biochemical mechanisms,
phenols normally behave as antioxidant molecules, which may account
for the aforementioned protective actions; but under some
circumstances they may also behave as pro-oxidant agents. GA has
been shown to prevent oxidative stress, but also to increase ROS
production, depending on the experimental conditions (11,12).
This is notable, since ROS are involved in the regulation of many
physiological and pathological processes, including cell
proliferation, invasion, cell death, tumor hypoxia, and drug
resistance (13,14). A number of these effects can be
explained by ROS-mediated upregulation or downregulation of
critical protein kinase activities, such as PI3K/Akt, MEK/ERK and
p38-MAPK (15–17). With these antecedents, we aimed to
analyze the capacity of GA to improve the anti-proliferative action
of PTX in ovarian carcinoma cells. Two cell models were used,
namely the A2780 cell line and a doxorubicin-resistant variant
A2780AD, which overexpresses P-glycoprotein (18,19).
Our results indicated that A2780AD cells are less sensitive to PTX
than A2780 cells, and that the cytostatic action of PTX is
increased in both cell models via co-treatment with GA, which
potentiates the PTX-induced G2/M phase arrest. Using the
drug-resistant cell line, we also demonstrated that proliferation
inhibition and G2/M phase arrest are mediated by
GA-provoked ROS overproduction, and by ROS-mediated inhibition of
PTX-provoked ERK activation.
Materials and methods
Reagents and antibodies
All components for cell culture were obtained from
Invitrogen (Thermo Fisher Scientific, Inc., Waltham, MA, USA).
2′7′-Dichlorodihydrofluorescein diacetate (H2DCFDA) and
propidium iodide (PI) were obtained from Molecular Probes (Thermo
Fisher Scientific, Inc., Eugene, OR, USA). The protein kinase
inhibitors SB203580, PD98059 and SP600125, and the rabbit
polyclonal antibodies (pAbs) phospho-p38-MAPK (Thr180/Tyr182)
(catalog #9211), phospho-Akt (Ser473) (catalog #4060), and
phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (catalog #4370), were
obtained from Cell Signaling Technology (Danvers, MA, USA).
Peroxidase-conjugated immunoglobulin G antibodies were obtained
from Dako Diagnostics, S.A. (Barcelona, Spain). N-acetyl
cysteine (NAC), GA and PTX were obtained from Sigma-Aldrich Quimica
SL (Madrid, Spain). GA and PTX were dissolved in deuterated
dimethylsulfoxide (DMSO-D6) at 20 mM and the final concentration
was obtained by successive dilutions with culture medium.
Cells
The human ovarian carcinoma A2780 cells
(drug-sensitive) and A2780AD cells (multi-drug-resistant ovarian
cancer) included in this study were a generous gift from Dr P.
Giannakakou, Weill Cornell Medical College (New York, NY, USA).
Cell lines were maintained in RPMI-1640 medium supplemented with
10% fetal bovine serum (FBS), 2 mM L-glutamine, 40 µg/ml gentamicin
and penicillin-streptomycin (100 U/ml penicillin and 100 µg/ml
streptomycin) at 37°C in a humidified atmosphere of 5%
CO2.
Cell viability
To determine the cytotoxic effect of the compounds
on cell lines A2780 and A2780AD, 10,000 cells/well were seeded in a
96-well cell culture plate, and treated for 48 h with the desired
concentration of GA and PTX, alone or in combination. At the end of
treatment, 20 µl MTT (2.5 mg/ml) was added to each well and the
plate was incubated for 4 h at 37°C. The reaction was then stopped
by adding 100 µl of MTT solubilizer 10% SDS (sodium dodecyl
sulfate) and 45% DMF (N,N-dimethylformamide) pH 5.5 (20). The plate was incubated at 37°C
overnight to dissolve the formazan precipitates, and the absorbance
of each well was measured at a wavelength of 595/690 nm in an
Appliskan (Thermo Electro-corporation, Vantaa, Finland) plate
reader. The targets used were wells without cells and the growth
controls were wells with cells containing the same proportion of
DMSO present in the wells with the treatments. The results were
analyzed in the GraphPad Prism 5 software (GraphPad Software, Inc.,
La Jolla, CA, USA) and were expressed as the mean ± standard error
of several independent experiments.
ROS production
To measure the drug effects on the relative
intracellular ROS accumulation, at the end of the treatments (24
h), the cells were washed twice with PBS and incubated for 30 min
at 37°C with RPMI-1640 medium containing 5 µM H2DCFDA, a
non-specific ROS-sensitive probe. H2DCFDA in the cell is
cleaved by intracellular esterases, producing the
membrane-impermeable product H2DCF, which accumulates
into the cell. H2DCF is not a fluorescent molecule, but
is oxidized by intracellular ROS to give the fluorescent product
DCF (21). The resulting
fluorescence was then measured in a fluorimeter FluoroMax-2 (Horiba
Scientific, Minami-ku Kyoto, Japan).
Flow cytometry
The effects of the drug on cell cycle progression
were determined via flow cytometry. Following treatments (24 h),
the cells were washed with PBS and fixed in 70% ethanol at 4°C for
at least 1 h. The cells were then washed twice with PBS,
resuspended in 500 µl PBS containing 60 µl/ml DNase-free RNase A
and 50 µl/ml PI, and the fluorescence was measured using a Coulter
Epics XL flow cytometer (Beckman Coulter Inc., Brea, CA, USA), as
previously described (22). The
resulting flow cytometry histograms were analyzed with the
FlowLogic 7.0 program (Iniwai, Victoria, Canada), to obtain the
relative percentages of cells at the G1, S and
G2/M phases of the cell cycle. In addition to the
typical cell cycle phases, the flow cytometry histograms showed a
sub-G1 population. This suggests reduction and damage of
DNA related to cell death (23).
Western blotting
Following treatments (24 h), the cells were
collected and lysed in lysis buffer (Tris-HCl 20 mM, pH 7.5,
glycerol 10%, NaCl 137 mM, NP40 1%) containing a protease inhibitor
cocktail (Thermo Fisher Scientific, Inc.), and the proteins were
quantified. Equal amounts of protein were dissolved in 2X Laemmli
buffer with β-mercaptoethanol, heated to 95°C for 3 min and
resolved via 10% SDS-PAGE. After electrophoretic separation, the
proteins were transferred to a PDVF membrane using the
Trans-Blot® Turbo™ Transfer System (Bio-Rad, Hercules,
CA, USA), following which the membranes were blocked with 5% milk
in TBS-Tween 20 for 1 h, and then incubated overnight at 4°C with
the following primary antibodies: phospho-p38-MAPK (Thr180/Tyr182),
1:100 dilution; phospho-Akt (Ser473), 1:200 dilution;
phospho-p44/42 MAPK (Erk1/2) Thr202/Tyr204, dilution 1:1,000. The
membrane was then washed twice in TBS-Tween, and incubated for 1 h
at room temperature with the corresponding secondary antibody
(anti-rabbit IgG, HRP-linked, dilution 1:2,000). The proteins were
visualized using the ChemiDoc Touch Imaging System (Bio-Rad
Laboratories, Inc.), using NZY Supreme ECL HRP Substrate (NZYTech,
Lda., Lisbon, Portugal) as a developer. β-actin was used as the
loading control.
Statistical analysis
Data were analyzed using the GraphPad Prism 5.0
statistical program (GraphPad Software, Inc.), and statistical
differences between groups were evaluated using one-way analysis of
variance followed by the Dunnett's test. P<0.05 was considered
to indicate a statistically significant difference.
Results
Cell proliferation
As an initial approach, we aimed to comparatively
examine the effect of GA, in a concentration range of 10–200 µM, on
total cell proliferation activity, as measured by the number of
viable cells in sensitive (A2780) and multi-drug-resistant
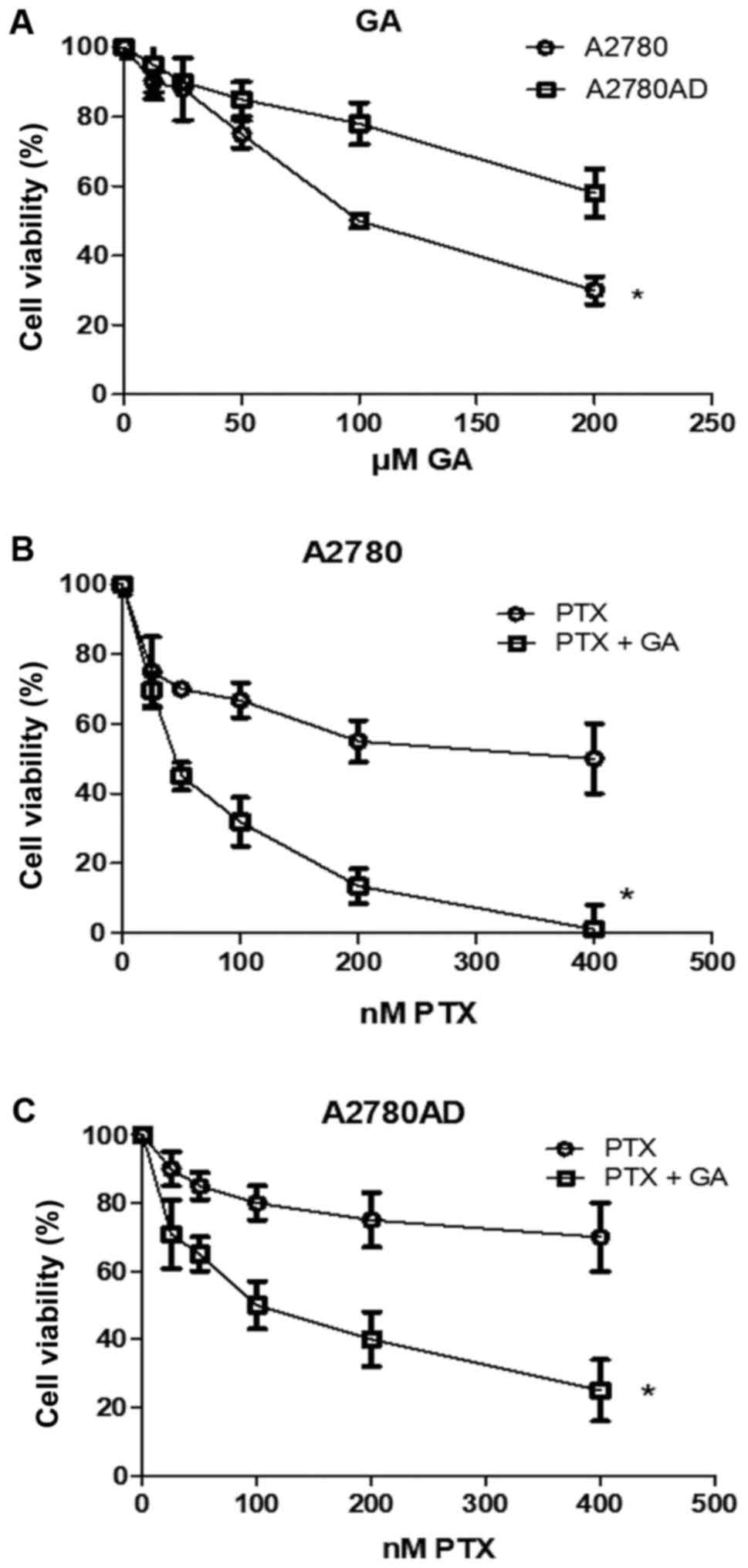
(A2780AD) ovarian carcinoma cell cultures. As indicated in Fig. 1A, GA caused a
concentration-dependent reduction in the percentage of viable
cells, an effect that was of lower intensity in the resistant cell
line. The approximate reduction rates were 25% with 50 µM GA in
A2780 cells, and 20% with 100 µM GA in A2780AD cells. These
concentrations were selected for the combination treatment assays
with the respective cell lines in the following determinations of
ROS generation, cell cycle and protein kinases.
Subsequently, we comparatively analyzed the effect
of PTX, at concentrations ranging from 10–400 nM, alone and in
combination with the previously indicated concentrations of GA. As
depicted in Fig. 1B and C, PTX
caused a drug-dependent decrease in the number of viable cells,
which was less pronounced in the resistant cell line. Thus,
treatment with 50 nM PTX caused an ~30% decrease in the number of
A2780 cells, whereas 200 nM caused only an ~20% decrease in the
number of A2780AD cells. In addition, we observed that PTX and GA
cooperated in more than an additive manner to inhibit cell
viability, even in the resistant A2780AD cell line. For example, 50
nM PTX plus 50 µM GA caused an almost 60% decrease in the number of
viable cells in the A2780 cell cultures; and the same result was
obtained using 200 nM PTX plus 100 µM GA in the A2780AD cell
cultures. Therefore, the concentrations of 50 and 200 nM PTX were
adopted for further experiments with the A2780 and A2780AD cell
lines, respectively.
ROS generation
As aforementioned, phenolic agents often provoke
oxidative stress in cultured tumor cells, which may be a
determinant of proliferation inhibition and cell death. The
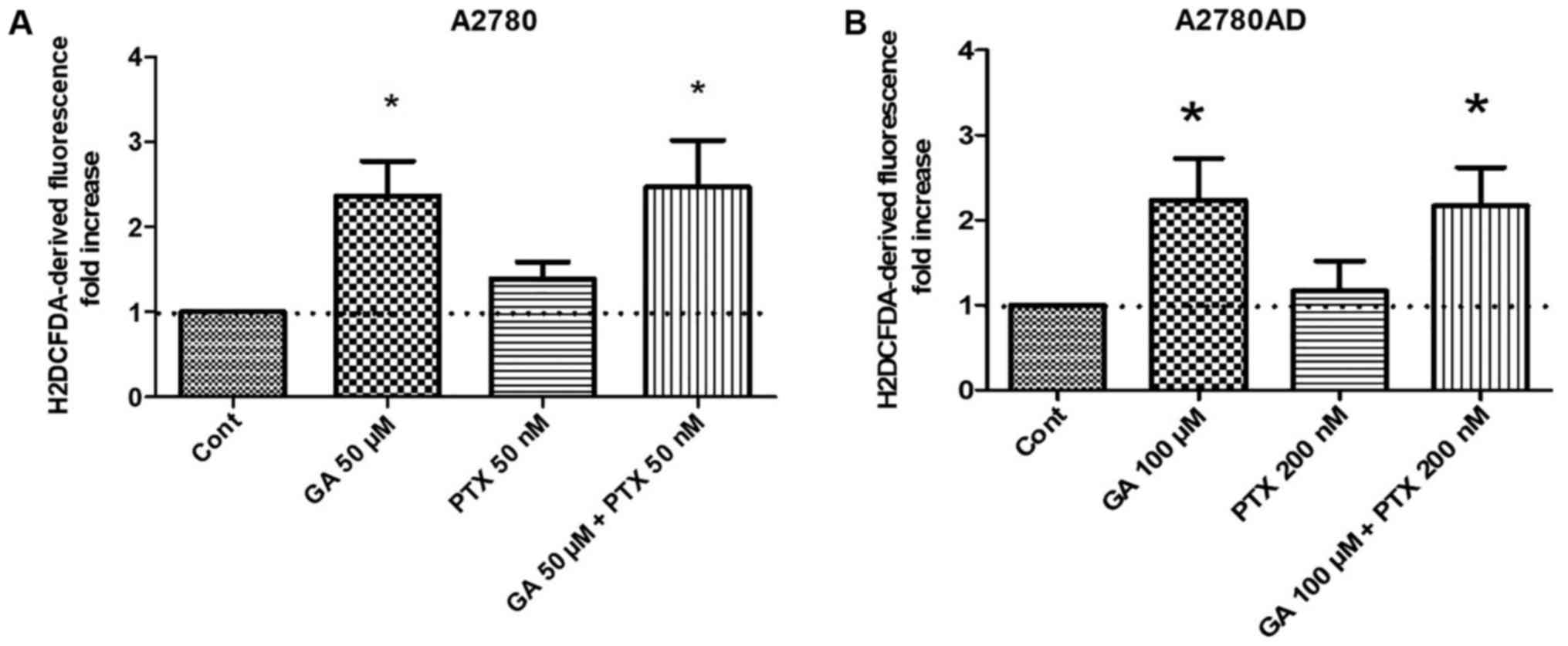
generation of oxidative stress by GA and PTX, evaluated alone and
in combination, was determined by measuring ROS production, using
an H2DCFDA ROS-sensitive probe. The results are
indicated in Fig. 2A and B.
Treatment with PTX alone did not significantly affect ROS
production, while treatment with GA, either alone or in combination
with PTX, caused an approximate 2-fold increase. At the drug
concentrations used, the results were approximately the same in the
two cell lines.
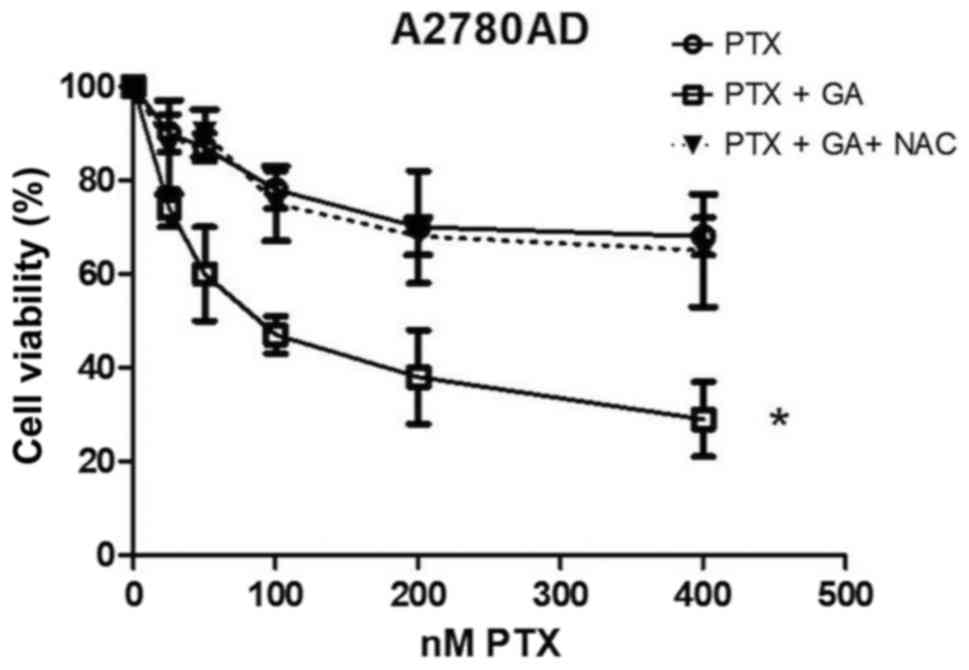
In order to investigate the role of ROS
overproduction in the inhibition of cell proliferation, we used the
antioxidant/ROS-scavenging agent NAC. The results presented in
Fig. 3 indicate that NAC markedly
attenuated the decrease in viability caused by the combination of
PTX plus GA in the A2780AD cell cultures, in such a manner that the
decrease in viability caused by the NAC + PTX + GA triple
combination was similar to that obtained with PTX alone. This seems
congruent with the observation that GA causes ROS overproduction,
while PTX does not. Taken together, the results in Figs. 2 and 3 indicate that ROS overproduction is an
essential determinant of the capacity of GA to potentiate PTX
toxicity in drug-resistant ovarian cancer cells.
Cell cycle distribution
PTX is a potent microtubule-targeting agent known to
cause mitotic cell cycle arrest (3). After establishing the inhibitory
action of PTX and GA on total cell proliferation activity, as well
as the protective action of NAC, we aimed to comparatively analyze
the effects of these agents on cell-cycle progression in
drug-resistant A2780AD cells. The flow cytometry assay results are
indicated in Fig. 4. As
hypothesized, treatment with PTX increased the proportion of cells
in the G2/M phase, a response that was potentiated by
co-treatment with GA. Of note, G2/M phase accumulation
was suppressed by the presence of NAC.
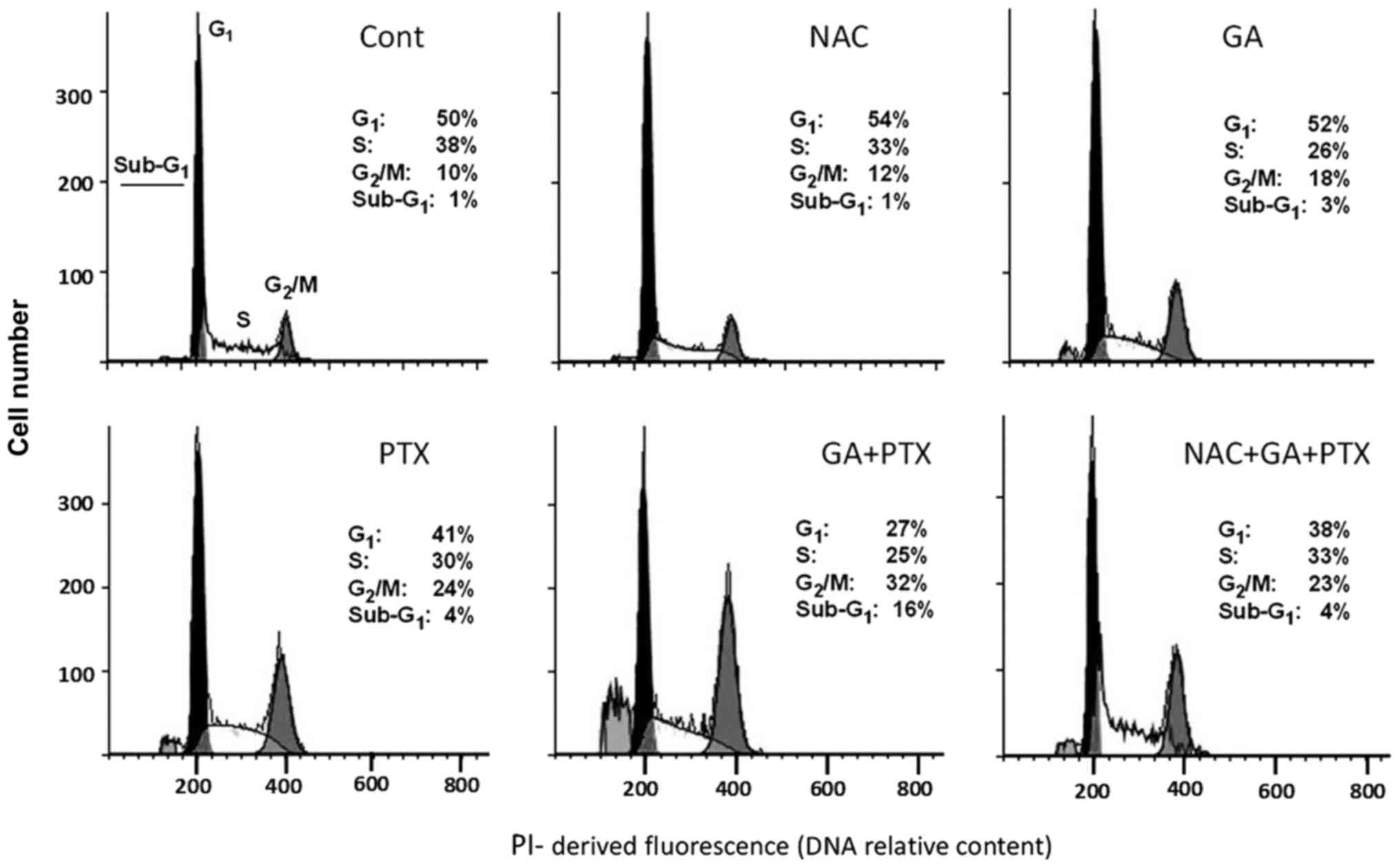 | Figure 4.Cell cycle disruption by antitumor
drugs and antioxidant agent. The figures show the percentages of
cells in the G0/G1, S and G2/M
cycle cycle phases, and of cells with sub-G1 DNA
content, in untreated A2780AD cell cultures (Cont) and cultures
treated for 24 h with NAC (5 mM), GA (100 µM), and PTX (200 nM),
either alone or in combination. NAC was applied 2 h before GA and
PTX. The histograms and cell frequencies are representative of one
of three determinations, with similar results. GA, gallic acid;
PTX, paclitaxel; NAC, N-acetyl-L-cysteine. |
In addition to discerning the distribution of the
typical cell cycle phases (G1, S and G2/M), a
flow cytometry assay can also reveal a subpopulation
(sub-G1) of cells with reduced DNA content, normally
interpreted as dead cells (23). In
the present study, this subpopulation was negligible in cells
treated with PTX and GA alone; but it reached a modest, albeit
significant, value upon treatment with GA plus PTX. As in the case
of G2/M, the sub-G1 fraction was suppressed
by co-treatment with NAC. Taken together, these results suggest
that the decrease in total cell proliferation in the PTX plus
GA-treated cultures (Fig. 1B and C)
is a consequence of both cell cycle blockade and induced cell
death.
Protein kinases
It is known that Akt and ERK normally function as
defensive protein kinases, which prevent apoptotic cell death while
favoring cell proliferation/cell cycle progression (24–26).
In addition, MAPK p38 is also activated in response to oxidative
stress (27,28), and may behave as either a positive
or negative regulator of apoptosis and the cell cycle, depending on
the cell line and the experimental conditions (29,30).
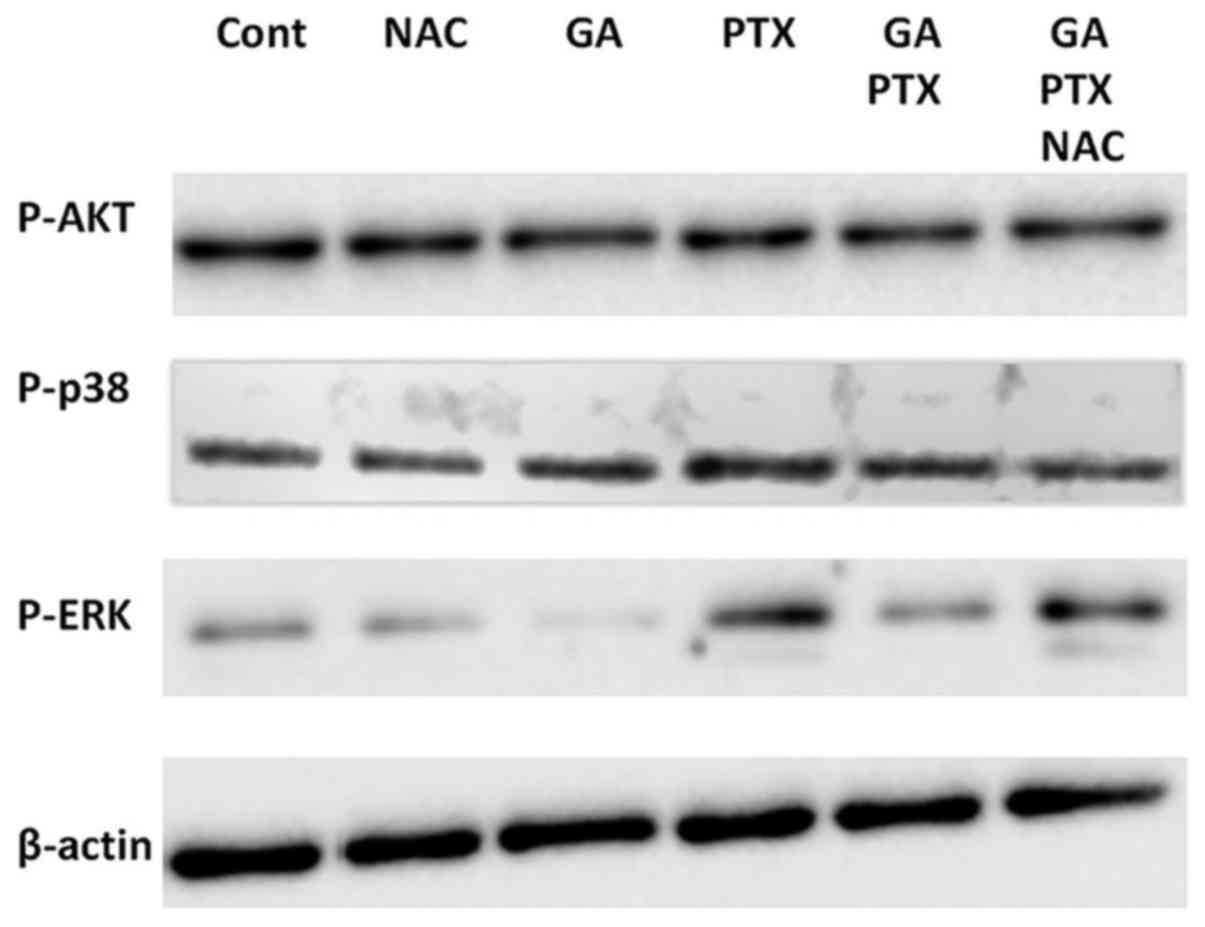
Thus, western blot assays were carried out in A2780AD cells in
order to investigate the phosphorylation/activation of these
kinases in response to GA and PTX, alone and in combination, and in
the absence or the presence of NAC. The results depicted in
Fig. 5 indicate that these
treatments did not significantly affect Akt and p38
phosphorylation. In contrast, we observed that PTX stimulated ERK
phosphorylation and that GA (which increased ROS production;
Fig. 2) decreased the basal
phosphorylation level of ERK and prevented the aforementioned
PTX-induced increase. We also observed that the inhibitory action
of GA in combination therapy was suppressed by the presence of NAC.
Taken together, these results suggest that ROS negatively regulated
ERK activation under experimental conditions used in the present
study.
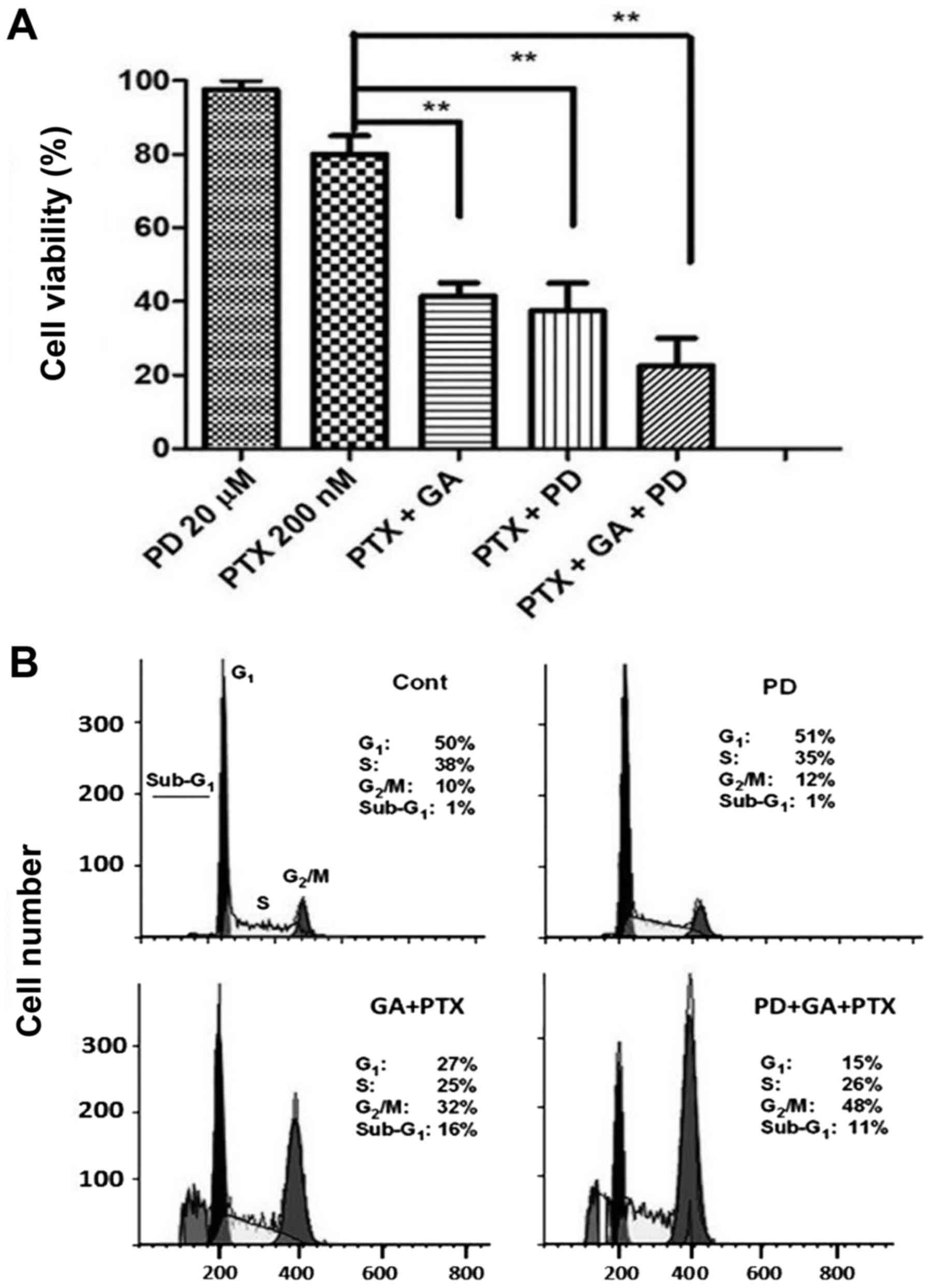
Finally, the functional importance of ERK activation
as a regulator of cell proliferation and cell cycle progression was
examined using the MEK/ERK inhibitor PD98059 (20 µM). It was
observed that PD98059, which was innocuous per se,
potentiated the PTX-induced decrease in cell viability, alone and
in combination with GA (Fig. 6A).
Accordingly, PD98059 potentiated the G2/M arrest caused
by GA plus PTX, although it did not potentiate, and even slightly
reduced, the size of the sub-G1 cell fraction (Fig. 6B). Taken together, these results
indicate that ERK activation exerts a defensive role, attenuating
the excessive cytostatic action induced by PTX in A2780AD
cells.
Discussion
The efficacy of chemotherapy is frequently hampered
by the limited potency of antitumor drugs when used alone, and by
the acquisition of a multi-drug-resistant phenotype after prolonged
exposure. The use of adjuvants, such as selected phenolic
compounds, may help to partially overcome these issues. To date,
there has been great interest in the study of these compounds,
particularly regarding their application in cancer therapies, alone
or as adjuvants, and for their chemosensitizing effects (31–33).
In the present study, we investigated the ability of
gallic acid (GA), a phenolic compound, to potentiate the
anti-proliferative action of PTX using two ovarian carcinoma cell
models: A2780 drug-sensitive cells and A2780AD cells, a
drug-resistant variant due to P-glycoprotein overexpression. Our
results showed that the anti-proliferative action of PTX was
reduced in A2780AD cells, concordant with the multi-drug-resistance
phenotype. In addition, the anti-proliferative action corresponded
to cell cycle arrest in the G2/M phase, which reflects
the action of PTX as a microtubule stabilizer. Notably,
co-treatment with GA potentiated the anti-proliferative action and
G2/M phase arrest induced by PTX in the A2780AD cells,
in which it was demonstrated that the efficacy of PTX monotherapy
was low. Furthermore, in the combined treatment, we observed an
accumulation of cells in the sub-G1 phase, suggesting
the induction of cell death. Prior reports demonstrated the
antitumor effect of GA in certain cancer cell lines, and indicate
that it could be a chemo-sensitizer and potentiate the cytotoxicity
of PTX in ovarian cancer cells with a resistance phenotype
(34,35).
ROS are a by-product of the respiratory chain, and
at low levels have positive effects on cells (mitogenic, signaling,
etc.). However, many agents, including phenolic compounds under
certain conditions, can overinduce ROS, which can have deleterious
effects on cells (36,37). It is known that GA has both
antioxidant and pro-oxidant activities, which are responsible for
inducing death in certain cancer cells (11). Our results showed that GA in A2780
and A2780AD cells induced the generation of ROS, presenting a
pro-oxidant effect, and that co-treatment with NAC greatly
attenuated the reduction in the number of viable A2780AD cells
caused by PTX plus GA; in fact A2780AD cells exhibited similar
reponse to treatment with PTX alone. This pro-oxidant effect of GA
has also been shown in A549 lung cancer cells, wherein GA inhibited
cell growth and increased the production of intracellular ROS
(38–40). Prior treatment of A2780AD cells with
the antioxidant NAC prevented the combined effect of GA plus PTX on
the G2/M phase, showing a similar effect in those cells
treated with only PTX.
The protective action of NAC confirms that ROS
effectively mediate the anti-proliferative action and
G2/M phase arrest observed with the combined
treatment.
It is known that ROS can regulate signaling pathways
that promote cell survival, including the mitogen-activated protein
kinase (MAPK) signaling pathway, which includes ERK and p38.
Reports indicate that the downregulation of these pathways is
important for inducing cell-cycle arrest and cell death (17,24).
We examined the actions of p38, Akt and ERK inhibitors, and the
results of western blot analysis revealed modulation only of the
ERKs. We observed pERK activation by PTX, but also pERK inhibition
by GA and the ability of GA to attenuate PTX-induced pERK
activation. Reports have indicated that propyl gallate induces
death in HeLa cells with concomitant inhibition of MAPK and
promotion of ROS levels (41). In a
study of osteosarcoma cells, it was observed that GA inhibited the
activation of pERK and pAKT, regulators of the MAPK pathways,
modulating cell proliferation, apoptosis, and angiogenesis
(42).
The use of a commercial inhibitor of ERK (PD98059)
on PTX-resistant A2780AD cells showed that, with the inhibition of
ERKs, the effects induced by GA plus PTX, namely inhibition of cell
proliferation and G2/M cell cycle arrest, were
potentiated. Taken together, the results appear to suggest that the
activation of ERKs by PTX acts as a defensive factor, attenuating
excessive cytostatic action. Other reports have associated PTX with
the activation of cell survival pathways, such as the MAPK p38/AKT
and ERK signaling pathways, and, therefore, the inhibition of
apoptosis. Furthermore, PTX promotes the expression of various
survival factors that induce the phosphorylation and stabilization
of surviving proteins (17,43–45).
Earlier studies indicated that ERK activation
negatively regulates apoptosis in various cancer cell lines
(39,40,46).
In particular, co-treatment with MEK/ERK inhibitors enhanced
PTX-induced apoptosis in breast, ovarian, lung and prostate cancer
cells; and in the case of prostate cancer, apoptosis potentiation
was accompanied by Bcl-2 inactivation and increased Bax expression
(44,47,48).
Thus, co-treatment with MEK/ERK inhibitors could be a clinically
useful strategy against tumours with constitutively high or
therapeutically induced ERK activation (42,49).
In conclusion, ROS overproduction negatively
regulates ERK activation and it is also an essential determinant of
the capacity of GA to potentiate PTX toxicity. Thus, the combined
therapy of PTX plus GA may represent a novel strategy for combating
cancer resistance, requiring lower doses as compared with PTX
alone. The use of GA could be effective as an adjuvant in
combination with PTX in ovarian cancer treatment.
Acknowledgements
The authors would like to acknowledge Silvia
Marquina for her technical help.
Funding
Jessica Nayelli Sánchez Carranza acknowledges
fellowship 351450 from CONACYT and the support from CONACYT
(CB240801, LN 279905), CONACYT-FOMIX (224038). Partial support from
UAEM (Grants SI-DGDI-UAEM/13/289) is acknowledged.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LGM, PA and JNSC formulated the original ideas and
working hypothesis, together with JFD designed the research study.
JNSC participated in the entire experimental process of the study;
MRH and IB supervised ROS production and antiproliferative studies;
LA and ARE supervised the western blot assays; PL, MRH and PA
participated in the flow cytometry analysis. All authors analyzed
and interpreted the data. PA, LGM and JNSC wrote the manuscript and
JFD and LA provided important reviews and considerations. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fuchs DA and Johnson RK: Cytologic
evidence that taxol, an antineoplastic agent from Taxus
brevifolia, acts as a mitotic spindle poison. Cancer Treat Rep.
62:1219–1222. 1978.PubMed/NCBI
|
|
3
|
Jordan MA, Toso RJ, Thrower D and Wilson
L: Mechanism of mitotic block and inhibition of cell proliferation
by taxol at low concentrations. Proc Natl Acad Sci USA.
90:9552–9556. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jordan MA and Wilson L: Microtubules as a
target for anticancer drugs. Nat Rev Cancer. 4:253–265. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Conte PF, Cianci C and Gadducci A: Up date
in the management of advanced ovarian carcinoma. Crit Rev Oncol
Hematol. 32:49–58. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kumar S, Mahdi H, Bryant C, Shah JP, Garg
G and Munkarah A: Clinical trials and progress with paclitaxel in
ovarian cancer. Int J Womens Health. 2:411–427. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McGrail DJ, Khambhati NN, Qi MX, Patel KS,
Ravikumar N, Brandenburg CP and Dawson MR: Alterations in ovarian
cancer cell adhesion drive taxol resistance by increasing
microtubule dynamics in a FAK-dependent manner. Sci Rep.
5:95292015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Link A, Balaguer F and Goel A: Cancer
chemoprevention by dietary polyphenols: Promising role for
epigenetics. Biochem Pharmacol. 80:1771–1792. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rattanata N, Klaynongsruang S, Daduang S,
Tavichakorntrakool R, Limpaiboon T, Lekphrom R, Boonsiri P and
Daduang J: Inhibitory effects of gallic acid isolated from
Caesalpinia mimosoides Lamk on cholangiocarcinoma cell lines
and foodborne pathogenic bacteria. Asian Pac J Cancer Prev.
17:1341–1345. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Inoue M, Suzuki R, Koide T, Sakaguchi N,
Ogihara Y and Yabu Y: Antioxidant, gallic acid, induces apoptosis
in HL-60RG cells. Biochem Biophys Res Commun. 204:898–904. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sakagami H and Satoh K: Prooxidant action
of two antioxidants: Ascorbic acid and gallic acid. Anticancer Res.
17A:221–224. 1997.
|
|
12
|
Strlic M, Radovic T, Kolar J and Pihlar B:
Anti- and prooxidative properties of gallic acid in fenton-type
systems. J Agric Food Chem. 50:6313–6317. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fleury C, Mignotte B and Vayssière JL:
Mitochondrial reactive oxygen species in cell death signaling.
Biochimie. 84:131–141. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wartenberg M, Hoffmann E, Schwindt H,
Grünheck F, Petros J, Arnold JR, Hescheler J and Sauer H: Reactive
oxygen species-linked regulation of the multidrug resistance
transporter P-glycoprotein in Nox-1 overexpressing prostate tumor
spheroids. FEBS Lett. 579:4541–4549. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McCubrey JA, Lahair MM and Franklin RA:
Reactive oxygen species-induced activation of the MAP kinase
signaling pathways. Antioxid Redox Signal. 8:1775–1789. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
MacKeigan JP, Taxman DJ, Hunter D, Earp HS
III, Graves LM and Ting JP: Inactivation of the antiapoptotic
phosphatidylinositol 3-kinase-Akt pathway by the combined treatment
of taxol and mitogen-activated protein kinase kinase inhibition.
Clin Cancer Res. 8:2091–2099. 2002.PubMed/NCBI
|
|
18
|
Kowalski RJ, Giannakakou P, Gunasekera SP,
Longley RE, Day BW and Hamel E: The microtubule-stabilizing agent
discodermolide competitively inhibits the binding of paclitaxel
(Taxol) to tubulin polymers, enhances tubulin nucleation reactions
more potently than paclitaxel, and inhibits the growth of
paclitaxel-resistant cells. Mol Pharmacol. 52:613–622. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bakker M, Renes J, Groenhuijzen A, Visser
P, Timmer-Bosscha H, Müller M, Groen HJ, Smit EF and de Vries EG:
Mechanisms for high methoxymorpholino doxorubicin cytotoxicity in
doxorubicin-resistant tumor cell lines. Int J Cancer. 73:362–366.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Buey RM, Barasoain I, Jackson E, Meyer A,
Giannakakou P, Paterson I, Mooberry S, Andreu JM and Díaz JF:
Microtubule interactions with chemically diverse stabilizing
agents: Thermodynamics of binding to the paclitaxel site predicts
cytotoxicity. Chem Biol. 12:1269–1279. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Eruslanov E and Kusmartsev S:
Identification of ROS using oxidized DCFDA and flow-cytometry.
Methods Mol Biol. 594:57–72. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Andreu JM and Barasoain I: The interaction
of Baccatin III with the Taxol binding site of microtubules
determined by a homogeneous assay with fluorescent taxoid.
Biochemistry. 40:11975–11984. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ormerod MG, Collins MKL,
Rodriguez-Tarduchy G and Robertson D: Apoptosis in
interleukin-3-dependent haemopoietic cells. Quantification by two
flow cytometric methods. J Immunol Methods. 153:57–65. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Martindale JL and Holbrook NJ: Cellular
response to oxidative stress: Signaling for suicide and survival. J
Cell Physiol. 192:1–15. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Platanias LC: Map kinase signaling
pathways and hematologic malignancies. Blood. 101:4667–4679. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chang L and Karin M: Mammalian MAP kinase
signalling cascades. Nature. 410:37–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Johnson NL, Gardner AM, Diener KM,
Lange-Carter CA, Gleavy J, Jarpe MB, Minden A, Karin M, Zon LI and
Johnson GL: Signal transduction pathways regulated by
mitogen-activated/extracellular response kinase kinase kinase
induce cell death. J Biol Chem. 271:3229–3237. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wada T and Penninger JM: Mitogen-activated
protein kinases in apoptosis regulation. Oncogene. 23:2838–2849.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sarkar D, Su Z-Z, Lebedeva IV, Sauane M,
Gopalkrishnan RV, Valerie K, Dent P and Fisher PB: mda-7 (IL-24)
Mediates selective apoptosis in human melanoma cells by inducing
the coordinated overexpression of the GADD family of genes by means
of p38 MAPK. Proc Natl Acad Sci USA. 99:10054–10059. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sarwar T, Zafaryab M, Husain MA, Ishqi HM,
Rehman SU, Rizvi MM and Tabish M: Redox cycling of endogenous
copper by ferulic acid leads to cellular DNA breakage and
consequent cell death: A putative cancer chemotherapy mechanism.
Toxicol Appl Pharmacol. 289:251–261. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Karthikeyan S, Kanimozhi G, Prasad NR and
Mahalakshmi R: Radiosensitizing effect of ferulic acid on human
cervical carcinoma cells in vitro. Toxicol In Vitro. 25:1366–1375.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Delman DM, Fabian CJ, Kimler BF, Yeh H and
Petroff BK: Effects of flaxseed lignan secoisolariciresinol
diglucosideon preneoplastic biomarkers of cancer progression in a
model of simultaneous breast and ovarian cancer development. Nutr
Cancer. 67:857–864. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kaur M, Velmurugan B, Rajamanickam S,
Agarwal R and Agarwal C: Gallic acid, an active constituent of
grape seed extract, exhibits anti-proliferative, pro-apoptotic and
anti-tumorigenic effects against prostate carcinoma xenograft
growth in nude mice. Pharm Res. 26:2133–2140. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sánchez-Carranza JN, Alvarez L,
Marquina-Bahena S, Salas-Vidal E, Cuevas V, Jiménez EW, Veloz G RA,
Carraz M and González-Maya L: Phenolic compounds isolated from
Caesalpinia coriaria induce S and G2/M phase cell cycle
arrest differentially and trigger cell death by interfering with
microtubule dynamics in cancer cell lines. Molecules. 22:6662017.
View Article : Google Scholar
|
|
36
|
Baran CP, Zeigler MM, Tridandapani S and
Marsh CB: The role of ROS and RNS in regulating life and death of
blood monocytes. Curr Pharm Des. 10:855–866. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wallach-Dayan SB, Izbicki G, Cohen PY,
Gerstl-Golan R, Fine A and Breuer R: Bleomycin initiates apoptosis
of lung epithelial cells by ROS but not by Fas/FasL pathway. Am J
Physiol Lung Cell Mol Physiol. 290:L790–L796. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ji BC, Hsu WH, Yang JS, Hsia TC, Lu CC,
Chiang JH, Yang JL, Lin CH, Lin JJ, Suen LJ, et al: Gallic acid
induces apoptosis via caspase-3 and mitochondrion-dependent
pathways in vitro and suppresses lung xenograft tumor growth in
vivo. J Agric Food Chem. 57:7596–7604. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Park WH and Kim SH: MAPK inhibitors
augment gallic acid-induced A549 lung cancer cell death through the
enhancement of glutathione depletion. Oncol Rep. 30:513–519. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Park WH: The effect of MAPK inhibitors and
ROS modulators on cell growth and death of
H2O2-treated HeLa cells. Mol Med Rep.
8:557–564. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
You BR and Park WH: The enhancement of
propyl gallate-induced HeLa cell death by MAPK inhibitors is
accompanied by increasing ROS levels. Mol Biol Rep. 38:2349–2358.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liang CZ, Zhang X, Li H, Tao YQ, Tao LJ,
Yang ZR, Zhou XP, Shi ZL and Tao HM: Gallic acid induces the
apoptosis of human osteosarcoma cells in vitro and in vivo via the
regulation of mitogen-activated protein kinase pathways. Cancer
Biother Radiopharm. 27:701–710. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Greenberg VL and Zimmer SG: Paclitaxel
induces the phosphorylation of the eukaryotic translation
initiation factor 4E-binding protein 1 through a Cdk1-dependent
mechanism. Oncogene. 24:4851–4860. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
MacKeigan JP, Collins TS and Ting JP-Y:
MEK inhibition enhances paclitaxel-induced tumor apoptosis. J Biol
Chem. 275:38953–38956. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Henson ES and Gibson SB: Surviving cell
death through epidermal growth factor (EGF) signal transduction
pathways: Implications for cancer therapy. Cell Signal.
18:2089–2097. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lee YJ, Kang IJ, Bünger R and Kang YH:
Enhanced survival effect of pyruvate correlates MAPK and NF-kappaB
activation in hydrogen peroxide-treated human endothelial cells. J
Appl Physiol 1985. 96:793–801; discussion 792. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
McDaid HM and Horwitz SB: Selective
potentiation of paclitaxel (taxol)-induced cell death by
mitogen-activated protein kinase kinase inhibition in human cancer
cell lines. Mol Pharmacol. 60:290–301. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zelivianski S, Spellman M, Kellerman M,
Kakitelashvilli V, Zhou X-W, Lugo E, Lee M-S, Taylor R, Davis TL,
Hauke R, et al: ERK inhibitor PD98059 enhances docetaxel-induced
apoptosis of androgen-independent human prostate cancer cells. Int
J Cancer. 107:478–485. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yu C, Wang S, Dent P and Grant S:
Sequence-dependent potentiation of paclitaxel-mediated apoptosis in
human leukemia cells by inhibitors of the mitogen-activated protein
kinase kinase/mitogen-activated protein kinase pathway. Mol
Pharmacol. 60:143–154. 2001. View Article : Google Scholar : PubMed/NCBI
|