Introduction
Gastrointestinal stromal tumors (GISTs) are the most
common type of mesenchymal tumor of the digestive tract. According
to worldwide reports, 60–70% of GISTs are detected in the stomach,
and 25–35% are found in the small or large intestine. Only a small
percentage of GISTs occur in other parts of the gastrointestinal
tract (1). For early-stage or
locally advanced GISTs, surgery is the optimal treatment method.
However, the prognosis for patients with GISTs is usually poor, as
GISTs have limited responses to traditional treatments such as
radiotherapy and chemotherapy (2,3).
Recent studies have identified oncogenic kinase
mutations in GISTs. The subsequent introduction of kinase
inhibitors as potential antineoplastic has provided a new insight
into the treatment of GISTs (4).
The KIT (also known as CD117) and platelet derived growth factor
receptor α (PDGFRA) genes, which encode 2 type III receptor
tyrosine kinases are recognized as the 2 most common mutations in
GISTs (5,6). Though progress has been made, the
pathogenesis of GISTs is not fully understood and there may be
other genetic regulators of the disease.
MicroRNAs (miRNA) are a type of hairpin-derived
small non-coding RNA, which can regulate gene expression at a
post-transcriptional level. More than 60% of protein-coding genes
have been computationally predicted as miRNA targets, based on
evolutionarily conserved base-pairing between mRNAs and the seed
sequences of miRNAs (7). It is
evident that miRNAs play essential roles in the regulation of
multiple cellular processes, including proliferation (8), apoptosis (9), differentiation (10) and the cell cycle (11). The abnormal expression of miRNAs
contributes to the pathogenesis of multiple human diseases
(12), including cancer (13,14). A
growing body of studies has uncovered a number of significant roles
of miRNAs in the development of human GISTs, including associations
with tumorigenesis, tumor progression, prognosis and drug
resistance (15–22). miR-221 and miR-222 induce GIST cell
apoptosis by negatively regulating the KIT/protein kinase B (AKT)
signaling pathway (18). miR-21
sensitizes GIST cells to imatinib treatment by supressing B-cell
lymphoma 2 (BCL-2) expression (17). Recently, dysregulated miR-182 was
observed in various types of malignant tumors (23–30).
However, little is known about the role of miR-182 in human
GISTs.
In the present study, it was found that miR-182 was
aberrantly upregulated in human GISTs compared with adjacent normal
tissues. Silencing of miR-182 via a miR-182 specific inhibitor,
suppressed cell proliferation and enhanced apoptosis. In addition,
miR-182 promoted GIST-T1 cell colony formation and migration.
Furthermore, cylindromatosis (CYLD) was identified as a direct
target of miR-182. The overexpression of miR-182 reduced CYLD
expression, resulting in enhanced NF-κB activation. It was also
revealed that the expression of CYLD significantly decreased in
association with the upregulation of miR-182 in human GISTs.
Materials and methods
Human study subjects
The human GISTs and adjacent normal tissues used in
the present study were obtained from Shanghai Renji Hospital after
informed consent was obtained from the patients. The samples were
immediately snap-frozen in liquid nitrogen. All samples were
confirmed as GISTs by trained pathologists. The study was approved
by the Ethics Committee of Renji Hospital, Shanghai Jiao Tong
University (Shanghai, China).
Cell culture and transfection
The human GIST-T1 cell line was a gift from the
Shanghai Cancer Institute. The GIST-T1 cell line is derived from a
GIST from the stomach of a Japanese woman, and has adherent cell
culture properties. The cells were cultured in Dulbecco's modified
Eagle's medium (DMEM) supplemented with 10% fetal bovine serum
(FBS), penicillin (100 U/ml) and streptomycin (100 µg/ml) (all from
Gibco; Thermo Fisher scientific, Inc., Waltham, MA, USA).
Lipofectamine® RNAiMAX and 2000 transfection reagents
(Invitrogen; Thermo Fisher Scientific, Inc.) were used for
transfection, following the manufacturer's instructions. The
miR-182 mimic and inhibitor, plus the corresponding negative
controls (NCs) were purchased from GenePharma (Shanghai GenePharma
Co., Ltd., Shanghai, China). A total of 100 nM NC mimic or miR-182
mimic and 200 nM NC inhibitor or miR-182 inhibitor were
transfected.
Cell proliferation assay (CCK-8
assay)
A total of 1×103 GIST-T1 cells were
seeded in a 96-well plate and incubated at 37°C until the cells
reached 30–40% confluence. Then, the cells were transfected with
miR-182 mimic, inhibitor or the corresponding NCs. Cell
proliferation was assessed at 24, 48 and 72 h as follows: 10 µl
CCK-8 solution (Dojindo Molecular Technologies, Inc., Kumamoto,
Japan) was added to each well and after 2 h of incubation at 37°C,
the optical density (OD) at 450 nm was read by a microplate
spectrophotometer. Four independent experiments were conducted.
Apoptosis assay
Annexin V-staining was performed using an Annexin
V-FITC apoptosis detection kit (BD Biosciences, Franklin Lakes, NJ,
USA) according to the manufacturer's instructions. Briefly, after
incubation the cells were harvested, washed with phosphate-buffered
saline (PBS) and stained with Annexin V-FITC and propidium iodide
in a binding buffer for 15 min at 37°C in the dark. The samples
were then analyzed by flow cytometry using a FACScan flow
cytometer.
Colony formation assay (CFA)
After transfection with the miR-182 mimic, inhibitor
or corresponding NCs, a total of 500 cells were plated in a 6-well
plate in complete culture medium. After 10–14 days, when the
colonies were visible to the naked eye, the cell culture was
terminated by removal of the medium and the cells were washed with
PBS. The colonies were fixed with methanol for 15 min, and then
dried and stained with Giemsa solution for 10 min. Images of the
stained plates were captured using a Nikon camera and the numbers
of colonies containing >50 cells were counted using
densitometric software (Clono-Counter). Each treatment was
performed in triplicate.
Cell migration assay
The ability of cells to migrate was determined using
a modified 24-well Boyden chamber (8-µm pore size; Corning Costar,
Cambridge, MA, USA). GIST-T1 cells were seeded in the upper chamber
of a Transwell at a density of 1×105 cells in 100 µl DMEM without
FBS, while DMEM with 10% FBS was added into the lower chamber.
After incubation for 24 h at 37°C, the cells that had migrated to
the lower surface of the filter were fixed with methanol for 10 min
and stained with crystal violet for 15 min. The membrane was then
washed with distilled water for 4 times. Five random microscopic
fields of each well were selected and migrated cells were counted
using a Leica DMIL LED inverted microscope (magnification, ×100).
The mean number of cells per field was recorded.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). The expression of
miR-182 was quantified using a TaqMan miRNA assay kit (Thermo
Fisher Scientific, Inc.). U6, a small nuclear RNA, was used as the
internal control. To determine the gene expression levels, RNA was
converted to cDNA using the PrimeScript RT Reagent kit (Takara Bio,
Inc., Otsu, Japan). RT-qPCR was then performed using a SYBR Premix
Ex Taq RT-PCR kit (Takara Bio, Inc.). GAPDH was used as the
internal control. The sequences of the PCR primers used were as
follows: GAPDH forward, 5′-GTCTCCTCTGACTTCAACAGCG-3′ and reverse,
5′-ACCACCCTGTTGCTGTAGCCAA-3′; human Ki-67 forward,
5′-GAAAGAGTGGCAACCTGCCTTC-3′ and reverse,
5′-GCACCAAGTTTTACTACATCTGCC-3′; human p65 forward,
5′-TGAACCGAAACTCTGGCAGCTG-3′ and reverse,
5′-TGAACCGAAACTCTGGCAGCTG-3′; human CYLD forward,
5′-GGTAATCCGTTGGATCGGTCAG-3′ and reverse
5′-AGTGCCTCTGAAGGTTCCATCC-3′ The thermocycling conditions were as
follows: Denatured at 95°C for 15 sec, followed by 40 cycles with
denaturation at 95°C for 5 sec and annealing at 60°C for 30
sec.
Western blotting
GIST-T1 cells were lysed by a radio
immunoprecipitation buffer containing proteinase and
pan-phosphatase inhibitors (Thermo Fisher Scientific, Inc.). The
protein concentration was determined by BCA quantification. A total
of 30 µg of proteins were loaded and separated by 10% SDS-PAGE gel.
The gel was then transferred to polyvinylidene difluoride (PVDF)
membranes at 180 mA for 2 h. The membranes were incubated with
blocking buffer (TBS containing 5% BSA and 0.1% Tween-20) at room
temperature for 1 h. Primary antibodies of phosphorylated (Ser) p65
(dilution 1:1,000; cat. no. 3033; Cell Signaling Technology, Inc.,
Danvers, MA, USA), p65 (dilution 1:1,000; cat. no. 8242; Cell
Signaling Technology, Inc.), β-tubulin (dilution 1:1,000; cat. no.
5666; Cell Signaling Technology, Inc.) and CYLD (dilution 1:1,000;
cat. no. 8462; Cell Signaling Technology, Inc.) were incubated at
4°C overnight. The membranes were detected by Pierce ECL Western
Blotting Substrate (Thermo Fisher Scientific, Inc.). Tanon 4100 Gel
Imaging Analysis System (Tanon Science & Technology Co., Ltd.,
Shanghai, China) was used for densitometry. All results were
normalized against β-tubulin.
Luciferase reporter assay
The 3′-untranslated region (UTR) of CYLD that
contains a predicted binding site of miR-182 validated by the web
tool TargetScanHuman 7.2 was cloned into the psiCHECK-2 vector
(Promega Corp., Madison, WI, USA). The primers used were as
follows: forward, 5′-CCGCTCGAGATGTCATGTTCCTCACCTCC-3′ and reverse,
5′-ATAAGAATGCGGCCGCCTGATACAATCCTAGGCACCT-3′. The GIST-T1 cells were
cultured in a 96-well plate to 80% confluence. They were then
transfected with 20 ng 3′-UTR luciferase reporter vector or the
empty vector with miR-182 mimic, inhibitor or the corresponding NCs
using Lipofectamine 2000, according to the manufacturer's
instructions. After 24 h the cells were lysed and subjected to a
luciferase reporter activity assay. The ratio of Renilla
luciferase activity to firefly luciferase activity was
calculated.
Statistical analysis
Three independent experiments were performed to
confirm the reproducibility of each experiment in vitro. The
data was presented as the mean ± standard error of the mean.
Differences between groups were analyzed for statistical
significance using the Student's t-test and two-way analysis of
variance followed by the Bonferroni post hoc test. The correlation
between miR-182 and CYLD was investigated using Spearman
correlation and linear regression. A P-value <0.05 was deemed to
indicate a statistically significant difference.
Results
miR-182 is ectopically expressed in
human GISTs
To study the role of miR-182 in human GISTs, miR-182
expression was quantified using a TaqMan microRNA assay in a cohort
of GIST patient samples. A total of 20 human GIST specimens and
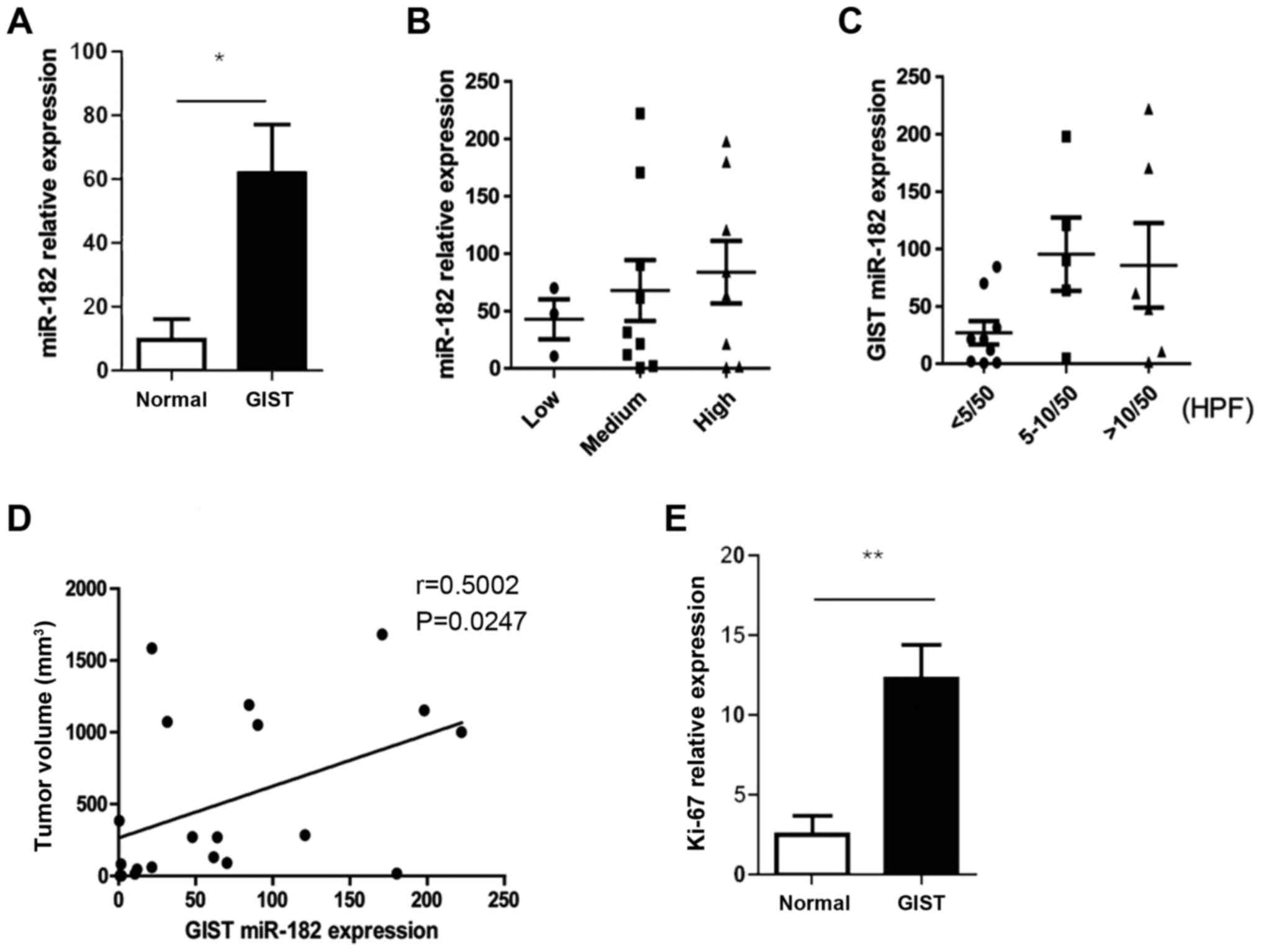
their corresponding adjacent normal tissues were analyzed (Table I). The results revealed that miR-182
was significantly upregulated in human GISTs compared with the
adjacent normal tissues (Fig. 1A).
In addition, the expression levels of miR-182 were strongly
associated with the clinicopathological characteristics, including
risk category, mitotic index and tumor size (Fig. 1B-D). It was also noted that the
expression levels of Ki-67, which is an indicator of abnormal cell
proliferation, were significantly elevated in GISTs compared with
the adjacent normal tissues (Fig.
1E).
 | Table I.Clinicopathological information of
human GIST patients. |
Table I.
Clinicopathological information of
human GIST patients.
| Patient ID | Sex | Age (yrs.) | Size
(cm3) | Location | Mitotic index | Riska |
|---|
| Patient 1 | Male | 57 | 269.5 | Stomach | <5/50
HPFb | Medium |
| Patient 2 | Female | 59 | 1050.0 |
Retroperitoneum | <5/50 HPF | High |
| Patient 3 | Male | 80 | 1190.0 | Stomach | >10/50 HPF | High |
| Patient 4 | Male | 66 | 18.0 | Stomach | >10/50 HPF | High |
| Patient 5 | Female | 62 | 1000.0 | Stomach | >10/50 HPF | High |
| Patient 6 | Female | 74 | 1152.0 | Stomach | >10/50 HPF | High |
| Patient 7 | Female | 52 | 270.0 | Stomach | <5/50 HPF | Medium |
| Patient 8 | Male | 52 | 283.5 | Stomach | 5-10/50 HPF | Medium |
| Patient 9 | Male | 72 | 45.0 | Stomach | 5-10/50 HP | Medium |
| Patient 10 | Female | 75 | 90.0 | Stomach | 5-10/50 HPF | Medium |
| Patient 11 | Female | 62 | 384.0 | Stomach | 5-10/50 HPF | High |
| Patient 12 | Male | 48 | 82.5 | Duodenum | <5/50 HPF | Medium |
| Patient 13 | Female | 53 | 1680.0 | Stomach | >10/50 HPF | High |
| Patient 14 | Male | 69 | 60.0 | Stomach | >5/50 HPF | Medium |
| Patient 15 | Female | 56 | 15.0 | Stomach | <5/50 HPF | Low |
| Patient 16 | Female | 62 | 130.0 | Small
intestine | ≤5/50 HPF | Medium |
| Patient 17 | Male | 86 | 1584.0 | Stomach | >10/50 HPF | High |
| Patient 18 | Male | 39 | 1072.5 | Stomach | <5/50 HPF | High |
| Patient 19 | Male | 65 | 2.5 | Stomach | <5/50 HPF | Low |
| Patient 20 | Female | 53 | 2.0 | Small
intestine | <2/50 HPF | Low |
miR-182 enhances GIST cell
proliferation
To further investigate the role of miR-182 in GISTs,
the effect of miR-182 on cell proliferation was evaluated by
manipulating miR-182 expression in the GIST-T1 cell line.
Overexpressing or silencing of miR-182 was performed by
transfection of the synthesized miR-182 mimic or inhibitor,
respectively. The corresponding non-specific oligonucleotides were
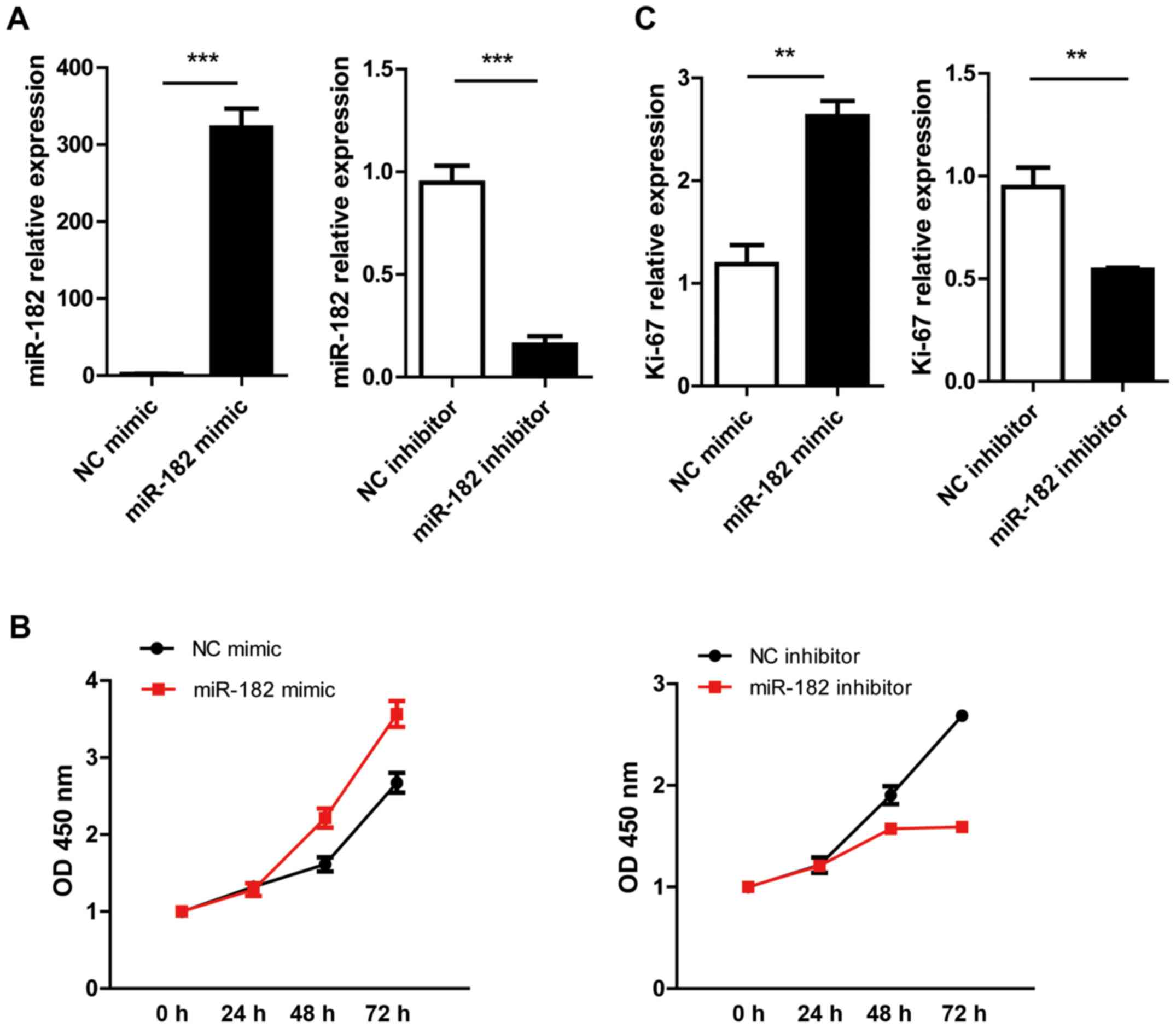
used as NCs. As shown by q-PCR, the expression levels of miR-182
were significantly increased in the miR-182 mimic-transfected group
compared with the NC mimic group. In the miR-182 inhibitor group,
miR-182 expression was 5-fold lower compared with the inhibitor NC
group (Fig. 2A). It was found that
miR-182 overexpression significantly upregulated Ki-67 expression.
The pro-proliferative function of the miR-182 mimic was further
verified by a CCK-8 assay. Transfection with the miR-182 inhibitor
suppressed cell growth and decreased Ki-67 expression (Fig. 2B and C).
miR-182 inhibits GIST cell
apoptosis
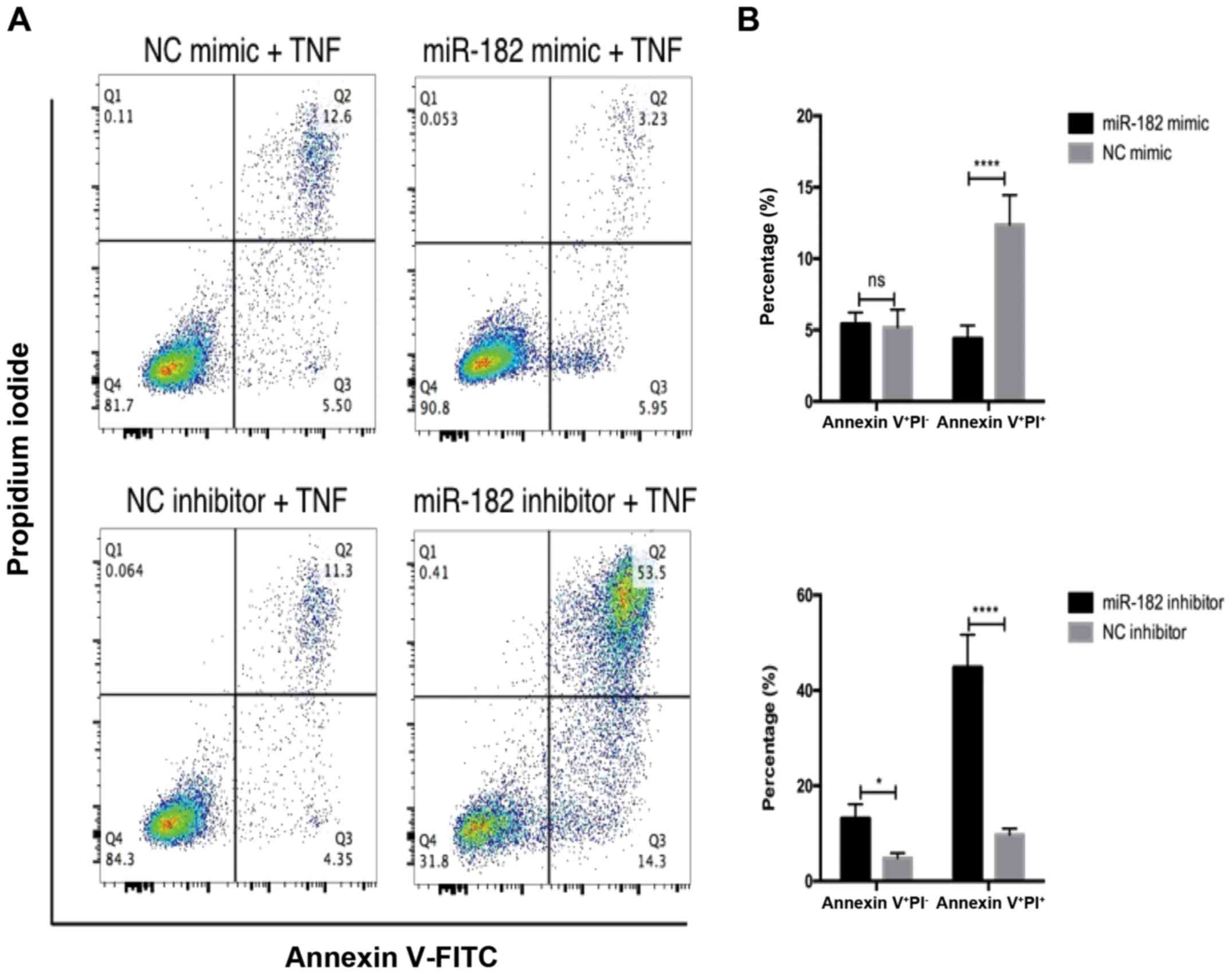
It was then investigated whether miR-182 influenced
GIST cell apoptosis. Flow cytometric analysis was performed to
analyze PI and Annexin V staining of GIST-T1 cells treated with
TNFα for 24 h. It was determined that overexpression of miR-182 did
not affect early apoptosis, as revealed by Annexin
V+PI−, however it did significantly suppress
late apoptosis (Annexin V+PI+) (Fig. 3A). Conversely, inhibition of miR-182
promoted both early and late apoptosis (Fig. 3B).
miR-182 stimulates colony formation
and migration of GIST cells
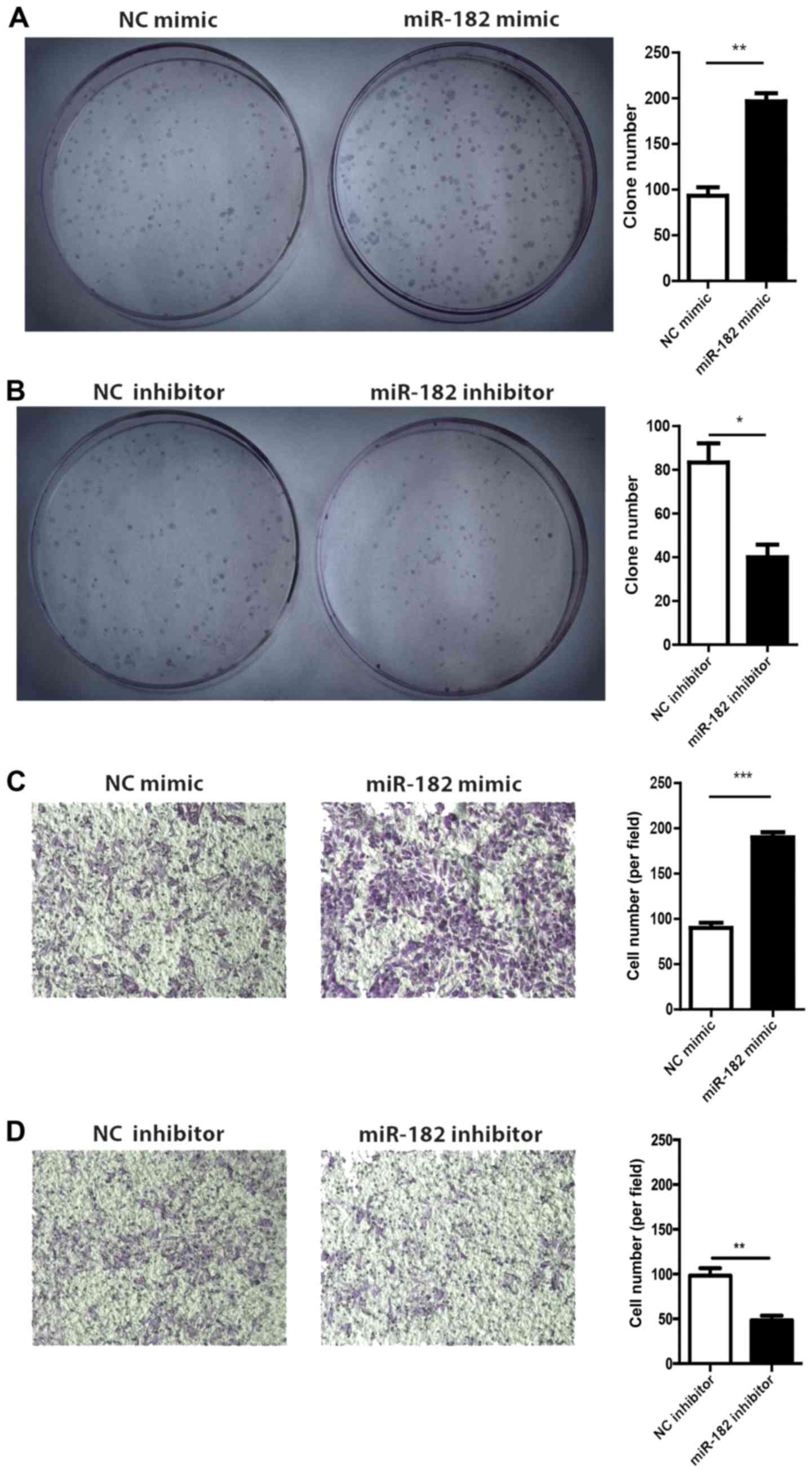
The role of miR-182 in GIST-T1 cell growth and
migration was evaluated by performing a CFA and Transwell migration
assay, respectively. The results of the CFA revealed that GIST-T1
cells transfected with miR-182 mimic exhibited a higher colony
formation rate compared with the mimic NC group (Fig. 4A). Conversely, the miR-182 inhibitor
suppressed colony formation (Fig.
4B). In the Transwell migration assay, overexpression of
miR-182 enhanced GIST-T1 cell migration and silencing of miR-182
blocked cell migration (Fig. 4C and
D), which indicated that miR-182 could promote tumor cell
migration.
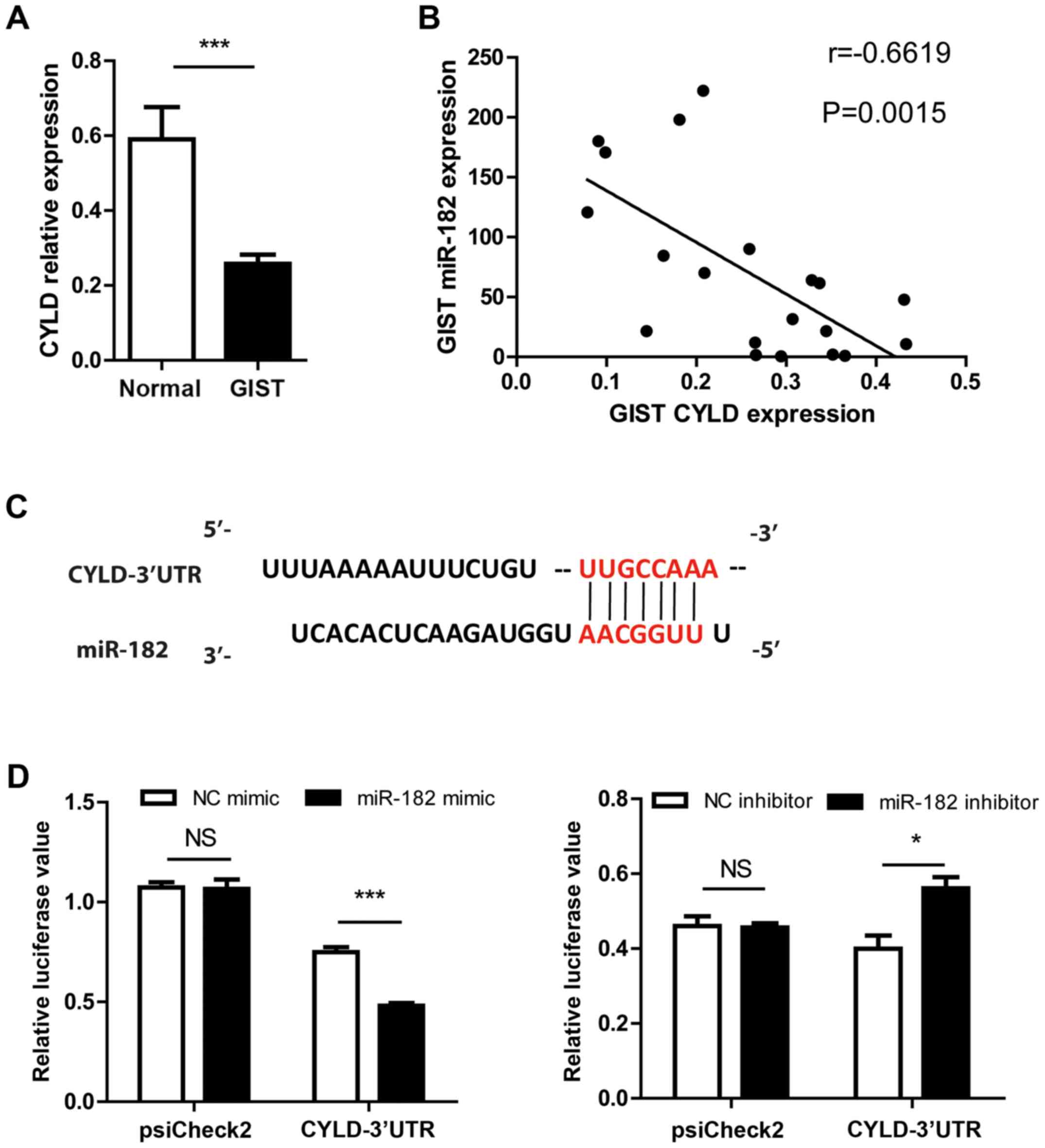
CYLD is a direct target of
miR-182
CYLD is recognized as a tumor suppressor, which is
involved in various types of carcinoma, including breast and lung
cancer (31–34). It was observed that the expression
of CYLD was downregulated in association with the upregulation of
miR-182 in human GISTs compared with adjacent normal tissues
(Fig. 5A and B). Sequence alignment
revealed that the 3′-UTR of CYLD mRNA contains a binding site for
the miR-182 seed region (Fig. 5C).
It is known that miRNA regulates gene expression by base-pairing
its target mRNAs with the seeding region, which leads to mRNA
degradation or translation repression (35). To further explore whether CYLD is a
target of miR-182, a luciferase reporter assay was performed. The
reporter plasmid psiCHECK2 contained the 3′UTR of CYLD and was
co-transfected with the miR-182 mimic, inhibitor or corresponding
NC into GIST-T1 cells. The luciferase assay revealed that miR-182
regulated luciferase activity in a CYLD 3′-UTR dependent manner
(Fig. 5D). Collectively, these
results demonstrated that CYLD is a direct target of miR-182.
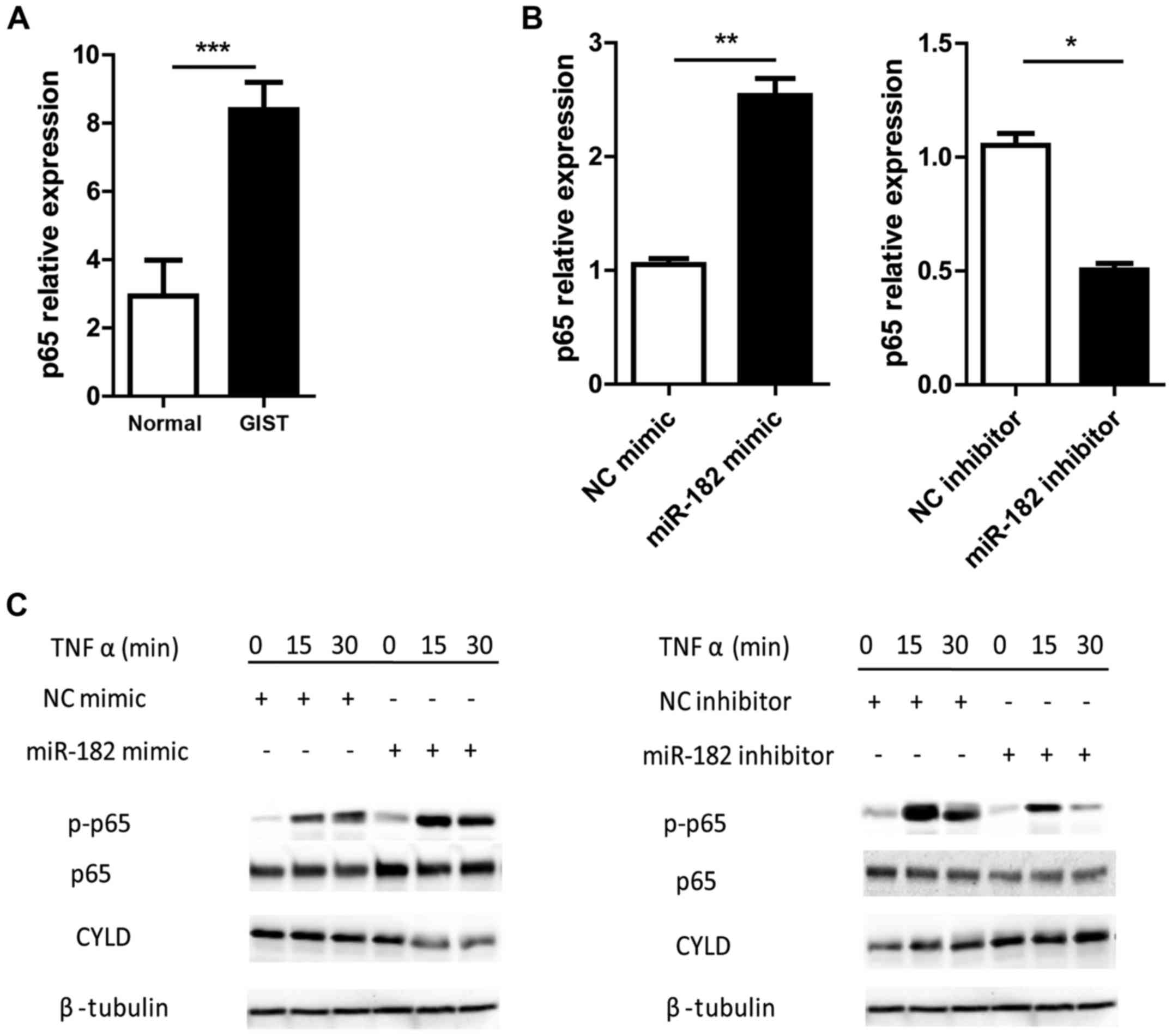
miR-182 enhances NF-κB signaling by
negatively regulating CYLD
A number of previous studies have revealed that CYLD
promotes cell survival and oncogenesis by negatively regulating
NF-κB signaling (32,36). Steady state NF-κB is sequestered by
I-κB in the cytoplasm. However, upon activation I-κB is
phosphorylated and undergoes degradation. The released NF-κB dimer
(p50-p65) is then phosphorylated and translocates into the nucleus
where it binds with the cognate response elements, inducing gene
transcription (37). Therefore, the
phosphorylation of p65 is recognized as an activation indicator of
the NF-κB signaling pathway. It was revealed that the expression
levels of p65 were significantly increased in human GISTs compared
with the adjacent normal tissues (Fig.
6A). Overexpression of miR-182 in GIST-T1 cells significantly
upregulated p65 expression, while inhibition of miR-182
downregulated its expression (Fig.
6B). To further investigate the role of miR-182 in the
regulation of NF-κB signaling, TNFα-induced NF-κB activation was
investigated in GIST-T1 cells with miR-182 intervention. GIST-T1
cells transfected with miR-182 mimics exhibited reduced protein
expression of CYLD and enhanced NF-κB activation, as determined by
p65 phosphorylation, compared with the mimic NC group. Conversely,
silencing miR-182 reduced p65 phosphorylation and increased CYLD
expression (Fig. 6C). These results
demonstrated that miR-182 promoted NF-κB signaling by negatively
regulating CYLD expression.
Discussion
Recently, a number of studies have demonstrated that
miRNAs are involved in human malignancies by regulating multiple
genes associated with various aspects of cancer biology (38,39).
Characterization of cancer-specific miRNAs as well as their
functional targets and target-mediated signaling pathways, would
improve understanding of miRNAs in tumorigenesis, which may be
important for the identification of novel therapeutic targets
(40,41). In the present study, it was
determined that miR-182 was ectopically expressed in human GISTs
compared with the adjacent normal tissues. miR-182 was originally
thought to be specifically expressed in sensory organs (42). However, recent studies have revealed
the oncogenic role of miR-182 in various types of human cancer
(43–51). Aberrant expression of miR-182
enhanced melanoma metastasis by negatively regulating FOXO3 and
microphthalmia-associated transcription factor expression (26). In the present study, it was observed
that silencing miR-182 significantly inhibited GIST-T1 cell growth
and invasion, which indicated that miR-182 may be developed into a
therapeutic treatment option for human GISTs. However, further
in vivo animal experiments are required to ascertain
this.
CYLD is a deubiquitinase, which can cleave lysine
63-linked ubiquitin chains from the target protein (31). It was originally identified as a
gene which was mutated in familial cylindromatosis, which
predisposes patients to skin cylindroma (52,53).
Recent studies have revealed that CYLD has a significant
suppressive role in cell survival and proliferation. The loss of
CYLD expression has been observed in various types of human cancer
(54,55). In the present study, it was revealed
that the expression of CYLD was significantly decreased in human
GISTs, and was associated with the upregulation of miR-182. A
luciferase reporter assay revealed that CYLD is a direct target of
miR-182. Inhibition of miR-182 enhanced CYLD expression in GIST-T1
cells, which resulted in reduced cell proliferation and increased
cell apoptosis.
NF-κB comprises a family of 5 transcription factors,
which can form distinct hetero- or homo-dimers and control
responsive gene expression (56).
Rigid regulation of NF-κB signaling is vital for various cellular
bioprocesses (57). Aberrant
activation of NF-κB is observed in multiple types of human cancer
where it promotes tumor cell survival through stimulation of
anti-apoptotic gene expression (58–61).
The results in the present study revealed that NF-κB was
upregulated in human GISTs compared with adjacent normal tissues.
CYLD is known to be a dominant negative regulator of NF-κB
signaling. GIST cells with increased CYLD protein levels exhibited
reduced p65 phosphorylation in response to TNFα stimulation.
Therefore, the present study uncovered a regulatory axis of
miR-182/CYLD/NF-κB which mediated cell survival in GISTs.
Reduction of apoptosis is a typical hallmark of
tumor progression, and one mechanism by which miRNAs influence
cancer development is through the mediation of the apoptotic
signaling pathway (62–65). In the present study, it was revealed
that miR-182 was involved in regulating GIST cell apoptosis.
Overexpression of miR-182 reduced TNFα-induced late apoptosis, as
determined by Annexin V and PI double-positive staining. However,
it had little effect on early apoptosis (Annexin
V+PI−) in the miR-182 mimic-treated cells.
This is not the first time that miRNA has selectively affected the
late-stage apoptosis of cancer cells. A previous study by Wang
et al demonstrated that miRNA-196b promoted pancreatic
cancer cell growth by inhibiting late apoptosis via negative
regulation of CADM1 expression (66). Further studies are required to
investigate the molecular mechanism of how miR-182 mediates late
apoptosis in GIST cells.
In conclusion, the results of the present study
demonstrated that miR-182 was aberrantly expressed in human GISTs.
It was also revealed that miR-182 promoted GIST cell growth and
invasion. In addition, tumor suppressor CYLD was identified as a
direct target of miR-182. These findings indicated that miR-182
functions as an oncomiR in GIST and may represent a promising
therapeutic target for human GIST treatment.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Shanghai Hospital Development Center (grant no. 16CR3001A) and the
State Key Laboratory of Oncogenes and Related Genes (grant no.
90-15-01)
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
HC conceived and designed the study. TL and FY
performed the experiments and analyzed the data. TL wrote the
manuscript. FY and HC reviewed and edited the manuscript. All
authors read and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
Renji Hospital, Shanghai Jiao Tong University (Shanghai, China) and
informed consent was obtained from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicting
financial interests.
References
|
1
|
Miettinen M, Sobin LH and Lasota J:
Gastrointestinal stromal tumors of the stomach: A
clinicopathologic, immunohistochemical, and molecular genetic study
of 1765 cases with long-term follow-up. Am J Surg Pathol. 29:52–68.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huang RX, Xiang P and Huang C:
Gastrointestinal stromal tumors: Current translational research and
management modalities. Eur Rev Med Pharmacol Sci. 18:3076–3085.
2014.PubMed/NCBI
|
|
3
|
Dematteo RP, Heinrich MC, El-Rifai WM and
Demetri G: Clinical management of gastrointestinal stromal tumors:
Before and after STI-571. Hum Pathol. 33:466–477. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Corless CL, Barnett CM and Heinrich MC:
Gastrointestinal stromal tumours: Origin and molecular oncology.
Nat Rev Cancer. 11:865–878. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hirota S, Isozaki K, Moriyama Y, Hashimoto
K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M,
et al: Gain-of-function mutations of c-kit in human
gastrointestinal stromal tumors. Science. 279:577–580. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Heinrich MC, Corless CL, Duensing A,
McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A,
Town A, et al: PDGFRA activating mutations in gastrointestinal
stromal tumors. Science. 299:708–710. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Friedman RC, Farh KK, Burge CB and Bartel
DP: Most mammalian mRNAs are conserved targets of microRNAs. Genome
Res. 19:92–105. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lenkala D, LaCroix B, Gamazon ER, Geeleher
P, Im HK and Huang RS: The impact of microRNA expression on
cellular proliferation. Hum Genet. 133:931–938. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hwang HW and Mendell JT: MicroRNAs in cell
proliferation, cell death, and tumorigenesis. Br J Cancer. 96
Suppl:R40–R44. 2007.PubMed/NCBI
|
|
10
|
Shivdasani RA: MicroRNAs: regulators of
gene expression and cell differentiation. Blood. 108:3646–3653.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bueno MJ and Malumbres M: MicroRNAs and
the cell cycle. Biochim Biophys Acta. 1812:592–601. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tüfekci KU, Oner MG, Meuwissen RL and Genç
S: The role of microRNAs in human diseases. Methods Mol Biol.
1107:33–50. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hayes J, Peruzzi PP and Lawler S:
MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol
Med. 20:460–469. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Reddy KB: MicroRNA (miRNA) in cancer.
Cancer Cell Int. 15:382015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim WK, Yang HK and Kim H: MicroRNA
involvement in gastrointestinal stromal tumor tumorigenesis. Curr
Pharm Des. 1227–1235. 2013.PubMed/NCBI
|
|
16
|
Fan R, Zhong J, Zheng S, Wang Z, Xu Y, Li
S, Zhou J and Yuan F: MicroRNA-218 inhibits gastrointestinal
stromal tumor cell and invasion by targeting KIT. Tumour Biol.
35:4209–4217. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cao CL, Niu HJ, Kang SP, Cong CL and Kang
SR: miRNA-21 sensitizes gastrointestinal stromal tumors (GISTs)
cells to Imatinib via targeting B-cell lymphoma 2 (Bcl-2). Eur Rev
Med Pharmacol Sci. 20:3574–3581. 2016.PubMed/NCBI
|
|
18
|
Ihle MA, Trautmann M, Kuenstlinger H, Huss
S, Heydt C, Fassunke J, Wardelmann E, Bauer S, Schildhaus HU,
Buettner R, et al: miRNA-221 and miRNA-222 induce apoptosis via the
KIT/AKT signalling pathway in gastrointestinal stromal tumours. Mol
Oncol. 9:1421–1433. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim WK, Park M, Kim YK, Tae YK, Yang HK,
Lee JM and Kim H: MicroRNA-494 downregulates KIT and inhibits
gastrointestinal stromal tumor cell proliferation. Clin Cancer Res.
17:7584–7594. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Akçakaya P, Caramuta S, Åhlen J, Ghaderi
M, Berglund E, Östman A, Bränström R, Larsson C and Lui WO:
microRNA expression signatures of gastrointestinal stromal tumours:
Associations with imatinib resistance and patient outcome. Br J
Cancer. 111:2091–2102. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Akçakaya P and Lui WO: MicroRNAs and
gastrointestinal stromal tumor. Adv Exp Med Biol. 889:51–70. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Haller F, von Heydebreck A, Zhang JD,
Gunawan B, Langer C, Ramadori G, Wiemann S and Sahin O:
Localization- and mutation-dependent microRNA (miRNA) expression
signatures in gastrointestinal stromal tumours (GISTs), with a
cluster of co-expressed miRNAs located at 14q32.31. J Pathol.
220:71–86. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chiang CH, Hou MF and Hung WC:
Up-regulation of miR-182 by β-catenin in breast cancer increases
tumorigenicity and invasiveness by targeting the matrix
metalloproteinase inhibitor RECK. Biochim Biophys Acta.
1830:3067–3076. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tang T, Wong HK, Gu W, Yu MY, To KF, Wang
CC, Wong YF, Cheung TH, Chung TK and Choy KW: MicroRNA-182 plays an
onco-miRNA role in cervical cancer. Gynecol Oncol. 129:199–208.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Guttilla IK and White BA: Coordinate
regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast
cancer cells. J Biol Chem. 284:23204–23216. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Segura MF, Hanniford D, Menendez S, Reavie
L, Zou X, Alvarez-Diaz S, Zakrzewski J, Blochin E, Rose A,
Bogunovic D, et al: Aberrant miR-182 expression promotes melanoma
metastasis by repressing FOXO3 and microphthalmia-associated
transcription factor. Proc Natl Acad Sci USA. 106:1814–1819. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu Z, Liu J, Segura MF, Shao C, Lee P,
Gong Y, Hernando E and Wei JJ: MiR-182 overexpression in
tumourigenesis of high-grade serous ovarian carcinoma. J Pathol.
228:204–215. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Moskwa P, Buffa FM, Pan Y, Panchakshari R,
Gottipati P, Muschel RJ, Beech J, Kulshrestha R, Abdelmohsen K,
Weinstock DM, et al: miR-182-mediated downregulation of BRCA1
impacts DNA repair and sensitivity to PARP inhibitors. Mol Cell.
41:210–220. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jiang L, Mao P, Song L, Wu J, Huang J, Lin
C, Yuan J, Qu L, Cheng SY and Li J: miR-182 as a prognostic marker
for glioma progression and patient survival. Am J Pathol.
177:29–38. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Song L, Liu L, Wu Z, Li Y, Ying Z, Lin C,
Wu J, Hu B, Cheng SY, Li M, et al: TGF-β induces miR-182 to sustain
NF-κB activation in glioma subsets. J Clin Invest. 122:3563–3578.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Massoumi R: CYLD: A deubiquitination
enzyme with multiple roles in cancer. Future Oncol. 7:285–297.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sun SC: CYLD: A tumor suppressor
deubiquitinase regulating NF-kappaB activation and diverse
biological processes. Cell Death Differ. 17:25–34. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hayashi M, Jono H, Shinriki S, Nakamura T,
Guo J, Sueta A, Tomiguchi M, Fujiwara S, Yamamoto-Ibusuki M,
Murakami K, et al: Clinical significance of CYLD downregulation in
breast cancer. Breast Cancer Res Treat. 143:447–457. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Deng LL, Shao YX, Lv HF, Deng HB and Lv
FZ: Over-expressing CYLD augments antitumor activity of TRAIL by
inhibiting the NF-κB survival signaling in lung cancer cells.
Neoplasma. 59:18–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Courtois G: Tumor suppressor CYLD:
Negative regulation of NF-kappaB signaling and more. Cell Mol Life
Sci. 65:1123–1132. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Oeckinghaus A and Ghosh S: The NF-kappaB
family of transcription factors and its regulation. Cold Spring
Harb Perspect Biol. 1:a0000342009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Calin GA and Croce CM: MicroRNA-cancer
connection: The beginning of a new tale. Cancer Res. 66:7390–7394.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Farazi TA, Hoell JI, Morozov P and Tuschl
T: MicroRNAs in human cancer. Adv Exp Med Biol. 774:1–20. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cheng CJ and Slack FJ: The duality of
oncomiR addiction in the maintenance and treatment of cancer.
Cancer J. 18:232–237. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kasinski AL and Slack FJ: Epigenetics and
genetics. MicroRNAs en route to the clinic: Progress in validating
and targeting microRNAs for cancer therapy. Nat Rev Cancer.
11:849–864. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wei Q, Lei R and Hu G: Roles of miR-182 in
sensory organ development and cancer. Thorac Cancer. 6:2–9. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hanke M, Hoefig K, Merz H, Feller AC,
Kausch I, Jocham D, Warnecke JM and Sczakiel G: A robust
methodology to study urine microRNA as tumor marker: microRNA-126
and microRNA-182 are related to urinary bladder cancer. Urol Oncol.
28:655–661. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang J, Li J, Shen J, Wang C, Yang L and
Zhang X: MicroRNA-182 downregulates metastasis suppressor 1 and
contributes to metastasis of hepatocellular carcinoma. BMC Cancer.
12:2272012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hirata H, Ueno K, Shahryari V, Deng G,
Tanaka Y, Tabatabai ZL, Hinoda Y and Dahiya R: MicroRNA-182-5p
promotes cell invasion and proliferation by down regulating FOXF2,
RECK and MTSS1 genes in human prostate cancer. PLoS One.
8:e555022013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lei R, Tang J, Zhuang X, Deng R, Li G, Yu
J, Liang Y, Xiao J, Wang HY, Yang Q, et al: Suppression of MIM by
microRNA-182 activates RhoA and promotes breast cancer metastasis.
Oncogene. 33:1287–1296. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yan D, Dong XD, Chen X, Yao S, Wang L,
Wang J, Wang C, Hu DN, Qu J and Tu L: Role of microRNA-182 in
posterior uveal melanoma: Regulation of tumor development through
MITF, BCL2 and cyclin D2. PLoS One. 7:e409672012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yang WB, Chen PH, Hsu T, Fu TF, Su WC,
Liaw H, Chang WC and Hung JJ: Sp1-mediated microRNA-182 expression
regulates lung cancer progression. Oncotarget. 5:740–753. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rasheed SA, Teo CR, Beillard EJ, Voorhoeve
PM and Casey PJ: MicroRNA-182 and microRNA-200a control G-protein
subunit α-13 (GNA13) expression and cell invasion synergistically
in prostate cancer cells. J Biol Chem. 288:7986–7995. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yang MH, Yu J, Jiang DM, Li WL, Wang S and
Ding YQ: microRNA-182 targets special AT-rich sequence-binding
protein 2 to promote colorectal cancer proliferation and
metastasis. J Transl Med. 12:1092014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang YQ, Guo RD, Guo RM, Sheng W and Yin
LR: MicroRNA-182 promotes cell growth, invasion, and
chemoresistance by targeting programmed cell death 4 (PDCD4) in
human ovarian carcinomas. J Cell Biochem. 114:1464–1473. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bowen S, Gill M, Lee DA, Fisher G,
Geronemus RG, Vazquez ME and Celebi JT: Mutations in the CYLD gene
in Brooke-Spiegler syndrome, familial cylindromatosis, and multiple
familial trichoepithelioma: Lack of genotype-phenotype correlation.
J Invest Dermatol. 124:919–920. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bignell GR, Warren W, Seal S, Takahashi M,
Rapley E, Barfoot R, Green H, Brown C, Biggs PJ, Lakhani SR, et al:
Identification of the familial cylindromatosis tumour-suppressor
gene. Nat Genet. 25:160–165. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
54
|
Nikolaou K, Tsagaratou A, Eftychi C,
Kollias G, Mosialos G and Talianidis I: Inactivation of the
deubiquitinase CYLD in hepatocytes causes apoptosis, inflammation,
fibrosis, and cancer. Cancer Cell. 21:738–750. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hellerbrand C, Bumes E, Bataille F,
Dietmaier W, Massoumi R and Bosserhoff AK: Reduced expression of
CYLD in human colon and hepatocellular carcinomas. Carcinogenesis.
28:21–27. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Gilmore TD: Introduction to NF-kappaB:
Players, pathways, perspectives. Oncogene. 25:6680–6684. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Schreck R, Albermann K and Baeuerle PA:
Nuclear factor kappa B: An oxidative stress-responsive
transcription factor of eukaryotic cells (a review). Free Radic Res
Commun. 17:221–237. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang CY, Mayo MW and Baldwin AS Jr: TNF-
and cancer therapy-induced apoptosis: Potentiation by inhibition of
NF-kappaB. Science. 274:784–787. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Dolcet X, Llobet D, Pallares J and
Matias-Guiu X: NF-κB in development and progression of human
cancer. Virchows Arch. 446:475–482. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Xia Y, Shen S and Verma IM: NF-κB, an
active player in human cancers. Cancer Immunol Res. 2:823–830.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Karin M, Cao Y, Greten FR and Li ZW:
NF-kappaB in cancer: From innocent bystander to major culprit. Nat
Rev Cancer. 2:301–310. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jovanovic M and Hengartner MO: miRNAs and
apoptosis: RNAs to die for. Oncogene. 25:6176–6187. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lima RT, Busacca S, Almeida GM, Gaudino G,
Fennell DA and Vasconcelos MH: MicroRNA regulation of core
apoptosis pathways in cancer. Eur J Cancer. 47:163–174. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Pileczki V, Cojocneanu-Petric R, Maralani
M, Neagoe IB and Sandulescu R: MicroRNAs as regulators of apoptosis
mechanisms in cancer. Clujul Med. 89:50–55. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Subramanian S and Steer CJ: MicroRNAs as
gatekeepers of apoptosis. J Cell Physiol. 223:289–298.
2010.PubMed/NCBI
|
|
66
|
Wang HL, Zhou R, Liu J, Chang Y, Liu S,
Wang XB, Huang MF and Zhao Q: MicroRNA-196b inhibits late apoptosis
of pancreatic cancer cells by targeting CADM1. Sci Rep.
7:114672017. View Article : Google Scholar : PubMed/NCBI
|