Introduction
Glioma is a tumor that originates in the glial cells
of the brain or the spine. Gliomas comprise ~30% of all brain and
central nervous system tumors, and 80% of all malignant brain
tumors (1). According to the
histopathological features and clinical presentation, primary brain
tumors can be graded from I to IV, of which glioblastoma multiforme
(GBM) is the most malignant brain glioma subtype (2). Despite several advances achieved
currently in multimodal treatments, the average lifespan expectancy
of patients with GBM is still <14 months (3). In the process of genesis, development
and malignant transformation, the expression of different signaling
molecules all can accelerate or delay the progress of the condition
of the patient. It has been reported that NF-κB and p38 signaling
pathways play crucial roles in the invasion and metastasis of
glioma cells (4–8), blocking of which are considered
treatment targets in the activated state in GBM (4,9).
Receptor-interacting protein 2 (RIP2), also called
RICK/CARDIAK, consists of an N-terminal serine/threonine kinase
domain and a CARD domain for protein-protein interaction (10). RIP2 is an inducible transcriptional
product of NF-κB activation, and serves as a positive regulator of
the NF-κB pathway by binding to the IKK complex (11). In addition, RIP2 has been associated
with activation of the c-Jun N-terminal kinase (JNK), extracellular
signal-regulated kinase (ERK) and p-38 pathways (12–14).
RIP2 expression is regulated at the transcriptional level. Its
upregulated expression has been associated with inflammatory
disease states and models such as peritoneal dialysis
(PD)-associated peritonitis, Crohn's disease, multiple sclerosis
(MS) and allergic asthma (15–18).
Recent research demonstrated that RIP2 is involved in the invasion
and metastasis of triple-negative breast cancer and bladder cancer
(19,20), which highlights a novel role of RIP2
in cancer. However, whether RIP2 plays a role in human glioblastoma
remains unclear.
Here, we showed the upregulation of RIP2 and NF-κB
and p38 pathway activation in human glioma tissues and cell lines.
In addition, the role of RIP2 in glioma cells was investigated. Our
results suggest that RIP2 may induce proliferation of glioma cells
by NF-κB pathway activation. Most recently, we demonstrated that
RIP2 physically and functionally interacts with tumor necrosis
factor receptor-associated factor 3 (TRAF3) (21), which is an important regulator in
carcinogenesis by negatively regulating mitogen-activated protein
kinase activation and alternative nuclear factor-κB signaling
(22,23). To further elucidate the effects of
RIP2 on the regulation of glioma cell proliferation, we started
from the interaction protein of RIP2 to explore the possible
mechanism of RIP2 in glioma. The present study identified that RIP2
and TRAF3 exist as a negative regulation link and TRAF3
functionally is a negative regulator involved in RIP2-induced
glioma cell growth.
Materials and methods
Patients and tissue samples
All the tissue samples were collected during surgery
from inpatients at the Department of Neurosurgery, The Second
Hospital of Hebei Medical University, and the study protocol was
approved by the Local and Medical Ethics Committee (The Second
Hospital of Hebei Medical Research Ethics Committee, approval no.
2012003). Written informed consent was obtained from all patients
or the next of kin, and the data for samples were analyzed
anonymously. Our clinical investigation was conducted according to
the principles expressed in the Declaration of Helsinki. Among the
34 samples, 28 cases were diagnosed as astrocytoma and graded
according to WHO's Histopathological Grading (2000): 8 cases of
grade II (mean age at diagnosis, 45.4±16.9 years; 4 males and 4
females), 11 cases of grade III (mean age at diagnosis, 46.6±17.8
years; 6 males and 5 females), 9 cases of grade IV (mean age at
diagnosis, 54.0±13.9 years; 3 males and 6 females). In addition, 6
normal control samples were obtained from resected tissues from
patients with traumatic brain injury treated by brain decompression
(mean age at diagnosis, 47.8±14.8 years; 2 males and 4 females).
Meanwhile, all the pathological results were diagnosed by two
senior physicians from the Department of Pathology. The fresh
tissue samples were stored in liquid nitrogen, and used for
analysis of the expression levels of RIP2, TRAF3 and Bcl-xL at the
mRNA or protein levels. The phosphorylation level of IκBα and p38
MAPK in the tissues was detected by western blot analysis.
Total RNA preparation and real-time quantitative PCR
(qPCR) assay. RNA pure reagent kit for rapid extraction of
ultrapure RNA (Biomed Technology Development Co., Ltd., Beijing,
China) was used to extract the total RNA from the clinical tissue
samples according to the manufacturer's instructions. Primer Script
Reverse Transcriptase (Takara Bio, Inc., Otsu, Japan) was used for
the reverse transcription of 1 µg total RNA of tissue samples
strictly following the operation manual enclosed with the kit.
qRT-PCR assay of RIP2, TRAF3 and Bcl-xL mRNA transcript was
performed with the SYBR-Green Master Mix (Fermentas, Burlington,
ON, Canada) within the 20 µl reaction system. The primer sequences
for RT-qPCR were: RIP2-qf, 5′-CCCTGCCAGCTCCTCAAGACA-3′; RIP2-qr,
5′-GGCTATACCAGGCTGCAGACGTT-3′; TRAF3-qf,
5′-AGGCGTGTAAATACCGGGAAGC-3′; TRAF3-qr,
5′-GGGCCTTGATCTGCTGGTTTGT-3′; Bcl-xL-qf,
5′-GAGGCAGGCGACGAGTTTGAAC-3′; Bcl-xL-qr,
5′-GCTGCGATCCGACTCACCAATAC-3′; GAPDH-qf,
5′-GAGTCAACGGATTTGGTCGT-3′; GAPDH-qr, 5′-TTGATTTTGGAGGGATCTCG-3′.
The endogenous control GAPDH was used to normalize the sample with
the 2−ΔΔCq method for relative quantification analysis
(24).
Cell lines
Human glioblastoma U87MG (HTB-14) and U251MG cells
(the Cell Center of Union Medical College, Beijing, China) were
used in the study. It has been shown that the DNA profile of U87MG
(HTB-14) cell line is different from that of the original cells
(25), but the gene expression
profile indicates that the U87MG (HTB-14) cell line is of CNS
origin and probably is a GBM cell line from an unknown patient.
Thus, the U87MG (HTB-14) cell line was used as a cell model of GBM.
To ensure the integrity of our research results, the U87 cell line
was authenticated by STR profiling (Shanghai Biowing Applied
Biotechnology Co., Ltd., Shanghai, China). Cells were cultured
respectively in RPMI-1640 medium and MEM/EBSS media (both from
HyClone; GE Healthcare, Logan, UT, USA) containing 10% fetal bovine
serum (Gibco: Thermo Fisher Scientific, Inc., Waltham, MA, USA), at
37°C with 5% CO2.
MTT assay
Cells were respectively seeded in 96-well plates at
5,000 cells/well in 10% FBS-supplemented RPMI-1640 and MEM media.
On the following day, the cell monolayers were incubated in
serum-free medium for 24 h, and then treated with various
concentrations of rhRIP2, as previously described (22). In another experiment, cells were
transfected with pCMV-Myc/TRAF3 or pCMV-Myc for 24 h, and then
treated with various concentrations of rhRIP2. After incubation for
48 h, cell growth was measured by
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
proliferation assay. Absorbance at 490 nm was measured using a
Bio-Rad model 550 microplate reader (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). The data represent the means of three
independent experiments.
Plasmids, siRNAs, and
transfection
The pCMV-Myc/TRAF3 and pCMV-HA/RIP2 expression
constructs have been described previously (21), as well as the cDNA target sequence
of siRNAs for RIP2 (26). All of
the plasmids were purified using the Plasmid Mini kit (Omega
Bio-Tek, Inc., Norcross, GA, USA). siRNAs were synthesized by the
Shanghai GenePharma Co., Ltd. (Shanghai, China) and transfected
into glioma cell lines using Lipofectamine 2000 (Invitrogen; Thermo
Fisher Scientific, Inc., Carlsbad, CA, USA).
Detection of apoptosis and flow
cytometric analysis
Apoptotic cells were quantified by measuring
externalized phosphatidylserine (PS) assessed by uptake of Annexin
V-EGFP and propidium iodide (PI). After various experimental
treatments, cells were stained using an Annexin V-EGFP apoptosis
detection kit (Nanjing KeyGen Biotech Co., Ltd., Nanjing, China).
Briefly, the harvested cells were rinsed once with
phosphate-buffered saline (PBS), and then resuspended in 500 µl of
1X binding buffer and 5 µl Annexin V-FITC and 5 µl of PI, and then
incubated at room temperature for 15 min in the dark. For flow
cytometric analysis, the cells were stained with Annexin/PI, and
the population of apoptotic cells was analyzed immediately by flow
cytometry (FACSCalibur; BD Biosciences, Franklin Lakes, NJ, USA).
Flow cytometric analysis was conducted at the flow cytometry
facility (FACSCalibur; BD Biosciences). Briefly, after 48–72 h of
transfection, the cells were fixed and then incubated in PBS
containing RNase A (100 µg/ml; Sigma-Aldrich: Merck KGaA,
Darmstadt, Germany) and PI (50 µg/ml; Sigma-Aldrich: Merck KGaA) at
room temperature for 30 min prior to analysis.
Western blot analysis
The ground clinical tissue samples or 48
h-transfected U87MG and U251MG cells were harvested and washed
twice with ice-cold PBS and lysed in RIPA lysis (Applygen
Technologies, Inc., Beijing, China) for 15 min on ice. Cell lysates
were then clarified by microcentrifugation at 12,000 × g for 10 min
at 4°C. Equal amounts of protein (40 µg) were quantified using the
Lowry Protein Assay kit (Nanjing KeyGen Biotech Co., Ltd.), and
separated by 12% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and electrophoretically transferred to
polyvinylidene fluoride (PVDF) membranes (EMD Millipore, Bedford,
MA, USA). Membranes were then blocked with PBST (PBS with 0.05%
Tween-20) containing 5% non-fat dry milk for 1 h at room
temperature. The specific antibodies (primary antibodies) against
Bcl-xL (cat. no. 2764) and TRAF3 (cat. no. 4729; both from Cell
Signaling Technology, Inc., Danvers, MA, USA), and RIP2 (cat. no.
ab75257) (Abcam, Cambridge, MA, USA) were prepared with 5% defatted
milk powder and diluted 1,000 times (1:1,000); polyclonal p100/p52
antibodies (cat. no. ab31409; Abcam) and the phosphorylated
anti-pP38 antibody (cat. no. ab4822) and p38 MAPK monoclonal
antibodies (cat. no. ab32142; Abcam) were prepared with 5% BSA and
diluted 1,000 times (1:1,000). GAPDH monoclonal antibodies (cat.
no. TA-08; Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd.,
Beijing, China) were prepared with 5% defatted milk powder and
diluted 2,000 times (1:2,000) overnight at 4°C, respectively. The
membranes were then washed with TBST for three times followed by
incubation with HRP-conjugated anti-mouse (cat. no. ZB-2305) or
HRP-conjugated anti-rabbit immunoglobulin G (IgG) (cat. no.
ZB-2301; Beijing Zhongshan Golden Bridge Biotechnology Co., Ltd.)
at room temperature for 1 h. Finally, the membranes were analyzed
using the enhanced chemiluminescence detection system (Amersham
Pharmacia Biotech; GE Healthcare, Oakville, Canada).
Statistical analysis
All data are expressed as mean ± SD. The difference
between two groups was analyzed using the Student's t-test, while
differences among more than two groups were analyzed using one-way
ANOVA method. Multiple comparisons between the groups was performed
using the Student-Newman-Keuls (SNK) method. Statistical analysis
was performed using SPSS 13.0 (SPSS, Inc., Chicago, IL, USA) and
MxPro 3000-qPCR statistical software, respectively. P<0.05 was
used to indicate a statistically significant difference.
Results
Analysis of RIP2 expression in normal
brain tissues and astrocytoma tissues of various grades
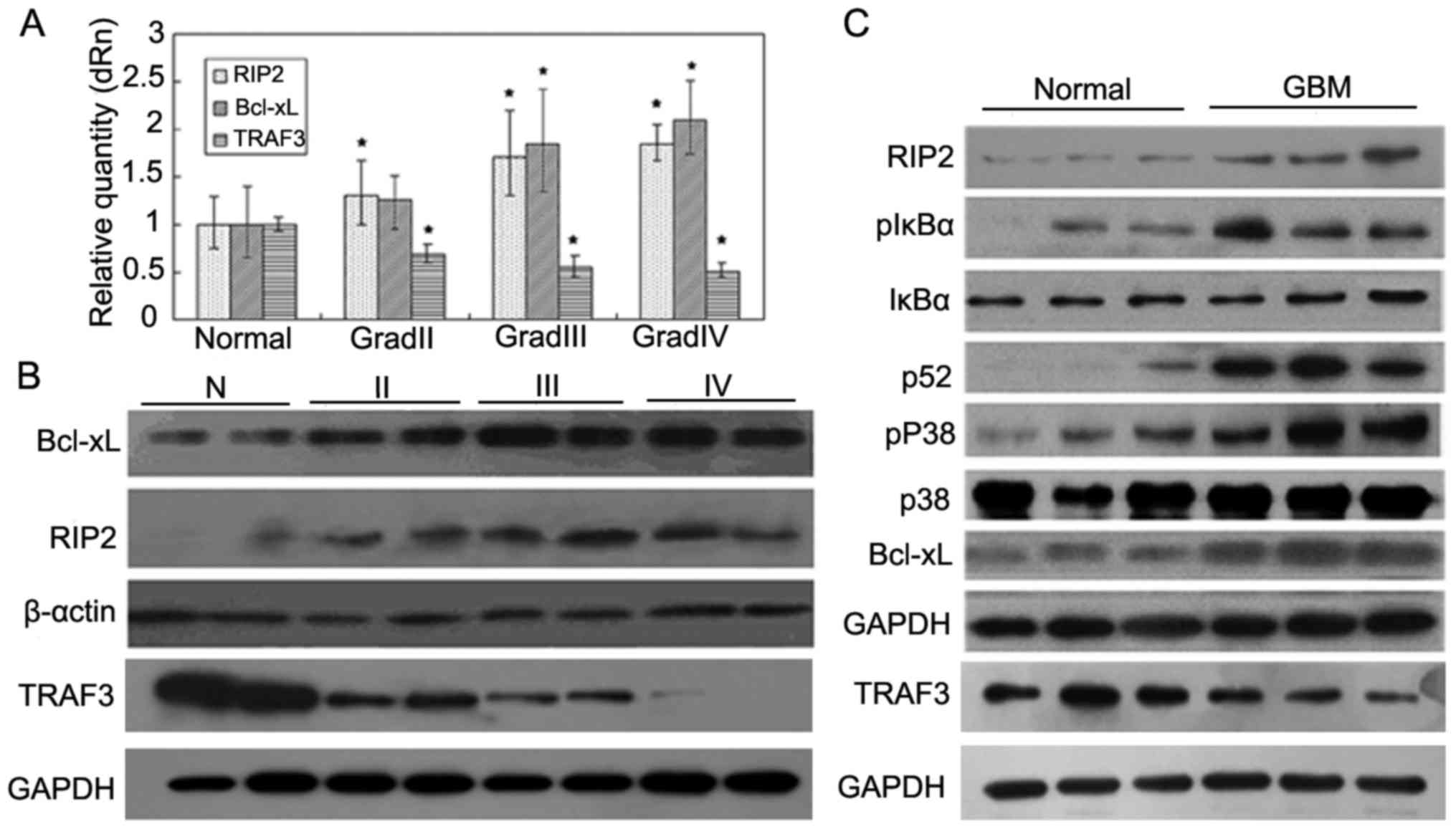
The expression of RIP2, TRAF3 and Bcl-xL was
detected in 34 tissue samples from control samples and patients
with glioma by qPCR. The results demonstrated that the expression
of RIP2 and Bcl-xL was markedly increased in the lower (II) and
higher (III, IV) grades of glioma in comparison with that in the
normal brain tissues (P<0.05; P<0.05), whereas the expression
of TRAF3 was decreased in the higher grades (III, IV) of glioma
(P<0.05) (Fig. 1A).
Furthermore, western blot analysis revealed that the
protein expression levels of both RIP2 and Bcl-xL in the glioma
patient tissues were also higher than these levels in the normal
tissues, and the expression of TRAF3 was decreased in the glioma
patient tissues, which was consistent with the qPCR results
(Fig. 1B). The results showed that
the mRNA and protein expression of RIP2 had a positive correlation
with the degree of tumor malignancy, whereas the expression of
TRAF3 had a negative correlation with the degree of tumor
malignancy.
As RIP2 is associated with activation of the NF-κB
and p38 pathways (12–14,21),
we evaluated the NF-κB signaling pathway including both canonical
NF-κB and alternative NF-κB as well as p38 MAPK activation in
glioma samples by western blot analysis. As shown in Fig. 1C, the phosphorylation levels of IκBα
and p38 MAPK in glioma tissues were markedly increased compared
with the levels in the normal brain tissues. In addition, we also
observed that the expression of p52 and Bcl-xL in glioma tissues
was also markedly increased (Fig.
1C) and positively correlated with the malignant degree of
glioma, which was concurrent with inhibition of the expression of
TRAF3 mRNA in glioma tissues. Taken together, the above results
indicate that RIP2 may take part in the regulation of NF-κB and p38
MAPK pathway-mediated anti-apoptosis by negative regulation of the
expression of TRAF3.
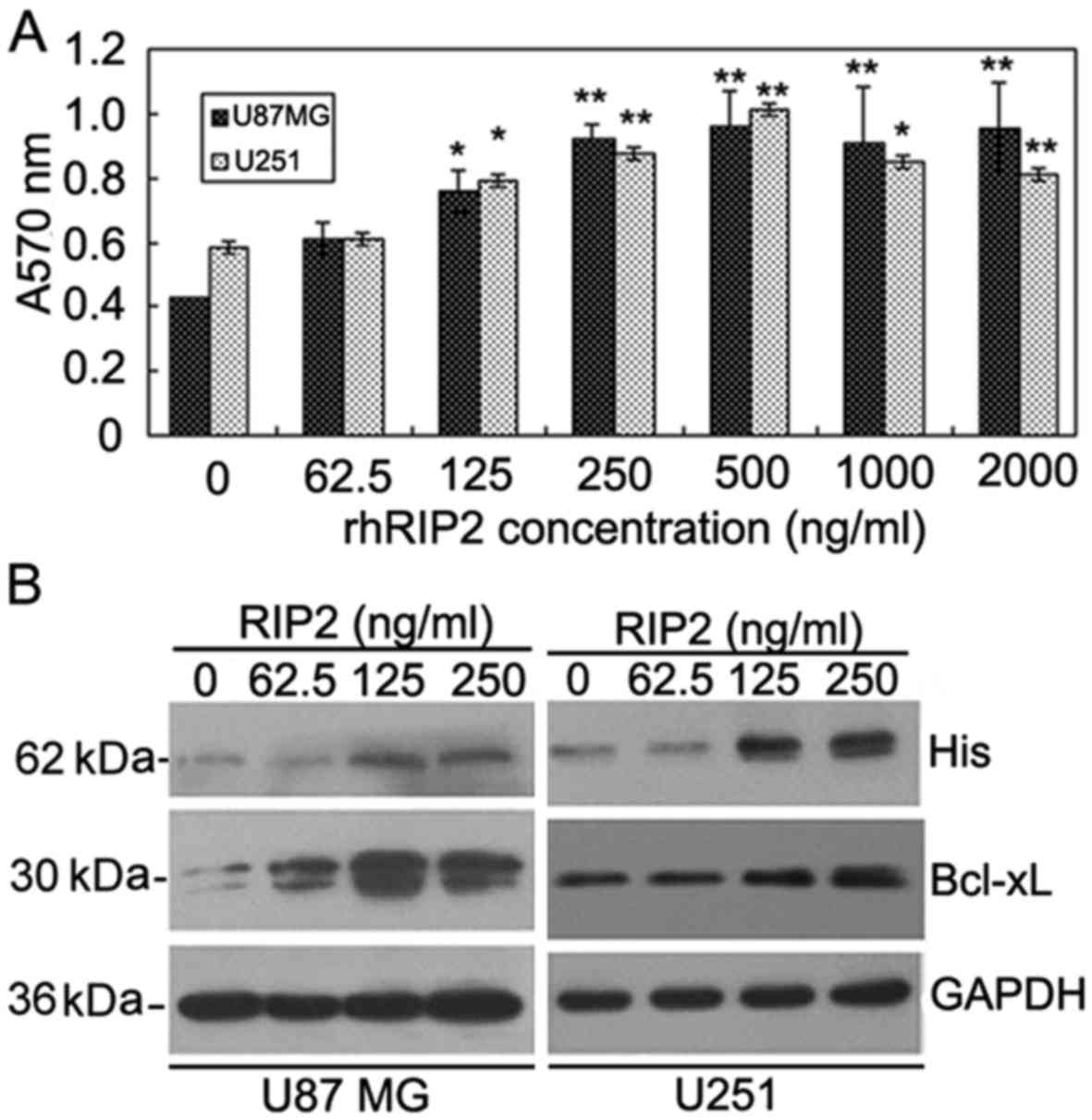
Exogenous RIP2 promotes glioma cell
proliferation
The effect of RIP2 on the U87MG and U251 cells was
examined by MTT assay. As shown in Fig.
2A, RIP2 stimulated the proliferation of U87MG and U251 cells
at a concentration of 62.5 ng/ml. The effect of rhRIP2 on the
proliferation of U87MG and U251 cells displayed a dose-dependent
manner from 62.5 to 500 ng/ml, which was decreased at
concentrations >1,000 ng/ml (Fig.
2A). The results demonstrated that RIP2 facilitated U87MG and
U251 cell proliferation.
We next investigated whether the entry of the RIP2
fusion protein into U87MG and U251 cells caused a specific
biological effect by regulation of an anti-apoptosis-related
protein. As shown in Fig. 2B, the
entry of RIP2 fusion protein into glioma cells was markedly
upregulated when U87MG and U251 cells were pretreated with rhRIP2
from 62.5 to 500 ng/ml. Moreover, we found that RIP2 induced
upregulation of the anti-apoptotic protein Bcl-xL in glioma cells
in a dose-dependent manner (Fig.
2B). These data indicated that the entry of RIP2 fusion protein
into glioma cells may facilitate cell proliferation by upregulating
the expression of Bcl-xL.
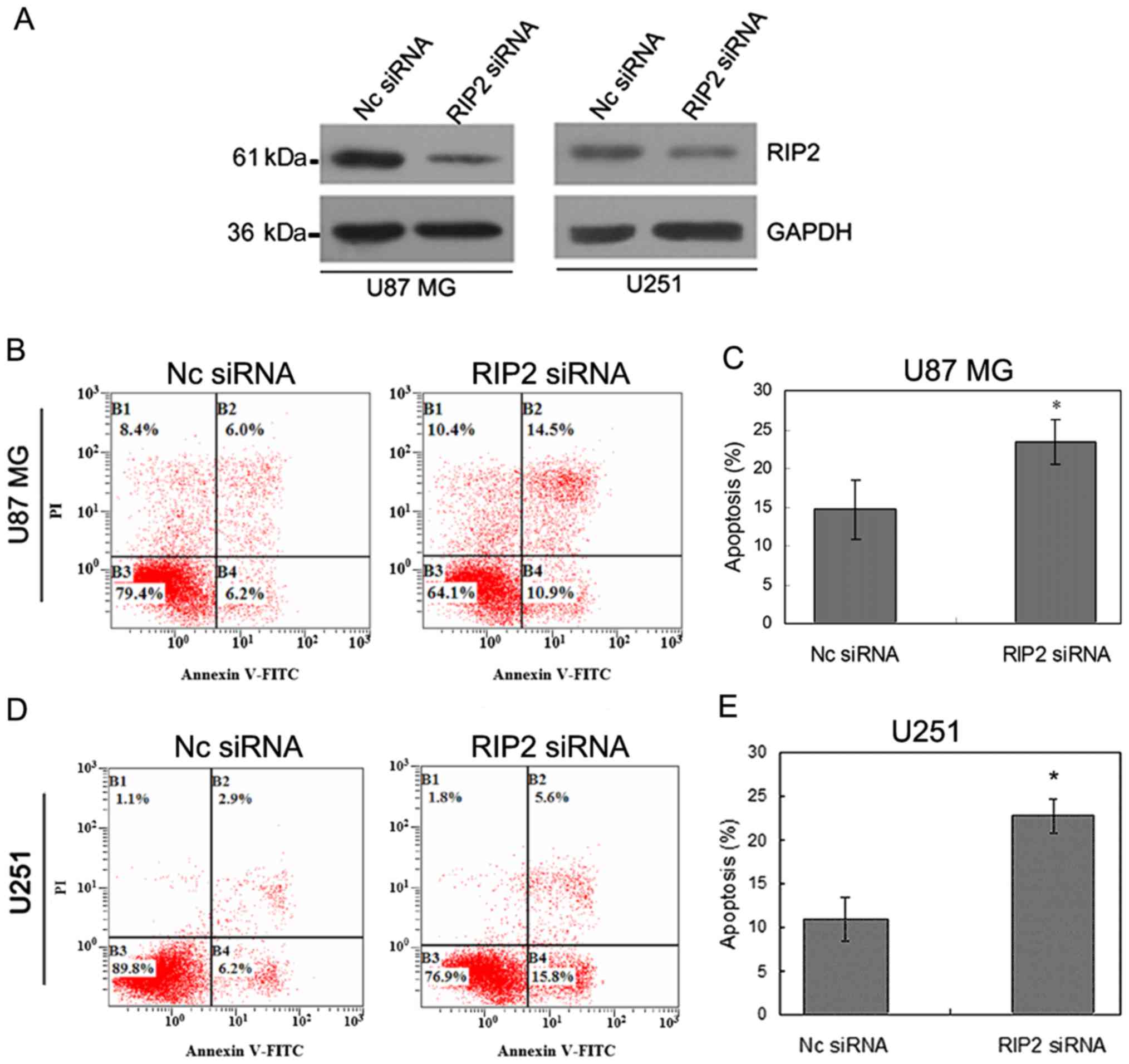
Knockdown of endogenous RIP2 promotes
glioma cell apoptosis
To assess whether the observed increase in
proliferation in response to exogenous RIP2 was due to the
anti-apoptotic role of RIP2 in glioma cells, apoptosis assay was
evaluated by Annexin V-FITC/PI double staining method after
knockdown of endogenous RIP2 (Fig.
3A) in the U87MG and U251 cells.
Apoptosis assay confirmed that the apoptotic rate
was increased by 18 and 21.83%, respectively, in the U87MG and U251
cells in comparison with the control group (Fig. 3B and D). ANOVA test indicated that
the percentage of apoptotic U87MG and U251 cells increased after
RIP2 knockdown as compared with Nc siRNA transfection (P<0.05,
Fig. 3C and E). The results
demonstrated that knockdown of endogenous RIP2 increased the
apoptosis in U87MG and U251 cells, which was consistent with the
effect of RIP2-promoting U87MG and U251 cell proliferation.
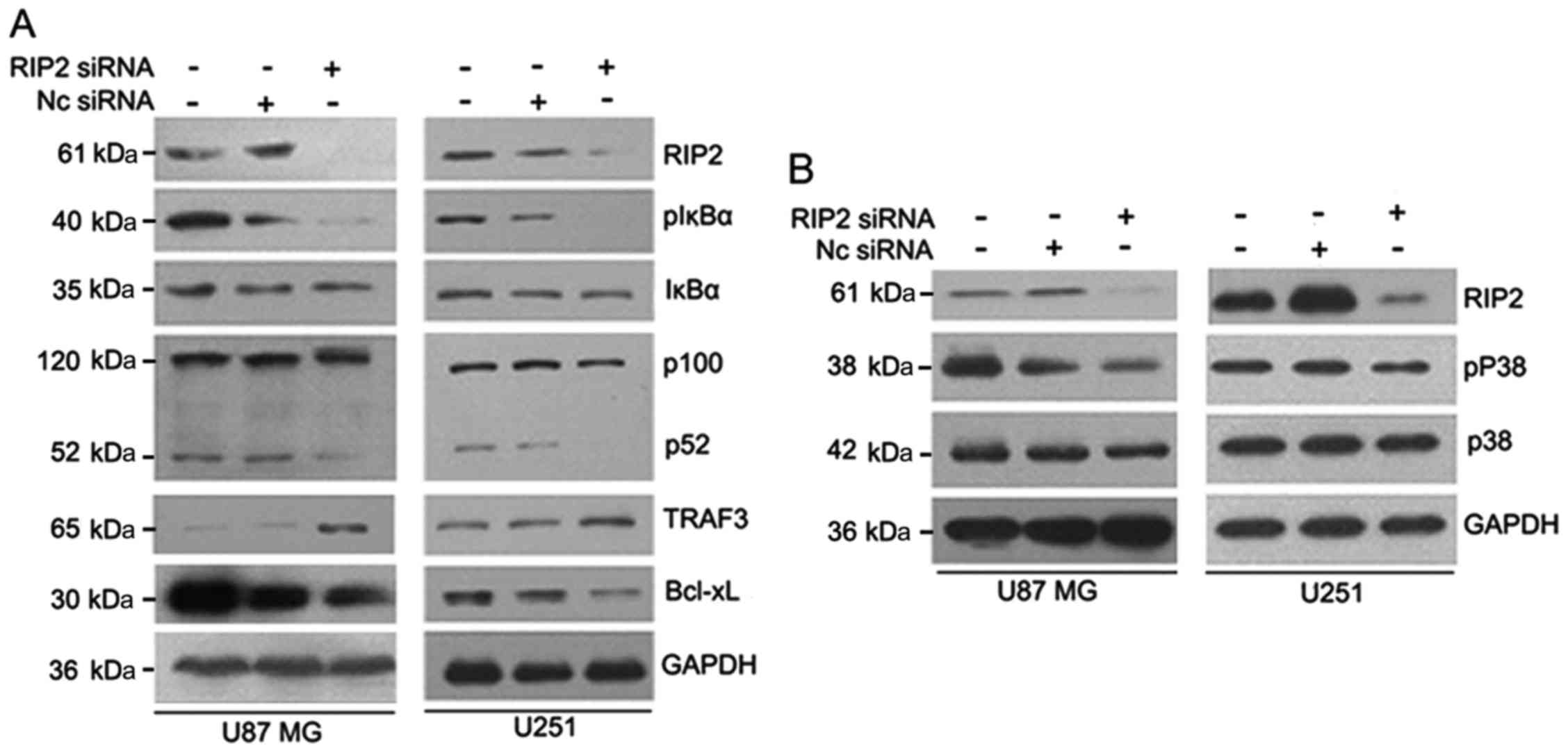
Knockdown of endogenous RIP2 inhibits NF-κB and p38
MAPK activation in glioma cells. Next, we aimed at identifying the
underlying molecular mechanisms responsible for the RIP2-stimulated
cell proliferation. The effects of the knockdown of endogenous RIP2
were examined in U87MG and U251 cells, since U87MG and U251 cells
express endogenous RIP2 at high levels. The loss of RIP2 expression
was confirmed by western blot analysis (Fig. 4A and B). Following, we firstly
focused on the effect of RIP2 on the activation of the canonical
NF-κB signaling pathway in glioma cells. Western blot analysis
indicated that knockdown of RIP2 markedly decreased the level of
IκBα phosphorylation at Ser32, while the protein level of total
IκBα was unchanged (Fig. 4A).
Given the identification of RIP2 as a novel binding
partner for TRAF3 and its involvement in the regulation of the
alternative NF-κB pathway in Ramos cells in our previous study, we
sought to determine whether RIP2 is involved in the TRAF3-regulated
alternative NF-κB pathway in glioma cells. The expression level of
TRAF3 and proteolytic processing of p100 to p52 were assessed. The
results showed that knockdown of RIP2 markedly inhibited the
processing of p100 to p52 and increased the expression level of
TRAF3. Meanwhile, RIP2 knockdown induced downregulation of
anti-apoptotic protein Bcl-xL in glioma cells (Fig. 4A). Moreover, the activation of the
p38 MAPK signaling pathway after RIP2 knockdown was also
investigated. As shown in Fig. 4B,
knockdown of RIP2 resulted in suppressed phosphorylation of p38
MAPK protein. Taken together, these findings suggest that RIP2 may
be involved in the development of glioma by activating the
canonical and alternative NF-κB pathways as well as the p38 MAPK
signaling pathway.
Endogenous TRAF3 overexpression
inhibits the RIP2-induced glioma cell proliferation
Given that RIP2 interacts with TRAF3, we
investigated whether TRAF3 affects RIP2-induced proliferation in
glioma cells. As U87MG and U251 cells express endogenous TRAF3 at
low levels, cells were transfected with pCMV-Myc/TRAF3 or pCMV-Myc
plasmid before treatment with various concentrations of rhRIP2. The
results showed that overexpression of TRAF3 effectively increased
its expression as compared with the negative control (pCMV-Myc)
(Fig. 5A). After confirmation of
the efficient overexpression of TRAF3, transfected cells were
stimulated with RIP2 for 48 h in order to investigate the role of
TRAF3 in the RIP2-induced proliferation effects on glioma cells.
MTT analysis indicated that TRAF3 overexpression decreased the
RIP2-induced proliferation effect on U87MG and U251 cells (Fig. 5B and C). These data showed that
TRAF3 negatively regulated the RIP2-induced proliferation effects
on glioma cells.
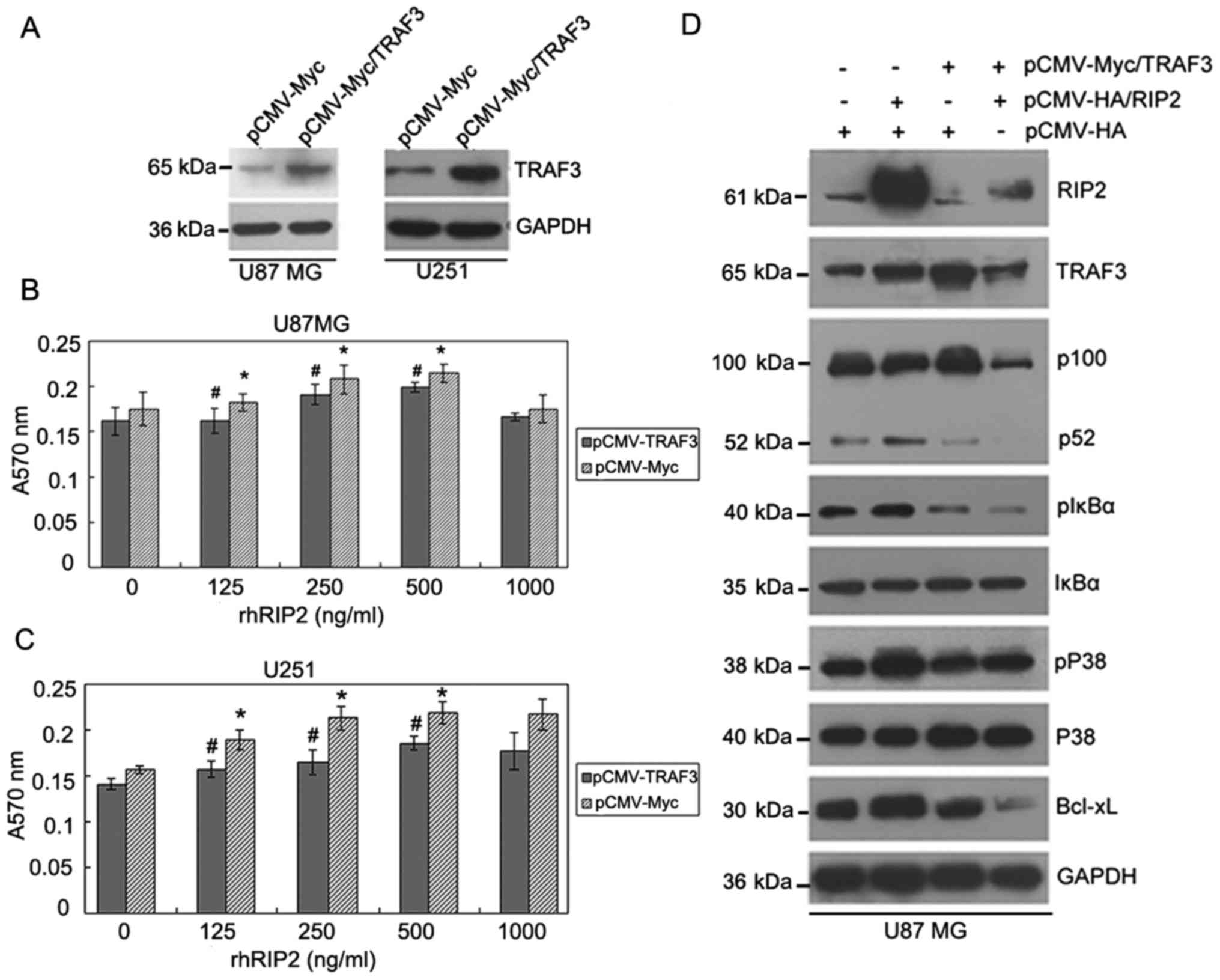 | Figure 5.A negative regulation link exists
between RIP2 and TRAF3 proteins in U87MG cells. (A) Overexpression
of TRAF3 decreased the RIP2-induced proliferation effect on U87MG
and U251 cells. The expression of TRAF3 protein of U87MG and U251
cells transfected with pCMV-Myc/TRAF3 or control pCMV-Myc plasmid
was determined by western blot analysis. GAPDH was used as loading
control. (B and C) Indicated amounts of rhRIP2 were added to
96-well plates containing 5,000 cells/well in transfected with
pCMV-Myc/TRAF3 or control pCMV-Myc cells. After incubation for 48
h, cell growth was measured by MTT assay. The data represent the
means of three independent experiments. *P<0.05; p-values were
calculated by comparing absorbance at 570 nm between rhRIP2
stimulation and control groups. #P<0.05; p-values
were calculated by comparing absorbance at 570 nm between
transfected with pCMV-Myc/TRAF3 and control pCMV-Myc. (D) U87MG
cells were respectively transfected with the control pCMV-HA,
pCMV-HA/RIP2, pCMV-Myc/TRAF3 plasmids or cotransfected with
pCMV-HA/RIP2 and pCMV-Myc/TRAF3 plasmids. The expression of RIP2
and TRAF3, phosphorylated IκBα, IκBα, p100, p52 and Bcl-xL were
analyzed by western blot analysis. Expression of GAPDH served as
loading control. RIP2, receptor-interacting protein 2; TRAF3, tumor
necrosis factor receptor-associated factor 3. |
Furthermore, we investigated how TRAF3
overexpression mediates the inhibition of RIP2-induced glioma cell
proliferation by western blot analysis. We found that the
expression of endogenous TRAF3 in U251 cells was higher than that
in U87MG cells. Upregulation of TRAF3 expression in U251 cells was
not as obvious as that in U87MG cells after transfection of the
TRAF3 expression plasmid. Therefore, representative U87MG cells
were selected to overexpress TRAF3, in order to better understand
its effect in RIP2-induced glioma cell proliferation. As shown in
Fig. 5D, markedly higher expression
of TRAF3 and RIP2 was observed in the U87MG cells which were
respectively transfected with TRAF3 and RIP2 expression plasmids.
Western blot analysis indicated that overexpression of endogenous
TRAF3 downregulated the expression of RIP2 and Bcl-xL. Meanwhile,
TRAF3 overexpression suppressed the activation of p100 and
phosphorylation of IκBα and p38 in the transfected U87 cells.
Conversely, overexpression of endogenous RIP2 downregulated the
expression of TRAF3 and increased the expression of Bcl-xL.
Accordingly, RIP2 overexpression induced the activation of p100 and
phosphorylation of IκBα and p38 in transfected U87MG cells.
Moreover, we examined the effect of TRAF3 and RIP2 cotransfection
in U87MG cells on the expression of each other. The result showed
that overexpression of RIP2 and TRAF3 could inhibit each other's
expression. We also observed that overexpression of endogenous
TRAF3 could inhibit RIP2-induced activation of p100 and
phosphorylation of IκBα and p38 by cotransfection with RIP2 and
TRAF3 in U87MG cells. Taken together, these results suggested that
RIP2 and TRAF3 proteins may exist in a negative regulation
link.
Discussion
RIP2 is a kinase with known central roles in
inflammation and immunity (27–29).
Recent study highlights RIP2 as a pro-metastasis kinase in patients
with advanced breast cancer and shows that targeted knockdown of
RIP2 may improve outcomes in advanced breast cancer patients
(19). Additionally, RIP2 may
mediate bladder cancer surveillance by involvement in the
development and recruitment of granulocytic myeloid-derived
suppressor cells (MDSCs) and highlight the contribution of MDSCs to
the development of metastases in bladder cancer (20). These results show a novel function
of RIP2 in cancer in addition to its known role in inflammation.
Moreover, research has found that RIP1, a member of the RIP kinase
family, is overexpressed in GBM, but not in low grade gliomas, and
that increased expression of RIP1 confers a worse prognosis
(30). However, whether or not the
expression level of RIP2 is associated with glioma progression
remains unknown. Glioma is one of the common tumors seriously
threatening human health. Despite several advances achieved
currently in multimodal treatments, the 5-year survival of glioma
patients ranks third, just after that of pancreatic and lung
cancers (3). Therefore, a greater
understanding of the biological mechanisms underlying glioma
pathogenesis may contribute to the development of targeted
therapies that can improve patient outcome.
Many previous studies have confirmed that the tumor
malignant degree is closely related with the activation of various
intracellular signaling pathways, such as NF-κB, PI3K/AKT/mTOR,
MAPK and JAK/STAT, among which much more attention has been paid to
the role of the NF-κB and MAPK pathways in the genesis and
progression of glioma (20).
Research has demonstrated that the NF-κB pathway is constitutively
activated and upregulated in glioma patient samples or glioma cells
in response to different stimuli (30,31).
Moreover, the invasion and metastasis of glioma cells require
specific intracellular signaling cascade activations, among which
the p38 signaling pathway is considered crucial (5–8), and
the levels of numerous NF-κB and p38 target genes are elevated in
glioma cells (8,32). However, whether or not activation of
the alternative NF-κB and p38 pathways is associated with the
genesis of glioma is unknown. Therefore, we explored the activation
status of alternative NF-κB and p38 pathways in addition to
detection of activation of the canonical NF-κB pathway in glioma
patient samples. Compared to the control samples, our analyses of
28 cases of glioma demonstrated that RIP2 expression was positively
correlated with the malignant degree of the glioma tissues. In
addition, we also found that the phosphorylation levels of IκBα and
p38 and the expression levels of p52 and Bcl-xL in glioma were
markedly higher than these levels in the normal brain tissues.
These findings indicated that the activation of the alternative
NF-κB and p38 pathways is correlated with the genesis of glioma.
These results imply that RIP2 expression may positively correlate
with canonical NF-κB and alternative NF-κB as well as p38 status.
Our previous study demonstrated that RIP2 interacts with TRAF3
(21), which acts as a negative
regulator of mitogen-activated protein kinase activation and
alternative NF-κB signaling (22,23).
Therefore, we also examined whether TRAF3 is associated with glioma
progression. In the present study, we showed that the expression of
TRAF3 is negatively correlated with the malignant grades of glioma,
which was concurrent with activation of the alternative NF-κB
pathway and p38 pathway status in glioma tissues. These results
suggest that RIP2 and TRAF3 may be involved in the genesis of
glioma.
To understand the role of RIP2 in glioma, we further
analyzed the functional significance of RIP2 upregulation in
vitro and in vivo. Our in vitro studies provided
several insights into the mechanism of RIP2 action. First, RIP2
promoted U87MG and U251 cell proliferation, and downregulation of
RIP2 induced cell apoptosis. In addition, RIP2 upregulated the
expression level of anti-apoptotic protein Bcl-xL in U87MG and U251
cells. Previous studies have demonstrated that RIP2 is involved in
multiple signaling pathways such as the NF-κB pathway and
mitogen-activated protein kinase (p38) pathways (12–14).
Collectively, together with the findings of activation of the NF-κB
and p38 pathways in glioma patient samples in this study, we
proposed that the anti-apoptotic activity of RIP2 in glioma may be
accomplished by regulating the NF-κB and p38 pathways. Using a
small interfering RNA strategy, we found that RIP2 was involved in
the positive regulation of canonical NF-κB and p38 pathways by
regulation of the phosphorylation of IκBα and p38. Given the
downregulation of TRAF3 expression and activation of alternative
NF-κB pathway in glioma tissues, we then further investigated the
effects of RIP2 knockdown on TRAF3 expression and alternative NF-κB
activation. Knockdown of RIP2 inhibited proteolytic processing of
p100 to p52 and promoted TRAF3 expression, suggesting that RIP2 may
have positive regulation on the activation of alternative NF-κB by
controlling the expression level of TRAF3.
TRAF3, a member of the TRAF family of cytoplasmic
adaptor proteins, is employed in signaling by the tumor necrosis
factor receptor (TNFR) superfamily and Toll-like receptors (TLRs)
(33,34). Recently, it was identified that
TRAF3 function mutations or complete gene deletions promoted
multiple myeloma cell survival by preventing the interaction of
TRAF3 with NIK and constitutive activation of NF-κB signaling in
malignant cells from patients (35,36).
Moreover, specific ablation of TRAF3 in B lymphocytes resulted in
severe peripheral B-cell hyperplasia (37). The above-mentioned results further
suggested that TRAF3 may play an important role in carcinogenesis.
In our previous study, the specific interaction between TRAF3 and
RIP2 was confirmed by co-immunoprecipitation and GST pull-down
assays, and RIP2 was found to be involved in the positive
regulation of the alternative NF-κB pathway by controlling the
expression level of TRAF3 in Ramos cells (21). To better clarify the relationship
between RIP2 and TRAF3 in the genesis of glioma, we explored the
effects of TRAF3 overexpression on RIP2-induced glioma cell
proliferation. Our experiments showed that TRAF3 overexpression
suppressed RIP2-induced glioma cell proliferation. Based on
previous and present experimental results, TRAF3 appears to play a
tumor suppressor-like role in cancers. Furthermore, we explored how
TRAF3 affects RIP2-induced glioma cell proliferation. Our study
showed that endogenous TRAF3 could inhibit RIP2-induced activation
of p100 and phosphorylation of IκBα and p38. We also discovered
that TRAF3 overexpression inhibited the expression of RIP2. These
findings led us to conclude that RIP2 and TRAF3 proteins exist in a
negative regulation link. In conclusion, the present study
established a negative regulation link between RIP2 and TRAF3 and
identified a new pathway for regulating glioma progression.
Acknowledgements
We thank Haoran Jing, Yao Chen and Lin Cai for
technical assistance. We further thank all participants recruited
in the present study.
Funding
This study was supported by grants from the National
Natural Science Foundation of China (nos. 31301168, 81170255, and
31271476).
Availability of data and materials
The datasets used or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
XC, DX, and YH conceived and designed the study. XC,
YY, WX, HK, MW and ML performed the experiments. HK, YH and WF
contributed to collection and analyze the samples from patients. XC
and YY wrote the paper. XC, YY, DX and YH reviewed and edited the
manuscript. All authors read and approved the manuscript and agree
to be accountable for all aspects of the research in ensuring that
the accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
The study protocol was approved by the Local and
Medical Ethics Committee (The Second Hospital of Hebei Medical
Research Ethics Committee, approval no. 2012003). Written informed
consent was obtained from all patients or the next of kin.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Furnari FB, Fenton T, Bachoo RM, Mukasa A,
Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, et al:
Malignant astrocytic glioma: Genetics, biology, and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ricard D, Idbaih A, Ducray F, Lahutte M,
Hoang-Xuan K and Delattre JY: Primary brain tumours in adults.
Lancet. 379:1984–1996. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sathornsumetee S and Rich JN: New
treatment strategies for malignant gliomas. Expert Rev Anticancer
Ther. 6:1087–1104. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nogueira L, Ruiz-Ontañon P,
Vazquez-Barquero A, Lafarga M, Berciano MT, Aldaz B, Grande L,
Casafont I, Segura V, Robles EF, et al: Blockade of the NFκB
pathway drives differentiating glioblastoma-initiating cells into
senescence both in vitro and in vivo. Oncogene. 30:3537–3548. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Woo JS, Kim SM, Jeong CH, Ryu CH and Jeun
SS: Lipoxygenase inhibitor MK886 potentiates TRAIL-induced
apoptosis through CHOP- and p38 MAPK-mediated up-regulation of
death receptor 5 in malignant glioma. Biochem Biophys Res Commun.
431:354–359. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim SM, Park JG, Baek WK, Suh MH, Lee H,
Yoo SK, Jung KH, Suh SI and Jang BC: Cadmium specifically induces
MKP-1 expression via the glutathione depletion-mediated p38 MAPK
activation in C6 glioma cells. Neurosci Lett. 440:289–293. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Posser T, de Aguiar CB, Garcez RC, Rossi
FM, Oliveira CS, Trentin AG, Neto VM and Leal RB: Exposure of C6
glioma cells to Pb(II) increases the phosphorylation of p38(MAPK)
and JNK1/2 but not of ERK1/2. Arch Toxicol. 81:407–414. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yoshino Y, Aoyagi M, Tamaki M, Duan L,
Morimoto T and Ohno K: Activation of p38 MAPK and/or JNK
contributes to increased levels of VEGF secretion in human
malignant glioma cells. Int J Oncol. 29:981–987. 2006.PubMed/NCBI
|
|
9
|
Zhang Z, Lv J, Lei X, Li S, Zhang Y, Meng
L, Xue R and Li Z: Baicalein reduces the invasion of glioma cells
via reducing the activity of p38 signaling pathway. PLoS One.
9:e903182014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang D, Lin J and Han J:
Receptor-interacting protein (RIP) kinase family. Cell Mol Immunol.
7:243–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yin X, Krikorian P, Logan T and Csizmadia
V: Induction of RIP-2 kinase by proinflammatory cytokines is
mediated via NF-kappaB signaling pathways and involves a novel
feed-forward regulatory mechanism. Mol Cell Biochem. 333:251–259.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Thome M, Hofmann K, Burns K, Martinon F,
Bodmer JL, Mattmann C and Tschopp J: Identification of CARDIAK, a
RIP-like kinase that associates with caspase-1. Curr Biol.
8:885–888. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Navas TA, Baldwin DT and Stewart TA: RIP2
is a Raf1-activated mitogen-activated protein kinase kinase. J Biol
Chem. 274:33684–33690. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jacquet S, Nishino Y, Kumphune S, Sicard
P, Clark JE, Kobayashi KS, Flavell RA, Eickhoff J, Cotten M and
Marber MS: The role of RIP2 in p38 MAPK activation in the stressed
heart. J Biol Chem. 283:11964–11971. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McCully ML, Baroja ML, Chau TA, Jain AK,
Barra L, Salgado A, Blake PG and Madrenas J: Receptor-interacting
protein 2 is a marker for resolution of peritoneal
dialysis-associated peritonitis. Kidney Int. 72:1273–1281. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hollenbach E, Vieth M, Roessner A, Neumann
M, Malfertheiner P and Naumann M: Inhibition of RICK/nuclear
factor-kappaB and p38 signaling attenuates the inflammatory
response in a murine model of Crohn disease. J Biol Chem.
280:14981–14988. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Humphries F, Yang S, Wang B and Moynagh
PN: RIP kinases: Key decision makers in cell death and innate
immunity. Cell Death Differ. 22:225–236. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Goh FY, Cook KL, Upton N, Tao L, Lah LC,
Leung BP and Wong WS: Receptor-interacting protein 2 gene silencing
attenuates allergic airway inflammation. J Immunol. 191:2691–2699.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Singel SM, Batten K, Cornelius C, Jia G,
Fasciani G, Barron SL, Wright WE and Shay JW: Receptor-interacting
protein kinase 2 promotes triple-negative breast cancer cell
migration and invasion via activation of nuclear factor-kappaB and
c-Jun N-terminal kinase pathways. Breast Cancer Res. 16:R282014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang H and Chin AI: Role of Rip2 in
development of tumor-infiltrating MDSCs and bladder cancer
metastasis. PLoS One. 9:e947932014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cai X, Du J, Liu Y, Xia W, Liu J, Zou M,
Wang Y, Wang M, Su H and Xu D: Identification and characterization
of receptor-interacting protein 2 as a TNFR-associated factor 3
binding partner. Gene. 517:205–211. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Häcker H, Redecke V, Blagoev B,
Kratchmarova I, Hsu LC, Wang GG, Kamps MP, Raz E, Wagner H, Häcker
G, et al: Specificity in Toll-like receptor signalling through
distinct effector functions of TRAF3 and TRAF6. Nature.
439:204–207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hildebrand JM, Yi Z, Buchta CM, Poovassery
J, Stunz LL and Bishop GA: Roles of tumor necrosis factor receptor
associated factor 3 (TRAF3) and TRAF5 in immune cell functions.
Immunol Rev. 244:55–74. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Allen M, Bjerke M, Edlund H, Nelander S
and Westermark B: Origin of the U87MG glioma cell line: Good news
and bad news. Sci Transl Med. 8:354re32016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cai X, Wang M, Kong H, Liu J, Liu Y, Xia
W, Zou M, Wang J, Su H and Xu D: Prokaryotic expression,
purification and functional characterization of recombinant human
RIP2. Mol Biol Rep. 40:59–65. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lupfer C, Thomas PG, Anand PK, Vogel P,
Milasta S, Martinez J, Huang G, Green M, Kundu M, Chi H, et al:
Receptor interacting protein kinase 2-mediated mitophagy regulates
inflammasome activation during virus infection. Nat Immunol.
14:480–488. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Magalhaes JG, Lee J, Geddes K, Rubino S,
Philpott DJ and Girardin SE: Essential role of Rip2 in the
modulation of innate and adaptive immunity triggered by Nod1 and
Nod2 ligands. Eur J Immunol. 41:1445–1455. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tigno-Aranjuez JT, Asara JM and Abbott DW:
Inhibition of RIP2's tyrosine kinase activity limits NOD2-driven
cytokine responses. Genes Dev. 24:2666–2677. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nogueira L, Ruiz-Ontañon P,
Vazquez-Barquero A, Moris F and Fernandez-Luna JL: The NFκB
pathway: A therapeutic target in glioblastoma. Oncotarget.
2:646–653. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tchoghandjian A, Jennewein C, Eckhardt I,
Rajalingam K and Fulda S: Identification of non-canonical NF-κB
signaling as a critical mediator of Smac mimetic-stimulated
migration and invasion of glioblastoma cells. Cell Death Dis.
4:e5642013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhao X, Laver T, Hong SW, Twitty GB Jr,
Devos A, Devos M, Benveniste EN and Nozell SE: An NF-κB p65-cIAP2
link is necessary for mediating resistance to TNF-α induced cell
death in gliomas. J Neurooncol. 102:367–381. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xie P, Kraus ZJ, Stunz LL and Bishop GA:
Roles of TRAF molecules in B lymphocyte function. Cytokine Growth
Factor Rev. 19:199–207. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Saha SK and Cheng G: TRAF3: A new
regulator of type I interferons. Cell Cycle. 5:804–807. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Annunziata CM, Davis RE, Demchenko Y,
Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W,
et al: Frequent engagement of the classical and alternative
NF-kappaB pathways by diverse genetic abnormalities in multiple
myeloma. Cancer Cell. 12:115–130. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Keats JJ, Fonseca R, Chesi M, Schop R,
Baker A, Chng WJ, Van Wier S, Tiedemann R, Shi CX, Sebag M, et al:
Promiscuous mutations activate the noncanonical NF-kappaB pathway
in multiple myeloma. Cancer Cell. 12:131–144. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xie P, Kraus ZJ, Stunz LL, Liu Y and
Bishop GA: TNF receptor-associated factor 3 is required for T
cell-mediated immunity and TCR/CD28 signaling. J Immunol.
186:143–155. 2011. View Article : Google Scholar : PubMed/NCBI
|